Ranitidine Injection: Package Insert / Prescribing Info
Package insert / product label
Generic name: ranitidine hydrochloride
Dosage form: injection, solution
Drug class: H2 antagonists
Medically reviewed by Drugs.com. Last updated on Nov 11, 2024.
On This Page
Ranitidine Injection Description
The active ingredient in ranitidine injection is ranitidine hydrochloride (HCl), a histamine H2-receptor antagonist. Chemically it is N[2-[[[5-[(dimethylamino)methyl]-2-furanyl]methyl]thio]ethyl]-N′-methyl-2-nitro-1,1-ethenediamine, hydrochloride. It has the following structure:
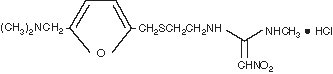
The empirical formula is C13H22N4O3S●HCl, representing a molecular weight of 350.87.
Ranitidine hydrochloride USP is a white to pale yellow, crystalline powder that is very soluble in water.
Ranitidine injection USP is a clear, colorless to yellow, nonpyrogenic liquid. The yellow color of the liquid tends to intensify without adversely affecting potency. The pH of the injection solution is 6.7 to 7.3.
Each 1 mL of aqueous solution contains ranitidine 25 mg (as the hydrochloride); phenol 5 mg as preservative; and 0.96 mg of monobasic potassium phosphate and 2.4 mg of dibasic sodium phosphate as buffers.
A pharmacy bulk package is a container of a sterile preparation for parenteral use that contains many single doses. The contents are intended for use in a pharmacy admixture program and are restricted to the preparation of admixtures for intravenous (IV) infusion.
Ranitidine Injection - Clinical Pharmacology
Ranitidine hydrochloride is a competitive, reversible inhibitor of the action of histamine at the histamine H2-receptors, including receptors on the gastric cells. Ranitidine hydrochloride does not lower serum Ca++ in hypercalcemic states. Ranitidine hydrochloride is not an anticholinergic agent.
Absorption
Ranitidine hydrochloride is absorbed very rapidly after intramuscular (IM) injection. Mean peak levels of 576 ng/mL occur within 15 minutes or less following a 50 mg intramuscular dose. Absorption from intramuscular sites is virtually complete, with a bioavailability of 90% to 100% compared with intravenous (IV) administration. Following oral administration, the bioavailability of ranitidine tablets is 50%.
Metabolism
In humans, the N-oxide is the principal metabolite in the urine; however, this amounts to <4% of the dose. Other metabolites are the S-oxide (1%) and the desmethyl ranitidine (1%). The remainder of the administered dose is found in the stool. Studies in patients with hepatic dysfunction (compensated cirrhosis) indicate that there are minor, but clinically insignificant, alterations in ranitidine half-life, distribution, clearance, and bioavailability.
Excretion
Following intravenous injection, approximately 70% of the dose is recovered in the urine as unchanged drug. Renal clearance averages 530 mL/min, with a total clearance of 760 mL/min. The elimination half-life is 2.0 to 2.5 hours.
Four patients with clinically significant renal function impairment (creatinine clearance 25 to 35 mL/min) administered 50 mg of ranitidine intravenously had an average plasma half-life of 4.8 hours, a ranitidine clearance of 29 mL/min, and a volume of distribution of 1.76 L/kg. In general, these parameters appear to be altered in proportion to creatinine clearance (see DOSAGE AND ADMINISTRATION).
Geriatrics
The plasma half-life is prolonged and total clearance is reduced in the elderly population due to a decrease in renal function. The elimination half-life is 3.1 hours (see PRECAUTIONS: Geriatric Use and DOSAGE AND ADMINISTRATION: Dosage Adjustment for Patients With Impaired Renal Function).
Pediatrics
There are no significant differences in the pharmacokinetic parameter values for ranitidine in pediatric patients (from 1 month up to 16 years of age) and healthy adults when correction is made for body weight. The pharmacokinetics of ranitidine hydrochloride in pediatric patients are summarized in Table 1.
|
T½ = Terminal half-life; CLp = Plasma clearance of ranitidine. |
|||||
|
ECMO = extracorporeal membrane oxygenation. |
|||||
| Population (age) | n | Dose (mg/kg) | t½ (hours) | Vd (L/kg) | CLp (mL/min/kg) |
| Peptic ulcer disease (<6 years) (6 to 11.9 years) (>12 years) Adults | 6 11 6 6 | 1.25 or 2.5 1.25 or 2.5 1.25 or 2.5 2.5 | 2.2 2.1 1.7 1.9 | 1.29 1.14 0.98 1.04 | 11.41 8.96 9.89 8.77 |
| Peptic ulcer disease (3.5–16 years) | 12 | 0.13 to 0.80 | 1.8 | 2.3 | 795 mL/min/1.73/m2
|
| Children in intensive care (1 day–12.6 years) | 17 | 1.0 | 2.4 | 2 | 11.7 |
| Neonates receiving ECMO | 12 | 2 | 6.6 | 1.8 | 4.3 |
Plasma clearance in neonatal patients (less than 1 month of age) receiving ECMO was considerably lower (3 to 4 mL/min/kg) than observed in children or adults. The elimination half-life in neonates averaged 6.6 hours as compared to approximately 2 hours in adults and pediatric patients.
Pharmacodynamics
Serum concentrations necessary to inhibit 50% of stimulated gastric acid secretion are estimated to be 36 to 94 ng/mL. Following single intravenous or intramuscular 50 mg doses, serum concentrations of ranitidine are in this range for 6 to 8 hours.
Antisecretory Activity
Ranitidine injection inhibits basal gastric acid secretion as well as gastric acid secretion stimulated by betazole and pentagastrin, as shown in Table 2.
|
| Time After Dose, hours | % Inhibition of Gastric Acid Output by Intravenous Dose, mg |
||
| 20 mg | 60 mg | 100 mg |
||
| Betazole | Up to 2 | 93 | 99 | 99 |
| Pentagastrin | Up to 3 | 47 | 66 | 77 |
In a group of 10 known hypersecretors, ranitidine plasma levels of 71, 180, and 376 ng/mL inhibited basal acid secretion by 76%, 90%, and 99.5%, respectively.
It appears that basal- and betazole-stimulated secretions are most sensitive to inhibition by ranitidine hydrochloride, while pentagastrin-stimulated secretion is more difficult to suppress.
Pepsin
Ranitidine hydrochloride does not affect pepsin secretion. Total pepsin output is reduced in proportion to the decrease in volume of gastric juice.
Intrinsic Factor
Ranitidine hydrochloride has no significant effect on pentagastrin-stimulated intrinsic factor secretion.
Serum Gastrin
Ranitidine hydrochloride has little or no effect on fasting or postprandial serum gastrin.
Other Pharmacologic Actions
- Gastric bacterial flora - increase in nitrate-reducing organisms, significance not known.
- Prolactin levels - no effect in recommended oral or intravenous dosage, but small, transient, dose-related increases in serum prolactin have been reported after intravenous bolus injections of 100 mg or more.
- Other pituitary hormones - no effect on serum gonadotropins, TSH, or GH. Possible impairment of vasopressin release.
- No change in cortisol, aldosterone, androgen, or estrogen levels.
- No antiandrogenic action.
- No effect on count, motility, or morphology of sperm.
Pediatrics
The ranitidine concentration necessary to suppress basal acid secretion by at least 90% has been reported to be 40 to 60 ng/mL in pediatric patients with duodenal or gastric ulcers.
In a study of 20 critically ill pediatric patients receiving ranitidine intravenous at 1 mg/kg every 6 hours, 10 patients with a baseline pH ≥ 4 maintained this baseline throughout the study. Eight of the remaining 10 patients with a baseline of pH ≤ 2 achieved pH ≥ 4 throughout varying periods after dosing. It should be noted, however, that because these pharmacodynamic parameters were assessed in critically ill pediatric patients, the data should be interpreted with caution when dosing recommendations are made for a less seriously ill pediatric population.
In another small study of neonatal patients (n = 5) receiving ECMO, gastric pH < 4 pretreatment increased to > 4 after a 2 mg/kg dose and remained above 4 for at least 15 hours.
Active Duodenal Ulcer
In a multicenter, double-blind, controlled, US study of endoscopically diagnosed duodenal ulcers, earlier healing was seen in the patients treated with oral ranitidine hydrochloride as shown in Table 3.
|
*All patients were permitted antacids as needed for relief of pain. |
||||
|
†P <0.0001 |
||||
| Oral Ranitidine Hydrochloride*
| Oral Placebo*
|
|||
| Number Entered | Healed/ Evaluable | Number Entered | Healed/ Evaluable |
|
| Outpatients Week 2 | 195 | 69/182 (38%)† | 188 | 31/164 (19%) |
| Week 4 | 137/187 (73%)† | 76/168 (45%) |
||
In these studies, patients treated with oral ranitidine hydrochloride reported a reduction in both daytime and nocturnal pain, and they also consumed less antacid than the placebo-treated patients.
| Ulcer Healed | Ulcer Not Healed |
|
| Oral Ranitidine Hydrochloride | 0.06 | 0.71 |
| Oral placebo | 0.71 | 1.43 |
Pathological Hypersecretory Conditions (such as Zollinger-Ellison syndrome)
Ranitidine hydrochloride inhibits gastric acid secretion and reduces occurrence of diarrhea, anorexia, and pain in patients with pathological hypersecretion associated with Zollinger-Ellison syndrome, systemic mastocytosis, and other pathological hypersecretory conditions (e.g., postoperative, "short-gut" syndrome, idiopathic). Use of oral ranitidine hydrochloride was followed by healing of ulcers in 8 of 19 (42%) patients who were intractable to previous therapy.
In a retrospective review of 52 Zollinger-Ellison patients given ranitidine hydrochloride as a continuous intravenous infusion for up to 15 days, no patients developed complications of acid-peptic disease such as bleeding or perforation. Acid output was controlled to ≤10 mEq/h.
Indications and Usage for Ranitidine Injection
Ranitidine injection is indicated in some hospitalized patients with pathological hypersecretory conditions or intractable duodenal ulcers, or as an alternative to the oral dosage form for short-term use in patients who are unable to take oral medication.
Contraindications
Ranitidine injection is contraindicated for patients known to have hypersensitivity to the drug.
Precautions
General
- Symptomatic response to therapy with ranitidine hydrochloride does not preclude the presence of gastric malignancy.
- Since ranitidine hydrochloride is excreted primarily by the kidney, dosage should be adjusted in patients with impaired renal function (see DOSAGE AND ADMINISTRATION). Caution should be observed in patients with hepatic dysfunction since ranitidine hydrochloride is metabolized in the liver.
- In controlled studies in normal volunteers, elevations in SGPT have been observed when H2-antagonists have been administered intravenously at greater-than-recommended dosages for 5 days or longer. Therefore, it seems prudent in patients receiving intravenous ranitidine at dosages ≥100 mg 4 times daily for periods of 5 days or longer to monitor SGPT daily (from day 5) for the remainder of intravenous therapy.
- Bradycardia in association with rapid administration of ranitidine injection has been reported rarely, usually in patients with factors predisposing to cardiac rhythm disturbances. Recommended rates of administration should not be exceeded (see DOSAGE AND ADMINISTRATION).
- Rare reports suggest that ranitidine hydrochloride may precipitate acute porphyric attacks in patients with acute porphyria. Ranitidine hydrochloride should therefore be avoided in patients with a history of acute porphyria.
Laboratory Tests
False-positive tests for urine protein with MULTISTIX® may occur during therapy with ranitidine hydrochloride, and therefore testing with sulfosalicylic acid is recommended.
Drug Interactions
Ranitidine has been reported to affect the bioavailability of other drugs through several different mechanisms such as competition for renal tubular secretion, alteration of gastric pH, and inhibition of cytochrome P450 enzymes.
Procainamide
Ranitidine, a substrate of the renal organic cation transport system, may affect the clearance of other drugs eliminated by this route. High doses of ranitidine (e.g., such as those used in the treatment of Zollinger-Ellison syndrome) have been shown to reduce the renal excretion of procainamide and N-acetylprocainamide resulting in increased plasma levels of these drugs. Although this interaction is unlikely to be clinically relevant at usual ranitidine doses, it may be prudent to monitor for procainamide toxicity when administered with oral ranitidine at a dose exceeding 300 mg per day.
Warfarin
There have been reports of altered prothrombin time among patients on concomitant warfarin and ranitidine therapy. Due to the narrow therapeutic index, close monitoring of increased or decreased prothrombin time is recommended during concurrent treatment with ranitidine.
Ranitidine may alter the absorption of drugs in which gastric pH is an important determinant of bioavailability. This can result in either an increase in absorption (e.g., triazolam, midazolam, glipizide) or a decrease in absorption (e.g., ketoconazole, atazanavir, delavirdine, gefitinib). Appropriate clinical monitoring is recommended.
Atazanavir
Atazanavir absorption may be impaired based on known interactions with other agents that increase gastric pH. Use with caution. See atazanavir label for specific recommendations.
Delavirdine
Delavirdine absorption may be impaired based on known interactions with other agents that increase gastric pH. Chronic use of H2-receptor antagonists with delavirdine is not recommended.
Gefitinib
Gefitinib exposure was reduced by 44% with the coadministration of ranitidine and sodium bicarbonate (dosed to maintain gastric pH above 5.0). Use with caution.
Glipizide
In diabetic patients, glipizide exposure was increased by 34% following a single 150 mg dose of oral ranitidine. Use appropriate clinical monitoring when initiating or discontinuing ranitidine.
Ketoconazole
Oral ketoconazole exposure was reduced by up to 95% when oral ranitidine was coadministered in a regimen to maintain a gastric pH of 6 or above. The degree of interaction with usual dose of ranitidine (150 mg twice daily) is unknown.
Midazolam
Oral midazolam exposure in 5 healthy volunteers was increased by up to 65% when administered with oral ranitidine at a dose of 150 mg twice daily. However, in another interaction study in 8 volunteers receiving intravenous midazolam, a 300 mg oral dose of ranitidine increased midazolam exposure by about 9%. Monitor patients for excessive or prolonged sedation when ranitidine is coadministered with oral midazolam.
Triazolam
Triazolam exposure in healthy volunteers was increased by approximately 30% when administered with oral ranitidine at a dose of 150 mg twice daily. Monitor patients for excessive or prolonged sedation.
Carcinogenesis, Mutagenesis, Impairment of Fertility
There was no indication of tumorigenic or carcinogenic effects in life-span studies in mice and rats at oral dosages up to 2,000 mg/kg/day.
Ranitidine was not mutagenic in standard bacterial tests (Salmonella, Escherichia coli) for mutagenicity at concentrations up to the maximum recommended for these assays.
In a dominant lethal assay, a single oral dose of 1,000 mg/kg to male rats was without effect on the outcome of 2 matings per week for the next 9 weeks.
Teratogenic Effects
Reproduction studies have been performed in rats and rabbits at oral doses up to 160 times the human oral dose and have revealed no evidence of impaired fertility or harm to the fetus due to ranitidine. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Ranitidine is secreted in human milk. Caution should be exercised when ranitidine hydrochloride is administered to a nursing mother.
Pediatric Use
The safety and effectiveness of ranitidine injection have been established in the age-group of 1 month to 16 years for the treatment of duodenal ulcer. Use of ranitidine hydrochloride in this age-group is supported by adequate and well-controlled studies in adults, as well as additional pharmacokinetic data in pediatric patients, and an analysis of the published literature.
Safety and effectiveness in pediatric patients for the treatment of pathological hypersecretory conditions have not been established.
Limited data in neonatal patients (less than 1 month of age) receiving ECMO suggest that ranitidine hydrochloride may be useful and safe for increasing gastric pH for patients at risk of gastrointestinal hemorrhage.
Geriatric Use
Clinical studies of ranitidine injection did not include sufficient numbers of subjects aged 65 and over to determine whether they responded differently from younger subjects. However, in clinical studies of oral formulations of ranitidine hydrochloride, of the total number of subjects enrolled in US and foreign controlled clinical trials, for which there were subgroup analyses, 4,197 were 65 and over, while 899 were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
This drug is known to be substantially excreted by the kidney and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, caution should be exercised in dose selection, and it may be useful to monitor renal function (see CLINICAL PHARMACOLOGY: Pharmacokinetics: Geriatric Use and DOSAGE AND ADMINISTRATION: Dosage Adjustment for Patients With Impaired Renal Function).
Adverse Reactions/Side Effects
Transient pain at the site of intramuscular injection has been reported. Transient local burning or itching has been reported with intravenous administration of ranitidine hydrochloride.
The following have been reported as events in clinical trials or in the routine management of patients treated with oral or parenteral ranitidine hydrochloride. The relationship to therapy with ranitidine hydrochloride has been unclear in many cases. Headache, sometimes severe, seems to be related to administration of ranitidine hydrochloride.
Central Nervous System
Rarely, malaise, dizziness, somnolence, insomnia, and vertigo. Rare cases of reversible mental confusion, agitation, depression, and hallucinations have been reported, predominantly in severely ill elderly patients. Rare cases of reversible blurred vision suggestive of a change in accommodation have been reported. Rare reports of reversible involuntary motor disturbances have been received.
Cardiovascular
As with other H2-blockers, rare reports of arrhythmias such as tachycardia, bradycardia, asystole, atrioventricular block, and premature ventricular beats.
Gastrointestinal
Constipation, diarrhea, nausea/vomiting, abdominal discomfort/pain, and rare reports of pancreatitis.
Hepatic
In normal volunteers, SGPT values were increased to at least twice the pretreatment levels in 6 of 12 subjects receiving 100 mg intravenously 4 times daily for 7 days, and in 4 of 24 subjects receiving 50 mg intravenously 4 times daily for 5 days. There have been occasional reports of hepatocellular, cholestatic, or mixed hepatitis, with or without jaundice. In such circumstances, ranitidine should be immediately discontinued. These events are usually reversible, but in rare circumstances death has occurred. Rare cases of hepatic failure have also been reported.
Musculoskeletal
Rare reports of arthralgias and myalgias.
Hematologic
Blood count changes (leukopenia, granulocytopenia, and thrombocytopenia) have occurred in a few patients. These were usually reversible. Rare cases of agranulocytosis, pancytopenia, sometimes with marrow hypoplasia, and aplastic anemia and exceedingly rare cases of acquired immune hemolytic anemia have been reported.
Endocrine
Controlled studies in animals and humans have shown no stimulation of any pituitary hormone by ranitidine hydrochloride and no antiandrogenic activity, and cimetidine-induced gynecomastia and impotence in hypersecretory patients have resolved when ranitidine hydrochloride has been substituted. However, occasional cases of impotence and loss of libido have been reported in male patients receiving ranitidine hydrochloride, but the incidence did not differ from that in the general population. Rare cases of breast symptoms and conditions, including galactorrhea and gynecomastia, have been reported in both males and females.
Integumentary
Rash, including rare cases of erythema multiforme. Rare cases of alopecia and vasculitis.
Respiratory
A large epidemiological study suggested an increased risk of developing pneumonia in current users of histamine-2-receptor antagonists (H2RAs) compared to patients who had stopped H2RA treatment, with an observed adjusted relative risk of 1.63 (95% CI, 1.07 to 2.48). However, a causal relationship between use of H2RAs and pneumonia has not been established.
Other
Rare cases of hypersensitivity reactions (e.g., bronchospasm, fever, rash, eosinophilia), anaphylaxis, angioneurotic edema, acute interstitial nephritis, and small increases in serum creatinine.
To report SUSPECTED ADVERSE REACTIONS, contact Zydus Pharmaceuticals (USA) Inc. at 1-877-993-8779 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Related/similar drugs
Overdosage
There has been virtually no experience with overdosage with ranitidine injection and limited experience with oral doses of ranitidine. Reported acute ingestions of up to 18 g orally have been associated with transient adverse effects similar to those encountered in normal clinical experience (see ADVERSE REACTIONS). In addition, abnormalities of gait and hypotension have been reported.
When overdosage occurs, clinical monitoring and supportive therapy should be employed.
Studies in dogs receiving dosages of ranitidine hydrochloride in excess of 225 mg/kg/day have shown muscular tremors, vomiting, and rapid respiration. Single oral doses of 1,000 mg/kg in mice and rats were not lethal. Intravenous LD50 values in mice and rats were 77 and 83 mg/kg, respectively.
Ranitidine Injection Dosage and Administration
Parenteral Administration
In some hospitalized patients with pathological hypersecretory conditions or intractable duodenal ulcers, or in patients who are unable to take oral medication, ranitidine hydrochloride may be administered parenterally according to the following recommendations:
Intermittent Intravenous Injection
50 mg (2 mL) every 6 to 8 hours. Dilute ranitidine injection, 50 mg, in 0.9% sodium chloride injection or other compatible intravenous solution (see Stability) to a concentration no greater than 2.5 mg/mL (20 mL). Inject at a rate no greater than 4 mL/min (5 minutes).
50 mg (2 mL) every 6 to 8 hours. Dilute ranitidine injection, 50 mg, in 5% dextrose injection or other compatible intravenous solution (see Stability) to a concentration no greater than 0.5 mg/mL (100 mL). Infuse at a rate no greater than 5 to 7 mL/min (15 to 20 minutes).
In some patients it may be necessary to increase dosage. When this is necessary, the increases should be made by more frequent administration of the dose, but generally should not exceed 400 mg/day.
Continuous Intravenous Infusion
Add ranitidine injection to 5% dextrose injection or other compatible intravenous solution (see Stability). Deliver at a rate of 6.25 mg/hour (e.g., 150 mg [6 mL] of ranitidine injection in 250 mL of 5% dextrose injection at 10.7 mL/hour).
For Zollinger-Ellison patients, dilute ranitidine injection in 5% dextrose injection or other compatible intravenous solution (see Stability) to a concentration no greater than 2.5 mg/mL. Start the infusion at a rate of 1.0 mg/kg/hour. If after 4 hours either a measured gastric acid output is >10 mEq/hour or the patient becomes symptomatic, the dose should be adjusted upward in 0.5 mg/kg/hour increments, and the acid output should be remeasured. Dosages up to 2.5 mg/kg/hour and infusion rates as high as 220 mg/hour have been used.
Pediatric Use
While limited data exist on the administration of intravenous ranitidine to children, the recommended dose in pediatric patients is for a total daily dose of 2 to 4 mg/kg, to be divided and administered every 6 to 8 hours, up to a maximum of 50 mg given every 6 to 8 hours. This recommendation is derived from adult clinical studies and pharmacokinetic data in pediatric patients. Limited data in neonatal patients (less than 1 month of age) receiving ECMO have shown that a dose of 2 mg/kg is usually sufficient to increase gastric pH to >4 for at least 15 hours. Therefore, doses of 2 mg/kg given every 12 to 24 hours or as a continuous infusion should be considered.
Dosage Adjustment for Patients With Impaired Renal Function
The administration of ranitidine as a continuous infusion has not been evaluated in patients with impaired renal function. On the basis of experience with a group of subjects with severely impaired renal function treated with ranitidine hydrochloride, the recommended dosage in patients with a creatinine clearance <50 mL/min is 50 mg every 18 to 24 hours. Should the patient's condition require, the frequency of dosing may be increased to every 12 hours or even further with caution. Hemodialysis reduces the level of circulating ranitidine. Ideally, the dosing schedule should be adjusted so that the timing of a scheduled dose coincides with the end of hemodialysis.
Elderly patients are more likely to have decreased renal function, therefore caution should be exercised in dose selection, and it may be useful to monitor renal function (see CLINICAL PHARMACOLOGY: Pharmacokinetics: Geriatric Use and PRECAUTIONS: Geriatric Use).
Stability
Undiluted, ranitidine injection tends to exhibit a yellow color that may intensify over time without adversely affecting potency. Ranitidine injection is stable for 48 hours at room temperature when added to or diluted with most commonly used intravenous solutions, e.g., 0.9% sodium chloride injection, 5% dextrose injection, 10% dextrose injection, lactated ringer's injection, or 5% sodium bicarbonate injection.
Note
Parenteral drug products should be inspected visually for particulate matter and discoloration before administration whenever solution and container permit.
Directions for Dispensing
Pharmacy Bulk Package - Not for Direct Infusion
The pharmacy bulk package is for use in a pharmacy admixture service only under a laminar flow hood. The closure should be penetrated only once with a sterile transfer set or other sterile dispensing device, which allows measured distribution of the contents, and the contents dispensed in aliquots using aseptic technique. CONTENTS SHOULD BE USED AS SOON AS POSSIBLE FOLLOWING INITIAL CLOSURE PUNCTURE. DISCARD ANY UNUSED PORTION WITHIN 24 HOURS OF FIRST ENTRY. Following closure puncture, container should be maintained below 30°C (86°F) under a laminar flow hood until contents are dispensed.
How is Ranitidine Injection supplied
Ranitidine injection, USP 25 mg/mL, containing phenol 0.5% as preservative, in a 40-mL pharmacy bulk package.
| NDC Number
| Strength
| Pack Style
| Description
|
| 70710-1550-1 | 25 mg/mL | 1 vial per carton | 40-mL pharmacy bulk package |
Store between 4° and 25°C (39° and 77°F); excursions permitted to 30°C (86°F). Protect from light. Store vial in carton until time of use.
MULTISTIX® is the registered trademark of Bayer Healthcare LLC.
Liva Pharmaceuticals Limited
Vadodara, India
Distributed by:
Zydus Pharmaceuticals (USA) Inc.
Pennington, NJ 08534
2068236
Rev.: 01/19
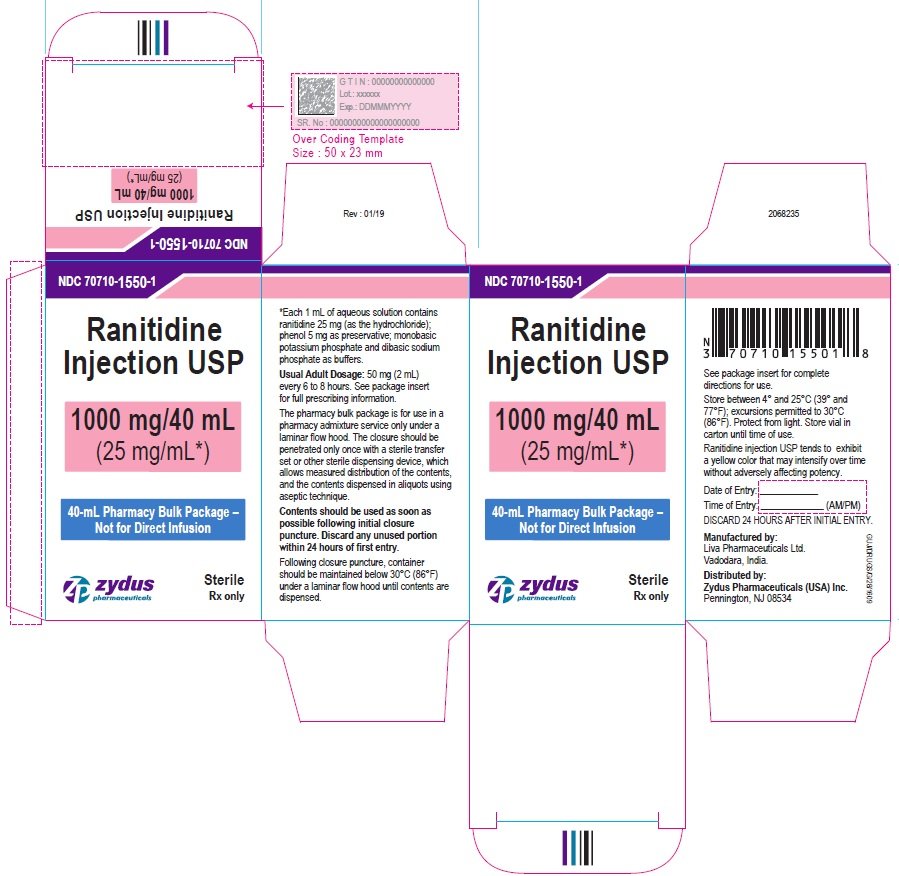
PRINCIPAL DISPLAY PANEL - 40 mL Vial Container Label
NDC 70710-1550-1
Ranitidine Injection USP
1000 mg/40 mL
(25 mg/mL*)
40-mL Pharmacy Bulk package – Not for Direct Infusion
Sterile
Rx only
Zydus pharmaceuticals

PRINCIPAL DISPLAY PANEL - 40 mL Vial Carton Label
NDC 70710-1550-1
Ranitidine Injection USP
1000 mg/40 mL
(25 mg/mL*)
40-mL Pharmacy Bulk Package – Not for Direct Infusion
Sterile
Rx only
Zydus pharmaceuticals
| RANITIDINE HYDROCHLORIDE
ranitidine hydrochloride injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Zydus Pharmaceuticals USA Inc. (156861945) |
| Registrant - Zydus Pharmaceuticals USA Inc. (156861945) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Zydus Lifesciences Limited | 873671928 | ANALYSIS(70710-1550) , MANUFACTURE(70710-1550) | |
More about ranitidine
- Check interactions
- Compare alternatives
- Reviews (163)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Drug class: H2 antagonists
- Breastfeeding
Patient resources
Professional resources
- Ranitidine prescribing information
- Ranitidine Capsule (FDA)
- Ranitidine Syrup (FDA)
- Ranitidine Tablets (FDA)
