Demadex: Package Insert / Prescribing Info
Package insert / product label
Generic name: torsemide
Dosage form: tablet
Drug class: Loop diuretics
Medically reviewed by Drugs.com. Last updated on Jan 6, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
DEMADEX (torsemide) tablets, for oral use
Initial U.S. Approval: 1993
Indications and Usage for Demadex
DEMADEX is a loop diuretic indicated for:
Demadex Dosage and Administration
Edema associated with:
- •
- Heart failure: Initial dose is 10 or 20 mg once daily. Titrate by factors of two; doses above 200 mg have not been studied. (2.1)
- •
- Chronic Renal Failure: Initial dose is 20 mg once daily. Titrate by factors of two; doses above 200 mg have not been studied. (2.1)
- •
- Hepatic Cirrhosis: Initial dose is 5 or 10 mg once daily. Titrate by factors of two; doses above 40 mg have not been studied. (2.1)
Hypertension:
- •
- The recommended initial dose is 5 mg once daily. After 4-6 weeks, increase to 10 mg once daily, if needed. If 10 mg is insufficient, consider adding another agent. (2.2)
Dosage Forms and Strengths
Tablets: 5 mg, 10 mg, 20 mg and 100 mg (3)
Contraindications
Hypersensitivity to DEMADEX or povidone, anuria, and hepatic coma. (4)
Warnings and Precautions
Adverse Reactions/Side Effects
The most common adverse reaction is excessive urination (6.7%). (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Meda Pharmaceuticals Inc. at 1-800-526-3840 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- •
- Non-steroidal anti-inflammatory drugs (NSAIDs): Reduced diuretic, natriuretic, and antihypertensive effects; risk of renal impairment. (7.1)
- •
- CYP2C9: Concomitant use with CYP2C9 inhibitors can decrease torsemide clearance. Torsemide may affect the efficacy and safety of sensitive CYP2C9 substrates or of substrates with a narrow therapeutic range, such as warfarin or phenytoin. (7.2)
- •
- Cholestyramine: Decreased exposure of DEMADEX. (7.3)
- •
- Organic anion drugs: may decrease diuretic activity of DEMADEX. (7.4)
- •
- Lithium: Risk of lithium toxicity. (7.5)
- •
- Renin-angiotensin inhibitors: Increased risk of hypotension and renal impairment. (7.7)
- •
- Radiocontrast agents: Increased risk of renal toxicity. (7.8)
- •
- Corticosteroids and ACTH: Increased risk of hypokalemia. (7.9)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2017
Full Prescribing Information
1. Indications and Usage for Demadex
1.1 Edema
DEMADEX is indicated for the treatment of edema associated with heart failure, renal disease or hepatic disease.
1.2 Hypertension
DEMADEX is indicated for the treatment of hypertension, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including the class to which this drug principally belongs. There are no controlled trials demonstrating risk reduction with DEMADEX.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (for example, patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
The antihypertensive effects of DEMADEX are on the average greater in black patients than in nonblack patients [see Clinical Pharmacology (12.2)]. Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
DEMADEX can be used alone or in combination with other antihypertensive agents.
2. Demadex Dosage and Administration
2.1 Treatment of Edema
Edema associated with heart failure
The recommended initial dose is 10 mg or 20 mg oral DEMADEX once daily. If the diuretic response is inadequate, titrate upward by approximately doubling until the desired diuretic response is obtained. Doses higher than 200 mg have not been adequately studied.
Edema associated with chronic renal failure
The recommended initial dose is 20 mg oral DEMADEX once daily. If the diuretic response is inadequate, titrate upward by approximately doubling until the desired diuretic response is obtained. Doses higher than 200 mg have not been adequately studied.
Edema associated with hepatic cirrhosis
The recommended initial dose is 5 mg or 10 mg oral DEMADEX once daily, administered together with an aldosterone antagonist or a potassium-sparing diuretic. If the diuretic response is inadequate, titrate upward by approximately doubling until the desired diuretic response is obtained. Doses higher than 40 mg have not been adequately studied in this population.
2.2 Treatment of Hypertension
The recommended initial dose is 5 mg once daily. If the 5 mg dose does not provide adequate reduction in blood pressure within 4 to 6 weeks, increase to 10 mg once daily. If the response to 10 mg is insufficient, add another antihypertensive agent to the treatment regimen.
3. Dosage Forms and Strengths
DEMADEX is available as white scored tablets in 5-, 10-, 20-, and 100-mg strengths.
4. Contraindications
DEMADEX is contraindicated in patients with known hypersensitivity to DEMADEX or to povidone.
DEMADEX is contraindicated in patients who are anuric.
DEMADEX is contraindicated in patients with hepatic coma.
5. Warnings and Precautions
5.1 Hypotension and Worsening Renal Function
Excessive diuresis may cause potentially symptomatic dehydration, blood volume reduction and hypotension and worsening renal function, including acute renal failure particularly in salt-depleted patients or those taking renin-angiotensin aldosterone inhibitors. Worsening of renal function can also occur with concomitant use of nephrotoxic drugs (e.g., aminoglycosides, cisplatin, and NSAIDs). Monitor volume status and renal function periodically.
5.2 Electrolyte and Metabolic Abnormalities
DEMADEX can cause potentially symptomatic hypokalemia, hyponatremia, hypomagnesemia, hypocalcemia, and hypochloremic alkalosis. Treatment with DEMADEX can cause an increase in blood glucose levels and hyperglycemia. Asymptomatic hyperuricemia can occur and gout may rarely be precipitated. Monitor serum electrolytes and blood glucose periodically.
6. Adverse Reactions/Side Effects
The following risks are discussed in more detail in other sections:
- •
- Hypotension and Worsening Renal Function [see Warnings and Precautions (5.1)]
- •
- Electrolyte and Metabolic Abnormalities [see Warnings and Precautions (5.2)]
- •
- Ototoxicity [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In pre-approval studies, DEMADEX has been evaluated for safety in approximately 4000 subjects; over 800 of these subjects received DEMADEX for at least 6 months, and over 380 were treated for more than 1 year. Among these subjects were 564 who received DEMADEX during United States-based trials in which 274 other subjects received placebo.
Discontinuation of therapy due to adverse reactions occurred in 3.5% of United States patients treated with DEMADEX and in 4.4% of patients treated with placebo.
In United States placebo-controlled trials excessive urination occurred in 6.7% of patients compared with 2.2% of patients receiving placebo. The daily doses of DEMADEX used in these trials ranged from 1.25 mg to 20 mg, with most patients receiving 5 mg to 10 mg; the duration of treatment ranged from 1 to 52 days, with a median of 41 days.
In the placebo-controlled hypertension studies excessive urination was dose related; 1% of patients receiving placebo, 4% of those treated with 5 mg of daily DEMADEX, and 15% of those treated with 10 mg. Excessive urination was generally not reported as an adverse event among patients who received DEMADEX for cardiac, renal, or hepatic failure.
There was no effect of age or sex on the incidence of adverse reactions.
Laboratory Parameters
Potassium
In controlled studies in the United States, DEMADEX was administered to hypertensive patients at doses of 5 mg or 10 mg daily. After 6 weeks at these doses, the mean decrease in serum potassium was approximately 0.1 mEq/L. The percentage of patients who had a serum potassium level below 3.5 mEq/L at any time during the studies was 1.5% on DEMADEX and 3% on placebo. In patients followed for 1 year, there was no progressive change in mean serum potassium levels. In patients with congestive heart failure, hepatic cirrhosis, or renal disease treated with DEMADEX at doses higher than those studied in United States antihypertensive trials, hypokalemia was observed with greater frequency, in a dose-related manner.
Blood Urea Nitrogen (BUN), Creatinine and Uric Acid
DEMADEX produces small dose-related increases in each of these laboratory values. In hypertensive patients who received 10 mg of DEMADEX daily for 6 weeks, the mean increase in blood urea nitrogen was 1.8 mg/dL (0.6 mmol/L), the mean increase in serum creatinine was 0.05 mg/dL (4 mmol/L), and the mean increase in serum uric acid was 1.2 mg/dL (70 mmol/L). Little further change occurred with long-term treatment, and all changes reversed when treatment was discontinued.
Glucose
Hypertensive patients who received 10 mg of daily DEMADEX experienced a mean increase in serum glucose concentration of 5.5 mg/dL (0.3 mmol/L) after 6 weeks of therapy, with a further increase of 1.8 mg/dL (0.1 mmol/L) during the subsequent year. In long-term studies in diabetics, mean fasting glucose values were not significantly changed from baseline.
6.2 Postmarketing Experience
The following adverse reactions have been identified during the post-approval use of DEMADEX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency reliably or establish a causal relationship to drug exposure.
Gastrointestinal System: Pancreatitis, abdominal pain
Nervous System: Paresthesia, confusion, visual impairment, loss of appetite
Hematologic: Leucopenia, thrombocytopenia, anemia
Hepatobiliary: Increase in liver transaminases, gamma-glutamyltransferase
Metabolism: Thiamine (vitamin B1) deficiency
Skin/hypersensitivity: Stevens-Johnson syndrome, toxic epidermal necrolysis, photosensitivity reaction, pruritus
Urogenital: Acute urinary retention
Related/similar drugs
7. Drug Interactions
7.1 Nonsteroidal Anti-inflammatory Drugs
Because DEMADEX and salicylates compete for secretion by renal tubules, patients receiving high doses of salicylates may experience salicylate toxicity when DEMADEX is concomitantly administered.
Concomitant use of nonsteroidal anti-inflammatory drugs (NSAIDs) and torsemide has been associated with the development of acute renal failure. The antihypertensive and diuretic effects of DEMADEX can be reduced by NSAIDs.
Partial inhibition of the natriuretic effect of DEMADEX by concomitant administration of indomethacin has been demonstrated for DEMADEX under conditions of dietary sodium restriction (50 mEq/day) but not in the presence of normal sodium intake (150 mEq/day).
7.2 Cytochrome P450 2C9 Inhibitors and Inducers
Torsemide is a substrate of CYP2C9. Concomitant use of CYP2C9 inhibitors (e.g., amiodarone, fluconazole, miconazole, oxandrolone) can decrease torsemide clearance and increase torsemide plasma concentrations. Concomitant use of CYP2C9 inducers (e.g., rifampin) increase torsemide clearance and decrease plasma torsemide concentrations. Monitor diuretic effect and blood pressure when used in combination with CYP2C9 inhibitor or inducer. Adjust torsemide dose if necessary.
Because of its inhibition of CYP2C9 metabolism, torsemide may affect the efficacy and safety of sensitive CYP2C9 substrates, such as celecoxib, or of substrates with a narrow therapeutic range, such as warfarin or phenytoin. Monitor patients and adjust dosages if necessary.
7.3 Cholestyramine
Concomitant use of torsemide and cholestyramine has not been studied in humans but, in a study in animals, coadministration of cholestyramine decreased the absorption of orally administered torsemide. If DEMADEX and cholestyramine should be coadministered, administer DEMADEX at least one hour before or 4 to 6 h after cholestyramine administration.
7.4 Organic Anion Drugs
Coadministration of organic anion drugs (e.g., probenecid) that undergo significant renal tubular secretion have the potential to reduce secretion of DEMADEX into the proximal tubule and thereby decreases the diuretic activity of DEMADEX. Monitor diuretic effect and blood pressure during coadministration.
7.5 Lithium
Like other diuretics, torsemide reduces the renal clearance of lithium, inducing a high risk of lithium toxicity. Monitor lithium levels periodically when torsemide is coadministered.
7.6 Ototoxic Drugs
Loop diuretics increase the ototoxic potential of other ototoxic drugs, including aminoglycoside antibiotics and ethacrynic acid. This effect has been reported with concomitant use of torsemide and gentamycin. Avoid concomitant use of DEMADEX and aminoglycoside antibiotics, if possible.
7.7 Renin-angiotensin Inhibitors
Coadministration of DEMADEX with ACE inhibitors or angiotensin receptor blockers can increase the risk of hypotension and renal impairment.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no available data on use of DEMADEX in pregnant women and the risk of major birth defects or miscarriage. In pregnant rats and rabbits dosed, on a mg/m2 basis, with 10 and 1.7 times a human dose of 20 mg/day, respectively, there was no fetotoxicity or teratogenicity. However, in pregnant rats and rabbits administered 50 and 6.8 times the human dose, respectively, decreases in body weight, decreased fetal resorption and delayed fetal ossification was observed.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major malformations and miscarriage in clinically recognized pregnancies is 2-4%, and 15-20%, respectively.
Data
There was no fetotoxicity or teratogenicity in rats treated with up to 5 mg/kg/day of torsemide (on a mg/kg basis, this is 15 times a human dose of 20 mg/day; on a mg/m2 basis, the animal dose is 10 times the human dose), or in rabbits, treated with 1.6 mg/kg/day (on a mg/kg basis, 5 times the human dose of 20 mg/kg/day; on a mg/m2 basis, 1.7 times this dose). Fetal and maternal toxicity (decrease in average body weight, increase in fetal resorption and delayed fetal ossification) occurred in rabbits and rats given doses 4 (rabbits) and 5 (rats) times larger.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Administration of another loop diuretic to premature infants has been associated with the precipitation of nephrocalcinosis/nephrolithiasis. Nephrocalcinosis/nephrolithiasis has also been observed in children under 4 years of age with no history of prematurity who have been treated chronically with the other loop diuretic. The other loop diuretic, when administered during the first weeks of life, has also been reported to increase the risk of persistent patent ductus arteriosus. The use of DEMADEX in such patients has not been studied.
8.5 Geriatric Use
Of the total number of patients who received DEMADEX in United States clinical studies, 24% were 65 or older while about 4% were 75 or older. No specific age-related differences in effectiveness or safety were observed between younger patients and elderly patients.
8.6 Use in Renal Impairment
In single-dose studies in patients with non-anuric renal failure, high doses of DEMADEX (20 mg to 200 mg) caused marked increases in water and sodium excretion. In patients with non-anuric renal failure, severe enough to require hemodialysis, chronic treatment with up to 200 mg of daily DEMADEX has not been shown to change steady-state fluid retention. When patients in a study of acute renal failure received total daily doses of 520 mg to 1200 mg of DEMADEX, 19% experienced seizures. Ninety-six patients were treated in this study; 6/32 treated with torsemide experienced seizures, 6/32 treated with comparably high doses of furosemide experienced seizures, and 1/32 treated with placebo experienced a seizure.
8.7 Use in Hepatic Impairment
DEMADEX can cause sudden alterations of fluid and electrolyte balance which may precipitate hepatic coma in patients with hepatic disease with cirrhosis and ascites. In these patients, diuresis with DEMADEX is best initiated in the hospital.
Diuretic treatment can cause or contribute to the development of hypovolemia, hypokalemia, metabolic alkalosis, hyponatremia or azotemia which can lead to new or worsening hepatic encephalopathy. Consider suspending or discontinuing DEMADEX [see Contraindications (4)].
To prevent hypokalemia and metabolic alkalosis, use an aldosterone antagonist or potassium-sparing drug with DEMADEX in patients with hepatic disease.
When given with aldosterone antagonists, DEMADEX also caused increases in sodium and fluid excretion in patients with edema or ascites due to hepatic cirrhosis. Urinary sodium excretion rate relative to the urinary excretion rate of DEMADEX is less in cirrhotic patients than in healthy subjects (possibly because of the hyperaldosteronism and resultant sodium retention that are characteristic of portal hypertension and ascites). However, because of the increased renal clearance of DEMADEX in patients with hepatic cirrhosis, these factors tend to balance each other, and the result is an overall natriuretic response that is similar to that seen in healthy subjects. Chronic use of any diuretic in hepatic disease has not been studied in adequate and well-controlled trials.
10. Overdosage
The signs and symptoms of overdosage can be anticipated to include those of excessive pharmacologic effect: dehydration, hypovolemia, hypotension, hyponatremia, hypokalemia, hypochloremic alkalosis, and hemoconcentration. Treatment of overdosage should consist of fluid and electrolyte replacement.
Laboratory determinations of serum levels of torsemide and its metabolites are not widely available.
No data are available to suggest physiological maneuvers (e.g., maneuvers to change the pH of the urine) that might accelerate elimination of torsemide and its metabolites. Torsemide is not dialyzable, so hemodialysis will not accelerate elimination.
11. Demadex Description
DEMADEX® (torsemide) is a diuretic of the pyridine-sulfonylurea class. Its chemical name is 1-isopropyl-3-[(4-m-toluidino-3-pyridyl) sulfonyl] urea and its structural formula is:
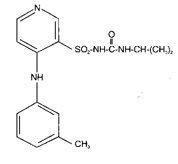
Its empirical formula is C16H20N4O3S, its pKa is 7.1, and its molecular weight is 348.43.
Torsemide is a white to off-white crystalline powder. The tablets for oral administration also contain lactose NF, crospovidone NF, povidone USP, microcrystalline cellulose NF, and magnesium stearate NF.
12. Demadex - Clinical Pharmacology
12.1 Mechanism of Action
Micropuncture studies in animals have shown that torsemide acts from within the lumen of the thick ascending portion of the loop of Henle, where it inhibits the Na+/K+/2Cl–-carrier system. Clinical pharmacology studies have confirmed this site of action in humans, and effects in other segments of the nephron have not been demonstrated. Diuretic activity thus correlates better with the rate of drug excretion in the urine than with the concentration in the blood.
Torsemide increases the urinary excretion of sodium, chloride, and water, but it does not significantly alter glomerular filtration rate, renal plasma flow, or acid-base balance.
12.2 Pharmacodynamics
With oral dosing, the onset of diuresis occurs within 1 hour and the peak effect occurs during the first or second hour and diuresis lasts about 6 to 8 hours. In healthy subjects given single doses, the dose-response relationship for sodium excretion is linear over the dose range of 2.5 mg to 20 mg. The increase in potassium excretion is negligible after a single dose of up to 10 mg and only slight (5 mEq to 15 mEq) after a single dose of 20 mg.
Edema
DEMADEX has been studied in controlled trials in patients with New York Heart Association Class II to Class IV heart failure. Patients who received 10 mg to 20 mg of daily DEMADEX in these studies achieved significantly greater reductions in weight and edema than did patients who received placebo.
Hypertension
In patients with essential hypertension, DEMADEX has been shown in controlled studies to lower blood pressure when administered once a day at doses of 5 mg to 10 mg. The antihypertensive effect is near maximal after 4 to 6 weeks of treatment, but it may continue to increase for up to 12 weeks. Systolic and diastolic supine and standing blood pressures are all reduced. There is no significant orthostatic effect, and there is only a minimal peak-trough difference in blood pressure reduction.
The antihypertensive effects of DEMADEX are, like those of other diuretics, on the average greater in black patients (a low-renin population) than in nonblack patients.
When DEMADEX is first administered, daily urinary sodium excretion increases for at least a week. With chronic administration, however, daily sodium loss comes into balance with dietary sodium intake. If the administration of DEMADEX is suddenly stopped, blood pressure returns to pretreatment levels over several days, without overshoot.
DEMADEX has been administered together with β-adrenergic blocking agents, ACE inhibitors, and calcium-channel blockers. Adverse drug interactions have not been observed, and special dosage adjustment has not been necessary.
12.3 Pharmacokinetics
Absorption
The bioavailability of DEMADEX tablets is approximately 80%, with small inter-subject variation; the 90% confidence interval is 75% to 89%. The drug is absorbed with little first-pass metabolism, and the serum concentration reaches its peak (Cmax) within 1 hour after oral administration. Cmax and area under the serum concentration-time curve (AUC) after oral administration are proportional to dose over the range of 2.5 mg to 200 mg. Simultaneous food intake delays the time to Cmax by about 30 minutes, but overall bioavailability (AUC) and diuretic activity are unchanged.
Distribution
The volume of distribution of torsemide is 12 to 15 liters in normal adults or in patients with mild to moderate renal failure or congestive heart failure. In patients with hepatic cirrhosis, the volume of distribution is approximately doubled. Torsemide is extensively bound to plasma protein (>99%).
Metabolism
Torsemide is metabolized by the hepatic cytochrome CYP2C9 and, to a minor extent, CYP2C8 and CYP2C18. Three main metabolites have been identified in humans. Metabolite M1 is formed by methyl-hydroxylation of torsemide, metabolite M3 is formed by ring hydroxylation of torsemide, and metabolite M5 is formed by oxidation of M1. The major metabolite in humans is the carboxylic acid derivative M5, which is biologically inactive. Metabolites M1 and M3 possess some pharmacological activity; however, their systemic exposures are much lower when compared to torsemide.
Elimination
In normal subjects the elimination half-life of torsemide is approximately 3.5 hours. Torsemide is cleared from the circulation by both hepatic metabolism (approximately 80% of total clearance) and excretion into the urine (approximately 20% of total clearance in patients with normal renal function).
Because torsemide is extensively bound to plasma protein (>99%), very little enters tubular urine via glomerular filtration. Most renal clearance of torsemide occurs via active secretion of the drug by the proximal tubules into tubular urine.
After a single oral dose, the amounts recovered in urine were: torsemide 21%, metabolite M1 12%, metabolite M3 2%, and metabolite M5 34%.
Renal Impairment
In patients with renal failure, renal clearance of torsemide is markedly decreased but total plasma clearance is not significantly altered. A smaller fraction of the administered dose is delivered to the intraluminal site of action, and the natriuretic action of any given dose of diuretic is reduced.
Hepatic Impairment
In patients with hepatic cirrhosis, the volume of distribution, plasma half-life, and renal clearance are all increased, but total clearance is unchanged.
Geriatric Patients
The renal clearance of torsemide is lower in elderly subjects as compared to younger adults, which is related to the decline in renal function that commonly occurs with aging. However, total plasma clearance and elimination half-life remain unchanged.
Heart Failure
In patients with decompensated congestive heart failure, hepatic and renal clearance are both reduced, probably because of hepatic congestion and decreased renal plasma flow, respectively. The total clearance of torsemide is approximately 50% of that seen in healthy volunteers, and the plasma half-life and AUC are correspondingly increased. Because of reduced renal clearance, a smaller fraction of any given dose is delivered to the intraluminal site of action, so at any given dose there is less natriuresis in patients with heart failure than in normal subjects.
Drug Interactions
Digoxin: Coadministration of digoxin is reported to increase the AUC for torsemide by 50%, but dose adjustment of DEMADEX is not necessary. Torsemide does not affect the pharmacokinetics of digoxin.
Spironolactone: In healthy subjects, coadministration of torsemide was associated with significant reduction in the renal clearance of spironolactone, with corresponding increases in the AUC. However, the pharmacokinetic profile and diuretic activity of torsemide are not altered by spironolactone.
Torsemide does not affect the protein binding of glyburide or warfarin.
Cimetidine: The pharmacokinetic profile and diuretic activity of torsemide are not altered by cimetidine.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No overall increase in tumor incidence was found when torsemide was given to rats and mice throughout their lives at doses up to 9 mg/kg/day (rats) and 32 mg/kg/day (mice). On a body-weight basis, these doses are 27 to 96 times a human dose of 20 mg; on a body-surface-area basis, they are 5 to 8 times this dose. In the rat study, the high-dose female group demonstrated renal tubular injury, interstitial inflammation, and a statistically significant increase in renal adenomas and carcinomas. The tumor incidence in this group was, however, not much higher than the incidence sometimes seen in historical controls. Similar signs of chronic non-neoplastic renal injury have been reported in high-dose animal studies of other diuretics such as furosemide and hydrochlorothiazide.
No mutagenic activity was detected in any of a variety of in vivo and in vitro tests of torsemide and its major human metabolite. The tests included the Ames test in bacteria (with and without metabolic activation), tests for chromosome aberrations and sister-chromatid exchanges in human lymphocytes, tests for various nuclear anomalies in cells found in hamster and murine bone marrow, tests for unscheduled DNA synthesis in mice and rats, and others.
In doses up to 25 mg/kg/day (75 times a human dose of 20 mg on a body-weight basis; 13 times this dose on a body-surface-area basis), torsemide had no adverse effect on the reproductive performance of male or female rats.
16. How is Demadex supplied
DEMADEX for oral administration is available as white, scored tablets as follows:
|
Dose |
Shape |
Debossing |
NDC 0037-xxxx-xx |
|
|
Side 1 |
Side 2 |
Bottle/100 |
||
|
5 mg |
elliptical |
5 |
5005 |
3505-01 |
|
10 mg |
elliptical |
10 |
5010 |
3510-01 |
|
20 mg |
elliptical |
20 |
5020 |
3520-01 |
|
100 mg |
capsule shaped |
100 |
5001 |
3500-01 |
Store at 15° to 30°C (59° to 86°F).
17. Patient Counseling Information
Symptomatic Hypotension: Advise patients receiving DEMADEX that lightheadedness can occur, especially during the first days of therapy, and that it should be reported to the prescribing physician. The patients should be told that if syncope occurs, DEMADEX should be discontinued until the physician has been consulted.
All patients should be cautioned that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope [see Warnings and Precautions (5.1)].
Non-Steroidal Anti-inflammatory Drugs (NSAID): Advise patients to discuss with their physician before taking NSAID medications concomitantly [see Drug Interactions (7.1)].
MEDA PHARMACEUTICALS and DEMADEX are registered trademarks of Meda AB. Any other trademarks are the property of their respective owners.
Manufactured By: Meda Manufacturing GmbH, Cologne, Germany
For: Meda Pharmaceuticals
Meda Pharmaceuticals Inc.
Somerset, NJ 08873-4120
Made in Germany
Printed in USA
© 2017 Meda Pharmaceuticals Inc.
IN-3500-02
Principal Display Panel - Package Label – 100 Count Bottle, 10 mg Tablets
NDC 0037-3510-01 100 Tablets
DEMADEX®
(torsemide)
10 mg
MEDA Rx only



| DEMADEX
torsemide tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| DEMADEX
torsemide tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| DEMADEX
torsemide tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Meda Pharmaceuticals (051229602) |
Frequently asked questions
More about Demadex (torsemide)
- Check interactions
- Compare alternatives
- Reviews (1)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: loop diuretics
- Breastfeeding
Patient resources
- Demadex drug information
- Demadex (Torsemide Intravenous) (Advanced Reading)
- Demadex (Torsemide Oral) (Advanced Reading)
