Myrbetriq: Package Insert / Prescribing Info
Package insert / product label
Generic name: mirabegron
Dosage form: tablet, film coated, extended release
Drug class: Urinary antispasmodics
Medically reviewed by Drugs.com. Last updated on Nov 20, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
MYRBETRIQ (mirabegron extended-release tablets), for oral use
MYRBETRIQ GRANULES (mirabegron for extended-release oral suspension)
Initial U.S. Approval: 2012
Recent Major Changes
Indications and Usage for Myrbetriq
MYRBETRIQ is a beta-3 adrenergic agonist indicated for the treatment of:
- •
- Overactive bladder (OAB) in adult patients with symptoms of urge urinary incontinence, urgency, and urinary frequency, either alone or in combination with the muscarinic antagonist solifenacin succinate. (1.1)
- •
- Neurogenic detrusor overactivity (NDO) in pediatric patients aged 3 years and older and weighing 35 kg or more. (1.2)
MYRBETRIQ Granules is a beta-3 adrenergic agonist indicated for the treatment of NDO in pediatric patients aged 3 years and older. (1.2)
Myrbetriq Dosage and Administration
- •
- MYRBETRIQ and MYRBETRIQ Granules are two different products and they are not substitutable on a milligram-per-milligram basis. Select the recommended product (MYRBETRIQ or MYRBETRIQ Granules) based on the indication and patient’s weight. Do not combine MYRBETRIQ and MYRBETRIQ Granules to achieve the total dose. A recommended dosage for MYRBETRIQ Granules for adults has not been determined. (2.1)
OAB in Adults - •
- The recommended starting dose of MYRBETRIQ is 25 mg orally once daily, either alone or in combination with solifenacin succinate 5 mg orally once daily. (2.2)
- •
- After 4 to 8 weeks, the MYRBETRIQ dose may be increased to 50 mg orally once daily. (2.2)
NDO in Pediatric Patients 3 Years and Older - •
- Pediatric Patients weighing less than 35 kg: Use MYRBETRIQ Granules: The recommended starting dose of MYRBETRIQ Granules is weight-based and administered as an extended-release oral suspension once daily. After 4 to 8 weeks, increase to the lowest effective dose without exceeding the maximum recommended dose. (2.3)
- •
- Pediatric Patients weighing 35 kg or more: Use MYRBETRIQ or MYRBETRIQ Granules:
- o
- The recommended starting dosage of MYRBETRIQ is 25 mg orally once daily. After 4 to 8 weeks, the MYRBETRIQ dose may be increased to 50 mg orally once daily. (2.3)
- o
- The recommended starting dosage of MYRBETRIQ Granules, administered as an extended-release oral suspension, is 6 mL (48 mg) orally once daily. After 4 to 8 weeks, increase to a maximum dosage of MYRBETRIQ Granules 10 mL (80 mg) orally once daily (2.3)
Adult or Pediatric Patients with Renal or Hepatic Impairment: Refer to the full prescribing information for recommended dosage. (2.4, 2.5)
Preparation for MYRBETRIQ Granules: Refer to the full prescribing information. (2.6)
Administration
- •
- MYRBETRIQ:
- •
- MYRBETRIQ Granules:
- o
- Pediatric patients: Take MYRBETRIQ Granules prepared as an extended-release oral suspension. Take with food. (2.7)
Dosage Forms and Strengths
Contraindications
Hypersensitivity to mirabegron or any inactive ingredients. (4)
Warnings and Precautions
- •
- Increases in Blood Pressure: Can increase blood pressure in adult or pediatric patients. Periodically monitor blood pressure, especially in hypertensive patients. MYRBETRIQ/MYRBETRIQ Granules are not recommended in patients with severe uncontrolled hypertension. (5.1)
- •
- Urinary Retention in Patients With Bladder Outlet Obstruction and in Patients Taking Muscarinic Antagonist Drugs for Overactive Bladder: Administer with caution in these patients because of risk of urinary retention. (5.2)
- •
- Angioedema: Angioedema of the face, lips, tongue, and/or larynx has been reported with mirabegron. (5.3, 6.2)
Adverse Reactions/Side Effects
- •
- Most commonly reported adverse reactions with MYRBETRIQ monotherapy in adult patients with OAB (> 2% and > placebo) were hypertension, nasopharyngitis, urinary tract infection, and headache. (6.1)
- •
- Most commonly reported adverse reactions with MYRBETRIQ, in combination with solifenacin succinate in adult patients with OAB (> 2% and > placebo and > comparator), were dry mouth, urinary tract infection, constipation, and tachycardia. (6.1)
- •
- Most commonly reported adverse reactions with MYRBETRIQ/MYRBETRIQ Granules in pediatric patients with NDO (≥ 3%) were UTI, nasopharyngitis, constipation, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Astellas Pharma US, Inc. at 1-800-727-7003 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- •
- Drugs Metabolized by CYP2D6: Mirabegron is a CYP2D6 inhibitor and, when used concomitantly with drugs metabolized by CYP2D6, especially narrow therapeutic index drugs, appropriate monitoring and possible dose adjustment of those drugs may be necessary. (5.4, 7.1, 12.3)
- •
- Digoxin: When initiating a combination of mirabegron and digoxin with or without solifenacin succinate, use the lowest dose of digoxin; monitor serum digoxin concentrations to titrate digoxin dose to desired clinical effect. (7.2, 12.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2021
Full Prescribing Information
1. Indications and Usage for Myrbetriq
1.1 Adult Overactive Bladder (OAB)
MYRBETRIQ Monotherapy
MYRBETRIQ® is indicated for the treatment of OAB in adult patients with symptoms of urge urinary incontinence, urgency, and urinary frequency.
MYRBETRIQ Combination Therapy with Solifenacin Succinate
MYRBETRIQ, in combination with the muscarinic antagonist solifenacin succinate, is indicated for the treatment of OAB in adult patients with symptoms of urge urinary incontinence, urgency, and urinary frequency.
1.2 Pediatric Neurogenic Detrusor Overactivity (NDO)
MYRBETRIQ Granules
MYRBETRIQ® Granules is indicated for the treatment of NDO in pediatric patients aged 3 years and older.
MYRBETRIQ
MYRBETRIQ is indicated for the treatment of NDO in pediatric patients aged 3 years and older and weighing 35 kg or more.
2. Myrbetriq Dosage and Administration
2.1 Important Dosage Information
MYRBETRIQ and MYRBETRIQ Granules are two different products and they are not substitutable on a milligram-per-milligram basis:
• Select the recommended product (MYRBETRIQ or MYRBETRIQ Granules) based on the indication and patient’s weight [see Indications and Usage (1) and Dosage and Administration (2.2, 2.3, 2.4, 2.5)].
• Do not combine MYRBETRIQ and MYRBETRIQ Granules to achieve the total dose.
• A recommended dosage for MYRBETRIQ Granules for adults has not been determined.
2.2 Recommended Dosage for Adult Patients with OAB
MYRBETRIQ Monotherapy
The recommended starting dosage of MYRBETRIQ is 25 mg orally once daily. If needed, increase to the maximum dosage of MYRBETRIQ 50 mg orally once daily after 4 to 8 weeks. For administration instructions, see Dosage and Administration (2.7).
MYRBETRIQ Combination Therapy with Solifenacin Succinate
The recommended starting dosage for combination treatment is MYRBETRIQ 25 mg orally once daily and solifenacin succinate 5 mg orally once daily. If needed, increase to the maximum dosage of MYRBETRIQ 50 mg orally once daily after 4 to 8 weeks. Refer to the Prescribing Information for solifenacin succinate for additional information. For administration instructions, see Dosage and Administration (2.7).
2.3 Recommended Dosage for Pediatric Patients Aged 3 Years and Older with NDO
For pediatric patients 3 years of age and older, select the appropriate product (MYRBETRIQ or MYRBETRIQ Granules) based on the patient’s weight.
Pediatric Patients weighing less than 35 kg: Use MYRBETRIQ Granules
The recommended starting and maximum doses of MYRBETRIQ Granules, administered as extended-release oral suspension once daily [see Dosage and Administration (2.6)], are shown in Table 1. The recommended dosages are determined based on patient weight. Evaluate patients periodically for potential dosage adjustment. For administration instructions, see Dosage and Administration (2.7).
|
Body Weight Range |
Starting Dose |
Maximum Volume |
|
11 kg to less than 22 kg |
3 mL (24 mg) |
6 mL (48 mg) |
|
22 kg to less than 35 kg |
4 mL (32 mg) |
8 mL (64 mg) |
|
Greater than or equal to 35 kg |
Refer to information in next section |
|
Pediatric Patients weighing 35 kg or more: Use MYRBETRIQ or MYRBETRIQ Granules
The recommended starting dosage of MYRBETRIQ is 25 mg orally once daily. If needed, increase to a maximum dosage of MYRBETRIQ 50 mg orally once daily after 4 to 8 weeks. For administration instructions, see Dosage and Administration (2.7).
The recommended starting dosage of MYRBETRIQ Granules is 6 mL (48 mg) orally once daily. If needed, increase to a maximum dosage of MYRBETRIQ Granules 10 mL (80 mg) orally once daily after 4 to 8 weeks. For administration instructions, see Dosage and Administration (2.7).
2.4 Recommended Dosage in Adult Patients with Renal or Hepatic Impairment
Dosage in Adults with Renal Impairment
The recommended dosage of MYRBETRIQ (administered orally once daily) in adult patients with renal impairment is described in Table 2 [see Use in Specific Populations (8.6)]. For administration instructions, see Dosage and Administration (2.7).
|
||
|
Estimated GFR* |
Starting Dose |
Maximum Dose |
|
eGFR 30 to 89 mL/min/1.73 m2 |
25 mg |
50 mg |
|
eGFR 15 to 29 mL/min/1.73 m2 |
25 mg |
25 mg |
|
eGFR < 15 mL/min/1.73 m2 or requiring dialysis |
Not recommended |
|
Dosage in Adults with Hepatic Impairment
The recommended dosage of MYRBETRIQ (administered orally once daily) in adult patients with hepatic impairment is described in Table 3 [see Use in Specific Populations (8.7)]. For administration instructions, see Dosage and Administration (2.7).
|
Hepatic Impairment Classification |
Starting Dose |
Maximum Dose |
|
Child-Pugh Class A (Mild hepatic impairment) |
25 mg |
50 mg |
|
Child-Pugh Class B (Moderate hepatic impairment) |
25 mg |
25 mg |
|
Child-Pugh Class C (Severe hepatic impairment) |
Not Recommended |
|
2.5 Recommended Dosage in Pediatric Patients with Renal or Hepatic Impairment
For pediatric patients 3 years of age and older, select the appropriate product (MYRBETRIQ or MYRBETRIQ Granules) based on the patient’s weight.
Pediatric Patients Weighing Less Than 35 kg with Renal or Hepatic Impairment: Use MYRBETRIQ Granules
Dosage in Pediatric Patients with Renal Impairment
The recommended dosage of MYRBETRIQ Granules in pediatric patients with renal impairment (administered orally once daily) is described in Table 4 [see Use in Specific Populations (8.6)]. For administration instructions, see Dosage and Administration (2.7).
|
|||
|
Estimated GFR* |
Body Weight Range |
Starting Dose |
Maximum Dose |
|
eGFR 30 to 89 mL/min/1.73 m2 |
11 kg to less than 22 kg |
3 mL (24 mg) |
6 mL (48 mg) |
|
22 kg to less than 35 kg |
4 mL (32 mg) |
8 mL (64 mg) |
|
|
eGFR 15 to 29 mL/min/1.73 m2 |
11 kg to less than 22 kg |
3 mL (24 mg) |
3 mL (24 mg) |
|
22 kg to less than 35 kg |
4 mL (32 mg) |
4 mL (32 mg) |
|
|
eGFR < 15 mL/min/1.73 m2 or undergoing dialysis |
Use is Not Recommended |
||
Dosage in Pediatric Patients with Hepatic Impairment
The recommended dosage of MYRBETRIQ Granules in pediatric patients with hepatic impairment (administered orally once daily) is described in Table 5 [see Use in Specific Populations (8.7)]. For administration instructions, see Dosage and Administration (2.7).
|
Hepatic Impairment Classification |
Body Weight Range |
Starting Dose |
Maximum Dose |
|
Child-Pugh Class A (Mild hepatic impairment) |
11 kg to less than 22 kg |
3 mL (24 mg) |
6 mL (48 mg) |
|
22 kg to less than 35 kg |
4 mL (32 mg) |
8 mL (64 mg) |
|
|
Child-Pugh Class B (Moderate hepatic impairment) |
11 kg to less than 22 kg |
3 mL (24 mg) |
3 mL (24 mg) |
|
22 kg to less than 35 kg |
4 mL (32 mg) |
4 mL (32 mg) |
|
|
Child-Pugh Class C (Severe hepatic impairment) |
Use is Not Recommended |
||
Pediatric Patients weighing 35 kg or more with renal or hepatic impairment: Use MYRBETRIQ or MYRBETRIQ Granules
Dosage in Pediatric Patients with Renal Impairment
The recommended dosage of MYRBETRIQ in pediatric patients with renal impairment weighing 35 kg or more (administered orally once daily) is described in Table 2 (above). Note that the dosage is the same as for adult patients with renal impairment [see Dosage and Administration (2.4) and Use in Specific Populations (8.6)]. For administration instructions, see Dosage and Administration (2.7).
The recommended dosage of MYRBETRIQ Granules in pediatric patients with renal impairment weighing 35 kg or more (administered orally once daily) is described in Table 6 [see Use in Specific Populations (8.6)]. For administration instructions, see Dosage and Administration (2.7).
|
||
|
Estimated GFR* |
Starting Dose |
Maximum Dose |
|
eGFR 30 to 89 mL/min/1.73 m2 |
6 mL (48 mg) |
10 mL (80 mg) |
|
eGFR 15 to 29 mL/min/1.73 m2 |
6 mL (48 mg) |
6 mL (48 mg) |
|
eGFR < 15 mL/min/1.73 m2 or undergoing dialysis |
Use is Not Recommended |
|
Dosage in Pediatric Patients with Hepatic Impairment
The recommended dosage of MYRBETRIQ in pediatric patients with hepatic impairment weighing 35 kg or more (administered orally once daily) is described in Table 3 (above). Note that the dosage is the same as for adult patients with hepatic impairment [see Dosage and Administration (2.4) and Use in Specific Populations (8.6)]. For administration instructions, see Dosage and Administration (2.7).
The recommended dosage of MYRBETRIQ Granules in pediatric patients with hepatic impairment weighing 35 kg or more (administered orally once daily) is described in Table 7 [see Use in Specific Populations (8.7)]. For administration instructions, see Dosage and Administration (2.7).
|
Hepatic Impairment Classification |
Starting Dose |
Maximum Dose |
|
Child-Pugh Class A (Mild hepatic impairment) |
6 mL (48 mg) |
10 mL (80 mg) |
|
Child-Pugh Class B (Moderate hepatic impairment) |
6 mL (48 mg) |
6 mL (48 mg) |
|
Child-Pugh Class C (Severe hepatic impairment) |
Use is Not Recommended |
|
2.6 Preparation and Storage Instructions for MYRBETRIQ Granules
The required dose for MYRBETRIQ Granules (mirabegron for extended-release oral suspension) is calculated based on the weight of the patient. Prepare oral suspension at the time of dispensing.
Keep the bottle in the pouch up until the time of reconstitution.
• Discard the pouch and desiccant prior to reconstitution. Do not dispense.
• Tap the closed bottle several times to loosen the granules.
• Measure 100 mL of water, add the total amount to the bottle, and immediately shake vigorously for 1 minute, then let it stand for 10 to 30 minutes. Shake vigorously again for 1 minute.
• If granules have not dispersed, shake vigorously for another 1 minute.
• Record the 28-day expiration date on the container and carton based on the reconstitution date.
• Give the patient an appropriate dosing device.
• Store the reconstituted suspension at 20°C to 25°C (68°F to 77°F) for up to 28 days.
• Discard the unused portion after 28 days [see How Supplied/Storage and Handling (16.2)].
After reconstitution with 100 mL water, the suspension contains 8 mg/mL of mirabegron.
2.7 Administration Instructions
Administration instructions for MYRBETRIQ and MYRBETRIQ Granules differ based on the patient population.
MYRBETRIQ
Adult patients: Swallow MYRBETRIQ whole with water. Do not chew, divide, or crush. Take with or without food.
Pediatric patients: Swallow MYRBETRIQ whole with water. Do not chew, divide, or crush. Take with food [see Use in Specific Populations (8.4)].
MYRBETRIQ Granules
Adult patients: A recommended dosage for MYRBETRIQ Granules for adults has not been determined.
Pediatric patients: Take MYRBETRIQ Granules prepared as an extended-release oral suspension [see Dosage and Administration (2.6)]. Take with food to reduce potential exposure-related risks [see Use in Specific Populations (8.4)].
3. Dosage Forms and Strengths
MYRBETRIQ (mirabegron extended-release tablets) are supplied in two different strengths as described below:
• 25 mg oval, brown, film-coated tablet, debossed with the  (Astellas logo) and “325”
(Astellas logo) and “325”
• 50 mg oval, yellow, film-coated tablet, debossed with the  (Astellas logo) and “355”
(Astellas logo) and “355”
MYRBETRIQ Granules (mirabegron for extended-release oral suspension): Each bottle is filled with approximately 8.3 g of yellowish white granules, which contain 830 mg of mirabegron. After reconstitution with 100 mL water, the oral suspension is pale brownish yellow to yellow with 8 mg/mL of mirabegron.
4. Contraindications
MYRBETRIQ/MYRBETRIQ Granules is contraindicated in patients with known hypersensitivity reactions to mirabegron or any inactive ingredients of the tablet or oral suspension [see Adverse Reactions (6.1, 6.2)].
5. Warnings and Precautions
5.1 Increases in Blood Pressure
Increases in Blood Pressure in Adults
MYRBETRIQ/MYRBETRIQ Granules can increase blood pressure. Periodic blood pressure determinations are recommended, especially in hypertensive patients. MYRBETRIQ/MYRBETRIQ Granules is not recommended for use in patients with severe uncontrolled hypertension (defined as systolic blood pressure greater than or equal to 180 mm Hg and/or diastolic blood pressure greater than or equal to 110 mm Hg) [see Clinical Pharmacology (12.2)].
In two, randomized, placebo-controlled, healthy adult volunteer studies, MYRBETRIQ was associated with dose-related increases in supine blood pressure. In these studies, at the maximum recommended dose of 50 mg, the mean maximum increase in systolic/diastolic blood pressure was approximately 3.5/1.5 mm Hg greater than placebo.
In contrast, in adult OAB patients in clinical trials, MYRBETRIQ, taken as monotherapy or in combination with solifenacin succinate 5 mg, the mean increase in systolic and diastolic blood pressure at the maximum recommended mirabegron dose of 50 mg was approximately 0.5 to 1 mm Hg greater than placebo. Worsening of pre-existing hypertension was reported infrequently in patients taking MYRBETRIQ.
Increases in Blood Pressure in Pediatric Patients 3 Years and Older
MYRBETRIQ/MYRBETRIQ Granules can increase blood pressure in pediatric patients. Blood pressure increases may be larger in children (3 to less than 12 years of age) than in adolescents (12 to less than 18 years of age). Periodic blood pressure determinations are recommended. MYRBETRIQ/MYRBETRIQ Granules is not recommended for use in pediatric patients with severe uncontrolled hypertension, defined as a systolic and/or diastolic blood pressure above the 99th percentile plus 5 mm Hg for age, sex, and stature using appropriate reference values [see Adverse Reactions (6.1)].
5.2 Urinary Retention in Patients with Bladder Outlet Obstruction and in Patients Taking Muscarinic Antagonist Medications for OAB
In patients taking MYRBETRIQ, urinary retention has been reported to occur in patients with bladder outlet obstruction (BOO) and in patients taking muscarinic antagonist medications for the treatment of OAB. A controlled clinical safety study in patients with BOO did not demonstrate increased urinary retention in patients treated with mirabegron; however, MYRBETRIQ should still be administered with caution to patients with clinically significant BOO. For example, monitor these patients for signs and symptoms of urinary retention. MYRBETRIQ should also be administered with caution to patients taking muscarinic antagonist medications for the treatment of OAB, including solifenacin succinate [see Clinical Pharmacology (12.2)].
5.3 Angioedema
Angioedema of the face, lips, tongue, and/or larynx has been reported with MYRBETRIQ/MYRBETRIQ Granules. In some cases, angioedema occurred after the first dose, however, cases have been reported to occur hours after the first dose or after multiple doses. Angioedema, associated with upper airway swelling, may be life-threatening. If involvement of the tongue, hypopharynx, or larynx occurs, promptly discontinue MYRBETRIQ/MYRBETRIQ Granules and provide appropriate therapy and/or measures necessary to ensure a patent airway [see Adverse Reactions (6.2)].
5.4 Patients Taking Drugs Metabolized by CYP2D6
Since MYRBETRIQ/MYRBETRIQ Granules is a moderate CYP2D6 inhibitor, the systemic exposure to CYP2D6 substrates is increased when coadministered with MYRBETRIQ/MYRBETRIQ Granules. Therefore, appropriate monitoring and dose adjustment may be necessary, especially with narrow therapeutic index drugs metabolized by CYP2D6 [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in more detail in other sections of the labeling.
- •
- Hypertension [see Warnings and Precautions (5.1)]
- •
- Urinary Retention [see Warnings and Precautions (5.2)]
- •
- Angioedema [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
MYRBETRIQ Monotherapy for Adult OAB
In three, 12-week, double-blind, placebo-controlled, safety and efficacy studies in patients with OAB (Studies 1, 2, and 3), MYRBETRIQ was evaluated for safety in 2736 patients [see Clinical Studies (14.1)]. Study 1 also included an active control. For the combined Studies 1, 2, and 3, 432 patients received MYRBETRIQ 25 mg, 1375 received MYRBETRIQ 50 mg, and 929 received MYRBETRIQ 100 mg once daily. In these studies, the majority of the patients were Caucasian (94%) and female (72%) with a mean age of 59 years (range 18 to 95 years).
MYRBETRIQ was also evaluated for safety in 1632 patients who received MYRBETRIQ 50 mg once daily (n=812 patients) or MYRBETRIQ 100 mg (n=820 patients) in a 1-year, randomized, fixed-dose, double-blind, active-controlled, safety study in patients with OAB (Study 4). Of these patients, 731 received MYRBETRIQ in a previous 12-week study. In Study 4, 1385 patients received MYRBETRIQ continuously for at least 6 months, 1311 patients received MYRBETRIQ for at least 9 months, and 564 patients received MYRBETRIQ for at least 1 year.
The most frequent adverse events (0.2%) leading to discontinuation in Studies 1, 2, and 3 for the 25 mg or 50 mg dose were nausea, headache, hypertension, diarrhea, constipation, dizziness, and tachycardia.
Atrial fibrillation (0.2%) and prostate cancer (0.1%) were reported as serious adverse events by more than 1 patient and at a rate greater than placebo.
Table 8 lists the adverse reactions, derived from all adverse events, that were reported in Studies 1, 2, and 3 at an incidence greater than placebo and in 1% or more of patients treated with MYRBETRIQ 25 mg or 50 mg once daily for up to 12 weeks. The most commonly reported adverse reactions (greater than 2% of MYRBETRIQ patients and greater than placebo) were hypertension, nasopharyngitis, urinary tract infection, and headache.
|
|||
|
Adverse Reaction |
Placebo (%) |
MYRBETRIQ 25 mg (%) |
MYRBETRIQ 50 mg (%) |
|
Number of Patients |
1380 |
432 |
1375 |
|
Hypertension* |
7.6 |
11.3 |
7.5 |
|
Nasopharyngitis |
2.5 |
3.5 |
3.9 |
|
Urinary Tract Infection |
1.8 |
4.2 |
2.9 |
|
Headache |
3.0 |
2.1 |
3.2 |
|
Constipation |
1.4 |
1.6 |
1.6 |
|
Upper Respiratory Tract Infection |
1.7 |
2.1 |
1.5 |
|
Arthralgia |
1.1 |
1.6 |
1.3 |
|
Diarrhea |
1.3 |
1.2 |
1.5 |
|
Tachycardia |
0.6 |
1.6 |
1.2 |
|
Abdominal Pain |
0.7 |
1.4 |
0.6 |
|
Fatigue |
1.0 |
1.4 |
1.2 |
Other adverse reactions reported by less than 1% of patients treated with MYRBETRIQ in Studies 1, 2, or 3 included:
Cardiac disorders: palpitations, blood pressure increased [see Clinical Pharmacology (12.2)]
Eye disorders: glaucoma [see Clinical Pharmacology (12.2)]
Gastrointestinal disorders: dyspepsia, gastritis, abdominal distension
Infections and Infestations: sinusitis, rhinitis
Investigations: GGT increased, AST increased, ALT increased, LDH increased
Renal and urinary disorders: nephrolithiasis, bladder pain
Reproductive system and breast disorders: vulvovaginal pruritus, vaginal infection
Skin and subcutaneous tissue disorders: urticaria, leukocytoclastic vasculitis, rash, pruritus, purpura, lip edema
Table 9 lists the rates of the most commonly reported adverse reactions, derived from all adverse events in patients treated with MYRBETRIQ 50 mg for up to 52 weeks in Study 4. The most commonly reported adverse reactions (> 3% of MYRBETRIQ patients) were hypertension, urinary tract infection, headache, and nasopharyngitis.
|
Adverse Reaction |
MYRBETRIQ 50 mg (%) |
Active Control (%) |
|
Number of Patients |
812 |
812 |
|
Hypertension |
9.2 |
9.6 |
|
Urinary Tract Infection |
5.9 |
6.4 |
|
Headache |
4.1 |
2.5 |
|
Nasopharyngitis |
3.9 |
3.1 |
|
Back Pain |
2.8 |
1.6 |
|
Constipation |
2.8 |
2.7 |
|
Dry Mouth |
2.8 |
8.6 |
|
Dizziness |
2.7 |
2.6 |
|
Sinusitis |
2.7 |
1.5 |
|
Influenza |
2.6 |
3.4 |
|
Arthralgia |
2.1 |
2.0 |
|
Cystitis |
2.1 |
2.3 |
In Study 4, in patients treated with MYRBETRIQ 50 mg once daily, adverse reactions leading to discontinuation reported by more than 2 patients and at a rate greater than active control included: constipation (0.9%), headache (0.6%), dizziness (0.5%), hypertension (0.5%), dry eyes (0.4%), nausea (0.4%), vision blurred (0.4%), and urinary tract infection (0.4%). Serious adverse events reported by at least 2 patients and exceeding active control included cerebrovascular accident (0.4%) and osteoarthritis (0.2%). Serum ALT/AST increased from baseline by greater than 10-fold in 2 patients (0.3%) taking MYRBETRIQ 50 mg; and these markers subsequently returned to baseline while both patients continued MYRBETRIQ.
In Study 4, serious adverse events of neoplasm were reported by 0.1%, 1.3%, and 0.5% of patients treated with MYRBETRIQ 50 mg, MYRBETRIQ 100 mg, and active control once daily, respectively. Neoplasms reported by 2 patients treated with MYRBETRIQ 100 mg included breast cancer, lung neoplasm malignant, and prostate cancer. A causal relationship between mirabegron and these reported neoplasms has not been established.
In a separate clinical study in Japan, a single case was reported as Stevens-Johnson syndrome with increased serum ALT, AST, and bilirubin in a patient taking MYRBETRIQ 100 mg as well as an herbal medication (Kyufu Gold).
MYRBETRIQ Combination Therapy with Solifenacin Succinate for Adult OAB
In three, 12-week, double-blind, randomized, active-controlled safety and efficacy studies in patients with OAB (Studies 5, 6, and 7), combination treatment of MYRBETRIQ and solifenacin succinate was evaluated for safety in 6818 patients [see Clinical Studies (14.2)]. Studies 5 and 6 also included a placebo control. For the combined Studies 5, 6, and 7, 997 patients received combination treatment with MYRBETRIQ 25 mg and solifenacin succinate 5 mg, and 1706 patients received combination treatment with MYRBETRIQ 50 mg and solifenacin succinate 5 mg. In these studies, the majority of the patients were Caucasian (88%) and female (77%) with a mean age of 57 years (range 18 to 89 years).
MYRBETRIQ 50 mg and solifenacin succinate 5 mg coadministration was also evaluated for safety in 1814 patients in a 52-week, double-blind, randomized, active-controlled study in patients with OAB (Study 8) [see Clinical Studies (14.2)].
In Studies 5, 6, and 7, the most commonly reported adverse reactions (greater than 2% of patients treated with combination therapy of MYRBETRIQ and solifenacin succinate 5 mg, and greater than placebo and/or MYRBETRIQ or solifenacin succinate comparator at the same dose as in the combination treatment) were dry mouth, urinary tract infection, constipation, and tachycardia. The most frequent adverse reactions (≥ 0.2%) leading to discontinuation in the coadministration trials were dry mouth and urinary retention.
Table 10 lists the adverse reactions, derived from all adverse events that were reported in Studies 5, 6, and 7 in 1% or more of patients treated with MYRBETRIQ 25 mg or 50 mg coadministered with solifenacin succinate 5 mg and at an incidence greater than placebo and mirabegron or solifenacin succinate comparator at the same dose as in the combination treatment when administered once daily for up to 12 weeks.
|
||||||
|
Adverse Reaction |
Placebo (%) |
MYRBETRIQ 25 mg (%) |
MYRBETRIQ 50 mg (%) |
Solifenacin Succinate 5 mg (%) |
MYRBETRIQ 25 mg + Solifenacin Succinate 5 mg (%) |
MYRBETRIQ 50 mg + Solifenacin Succinate 5 mg* (%) |
|
Number of Patients |
510 |
500 |
500 |
1288 |
997 |
1706 |
|
Dry Mouth |
2.2 |
3.8 |
3.6 |
6.5 |
9.3 |
7.2 |
|
Urinary Tract Infections† |
5.3 |
4.0 |
4.2 |
3.6 |
7.0 |
4.0 |
|
Constipation |
1.2 |
1.2 |
2.8 |
2.4 |
4.2 |
3.9 |
|
Tachycardia |
0.8 |
1.6 |
1.6 |
0.7 |
2.2 |
0.9 |
|
Dyspepsia |
0.6 |
0.4 |
0.2 |
0.7 |
1.1 |
1.3 |
|
Dizziness |
0.4 |
0.8 |
1.2 |
1.2 |
1.3 |
0.4 |
|
Vision Blurred |
0.4 |
0.2 |
0.2 |
0.9 |
0.7 |
1.1 |
|
Arthralgia |
0.8 |
0.8 |
0.8 |
0.8 |
0.5 |
1.1 |
In Study 8, the most common adverse reactions (more than 2% of patients treated with coadministration of MYRBETRIQ and solifenacin succinate and exceeding comparator rate) were UTI, dry mouth, constipation, and headache. The most frequent adverse reactions leading to discontinuation in the trial were constipation (0.2%), urinary retention (0.2%), urinary hesitation (0.2%), and vision blurred (0.2%).
In Study 8, serious adverse events of neoplasm were reported by 0.7%, 0.3%, and 0% of patients who received coadministration of MYRBETRIQ 50 mg and solifenacin succinate 5 mg, MYRBETRIQ 50 mg monotherapy, and solifenacin succinate 5 mg monotherapy, respectively. Neoplasms reported by more than 1 patient who received coadministration with MYRBETRIQ 50 mg and solifenacin succinate 5 mg included basal cell carcinoma (n=3), breast cancer (n=2), melanoma (n=2), and squamous cell carcinoma (n=2). A causal relationship between the coadministration of mirabegron and solifenacin succinate and these reported neoplasms has not been established.
Table 11 lists the adverse reactions, derived from all adverse events that were reported at an incidence greater than comparator and in 2% or more of patients treated with MYRBETRIQ 50 mg coadministered with solifenacin succinate 5 mg once daily for up to 52 weeks in Study 8.
|
|||
|
Adverse Reaction |
MYRBETRIQ 50 mg (%) |
Solifenacin Succinate 5 mg (%) |
MYRBETRIQ 50 mg + Solifenacin Succinate 5 mg (%) |
|
Number of Patients |
305 |
303 |
1206 |
|
Urinary Tract Infections* |
6.2 |
5.9 |
8.4 |
|
Dry Mouth |
3.9 |
5.9 |
6.1 |
|
Constipation |
1.0 |
2.3 |
3.3 |
|
Headache |
1.6 |
1.7 |
2.9 |
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
The safety of MYRBETRIQ/MYRBETRIQ Granules was evaluated in a 52-week, open-label, baseline-controlled, multicenter, dose titration study (Study 9) [see Clinical Studies (14.3)]. The study included 86 pediatric patients 3 to 17 years of age with neurogenic detrusor overactivity (NDO); 55% were female, 72% were White. Treatment was initiated at the weight-based starting recommended dose and was increased to a dose equivalent of MYRBETRIQ 50 mg daily dose in adults by Week 8. Subsequent to the dose titration period, patients continued their optimized dose for the duration of the 52-week study (mean exposure duration 303 days, range 1 to 390 days).
The most commonly reported adverse reactions were UTI, nasopharyngitis, constipation, and headache.
Table 12 lists the adverse reactions that were reported in 2% or more of patients treated with MYRBETRIQ/MYRBETRIQ Granules for oral suspension in Study 9.
|
|
|
Adverse Reaction |
Percentage (%) of Patients Reporting Adverse Reactions N=86 |
|
Number of Patients |
51 (59.3) |
|
Urinary Tract Infection* |
24.4 |
|
Nasopharyngitis |
5.8 |
|
Constipation |
4.7 |
|
Headache |
3.5 |
|
Nausea |
2.3 |
|
Gastroenteritis |
2.3 |
|
Rhinitis |
2.3 |
|
Cough |
2.3 |
Increased Blood Pressure in Pediatric Patients with NDO Treated with MYRBETRIQ/MYRBETRIQ Granules:
Mean systolic and diastolic blood pressures increased in Study 9 by 4.3 mm Hg and 1.7 mm Hg, respectively, in patients less than 12 years of age on MYRBETRIQ/MYRBETRIQ Granules at a dose equivalent of MYRBETRIQ 50 mg daily dose in adults. The blood pressure increases were larger in patients less than 8 years of age with mean systolic and diastolic blood pressure increases of 5.9 mm Hg and 2.3 mm Hg, respectively. Ten (24%) patients less than 12 years of age who were normotensive at baseline had at least one blood pressure measured at or above the 95th percentile for age, sex, and stature during Study 9. Stage 1 hypertension, defined as repeated blood pressure measurements at or above the 95th percentile for age, sex, and stature, was sustained in six of these 10 patients (60%) at the end of the study.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of MYRBETRIQ/MYRBETRIQ Granules. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following events have been reported in association with mirabegron use in worldwide postmarketing experience:
Cardiac disorders: atrial fibrillation
Gastrointestinal disorders: nausea, constipation, diarrhea
Nervous system disorders: dizziness, headache
There have been postmarketing reports of confusion, hallucinations, insomnia, and anxiety in patients taking mirabegron. The majority of these patients had pre-existing medical conditions or concomitant medications that may cause confusion, hallucinations, insomnia, and anxiety. A causal relationship between mirabegron and these disorders has not been established.
Skin and subcutaneous tissue disorders: angioedema of the face, lips, tongue, and larynx, with or without respiratory symptoms [see Warnings and Precautions (5.3)]; pruritus
Renal and urinary disorders: urinary retention [see Warnings and Precautions (5.2)]
Related/similar drugs
7. Drug Interactions
Drug interaction studies were conducted in adult patients to investigate the effect of coadministered drugs on the pharmacokinetics of mirabegron and the effect of mirabegron on the pharmacokinetics of coadministered drugs (e.g., ketoconazole, rifampin, solifenacin succinate, tamsulosin, and oral contraceptives) [see Clinical Pharmacology (12.3)]. No dose adjustment is recommended when these drugs are coadministered with mirabegron.
The following are drug interactions for which monitoring is recommended:
7.1 Drugs Metabolized by CYP2D6
Since mirabegron is a moderate CYP2D6 inhibitor, the systemic exposure of drugs metabolized by CYP2D6 enzyme is increased when coadministered with mirabegron. Therefore, appropriate monitoring and dose adjustment may be necessary when MYRBETRIQ/MYRBETRIQ Granules is coadministered with these drugs, especially with narrow therapeutic index CYP2D6 substrates [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)].
7.2 Digoxin
When given in combination, 100 mg mirabegron increased mean digoxin Cmax from 1.01 to 1.3 ng/mL (29%) and AUC from 16.7 to 19.3 ng.h/mL (27%). Concomitant administration of 0.25 mg digoxin with a combination of 5 mg solifenacin and 50 mg mirabegron increased digoxin AUCtau and Cmax by approximately 10% and 14%, respectively. For patients who are initiating a combination of mirabegron and digoxin, the lowest dose for digoxin should initially be considered. Serum digoxin concentrations should be monitored and used for titration of the digoxin dose to obtain the desired clinical effect [see Clinical Pharmacology (12.3)].
7.3 Warfarin
The mean Cmax of S- and R-warfarin was increased by approximately 4% and AUC by approximately 9% when administered as a single dose of 25 mg after multiple doses of 100 mg mirabegron. Following a single dose administration of 25 mg warfarin, mirabegron had no effect on the warfarin pharmacodynamic endpoints such as International Normalized Ratio (INR) and prothrombin time. However, the effect of mirabegron on multiple doses of warfarin and on warfarin pharmacodynamic end points such as INR and prothrombin time has not been fully investigated [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no studies with the use of MYRBETRIQ/MYRBETRIQ Granules in pregnant women or adolescents to inform a drug-associated risk of major birth defects, miscarriages, or adverse maternal or fetal outcomes. Mirabegron administration to pregnant animals during organogenesis resulted in reversible skeletal variations (in rats) at 22-fold (via AUC) the maximum recommended human dose (MRHD) of 50 mg/day and decreased fetal body weights (in rabbits) at 14-fold the MRHD. At maternally-toxic exposures in rats (96-fold), decreased fetal weight and increased fetal mortality were observed and, in rabbits (36-fold), cardiac findings (fetal cardiomegaly and fetal dilated aortae) were observed [see Data].
The estimated background risks of major birth defects and miscarriage for the indicated populations are unknown. In the U.S. general population, the estimated background risk of major birth defects or miscarriage in clinically recognized pregnancies are 2-4% and 15-20%, respectively.
Data
Animal Data
No embryo-fetal lethality or morphological fetal developmental abnormalities were produced in pregnant rats following daily oral administration of mirabegron during the period of organogenesis (Days 7 to 17 of gestation) at 0, 10, 30, 100, or 300 mg/kg, doses which were associated with systemic exposures (AUC) 0, 1, 6, 22, and 96-fold the MRHD. Skeletal variations (wavy ribs, delayed ossification) were observed in fetuses at doses 22-fold the systemic exposure at the MRHD and were reversible during development. Exposures 96-fold the MRHD were maternally-toxic (mortality, decreased body weight gain) and associated with fetal growth reduction.
Pregnant rabbits were treated with daily oral doses of mirabegron at 0, 3, 10, or 30 mg/kg/day during the period of organogenesis (Days 6 to 20 of gestation), which resulted in plasma exposures that were 0, 1, 14, or 36-fold the MRHD based on AUC. At 10 mg/kg/day (14-fold the MRHD) and higher, fetal body weights were reduced. At 30 mg/kg/day, maternal toxicity (increased heart rate, mortality, reduced body weight gain, reduced food consumption) occurred, and fetal deaths, fetal cardiomegaly and fetal dilated aortae were observed at systemic exposure levels (AUC) 36-fold the MRHD.
In a pre- and postnatal developmental study, rats were treated with daily oral doses of mirabegron at 0, 10, 30, or 100 mg/kg/day (0, 1, 6, or 22-fold the MRHD) from day 7 of gestation until day 20 after birth. Decreased maternal body weight was observed along with decreased pup survival in the first few days after birth (92.7% survival) compared to the control group (98.8% survival), at 100 mg/kg/day (22-fold the MRHD). Pup body weight gain was reduced until postnatal day 7 but not further affected throughout the remainder of the lactation period. In utero and lactational exposure did not affect developmental milestones, behavior, or fertility of offspring. No effects were observed at 30 mg/kg/day.
8.2 Lactation
Risk Summary
There are no data on the presence of mirabegron in human milk, the effects on the breastfed child, or the effects on milk production. Mirabegron-related material was present in rat milk and in the stomach of nursing pups following administrations of a single 10 mg/kg oral dose of 14C-labeled mirabegron to lactating rats. When a drug is present in animal milk, it is likely that the drug will be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for MYRBETRIQ/MYRBETRIQ Granules and any potential adverse effects on the breastfed child from mirabegron or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness have been established only for the following pediatric indications:
- •
- MYRBETRIQ: Treatment of neurogenic detrusor overactivity (NDO) in pediatric patients 3 years of age and older and weighing 35 kg or more.
- •
- MYRBETRIQ Granules: Treatment of neurogenic detrusor overactivity (NDO) in pediatric patients 3 years of age and older.
The safety and effectiveness of MYRBETRIQ/MYRBETRIQ Granules in pediatric patients aged 3 years and older have been established for the treatment of neurogenic detrusor overactivity (NDO) and the information on this use is discussed throughout the labeling. Use of MYRBETRIQ/MYRBETRIQ Granules for this indication is supported by evidence from a 52-week, open-label, baseline-controlled, multicenter, dose titration trial in pediatric patients 3 years of age and older with NDO (Study 9) [see Adverse Reactions (6.1), Clinical Studies (14.3)]. Results showed an improvement from baseline in maximum cystometric (bladder) capacity (MCC) with MYRBETRIQ/MYRBETRIQ Granules use [see Clinical Studies (14.3)]. The most commonly reported adverse reactions in Study 9 (≥ 3%) were UTI, nasopharyngitis, constipation, and headache. Increased mean systolic and diastolic blood pressures with use of MYRBETRIQ/MYRBETRIQ Granules occurred in patients less than 12 years of age with larger increases in patients younger than 8 years of age [see Adverse Reactions (6.1)].
Take MYRBETRIQ/MYRBETRIQ Granules with food to reduce potential exposure-related risks, such as increased heart rate, as predicted by modeling of vital signs data in Study 9 [see Clinical Pharmacology (12.3)].
8.5 Geriatric Use
Of 5648 patients who received MYRBETRIQ monotherapy in the phase 2 and 3 studies for OAB, 2029 (35.9%) were 65 years of age or older, and 557 (9.9%) were 75 years of age or older. No overall differences in safety or effectiveness were observed between patients younger than 65 years of age and those 65 years of age or older in these studies.
8.6 Renal Impairment
MYRBETRIQ/MYRBETRIQ Granules have not been studied in patients with End-Stage Renal Disease (eGFR < 15 mL/min/1.73 m2) or patients requiring hemodialysis and, therefore, is not recommended for use in these patient populations. No dose adjustment is necessary in patients with mild or moderate renal impairment (eGFR 30 to 89 mL/min/1.73 m2).
In adult patients with severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2), the daily dose of MYRBETRIQ should not exceed 25 mg. In pediatric patients with severe renal impairment, the daily dose of MYRBETRIQ/MYRBETRIQ Granules should not exceed the recommended starting dose [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
MYRBETRIQ/MYRBETRIQ Granules have not been studied in patients with severe hepatic impairment (Child-Pugh Class C) and, therefore, is not recommended for use in this patient population. No dose adjustment is necessary in patients with mild hepatic impairment (Child-Pugh Class A).
In adult patients with moderate hepatic impairment (Child-Pugh Class B), the daily dose of MYRBETRIQ should not exceed 25 mg. In pediatric patients with moderate hepatic impairment (Child-Pugh Class B), the daily dose of MYRBETRIQ/MYRBETRIQ Granules should not exceed the recommended starting dose [see Clinical Pharmacology (12.3)].
10. Overdosage
Mirabegron has been administered to healthy volunteers at single doses up to 400 mg. At this dose, adverse events reported included palpitations (1 of 6 subjects) and increased pulse rate exceeding 100 beats per minute (bpm) (3 of 6 subjects). Multiple doses of mirabegron up to 300 mg daily for 10 days showed increases in pulse rate and systolic blood pressure when administered to healthy volunteers. Treatment for overdosage should be symptomatic and supportive. In the event of overdosage, pulse rate, blood pressure and ECG monitoring is recommended.
11. Myrbetriq Description
MYRBETRIQ (mirabegron extended-release tablets) for oral use and MYRBETRIQ Granules (mirabegron for extended-release oral suspension) are beta-3 adrenergic agonists.
The chemical name of mirabegron is 2-(2-aminothiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2-phenylethyl]amino}ethyl)phenyl]acetamide having an empirical formula of C21H24N4O2S and a molecular weight of 396.51. The structural formula of mirabegron is:
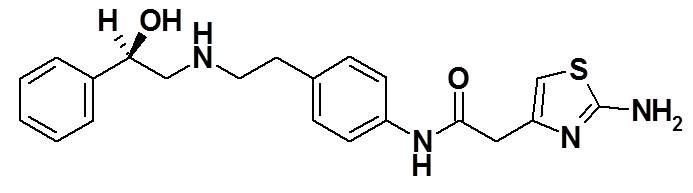
Mirabegron is a white powder. It is practically insoluble in water (0.082 mg/mL). It is soluble in methanol and dimethyl sulfoxide.
Each MYRBETRIQ (mirabegron extended-release tablets) for oral use contains either 25 mg or 50 mg of mirabegron and the following inactive ingredients: butylated hydroxytoluene, hydroxypropyl cellulose, hypromellose, magnesium stearate, polyethylene glycol, polyethylene oxide, red ferric oxide (25 mg tablet only), and yellow ferric oxide.
Each bottle of MYRBETRIQ Granules (mirabegron for extended-release oral suspension) contains approximately 8.3 g of granules, which contain 830 mg of mirabegron, and the following inactive ingredients: acesulfame potassium, diluted hydrochloric acid, ethylparaben, hypromellose, magnesium stearate, mannitol, methylparaben, silicon dioxide, simethicone, sodium polystyrene sulfonate, and xanthan gum. After reconstituted with 100 mL water, the suspension contains 8 mg/mL of mirabegron.
12. Myrbetriq - Clinical Pharmacology
12.1 Myrbetriq Mechanism of Action
Mirabegron is an agonist of the human beta-3 adrenergic receptor (AR) as demonstrated by in vitro laboratory experiments using the cloned human beta-3 AR. Mirabegron relaxes the detrusor smooth muscle during the storage phase of the urinary bladder fill-void cycle by activation of beta-3 AR which increases bladder capacity. Although mirabegron showed very low intrinsic activity for cloned human beta-1 AR and beta-2 AR, results in humans indicate that beta-1 AR stimulation occurred at a mirabegron dose of 200 mg.
12.2 Pharmacodynamics
Urodynamics
The effects of mirabegron on maximum urinary flow rate and detrusor pressure at maximum flow rate were assessed in a urodynamic study consisting of 200 male patients with lower urinary tract symptoms (LUTS) and BOO. Administration of mirabegron once daily for 12 weeks did not adversely affect the mean maximum flow rate or mean detrusor pressure at maximum flow rate in this study. Nonetheless, mirabegron should be administered with caution to patients with clinically significant BOO [see Warnings and Precautions (5.2)].
Cardiac Electrophysiology
The effect of multiple doses of mirabegron 50 mg, 100 mg, and 200 mg (four times the maximum recommended dose) once daily on QTc interval was evaluated in a randomized, placebo- and active-controlled (moxifloxacin 400 mg), four-treatment arm, parallel crossover study in 352 healthy subjects. In a study with demonstrated ability to detect small effects, the upper bound of the one-sided 95% confidence interval for the largest placebo-adjusted, baseline-corrected QTc based on individual correction method (QTcI) was below 10 msec. For the 50 mg mirabegron dose group (the maximum approved dosage), the mean difference from placebo on QTcI interval at 4 to 5 hours post-dose was 3.7 msec (upper bound of the 95% CI 5.1 msec).
For the mirabegron 100 mg and 200 mg dose groups (dosages greater than the maximum approved dose and resulting in substantial multiples of the anticipated maximum blood levels at 50 mg), the mean differences from placebo in QTcI interval at 4 to 5 hours post dose were 6.1 msec (upper bound of the 95% CI 7.6 msec) and 8.1 msec (upper bound of the 95% CI 9.8 msec), respectively. At the mirabegron 200 mg dose, in females, the mean effect was 10.4 msec (upper bound of the 95% CI 13.4 msec).
In this thorough QT study, mirabegron increased heart rate on ECG in a dose-dependent manner. Maximum mean increases from baseline in heart rate for the 50 mg, 100 mg, and 200 mg dose groups compared to placebo were 6.7 bpm, 11 bpm, and 17 bpm, respectively. In the clinical efficacy and safety studies, the change from baseline in mean pulse rate for mirabegron 50 mg was approximately 1 bpm. In this thorough QT study, mirabegron also increased blood pressure in a dose-dependent manner (see Effects on Blood Pressure).
Effects on Blood Pressure
In a study of 352 healthy subjects assessing the effect of multiple daily doses of 50 mg, 100 mg, and 200 mg (four times the maximum recommended dose) of mirabegron for 10 days on the QTc interval, the maximum mean increase in supine systolic blood pressure (SBP)/diastolic blood pressure (DBP) at the maximum recommended dose of 50 mg was approximately 4.0/1.6 mm Hg greater than placebo [see Warnings and Precautions (5.1)]. The 24-hour average increases in SBP compared to placebo were 3.0, 5.5, and 9.7 mm Hg at mirabegron doses of 50 mg, 100 mg, and 200 mg, respectively. Increases in DBP were also dose-dependent, but were smaller than SBP.
In another study in 96 healthy subjects to assess the impact of age on pharmacokinetics of multiple daily doses of 50 mg, 100 mg, 200 mg, and 300 mg (six times the maximum recommended dose) of mirabegron for 10 days, SBP also increased in a dose-dependent manner. The mean maximum increases in SBP were approximately 2.5, 4.5, 5.5, and 6.5 mm Hg for mirabegron exposures associated with doses of 50 mg, 100 mg, 200 mg, and 300 mg, respectively.
In three, 12-week, double-blind, placebo-controlled, safety and efficacy studies (Studies 1, 2, and 3) in patients with OAB receiving mirabegron 25 mg, 50 mg, or 100 mg (two times the maximum recommended dose) once daily, mean increases in SBP/DBP compared to placebo of approximately 0.5 – 1 mm Hg were observed. Morning SBP increased by at least 15 mm Hg from baseline in 5.3%, 5.1%, and 6.7% of placebo, mirabegron 25 mg and mirabegron 50 mg patients, respectively. Morning DBP increased by at least 10 mm Hg in 4.6%, 4.1%, and 6.6% of placebo, mirabegron 25 mg, and mirabegron 50 mg patients, respectively. Both SBP and DBP increases were reversible upon discontinuation of treatment.
In a 12-week, double-blind, placebo-controlled, safety and efficacy study (Study 6) in patients with OAB receiving mirabegron 25 mg or 50 mg once daily coadministered with solifenacin succinate 5 mg, no consistent differences in 24‑hour mean SBP/DBP were observed compared to placebo, mirabegron or solifenacin succinate monotherapy as assessed with 24-hour Ambulatory Blood Pressure Monitoring (ABPM). Similar frequencies of categorical changes were observed for combination treatment versus placebo in 24‑hour mean SBP/DBP.
Effect on Intraocular Pressure (IOP)
Mirabegron 100 mg once daily did not increase IOP in healthy subjects after 56 days of treatment. In a phase 1 study assessing the effect of mirabegron on IOP using Goldmann applanation tonometry in 310 healthy subjects, a dose of mirabegron 100 mg was non-inferior to placebo for the primary endpoint of the treatment difference in mean change from baseline to day 56 in subject average IOP; the upper bound of the two-sided 95% CI of the treatment difference between mirabegron 100 mg and placebo was 0.3 mm Hg.
12.3 Pharmacokinetics
Absorption
MYRBETRIQ Monotherapy for Adult OAB
After oral administration of mirabegron in healthy volunteers, mirabegron was absorbed to reach maximum plasma concentrations (Cmax) at approximately 3.5 hours. The absolute bioavailability increased from 29% at a dose of 25 mg to 35% at a dose of 50 mg. Mean Cmax and AUC increased more than dose proportionally. This relationship was more apparent at doses above 50 mg. In the overall population of males and females, a 2-fold increase in dose from 50 mg to 100 mg mirabegron increased Cmax and AUCtau by approximately 2.9- and 2.6-fold, respectively, whereas a 4-fold increase in dose from 50 to 200 mg mirabegron increased Cmax and AUCtau by approximately 8.4- and 6.5-fold. Steady-state concentrations were achieved within 7 days of once daily dosing with mirabegron. After once daily administration, plasma exposure of mirabegron at steady-state was approximately double that seen after a single dose.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
The median Tmax of mirabegron following oral administration of a single dose of MYRBETRIQ or mirabegron for extended-release oral suspension in pediatric patients under fed state was 4-5 hours. Population pharmacokinetic analysis predicted that the median Tmax of MYRBETRIQ or mirabegron for extended-release oral suspension at steady-state was 3-4 hours.
-
Effect of Food
MYRBETRIQ Monotherapy for Adult OAB
There were no clinically significant differences in mirabegron pharmacokinetics when administered with or without food in adult patients.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
In the fasted state, steady-state mirabegron AUC increased by 120% relative to the fed state in pediatric patients
receiving MYRBETRIQ. Fasted Cmax and AUC increased by 170% and 80%, respectively, compared to the fed state
following administration of MYRBETRIQ Granules in healthy volunteers.
Distribution
MYRBETRIQ Monotherapy for Adult OAB
Mirabegron is extensively distributed in the body. The volume of distribution at steady-state (Vss) is approximately 1670 L following intravenous administration. Mirabegron is bound (approximately 71%) to human plasma proteins, and shows moderate affinity for albumin and alpha-1 acid glycoprotein. Mirabegron distributes to erythrocytes. Based on an in vitro study, erythrocyte concentrations of 14C-mirabegron were about 2-fold higher than in plasma.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
Mirabegron volume of distribution was relatively large in pediatric patients (the range of mean Vz/F under fed state in pediatric patients across studies: 4895-13726 L) and increased with increasing body weight.
Elimination
MYRBETRIQ Monotherapy for Adult OAB
The terminal elimination half-life (t1/2) of mirabegron is approximately 50 hours in patients.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
The mean terminal elimination half-life (t1/2) of mirabegron is approximately 26 to 31 hours in pediatric patients.
-
Metabolism
Mirabegron is metabolized via multiple pathways involving dealkylation, oxidation, (direct) glucuronidation, and amide hydrolysis. Mirabegron is the major circulating component following a single dose of 14C-mirabegron. Two major metabolites were observed in human plasma and are phase 2 glucuronides representing 16% and 11% of total exposure, respectively. These metabolites are not pharmacologically active toward beta-3 adrenergic receptor. Although, in vitro studies suggest a role for CYP2D6 and CYP3A4 in the oxidative metabolism of mirabegron, in vivo results indicate that these isozymes play a limited role in the overall elimination. In healthy subjects who were genotypically poor metabolizers of CYP2D6, mean Cmax and AUCtau were approximately 16% and 17% higher than in extensive metabolizers of CYP2D6, respectively. In vitro and ex vivo studies have shown the involvement of butylcholinesterase, uridine diphospho-glucuronosyltransferases (UGT), and possibly alcohol dehydrogenase in the metabolism of mirabegron, in addition to CYP3A4 and CYP2D6.
Excretion
MYRBETRIQ Monotherapy for Adult OAB
Total body clearance (CLtot) from plasma is approximately 57 L/h following intravenous administration. Renal clearance (CLR) is approximately 13 L/h, which corresponds to nearly 25% of CLtot. Renal elimination of mirabegron is primarily through active tubular secretion along with glomerular filtration. The urinary elimination of unchanged mirabegron is dose-dependent and ranges from approximately 6.0% after a daily dose of 25 mg to 12.2% after a daily dose of 100 mg. Following the administration of 160 mg 14C-mirabegron solution to healthy volunteers, approximately 55% of the radioactivity dose was recovered in the urine and 34% in the feces. Approximately 25% of unchanged mirabegron was recovered in urine and 0% in feces.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
Population pharmacokinetic model predicted that mirabegron clearance in pediatric patients increased with body weight.
Specific Populations
-
Geriatric Patients
The Cmax and AUC of mirabegron following multiple oral doses in elderly volunteers (≥ 65 years) were similar to those in younger volunteers (18 to 45 years) [see Use in Specific Populations (8.5)].
Pediatric Patients
In patients 3 to less than 18 years of age, age was not predicted to affect mirabegron pharmacokinetic parameters after accounting for differences in body weight [see Use in Specific Populations (8.4)].
Gender
MYRBETRIQ Monotherapy for Adult OAB
The Cmax and AUC of mirabegron were approximately 40% to 50% higher in females than in males. When corrected for differences in body weight, the mirabegron systemic exposure was 20%-30% higher in females compared to males.
MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
Gender has no meaningful impact on mirabegron pharmacokinetics in the pediatric population from 3 to less than 18 years of age.
Race
The pharmacokinetics of mirabegron were comparable between Caucasians and African-American Blacks. Cross studies comparison showed that the exposure in Japanese subjects were higher than that in North American subjects. However, when the Cmax and AUC were normalized for dose and body weight, the difference was smaller.
Patients with Renal Impairment
Following single-dose administration of 100 mg mirabegron in adult volunteers with mild renal impairment (eGFR 60 to 89 mL/min/1.73 m2 as estimated by MDRD), mean mirabegron Cmax and AUC were increased by 6% and 31% relative to adult volunteers with normal renal function. In adult volunteers with moderate renal impairment (eGFR 30 to 59 mL/min/1.73 m2), Cmax and AUC were increased by 23% and 66%, respectively. In adult volunteers with severe renal impairment (eGFR 15 to 29 mL/min/1.73 m2), mean Cmax and AUC values were 92% and 118% higher compared to healthy subjects with normal renal function. Mirabegron has not been studied in adult patients with End-Stage Renal Disease (ESRD) (eGFR less than 15 mL/min/1.73 m2) or adult patients requiring dialysis.
Patients with Hepatic Impairment
Following single-dose administration of 100 mg mirabegron in adult volunteers with mild hepatic impairment (Child-Pugh Class A), mean mirabegron Cmax and AUC were increased by 9% and 19%, relative to adult volunteers with normal hepatic function. In adult volunteers with moderate hepatic impairment (Child-Pugh Class B), mean Cmax and AUC values were 175% and 65% higher. Mirabegron has not been studied in adult patients with severe hepatic impairment (Child-Pugh Class C).
Drug Interaction Studies
-
In Vitro Studies
Effect of Other Drugs on Mirabegron
Mirabegron is transported and metabolized through multiple pathways. Mirabegron is a substrate for CYP3A4, CYP2D6, butyrylcholinesterase, UGT, the efflux transporter P-glycoprotein (P-gp), and the influx organic cation transporters (OCT) OCT1, OCT2, and OCT3. Sulfonylurea hypoglycemic agents glibenclamide (a CYP3A4 substrate), gliclazide (a CYP2C9 and CYP3A4 substrate), and tolbutamide (a CYP2C9 substrate) did not affect the in vitro metabolism of mirabegron.
Effect of Mirabegron on Other Drugs
Studies of mirabegron using human liver microsomes and recombinant human CYP enzymes showed that mirabegron is a moderate and time-dependent inhibitor of CYP2D6 and a weak inhibitor of CYP3A. Mirabegron is unlikely to inhibit the metabolism of coadministered drugs metabolized by the following cytochrome P450 enzymes: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2E1 because mirabegron did not inhibit the activity of these enzymes at clinically relevant concentrations. Mirabegron did not induce CYP1A2 or CYP3A. Mirabegron inhibited P-gp-mediated drug transport at high concentrations. Mirabegron is predicted not to cause clinically relevant inhibition of OCT-mediated drug transport. Mirabegron did not affect the metabolism of glibenclamide or tolbutamide.
Effect of Alcohol on Mirabegron
The addition of alcohol (5, 10, 20, and 40%) increases the dissolution rate of mirabegron from MYRBETRIQ Granules at pH 6.8. The clinical impact on the systemic exposure of mirabegron has not been evaluated. The addition of alcohol does not increase the dissolution rate of MYRBETRIQ Granules at pH 1.0 or MYRBETRIQ (extended-release tablets) regardless of pH.
In Vivo Studies
MYRBETRIQ Monotherapy for Adult OAB
The effect of coadministered drugs on the pharmacokinetics of mirabegron and the effect of mirabegron on the pharmacokinetics of coadministered drugs was studied after single and multiple doses of mirabegron. Most drug-drug interactions (DDI) were studied using mirabegron 100 mg extended-release tablets. However, interaction studies of mirabegron with metoprolol and with metformin were studied using mirabegron 160 mg immediate-release (IR) tablets.
The effect of ketoconazole, rifampicin, solifenacin succinate, tamsulosin, and metformin on systemic mirabegron exposure is shown in Figure 1.
The effect of mirabegron on metoprolol, desipramine, combined oral contraceptive-COC (ethinyl estradiol-EE, levonorgestrel-LNG), solifenacin succinate, digoxin, warfarin, tamsulosin, and metformin is shown in Figure 2.
In these studies, the largest increase in mirabegron systemic exposure was seen in the ketoconazole DDI study. As a potent CYP3A4 inhibitor, ketoconazole increased mirabegron Cmax by 45% and mirabegron AUC by 80% after multiple dose administration of 400 mg of ketoconazole for 9 days prior to the administration of a single dose of 100 mg mirabegron in 23 male and female healthy subjects.
As a moderate CYP2D6 inhibitor, mirabegron increased the systemic exposure to metoprolol and desipramine:
• Mirabegron increased the Cmax of metoprolol by 90% and metoprolol AUC by 229% after multiple doses of 160 mg mirabegron IR tablets once daily for 5 days and a single dose of 100 mg metoprolol tablet in 12 healthy male subjects administered before and concomitantly with mirabegron.
• Mirabegron increased the Cmax of desipramine by 79% and desipramine AUC by 241% after multiple dose administration of 100 mg mirabegron once daily for 18 days and a single dose of 50 mg desipramine before and concomitantly with mirabegron in 28 male and female healthy subjects.
The effect on the pharmacokinetics of coadministered digoxin and tamsulosin was studied after multiple doses of a combination of mirabegron and solifenacin succinate. Concomitant administration of 0.25 mg digoxin with a combination of 5 mg solifenacin succinate and 50 mg mirabegron increased digoxin AUCtau and Cmax by approximately 10% and 14%, respectively. Concomitant administration of 0.4 mg tamsulosin with a combination of 5 mg solifenacin succinate and 50 mg mirabegron increased tamsulosin AUCtau and Cmax by 47.5% and 54.3%, respectively. The observed changes in the pharmacokinetics of tamsulosin are in line with cytochrome P450 inhibition as shown by coadministration with mirabegron alone.
Figures 1 and 2 show the magnitude of these interactions on the pharmacokinetic parameters and the recommendations for dose adjustment, if any:

- (1) Although no dose adjustment is recommended with solifenacin succinate or tamsulosin based on the lack of pharmacokinetic interaction, MYRBETRIQ should be administered with caution to patients taking muscarinic antagonist medications for the treatment of OAB and in patients with clinically significant BOO because of the risk of urinary retention [see Warnings and Precautions (5.2)].
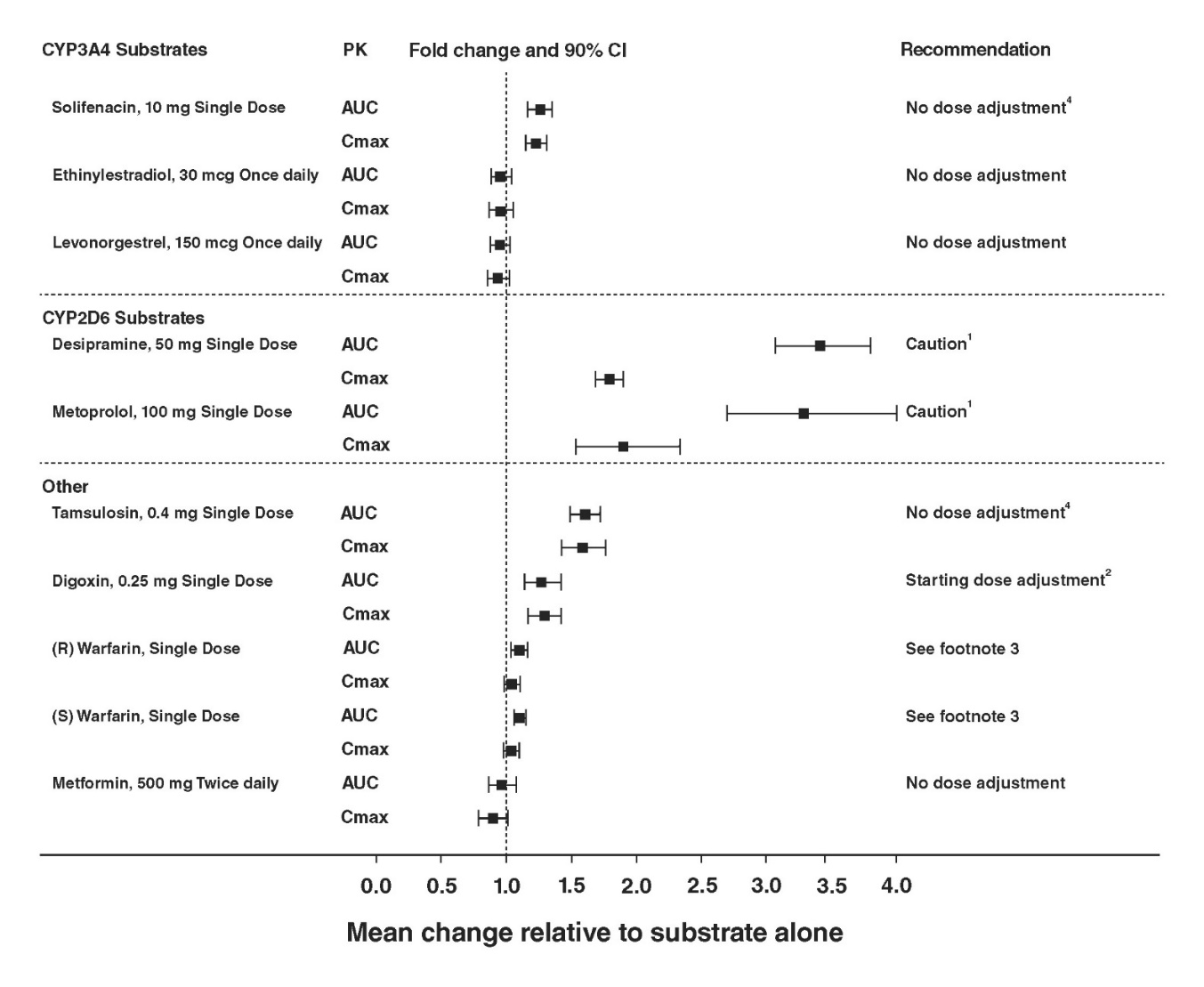
- (1) Since mirabegron is a moderate CYP2D6 inhibitor, the systemic exposure to CYP2D6 substrates such as metoprolol and desipramine is increased when coadministered with mirabegron. Therefore, appropriate monitoring and dose adjustment may be necessary, especially with narrow therapeutic index CYP2D6 substrates, such as thioridazine, flecainide, and propafenone [see Warnings and Precautions (5.4) and Drug Interactions (7.1)].
(2) For patients who are initiating a combination of mirabegron and digoxin, the lowest dose for digoxin should initially be prescribed. Serum digoxin concentrations should be monitored and used for titration of the digoxin dose to obtain the desired clinical effect [see Drug Interactions (7.2)]. The same approach for the dose of digoxin should be followed when digoxin is coadministered with mirabegron and solifenacin succinate.
(3) Warfarin was administered as a single 25 mg dose of the racemate (a mixture of R-warfarin and S-warfarin). Based on this single-dose study, mirabegron had no effect on the warfarin pharmacodynamic endpoints such as INR and prothrombin time. However, the effect of mirabegron on multiple doses of warfarin and on warfarin pharmacodynamic end points such as INR and prothrombin time has not been fully investigated [see Drug Interactions (7.3)].
(4) Although no dose adjustment is recommended with solifenacin succinate or tamsulosin based on the lack of pharmacokinetic interaction, MYRBETRIQ should be administered with caution to patients taking muscarinic antagonist medications for the treatment of OAB and in BOO because of the risk of urinary retention [see Warnings and Precautions (5.2)].
Based on the lack of relevant pharmacokinetic interaction, no dose adjustment for tamsulosin is recommended when coadministered with mirabegron and solifenacin succinate.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Long-term carcinogenicity studies were conducted in rats and mice dosed orally with mirabegron for two years. Male rats were dosed at 0, 12.5, 25, or 50 mg/kg/day and female rats and both sexes of mice were dosed at 0, 25, 50, or 100 mg/kg/day. Mirabegron showed no carcinogenic potential at systemic exposures (AUC) 38 to 45-fold higher than the MRHD in rats and 21 to 38-fold higher than the MRHD in mice than the human systemic exposure at the 50 mg dose.
Mutagenesis
Mirabegron was not mutagenic in the Ames bacterial reverse mutation assay, did not induce chromosomal aberrations in human peripheral blood lymphocytes at concentrations that were not cytotoxic, and was not clastogenic in the rat micronucleus assay.
Impairment of Fertility
Fertility studies in rats showed that mirabegron had no effect on either male or female fertility at non-lethal doses up to 100 mg/kg/day. Systemic exposures (AUC) at 100 mg/kg in female rats was estimated to be 22-fold the MRHD in women and 93-fold the MRHD in men.
14. Clinical Studies
14.1 MYRBETRIQ Monotherapy for Adult OAB
MYRBETRIQ was evaluated in three, 12-week, double-blind, randomized, placebo-controlled, parallel group, multicenter clinical trials in patients with overactive bladder with symptoms of urge urinary incontinence, urgency, and urinary frequency (Studies 1, 2, and 3). Entry criteria required that patients had symptoms of overactive bladder for at least 3 months duration, at least 8 micturitions per day, and at least 3 episodes of urgency with or without incontinence over a 3-day period. The majority of patients were Caucasian (94%) and female (72%) with a mean age of 59 years (range 18 – 95 years). The population included both naïve patients who had not received prior muscarinic antagonist pharmacotherapy for overactive bladder (48%) and those who had received prior muscarinic antagonist pharmacotherapy for OAB (52%).
In Study 1 (NCT00689104), patients were randomized to placebo, MYRBETRIQ 50 mg, MYRBETRIQ 100 mg, or an active control once daily. In Study 2 (NCT00662909), patients were randomized to placebo, MYRBETRIQ 50 mg or MYRBETRIQ 100 mg once daily. In Study 3 (NCT00912964), patients were randomized to placebo, MYRBETRIQ 25 mg or MYRBETRIQ 50 mg once daily.
The co-primary efficacy endpoints in all 3 trials were (1) change from baseline to end of treatment (Week 12) in mean number of incontinence episodes per 24 hours and (2) change from baseline to end of treatment (Week 12) in mean number of micturitions per 24 hours, based on a 3-day micturition diary. An important secondary endpoint was the change from baseline to end of treatment (Week 12) in mean volume voided per micturition.
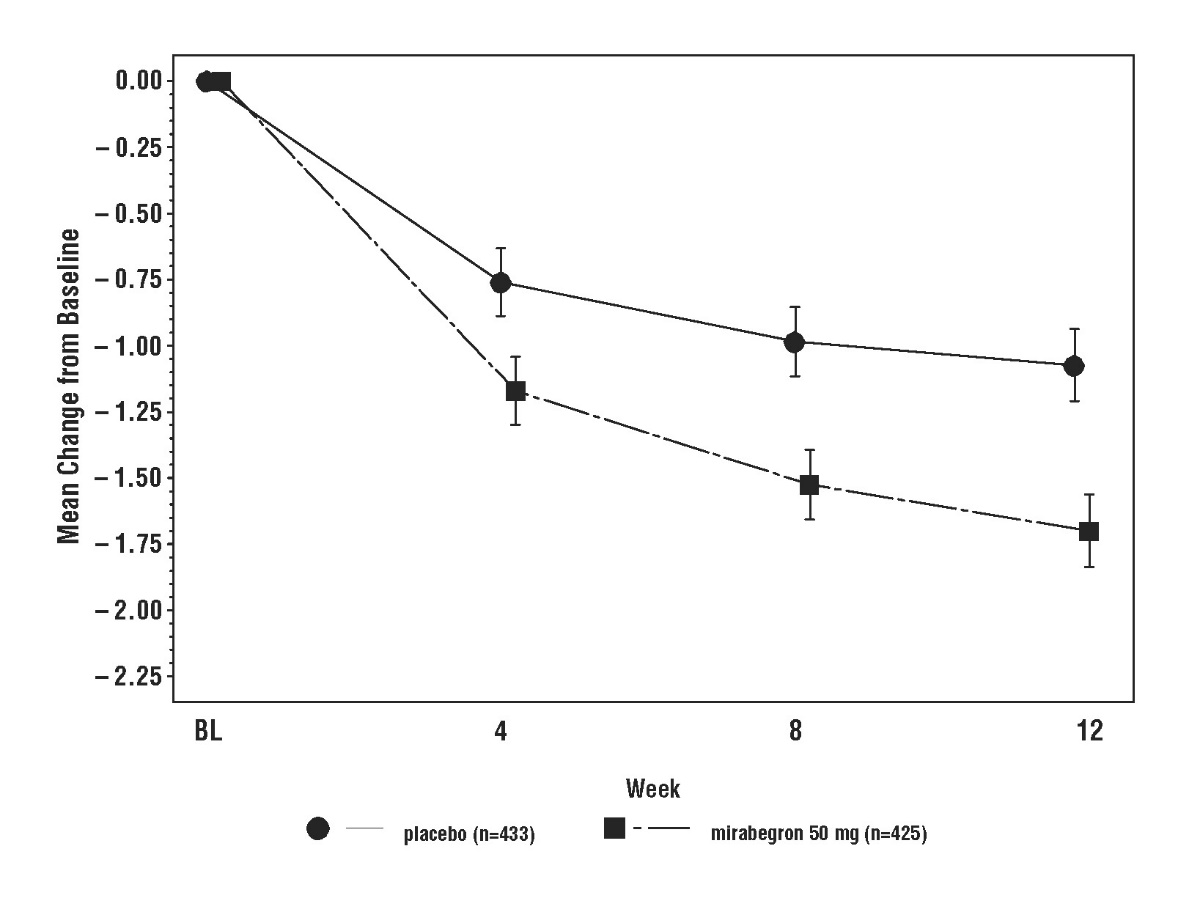
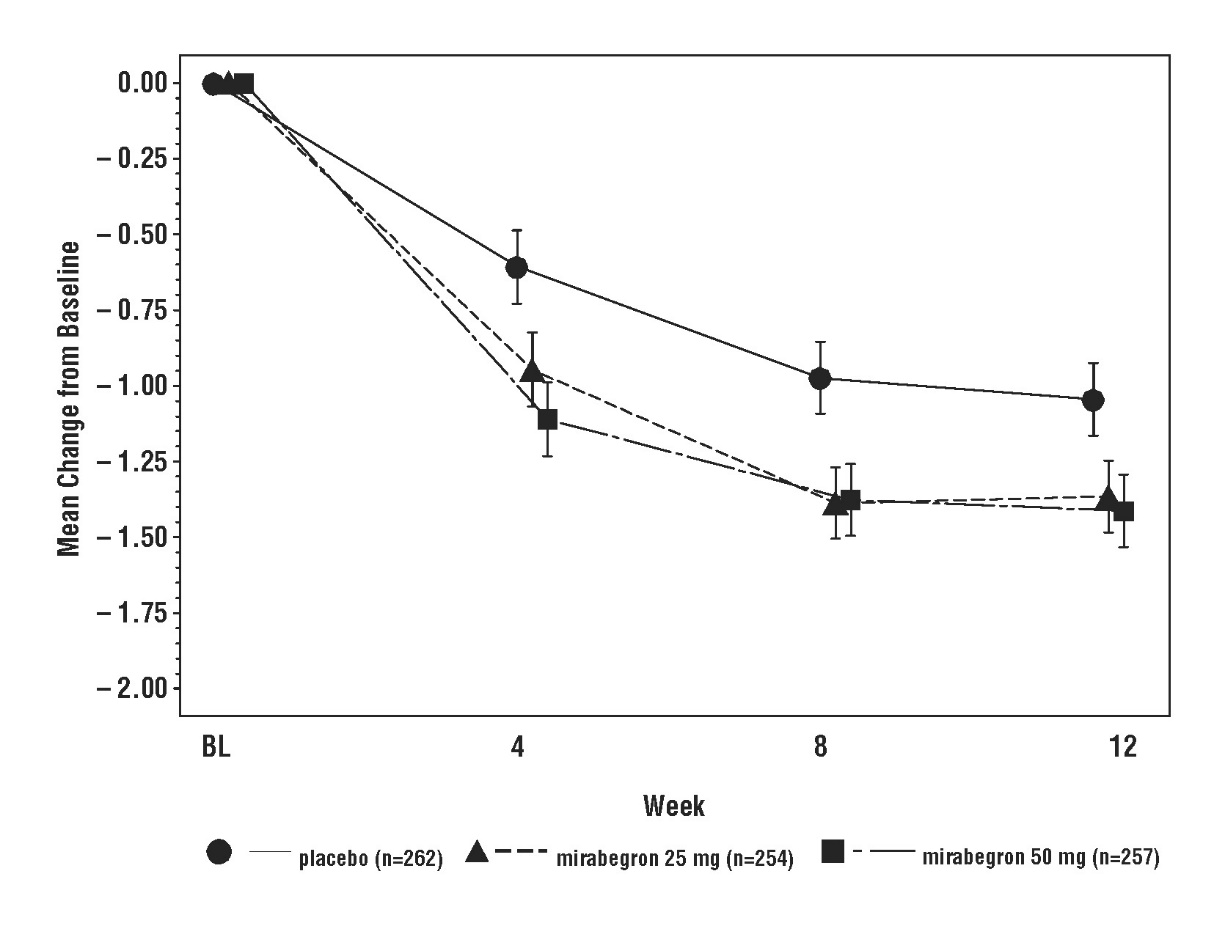
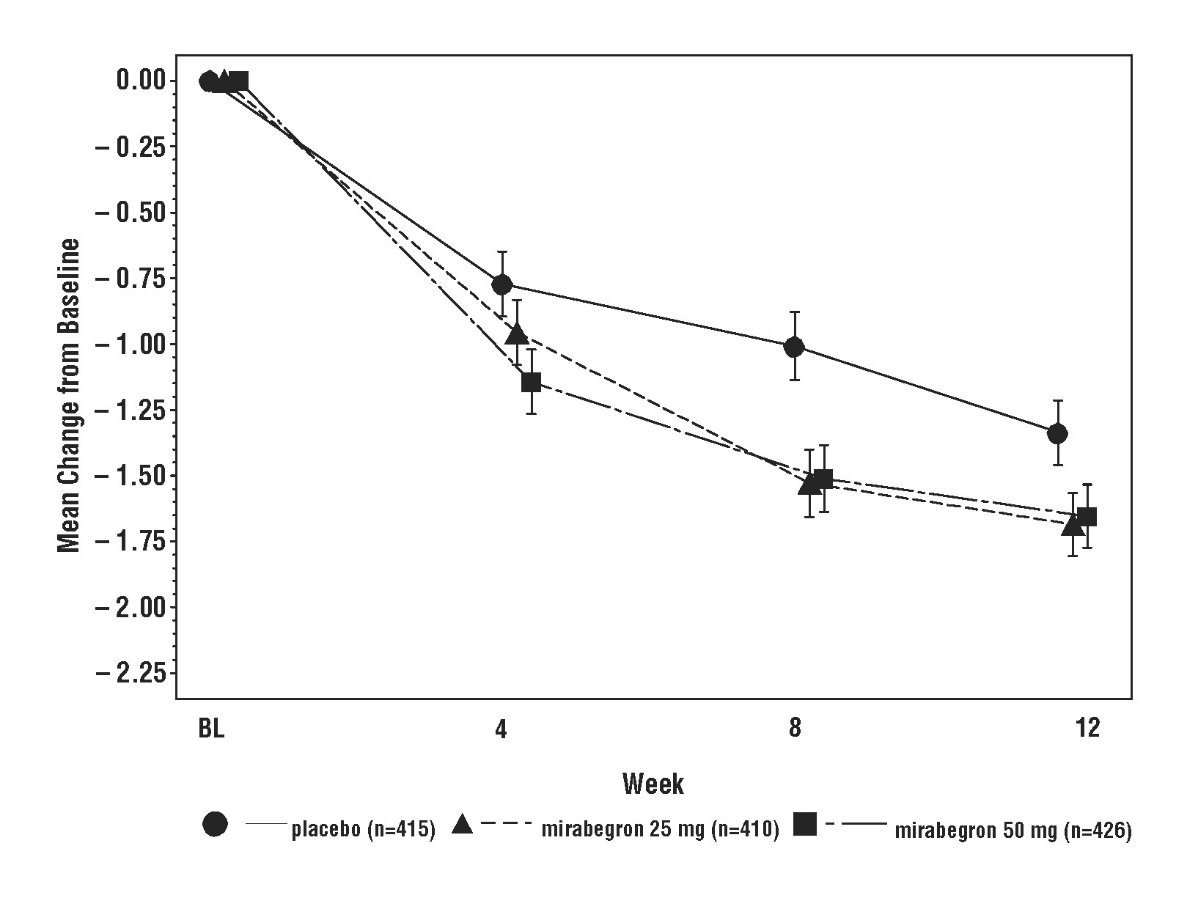
Results for the co-primary endpoints and mean volume voided per micturition from Studies 1, 2, and 3 are shown in Table 13.
|
|||||||
|
Parameter |
Study 1 |
Study 2 |
Study 3 |
||||
|
Placebo |
MYRBETRIQ 50 mg |
Placebo |
MYRBETRIQ 50 mg |
Placebo |
MYRBETRIQ 25 mg |
MYRBETRIQ 50 mg |
|
|
Number of Incontinence Episodes per 24 Hours† |
|||||||
|
n |
291 |
293 |
325 |
312 |
262 |
254 |
257 |
|
Baseline (mean) |
2.67 |
2.83 |
3.03 |
2.77 |
2.43 |
2.65 |
2.51 |
|
Change from baseline (adjusted mean‡) |
-1.17 |
-1.57 |
-1.13 |
-1.47 |
-0.96 |
-1.36 |
-1.38 |
|
Difference from placebo (adjusted mean‡) |
-- |
-0.41 |
-- |
-0.34 |
-- |
-0.40 |
-0.42 |
|
95% Confidence Interval |
-- |
(-0.72, -0.09) |
-- |
(-0.66, -0.03) |
-- |
(-0.74, -0.06) |
(-0.76, -0.08) |
|
p-value |
-- |
0.003§ |
-- |
0.026§ |
-- |
0.005§ |
0.001§ |
|
Number of Micturitions per 24 Hours |
|||||||
|
n |
480 |
473 |
433 |
425 |
415 |
410 |
426 |
|
Baseline (mean) |
11.71 |
11.65 |
11.51 |
11.80 |
11.48 |
11.68 |
11.66 |
|
Change from baseline (adjusted mean‡) |
-1.34 |
-1.93 |
-1.05 |
-1.66 |
-1.18 |
-1.65 |
-1.60 |
|
Difference from placebo (adjusted mean‡) |
-- |
-0.60 |
-- |
-0.61 |
-- |
-0.47 |
-0.42 |
|
95% Confidence Interval |
-- |
(-0.90, -0.29) |
-- |
(-0.98, -0.24) |
-- |
(-0.82, -0.13) |
(-0.76, -0.08) |
|
p-value |
-- |
< 0.001§ |
-- |
0.001§ |
-- |
0.007§ |
0.015§ |
|
Volume Voided (mL) per Micturition |
|||||||
|
n |
480 |
472 |
433 |
424 |
415 |
410 |
426 |
|
Baseline (mean) |
156.7 |
161.1 |
157.5 |
156.3 |
164.0 |
165.2 |
159.3 |
|
Change from baseline (adjusted mean‡) |
12.3 |
24.2 |
7.0 |
18.2 |
8.3 |
12.8 |
20.7 |
|
Difference from placebo (adjusted mean‡) |
-- |
11.9 |
-- |
11.1 |
-- |
4.6 |
12.4 |
|
95% Confidence Interval |
-- |
(6.3, 17.4) |
-- |
(4.4, 17.9) |
-- |
(-1.6, 10.8) |
(6.3, 18.6) |
|
p-value |
-- |
< 0.001§ |
-- |
0.001§ |
-- |
0.15 |
< 0.001§ |
MYRBETRIQ 25 mg was effective in treating the symptoms of OAB within 8 weeks and MYRBETRIQ 50 mg was effective in treating the symptoms of OAB within 4 weeks. Efficacy of both 25 mg and 50 mg doses of MYRBETRIQ was maintained through the 12-week treatment period.
Figures 3 through 8 show the co-primary endpoints, mean change from baseline (BL) over time in number of incontinence episodes per 24 hours, and mean change from baseline over time in number of micturitions per 24 hours, in Studies 1, 2, and 3.
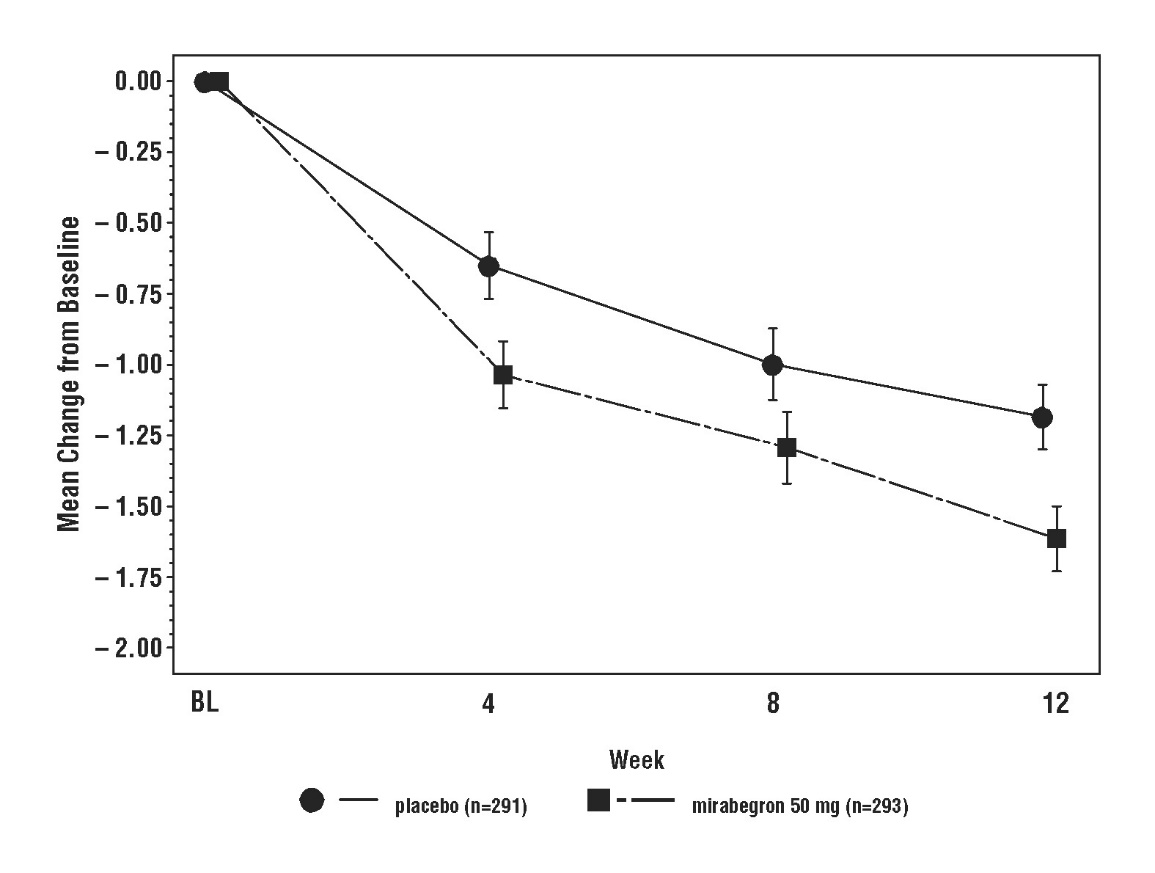
Figure 3: Mean (SE) Change from Baseline in Mean Number of Incontinence Episodes per 24 Hours – Study 1
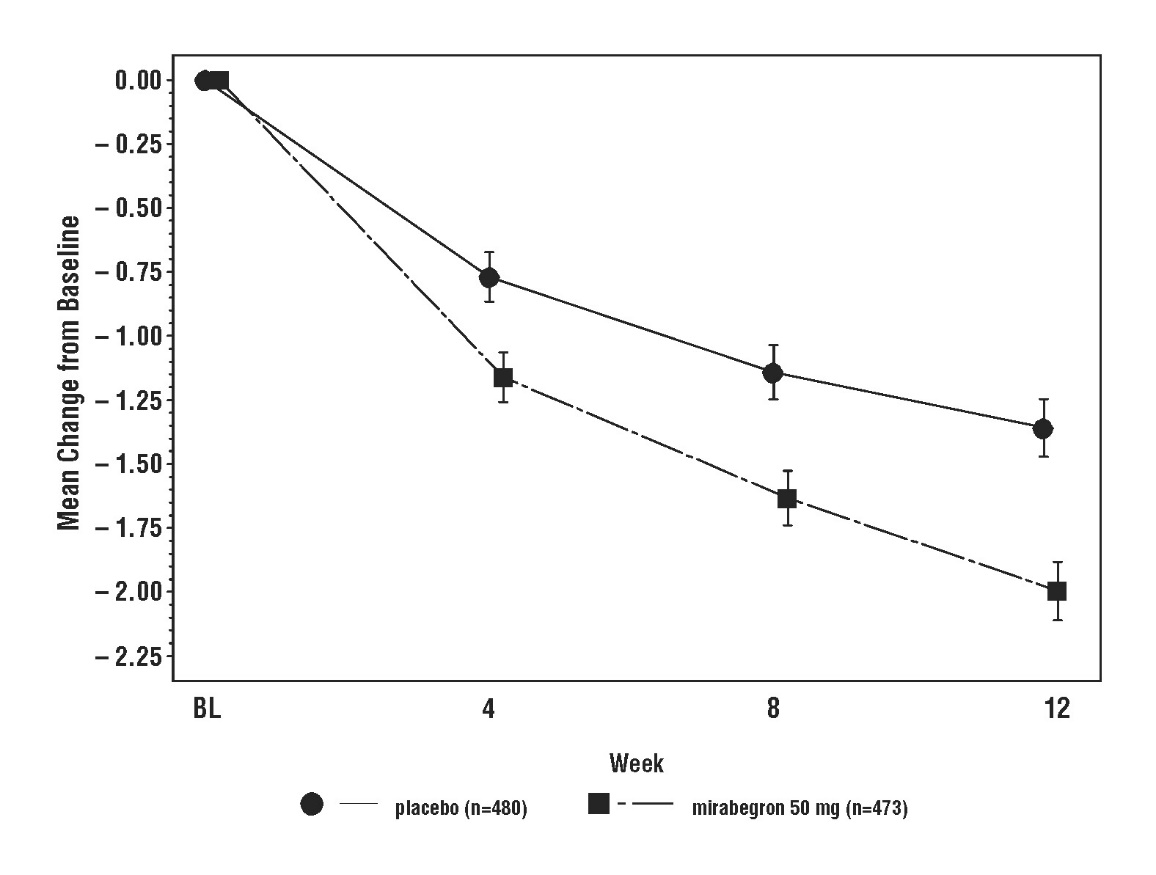
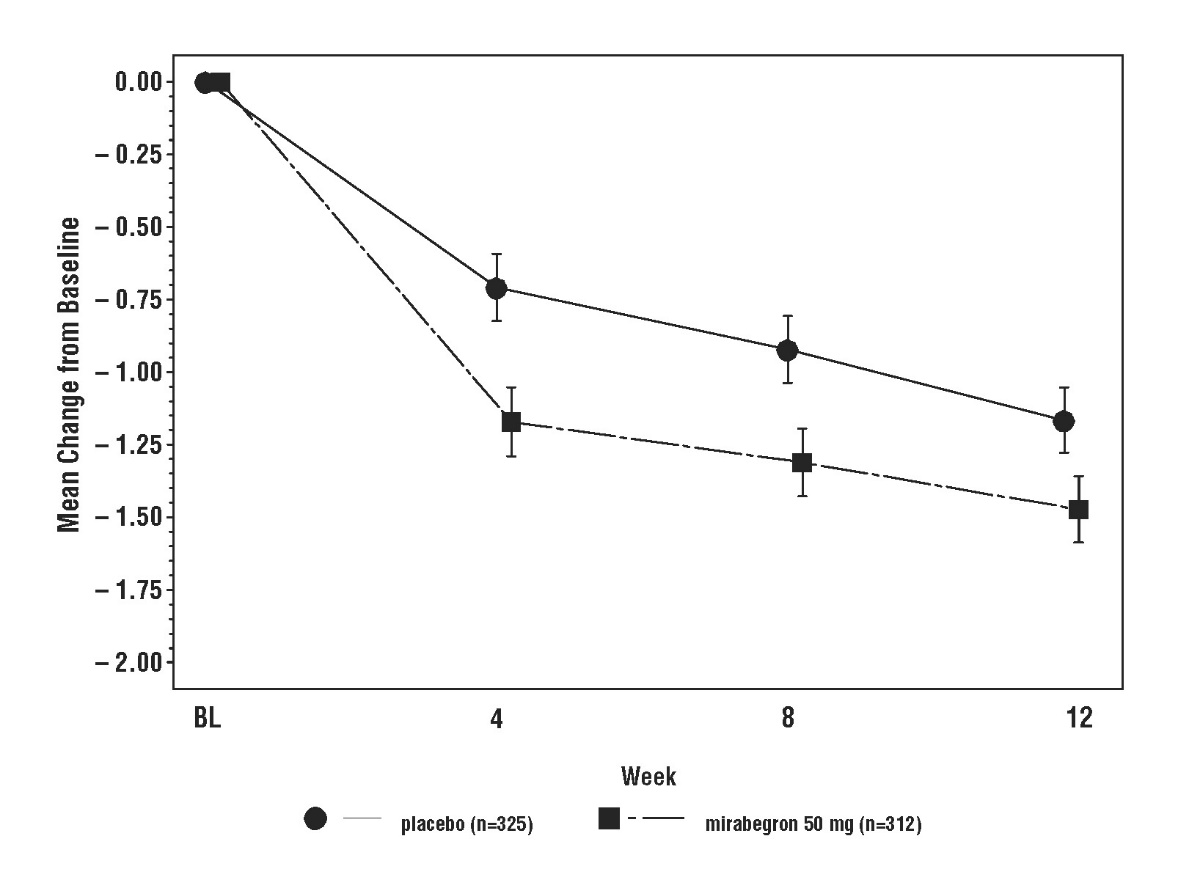
Figure 5: Mean (SE) Change from Baseline in Mean Number of Incontinence Episodes per 24 Hours – Study 2
14.2 MYRBETRIQ Combination Therapy for Adult OAB
Coadministration of MYRBETRIQ with Solifenacin Succinate
Coadministration of MYRBETRIQ with solifenacin succinate was evaluated in a 12-week, double-blind, randomized, placebo-controlled, parallel group, multicenter clinical trial in patients with OAB with symptoms of urge urinary incontinence, urgency, and urinary frequency (Study 6). Entry criteria required that patients had symptoms of OAB for at least 3 months duration, on average at least 8 micturitions and at least 1 urgency episode per day, and at least 3 episodes of incontinence, over a 7-day period. The majority of patients were Caucasian (80%) and female (77%) with a mean age of 57 years (range 18 to 86 years). The population included both naïve patients who had not received prior pharmacotherapy for OAB (54%) and those who had received prior pharmacotherapy for OAB (46%).
In Study 6 (NCT01972841), patients were randomized to placebo, solifenacin succinate 5 mg, MYRBETRIQ 25 mg, MYRBETRIQ 50 mg, solifenacin succinate 5 mg plus MYRBETRIQ 25 mg, or solifenacin succinate 5 mg plus MYRBETRIQ 50 mg once daily.
The co-primary efficacy endpoints in Study 6 were (1) change from baseline to end of treatment (week 12) in mean number of incontinence episodes per 24 hours and (2) change from baseline to end of treatment (week 12) in mean number of micturitions per 24 hours, based on a 7-day micturition diary. An important secondary endpoint was the change from baseline to end of treatment (week 12) in mean volume voided per micturition.
Results for the co-primary endpoints and mean volume voided per micturition for the overall patient population from Study 6 are shown in Table 14.
|
Parameter |
Placebo |
MYRBETRIQ 25 mg |
MYRBETRIQ 50 mg |
Solifenacin Succinate 5 mg |
MYRBETRIQ 25 mg + Solifenacin Succinate 5 mg |
MYRBETRIQ 50 mg + Solifenacin Succinate 5 mg |
|
Number of Incontinence Episodes per 24 Hours |
||||||
|
n |
412 |
409 |
406 |
413 |
823 |
816 |
|
Baseline (mean) |
3.40 |
3.42 |
3.16 |
3.59 |
3.21 |
3.15 |
|
Change from baseline (adjusted mean†) |
-1.34 |
-1.70 |
-1.76 |
-1.79 |
-2.04 |
-1.98 |
|
Difference from Solifenacin Succinate (adjusted mean†) |
-- |
-- |
-- |
-- |
-0.25 |
-0.20 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(-0.49, -0.01) |
(-0.44, 0.04) |
|
Difference from MYRBETRIQ (at the same MYRBETRIQ dose, adjusted mean†) |
-- |
-- |
-- |
-- |
-0.34 |
-0.23 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(-0.58, -0.10) |
(-0.47, 0.01) |
|
Number of Micturitions per 24 Hours |
||||||
|
n |
412 |
409 |
406 |
413 |
823 |
816 |
|
Baseline (mean) |
10.97 |
10.81 |
11.19 |
10.74 |
10.72 |
10.72 |
|
Change from baseline (adjusted mean†) |
-1.64 |
-2.00 |
-2.03 |
-2.20 |
-2.49 |
-2.59 |
|
Difference from Solifenacin Succinate (adjusted mean†) |
-- |
-- |
-- |
-- |
-0.29 |
-0.39 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(-0.57, -0.01) |
(-0.67, -0.11) |
|
Difference from MYRBETRIQ (at the same MYRBETRIQ dose, adjusted mean†) |
-- |
-- |
-- |
-- |
-0.48 |
-0.56 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(-0.76, -0.21) |
(-0.84, -0.28) |
|
Volume Voided (mL) per Micturition |
||||||
|
n |
413 |
407 |
408 |
411 |
821 |
821 |
|
Baseline (mean) |
157.82 |
152.46 |
155.35 |
151.86 |
159.19 |
153.83 |
|
Change from baseline (adjusted mean†) |
8.44 |
13.32 |
21.99 |
30.99 |
34.84 |
39.73 |
|
Difference from Solifenacin Succinate (adjusted mean†) |
-- |
-- |
-- |
-- |
3.85 |
8.75 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(-2.29, 10.00) |
(2.61, 14.89) |
|
Difference from MYRBETRIQ (at the same MYRBETRIQ dose, adjusted mean†) |
-- |
-- |
-- |
-- |
21.52 |
17.74 |
|
95% Confidence Interval |
-- |
-- |
-- |
-- |
(15.35, 27.68) |
(11.58, 23.90) |
|
ANCOVA: Analysis of covariance | ||||||
Figures 9 and 10 show the co-primary endpoints, mean change from baseline (BL) over time in number of incontinence episodes per 24 hours, and mean change from baseline over time in number of micturitions per 24 hours, in the overall patient population in Study 6.
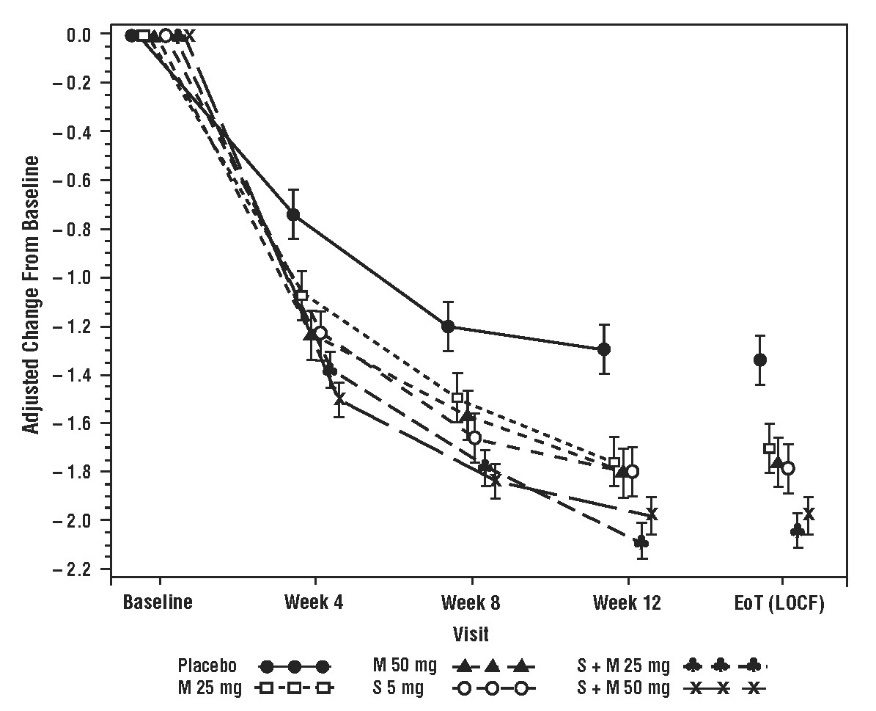
Figure 9: Mean Change from Baseline in Mean (± SE) Number of Incontinence Episodes per 24 Hours at Each Visit (FAS) – Study 6
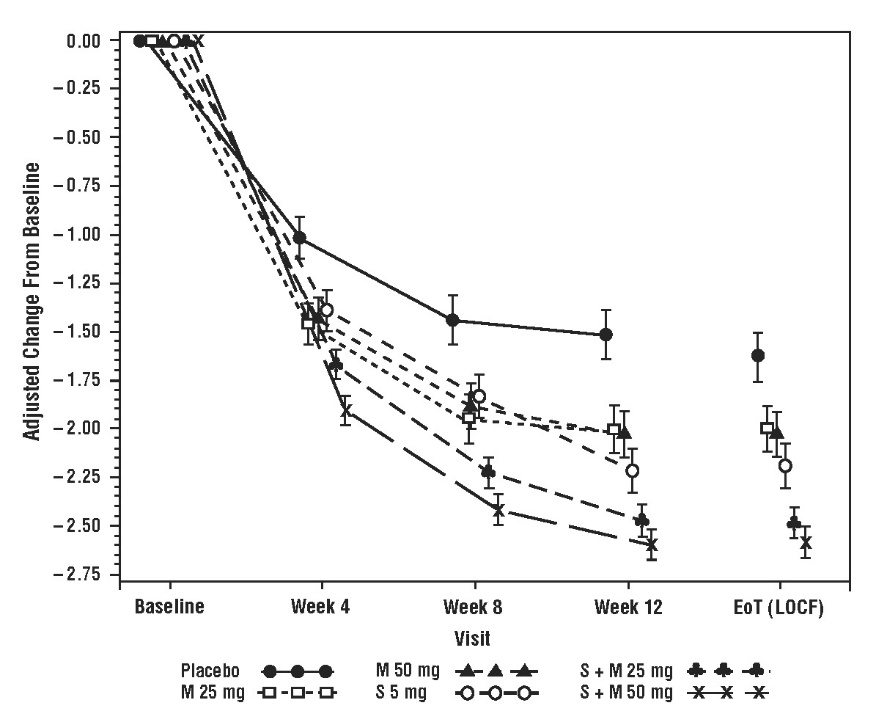
Figure 10: Mean Change from Baseline in Mean (± SE) Number of Micturitions per 24 Hours at Each Visit (FAS) – Study 6
MYRBETRIQ as Add-on Therapy to Solifenacin Succinate
MYRBETRIQ add-on therapy to solifenacin succinate was evaluated in one, 12-week, double-blind, randomized, active‑controlled, multicenter clinical trial in incontinent OAB patients who received solifenacin succinate for 4 weeks and required additional relief for their OAB symptoms (Study 7). Entry criteria required that patients had symptoms of OAB for at least 3 months duration (urge urinary incontinence, urgency, and urinary frequency), and at least 1 incontinence episode during a 3-day period after being treated with solifenacin succinate 5 mg for 4 weeks. The majority of patients were Caucasian (94%) and female (83%) with a mean age of 57 years (range 18 to 89 years). Patients were randomized to solifenacin succinate 5 mg, solifenacin succinate 10 mg, or solifenacin succinate 5 mg plus MYRBETRIQ 25 mg once daily. After 4 weeks, all patients in the combination treatment arm had a dose increase from MYRBETRIQ 25 mg to MYRBETRIQ 50 mg.
The primary efficacy endpoint in Study 7 (NCT01908829) was change from baseline to end of treatment (week 12) in mean number of incontinence episodes per 24 hours. Two important secondary endpoints were change from baseline to end of treatment in mean number of micturitions per 24 hours and change from baseline to end of treatment in mean volume voided per micturition. Results for the primary and additional endpoints from Study 7 are shown in Table 15.
|
||
|
Parameter |
Solifenacin Succinate 5 mg |
MYRBETRIQ 25 mg/50 mg + Solifenacin Succinate 5 mg |
|
Number of Incontinence Episodes per 24 Hours |
||
|
n |
704 |
706 |
|
Baseline (mean) |
3.15 |
3.24 |
|
Change from baseline (adjusted mean†) |
-1.53 |
-1.80 |
|
Difference (MYRBETRIQ + Solifenacin Succinate) from Solifenacin Succinate (adjusted mean‡) |
-0.26 |
-- |
|
95% Confidence Interval |
(-0.47, -0.05) |
|
|
Number of Micturitions per 24 Hours |
||
|
n |
704 |
706 |
|
Baseline (mean) |
8.90 |
9.13 |
|
Change from baseline (adjusted mean†) |
-1.14 |
-1.59 |
|
Difference (MYRBETRIQ + Solifenacin Succinate) from Solifenacin Succinate (adjusted mean‡) |
-0.45 (0.12) |
-- |
|
95% Confidence Interval |
(-0.67, -0.22) |
|
|
Volume Voided (mL) per Micturition |
||
|
n |
682 |
680 |
|
Baseline (mean) |
170.92 |
172.93 |
|
Change from baseline (adjusted mean†) |
16.52 |
28.05 |
|
Difference (MYRBETRIQ + Solifenacin Succinate) from Solifenacin Succinate (adjusted mean‡) |
11.52 |
|
|
95% Confidence Interval |
(6.06, 16.99) | |
|
ANCOVA: Analysis of covariance | ||
Long-term Coadministration of MYRBETRIQ with Solifenacin Succinate
Long-term efficacy of coadministration of MYRBETRIQ 50 mg with solifenacin succinate 5 mg was evaluated in a 52-week, double-blind, randomized, active-controlled, parallel group, multicenter clinical trial in patients with OAB Study 8 (NCT02045862). The primary objective of this study was to evaluate the safety and tolerability of long-term combination treatment and the evaluation of efficacy was the secondary objective of the study. Entry criteria included patients who had completed Study 6 or Study 7 or new patients. All patients had symptoms of OAB for at least 3 months duration, on average at least 8 micturitions and at least 1 urgency episode per day, and at least 3 episodes of incontinence over a 7-day period. Patients were randomized to solifenacin succinate 5 mg, MYRBETRIQ 50 mg, or solifenacin succinate 5 mg plus MYRBETRIQ 50 mg once daily.
Primary efficacy variables were change from baseline to end of treatment in mean number of incontinence episodes per 24 hours and change from baseline to end of treatment in mean number of micturitions per 24 hours. Combination treatment with MYRBETRIQ and solifenacin succinate demonstrated statistically significant greater improvements from baseline compared to MYRBETRIQ 50 mg and solifenacin succinate 5 mg for both efficacy endpoints. The improvements from baseline observed with coadministration of MYRBETRIQ 50 mg and solifenacin succinate 5 mg compared to MYRBETRIQ 50 mg and solifenacin succinate 5 mg were demonstrated at 3 months and were maintained throughout the 1‑year treatment period. Also, for the secondary efficacy variable of change from baseline to end of treatment in mean volume voided (MVV) per micturition, the increase in MVV was statistically significantly greater for combination treatment compared to the MYRBETRIQ 50 mg and solifenacin succinate 5 mg groups.
14.3 MYRBETRIQ/MYRBETRIQ Granules for Pediatric Neurogenic Detrusor Overactivity (NDO)
The efficacy of MYRBETRIQ/MYRBETRIQ Granules was evaluated in Study 9 (NCT02751931), a 52-week, open-label, baseline-controlled, multicenter, dose titration study in pediatric patients 3 years of age and older for the treatment of neurogenic detrusor overactivity (NDO). Study 9 included patients 3 to 17 years of age. Entry criteria required that patients had a diagnosis of neurogenic detrusor overactivity (NDO) with involuntary detrusor contractions with detrusor pressure increase greater than 15 cm H2O and that patients or their caregivers practiced clean intermittent catheterization (CIC). MYRBETRIQ/MYRBETRIQ Granules were administered orally once daily. All patients initially received a weight-based starting dose equivalent to 25 mg daily dose followed by dose titration to a dose equivalent of 50 mg daily dose. The duration of the dose titration period was up to 8 weeks and this period was followed by a dose maintenance period that continued for the duration of the 52-week study.
In Study 9, a total of 86 patients 3 to 17 years of age received MYRBETRIQ/MYRBETRIQ Granules. Of these, 71 patients completed treatment through week 24 and 70 completed 52 weeks of treatment. A total of 68 patients (43 patients 3 to less than 12 years of age and 25 patients 12 to 17 years of age) had valid urodynamic measurements for evaluation of efficacy. The study population included 39 males (45%) and 47 females (55%). The optimized maintenance dose within this study population included 94% of patients at the maximum dose, and 6% of patients at the starting dose.
The primary efficacy endpoint was change from baseline in the patients’ maximum cystometric (bladder) capacity (MCC) after 24 weeks of treatment with MYRBETRIQ/MYRBETRIQ Granules. As shown in Table 16, improvements in MCC were observed in patients 3 to less than 12 years of age and in patients 12 to 17 years of age. The magnitude of the observed changes from baseline in the primary and secondary efficacy endpoints were comparable between patients 3 to less than 12 years of age and patients 12 to 17 years of age.
|
|||
|
Parameter |
Children Aged 3 to Less than 12 Years (N=43)* Mean (SD) |
Adolescents Aged 12 to 17 Years (N=25)* Mean (SD) |
|
|
Maximum Cystometric Capacity (mL) |
|||
|
Baseline Week 24 Change from baseline 95% CI |
159 (95) 231 (129) 72 (87) (45, 99) |
239 (99) 352 (125) 113 (83) (79, 147) |
|
Secondary efficacy endpoints from Study 9 for MYRBETRIQ/MYRBETRIQ Granules in pediatric patients with neurogenic detrusor overactivity (NDO) are shown below in Table 17 and Table 18.
|
||
|
Parameter |
Children Aged 3 to Less than 12 Years (N=43)* Mean (SD) |
Adolescents Aged 12 to 17 Years (N=25)* Mean (SD) |
|
Bladder Compliance (mL/cm H2O)† |
||
| ||
|
16.0 (55.8) 14.6 (42.1) 95% CI: -0.3, 29.5 |
11.1 (10.7) 13.6 (15.0) 95% CI: 6.7, 20.4 |
|
|
Number of Overactive Detrusor Contractions (> 15 cm H2O)† |
||
| ||
|
3.0 (4.0) -1.9 (4.2) 95% CI: -3.3, -0.4 |
2.1 (3.1) -0.8 (3.9) 95% CI: -2.5, 0.9 |
|
|
Bladder Volume Prior To First Detrusor Contraction (> 15 cm H2O)† |
||
|
115 (83) 93 (88) 95% CI: 64, 122 |
177 (117) 121 (160) 95% CI: 54, 189 |
|
||
|
Parameter |
Children Aged 3 to Less than 12 Years (N=43)* Mean (SD) |
Adolescents Aged 12 to 17 Years (N=25)* Mean (SD) |
|
Maximum Catheterized Urine Volume per Day (mL)† |
||
| ||
|
304 (109) 50 (104) 95% CI: 17, 83 |
360 (111) 84 (122) 95% CI: 32, 137 |
|
|
Number of Leakage Episodes per Day† |
||
|
2.8 (3.7) -2.0 (3.2) 95% CI: -3.2, -0.7 |
1.8 (1.7) -1.0 (1.1) 95% CI: -1.5, -0.5 |
16. How is Myrbetriq supplied
16.1 MYRBETRIQ (mirabegron extended-release tablets)
MYRBETRIQ is supplied as oval, film-coated, extended-release tablets, available in bottles as follows:
|
Strength |
25 mg |
50 mg |
|
Shape/Color |
Oval/Brown |
Oval/Yellow |
|
Branding on Tablet | ||
|
Available |
bottles of approximately 960 tablets, NDC 55154-8712-8 |
bottles of approximately 1080 tablet, NDC 55154-8713-8 |
 logo, 325
logo, 325 logo, 355
logo, 355
Store and Dispense
Store at 20°C to 25°C (68°F to 77°F) with excursions permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient and/or caregiver to read the FDA-approved patient labeling (Patient Information).
Increases in Blood Pressure
Inform patients and/or their caregivers that MYRBETRIQ/MYRBETRIQ Granules may increase blood pressure. Advise patients, especially patients with hypertension, to periodically monitor their blood pressure and report increased measurement to their health care provider [see Warnings and Precautions (5.1)].
Urinary Retention
Inform patients and/or their caregivers that MYRBETRIQ may cause urinary retention in adult patients with bladder outlet obstruction and in patients taking muscarinic antagonist medications for the treatment of OAB. Advise patients to contact their physician if they experience these effects while taking MYRBETRIQ [see Warnings and Precautions (5.2)].
Angioedema
Inform patients and/or their caregivers that MYRBETRIQ/MYRBETRIQ Granules may cause angioedema. Advise patients and/or their caregivers to promptly discontinue MYRBETRIQ/MYRBETRIQ Granules and seek medical attention if angioedema associated with the upper airway swelling occurs as this may be life-threatening [see Warnings and Precautions (5.3)].
Drug Interactions
Advise patients to report their use of any other prescription or nonprescription medications or dietary supplements because co-administration with MYRBETRIQ/MYRBETRIQ Granules may require a dose adjustment and/or increased monitoring of these drugs [see Drug Interactions (7)].
Administration Instructions
MYRBETRIQ
Advise adult patients to swallow MYRBETRIQ whole with water and not to chew, divide, or crush. Advise adult patients to take MYRBETRIQ with or without food.
Advise pediatric patients and/or their caregivers to swallow MYRBETRIQ whole with water and not to chew, divide, or crush. Advise pediatric patients to take MYRBETRIQ with food.
MYRBETRIQ Granules
Advise pediatric patients and/or their caregivers to use an appropriate measuring device and instructions for measuring the correct dose of MYRBETRIQ Granules for extended-release oral suspension. Instruct patients or their caregivers that patients should take MYRBETRIQ Granules for extended-release oral suspension orally within 1 hour after preparation with food once daily and not save the dose for later.
The bottle should be shaken for 1 minute each day if the suspension will not be used for 2 or more days. When ready to use, shake the bottle vigorously for 1 minute then let it stand until the foam on top of the suspension is gone (approximately 1 to 2 minutes).
Missed Dose
Instruct patients and/or their caregivers to take any missed doses as soon as they remember, unless more than 12 hours have passed since the missed dose. If more than 12 hours have passed, the missed dose can be skipped and the next dose should be taken at the usual time.
Marketed and Distributed by:
Astellas Pharma US, Inc.
Northbrook, IL 60062
Distributed By:
Cardinal Health
Dublin, OH 43017
ER7606J Rev. A
ER7607J Rev. A
MYRBETRIQ is a registered trademark of Astellas Pharma Inc. All other trademarks are the property of their respective owners.
© 2012 – 2021 Astellas Pharma US, Inc.
|
Patient Information
|
|
|
MYRBETRIQ® (meer-BEH-trick) (mirabegron extended-release tablets) for oral use |
MYRBETRIQ® (meer-BEH-trick) GRANULES (mirabegron for extended-release oral suspension) |
|
What are MYRBETRIQ tablets and MYRBETRIQ GRANULES? Adults
Children
It is not known if MYRBETRIQ tablets and MYRBETRIQ GRANULES to treat NDO, are safe and effective in children under 3 years of age. |
|
|
Who should not take MYRBETRIQ tablets or MYRBETRIQ GRANULES? Do not take MYRBETRIQ tablets or MYRBETRIQ GRANULES if you are allergic to mirabegron or any of the ingredients in MYRBETRIQ tablets or MYRBETRIQ GRANULES. See the end of this Patient Information leaflet for a complete list of ingredients in MYRBETRIQ tablets and MYRBETRIQ GRANULES. |
|
|
Before you take MYRBETRIQ tablets or MYRBETRIQ GRANULES, tell your doctor about all of your medical conditions, including if you:
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. MYRBETRIQ tablets and MYRBETRIQ GRANULES may affect the way other medicines work, and other medicines may affect how MYRBETRIQ tablets and MYRBETRIQ GRANULES work. Especially tell your doctor if you take:
|
|
|
How should I take MYRBETRIQ tablets?
|
|
|
How should I take MYRBETRIQ GRANULES?
Note: You will receive MYRBETRIQ GRANULES from the pharmacy as a suspension. The suspension is prepared by your pharmacist. You will receive an oral dosing device with the suspension. If the suspension will not be used for 2 or more days, shake the bottle vigorously for 1 minute each day to make sure the granules are mixed well (dispersed). Talk to your pharmacist if you have any questions. |
|
|
What are the possible side effects of MYRBETRIQ tablets and MYRBETRIQ GRANULES? MYRBETRIQ tablets and MYRBETRIQ GRANULES may cause serious side effects, including:
Adults with Overactive Bladder The most common side effects of MYRBETRIQ tablets include:
The most common side effects of MYRBETRIQ tablets, when used with solifenacin succinate, include:
Children with Neurogenic Detrusor Overactivity The most common side effects of MYRBETRIQ tablets and MYRBETRIQ GRANULES include:
Tell your doctor if you have any side effect that bothers you, does not go away, or if you have swelling of the face, lips, tongue or throat, hives, skin rash or itching while taking MYRBETRIQ tablets or MYRBETRIQ GRANULES. These are not all the possible side effects of MYRBETRIQ tablets and MYRBETRIQ GRANULES. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
How should I store MYRBETRIQ tablets or MYRBETRIQ GRANULES?
Keep MYRBETRIQ tablets, MYRBETRIQ GRANULES, and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of MYRBETRIQ tablets and MYRBETRIQ GRANULES. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use MYRBETRIQ tablets or MYRBETRIQ GRANULES for a condition for which it was not prescribed. Do not give MYRBETRIQ tablets or MYRBETRIQ GRANULES to other people, even if they have the same symptoms you have. It may harm them. This leaflet summarizes the most important information about MYRBETRIQ tablets and MYRBETRIQ GRANULES. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about MYRBETRIQ tablets or MYRBETRIQ GRANULES that is written for health professionals. |
|
|
What are the ingredients in MYRBETRIQ tablets? Active ingredient: mirabegron Inactive ingredients: butylated hydroxytoluene, hydroxypropyl cellulose, hypromellose, magnesium stearate, polyethylene glycol, polyethylene oxide, red ferric oxide (25 mg MYRBETRIQ tablet only), and yellow ferric oxide. |
|
|
What are the ingredients in MYRBETRIQ GRANULES? Active ingredient: mirabegron Inactive ingredients: acesulfame potassium, diluted hydrochloric acid, ethylparaben, hypromellose, magnesium stearate, mannitol, methylparaben, silicon dioxide, simethicone, sodium polystyrene sulfonate, and xanthan gum. |
|
|
Marketed and Distributed by: Astellas Pharma US, Inc. Northbrook, IL 60062 MYRBETRIQ and VESIcare are registered trademarks of Astellas Pharma Inc. All other trademarks are the property of their respective owners. © 2012 – 2021 Astellas Pharma US, Inc. For more information, go to www.Myrbetriq.com or call 1-800-727-7003. Distributed By: Cardinal Health Dublin, OH 43017 ER7606J Rev. A ER7607J Rev. A |
|
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 4/2021


| MYRBETRIQ
mirabegron tablet, film coated, extended release |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| MYRBETRIQ
mirabegron tablet, film coated, extended release |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Cardinal Health 107, LLC (118546603) |
More about Myrbetriq (mirabegron)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (281)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Generic availability
- Support group
- FDA approval history
- Drug class: urinary antispasmodics
- Breastfeeding
- En español
