DermacinRx Cinlone-II CPI: Package Insert / Prescribing Info
Package insert / product label
Generic name: lidocaine and prilocaine, triamcinolone acetonide
Dosage form: kit
Drug class: Topical anesthetics
Medically reviewed by Drugs.com. Last updated on Mar 25, 2025.
On This Page
DermacinRx Cinlone-II CPI Description
Lidocaine and prilocaine cream, 2.5%/2.5% is an emulsion in which the oil phase is a eutectic mixture of lidocaine and prilocaine in a ratio of 1:1 by weight. This eutectic mixture has a melting point below room temperature and therefore both local anesthetics exist as a liquid oil rather than as crystals. It is packaged in 30 gram tubes and 5 gram tubes for hospital use. Lidocaine is chemically designated as acetamide, 2-(diethylamino)-N-(2,6-dimethylphenyl), has an octanol: water partition ratio of 43 at pH 7.4, and has the following structure:
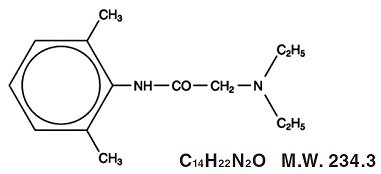
Prilocaine is chemically designated as propanamide, N-(2-methylphenyl)-2-(propylamino), has an octanol: water partition ratio of 25 at pH 7.4, and has the following structure:
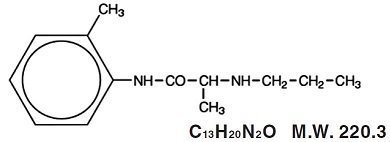
Each gram of lidocaine and prilocaine cream, 2.5%/2.5% contains lidocaine 25 mg, prilocaine 25 mg, polyoxyethylene fatty acid esters (as emulsifiers), carbomer 934P (as a thickening agent), sodium hydroxide to adjust to a pH approximating 9, and purified water. Lidocaine and prilocaine cream, 2.5%/2.5% contains no preservative, however it passes the USP antimicrobial effectiveness test due to the pH. The specific gravity of lidocaine and prilocaine cream, 2.5%/2.5% is approximately 1.
DermacinRx Cinlone-II CPI - Clinical Pharmacology
Mechanism of Action: Lidocaine and prilocaine cream, 2.5%/2.5%, applied to intact skin under occlusive dressing, provides dermal analgesia by the release of lidocaine and prilocaine from the cream into the epidermal and dermal layers of the skin and by the accumulation of lidocaine and prilocaine in the vicinity of dermal pain receptors and nerve endings. Lidocaine and prilocaine are amide-type local anesthetic agents. Both lidocaine and prilocaine stabilize neuronal membranes by inhibiting the ionic fluxes required for the initiation and conduction of impulses, thereby effecting local anesthetic action.
The onset, depth and duration of dermal analgesia on intact skin provided by lidocaine and prilocaine cream, 2.5%/2.5% depends primarily on the duration of application. To provide sufficient analgesia for clinical procedures such as intravenous catheter placement and venipuncture, lidocaine and prilocaine cream, 2.5%/2.5% should be applied under an occlusive dressing for at least 1 hour. To provide dermal analgesia for clinical procedures such as split skin graft harvesting, lidocaine and prilocaine cream, 2.5%/2.5% should be applied under occlusive dressing for at least 2 hours. Satisfactory dermal analgesia is achieved 1 hour after application, reaches maximum at 2 to 3 hours, and persists for 1 to 2 hours after removal. Absorption from the genital mucosa is more rapid and onset time is shorter (5 to 10 minutes) than after application to intact skin. After a 5 to 10 minute application of lidocaine and prilocaine cream, 2.5%/2.5% to female genital mucosa, the average duration of effective analgesia to an argon laser stimulus (which produced a sharp, pricking pain) was 15 to 20 minutes (individual variations in the range of 5 to 45 minutes).
Dermal application of lidocaine and prilocaine cream, 2.5%/2.5% may cause a transient, local blanching followed by a transient, local redness or erythema.
Pharmacokinetics: Lidocaine and prilocaine cream, 2.5%/2.5% is a eutectic mixture of lidocaine 2.5% and prilocaine 2.5% formulated as an oil in water emulsion. In this eutectic mixture, both anesthetics are liquid at room temperature (see DESCRIPTION) and the penetration and subsequent systemic absorption of both prilocaine and lidocaine are enhanced over that which would be seen if each component in crystalline form was applied separately as a 2.5% topical cream.
Absorption- The amount of lidocaine and prilocaine systemically absorbed from lidocaine and prilocaine cream, 2.5%/2.5% is directly related to both the duration of application and to the area over which it is applied. In two pharmacokinetic studies, 60 g of lidocaine and prilocaine cream, 2.5%/2.5% (1.5 g lidocaine and 1.5 g prilocaine) was applied to 400 cm2 of intact skin on the lateral thigh and then covered by an occlusive dressing. The subjects were then randomized such that one-half of the subjects had the occlusive dressing and residual cream removed after 3 hours, while the remainder left the dressing in place for 24 hours. The results from these studies are summarized below.
|
LIDOCAINE and PRILOCAINE CREAM, 2.5%/2.5% (g) |
Area |
Time on |
Drug Content |
Absorbed |
Cmax |
Tmax |
|
60 |
400 |
3 |
lidocaine 1500 |
54 |
0.12 |
4 |
|
prilocaine 1500 |
92 |
0.07 |
4 |
|||
|
60 |
400 |
24* |
lidocaine 1500 |
243 |
0.28 |
10 |
|
prilocaine 1500 |
503 |
0.14 |
10 |
* Maximum recommended duration of exposure is 4 hours.
When 60 g of lidocaine and prilocaine cream, 2.5%/2.5% was applied over 400 cm2 for 24 hours, peak blood levels of lidocaine are approximately 1/20 the systemic toxic level. Likewise, the maximum prilocaine level is about 1/36 the toxic level. In a pharmacokinetic study, lidocaine and prilocaine cream, 2.5%/2.5% was applied to penile skin in 20 adult male patients in doses ranging from 0.5 g to 3.3 g for 15 minutes. Plasma concentrations of lidocaine and prilocaine following lidocaine and prilocaine cream, 2.5%/2.5% application in this study were consistently low (2.5-16 ng/mL for lidocaine and 2.5-7 ng/mL for prilocaine). The application of lidocaine and prilocaine cream, 2.5%/2.5% to broken or inflamed skin, or to 2,000 cm2 or more of skin where more of both anesthetics are absorbed, could result in higher plasma levels that could, in susceptible individuals, produce a systemic pharmacologic response.
The absorption of lidocaine and prilocaine cream, 2.5%/2.5% applied to genital mucous membranes was studied in two open-label clinical trials. Twenty-nine patients received 10 g of lidocaine and prilocaine cream, 2.5%/2.5% applied for 10 to 60 minutes in the vaginal fornices. Plasma concentrations of lidocaine and prilocaine following lidocaine and prilocaine cream, 2.5%/2.5% application in these studies ranged from 148 to 641 ng/mL for lidocaine and 40 to 346 ng/mL for prilocaine and time to reach maximum concentration (tmax) ranged from 21 to 125 minutes for lidocaine and from 21 to 95 minutes for prilocaine. These levels are well below the concentrations anticipated to give rise to systemic toxicity (approximately 5000 ng/mL for lidocaine and prilocaine).
Distribution- When each drug is administered intravenously, the steady-state volume of distribution is 1.1 to 2.1 L/kg (mean 1.5, ±0.3 SD, n=13) for lidocaine and is 0.7 to 4.4 L/kg (mean 2.6, ±1.3 SD, n=13) for prilocaine. The larger distribution volume for prilocaine produces the lower plasma concentrations of prilocaine observed when equal amounts of prilocaine and lidocaine are administered. At concentrations produced by application of lidocaine and prilocaine cream, 2.5%/2.5%, lidocaine is approximately 70% bound to plasma proteins, primarily alpha-1-acid glycoprotein. At much higher plasma concentrations (1 to 4 mcg/mL of free base) the plasma protein binding of lidocaine is concentration dependent. Prilocaine is 55% bound to plasma proteins. Both lidocaine and prilocaine cross the placental and blood brain barrier, presumably by passive diffusion.
Metabolism- It is not known if lidocaine or prilocaine are metabolized in the skin. Lidocaine is metabolized rapidly by the liver to a number of metabolites including monoethylglycinexylidide (MEGX) and glycinexylidide (GX), both of which have pharmacologic activity similar to, but less potent than that of lidocaine. The metabolite, 2,6-xylidine, has unknown pharmacologic activity. Following intravenous administration, MEGX and GX concentrations in serum range from 11 to 36% and from 5 to 11% of lidocaine concentrations, respectively. Prilocaine is metabolized in both the liver and kidneys by amidases to various metabolites including ortho-toluidine and N-n-propylalanine. It is not metabolized by plasma esterases. The ortho-toluidine metabolite has been shown to be carcinogenic in several animal models (see Carcinogenesis subsection of PRECAUTIONS). In addition, ortho-toluidine can produce methemoglobinemia following systemic doses of prilocaine approximating 8 mg/kg (see ADVERSE REACTIONS). Very young patients, patients with glucose-6-phosphate dehydrogenase deficiencies and patients taking oxidizing drugs such as antimalarials and sulfonamides are more susceptible to methemoglobinemia (see Methemoglobinemia subsection of PRECAUTIONS).
Elimination- The terminal elimination half-life of lidocaine from the plasma following IV administration is approximately 65 to 150 minutes (mean 110, ±24 SD, n=13). More than 98% of an absorbed dose of lidocaine can be recovered in the urine as metabolites or parent drug. The systemic clearance is 10 to 20 mL/min/kg (mean 13, ±3 SD, n=13). The elimination half-life of prilocaine is approximately 10 to 150 minutes (mean 70, ±48 SD, n=13). The systemic clearance is 18 to 64 mL/min/kg (mean 38, ±15 SD, n=13). During intravenous studies, the elimination half-life of lidocaine was statistically significantly longer in elderly patients (2.5 hours) than in younger patients (1.5 hours). No studies are available on the intravenous pharmacokinetics of prilocaine in elderly patients.
Pediatrics- Some pharmacokinetic (PK) data are available in infants (1 month to <2 years old) and children (2 to <12 years old). One PK study was conducted in 9 full-term neonates (mean age: 7 days and mean gestational age: 38.8 weeks). The study results show that neonates had comparable plasma lidocaine and prilocaine concentrations and blood methemoglobin concentrations as those found in previous pediatric PK studies and clinical trials. There was a tendency towards an increase in methemoglobin formation. However, due to assay limitations and very little amount of blood that could be collected from neonates, large variations in the above reported concentrations were found.
Clinical Studies
Lidocaine and prilocaine cream, 2.5%/2.5% application in adults prior to IV cannulation or venipuncture was studied in 200 patients in four clinical studies in Europe. Application for at least 1 hour provided significantly more dermal analgesia than placebo cream or ethyl chloride. Lidocaine and prilocaine cream, 2.5%/2.5% was comparable to subcutaneous lidocaine, but was less efficacious than intradermal lidocaine. Most patients found lidocaine and prilocaine cream, 2.5%/2.5% treatment preferable to lidocaine infiltration or ethyl chloride spray.
Lidocaine and prilocaine cream, 2.5%/2.5% was compared with 0.5% lidocaine infiltration prior to skin graft harvesting in one open label study in 80 adult patients in England. Application of lidocaine and prilocaine cream, 2.5%/2.5% for 2 to 5 hours provided dermal analgesia comparable to lidocaine infiltration.
Lidocaine and prilocaine cream, 2.5%/2.5% application in children was studied in seven non-US studies (320 patients) and one US study (100 patients). In controlled studies, application of lidocaine and prilocaine cream, 2.5%/2.5% for at least 1 hour with or without presurgical medication prior to needle insertion provided significantly more pain reduction than placebo. In children under the age of seven years, lidocaine and prilocaine cream, 2.5%/2.5% was less effective than in older children or adults.
Lidocaine and prilocaine cream, 2.5%/2.5% was compared with placebo in the laser treatment of facial port-wine stains in 72 pediatric patients (ages 5-16). Lidocaine and prilocaine cream, 2.5%/2.5% was effective in providing pain relief during laser treatment.
Lidocaine and prilocaine cream, 2.5%/2.5% alone was compared to lidocaine and prilocaine cream, 2.5%/2.5% followed by lidocaine infiltration and lidocaine infiltration alone prior to cryotherapy for the removal of male genital warts. The data from 121 patients demonstrated that lidocaine and prilocaine cream, 2.5%/2.5% was not effective as a sole anesthetic agent in managing the pain from the surgical procedure. The administration of lidocaine and prilocaine cream, 2.5%/2.5% prior to lidocaine infiltration provided significant relief of discomfort associated with local anesthetic infiltration and thus was effective in the overall reduction of pain from the procedure only when used in conjunction with local anesthetic infiltration of lidocaine.
Lidocaine and prilocaine cream, 2.5%/2.5% was studied in 105 full term neonates (gestational age: 37 weeks) for blood drawing and circumcision procedures. When considering the use of lidocaine and prilocaine cream, 2.5%/2.5% in neonates, the primary concerns are the systemic absorption of the active ingredients and the subsequent formation of methemoglobin. In clinical studies performed in neonates, the plasma levels of lidocaine, prilocaine, and methemoglobin were not reported in a range expected to cause clinical symptoms.
Local dermal effects associated with lidocaine and prilocaine cream, 2.5%/2.5% application in these studies on intact skin included paleness, redness and edema and were transient in nature (see ADVERSE REACTIONS).
The application of lidocaine and prilocaine cream, 2.5%/2.5% on genital mucous membranes for minor, superficial surgical procedures (eg, removal of condylomata acuminata) was studied in 80 patients in a placebo-controlled clinical trial (60 patients received lidocaine and prilocaine cream, 2.5%/2.5% and 20 patients received placebo). Lidocaine and prilocaine cream, 2.5%/2.5% (5 to 10 g) applied between 1 and 75 minutes before surgery, with a median time of 15 minutes, provided effective local anesthesia for minor superficial surgical procedures. The greatest extent of analgesia, as measured by VAS scores, was attained after 5 to 15 minutes. The application of lidocaine and prilocaine cream, 2.5%/2.5% to genital mucous membranes as pretreatment for local anesthetic infiltration was studied in a double-blind, placebo-controlled study in 44 female patients (21 patients received lidocaine and prilocaine cream, 2.5%/2.5% and 23 patients received placebo) scheduled for infiltration prior to a surgical procedure of the external vulva or genital mucosa. Lidocaine and prilocaine cream, 2.5%/2.5% applied to the genital mucous membranes for 5 to 10 minutes resulted in adequate topical anesthesia for local anesthetic injection.
Individualization of Dose: The dose of lidocaine and prilocaine cream, 2.5%/2.5% which provides effective analgesia depends on the duration of the application over the treated area.
All pharmacokinetic and clinical studies employed a thick layer of lidocaine and prilocaine cream, 2.5%/2.5% (1 to 2 g/10 cm2). The duration of application prior to venipuncture was 1 hour. The duration of application prior to taking split thickness skin grafts was 2 hours. A thinner application has not been studied and may result in less complete analgesia or a shorter duration of adequate analgesia.
The systemic absorption of lidocaine and prilocaine is a side effect of the desired local effect. The amount of drug absorbed depends on surface area and duration of application. The systemic blood levels depend on the amount absorbed and patient size (weight) and rate of systemic drug elimination. Long duration of application, large treatment area, small patients, or impaired elimination may result in high blood levels. The systemic blood levels are typically a small fraction (1/20 to 1/36) of the blood levels which produce toxicity. Table 2 below gives maximum recommended doses, application areas and application times for infants and children.
TABLE 2 LIDOCAINE AND PRILOCAINE CREAM, 2.5%/2.5% MAXIMUM RECOMMENDED DOSE, APPLICATION AREA, AND APPLICATION TIME BY AGE AND WEIGHT*
For Infants and Children Based on Application to Intact Skin
|
Age and Body Weight Requirements |
Maximum total Dose of |
Maximum |
Maximum |
|
0 up to 3 months or <5 kg |
1g |
10 cm2 |
1 hour |
|
3 up to 12 months and >5 kg |
2g |
20 cm2 |
4 hours |
|
1 to 6 years and >10 kg |
10g |
100 cm2 |
4 hours |
|
7 to 12 years and >20 kg |
20g |
200 cm2 |
4 hours |
Please note: If a patient greater than 3 months old does not meet the minimum weight requirement, the maximum total dose of lidocaine and prilocaine cream, 2.5%/2.5% should be restricted to that which corresponds to the patient's weight.
* These are broad guidelines for avoiding systemic toxicity in applying lidocaine and prilocaine cream, 2.5%/2.5% to patients with normal intact skin and with normal renal and hepatic function.
** For more individualized calculation of how much lidocaine and prilocaine may be absorbed, physicians can use the following estimates of lidocaine and prilocaine absorption for children and adults:
The estimated mean (±SD) absorption of lidocaine is 0.045 (±0.016) mg/cm2/hr.
The estimated mean (±SD) absorption of prilocaine is 0.077 (±0.036) mg/cm2/hr.
An IV antiarrhythmic dose of lidocaine is 1 mg/kg (70 mg/70 kg) and gives a blood level of about 1 mcg/mL. Toxicity would be expected at blood levels above 5 mcg/mL. Smaller areas of treatment are recommended in a debilitated patient, a small child or a patient with impaired elimination. Decreasing the duration of application is likely to decrease the analgesic effect.
Indications and Usage for DermacinRx Cinlone-II CPI
Lidocaine and prilocaine cream, 2.5%/2.5% (a eutectic mixture of lidocaine 2.5% and prilocaine 2.5%) is indicated as a topical anesthetic for use on:
- normal intact skin for local analgesia.
- genital mucous membranes for superficial minor surgery and as pretreatment for infiltration anesthesia.
Lidocaine and prilocaine cream, 2.5%/2.5% is not recommended in any clinical situation when penetration or migration beyond the tympanic membrane into the middle ear is possible because of the ototoxic effects observed in animal studies (see WARNINGS).
Contraindications
Lidocaine and prilocaine cream, 2.5%/2.5% is contraindicated in patients with a known history of sensitivity to local anesthetics of the amide type or to any other component of the product.
Warnings
Application of lidocaine and prilocaine cream, 2.5%/2.5% to larger areas or for longer times than those recommended could result in sufficient absorption of lidocaine and prilocaine resulting in serious adverse effects (see Individualization of Dose).
Patients treated with class III anti-arrhythmic drugs (eg, amiodarone, bretylium, sotalol, dofetilide) should be under close surveillance and ECG monitoring considered, because cardiac effects may be additive.
Studies in laboratory animals (guinea pigs) have shown that lidocaine and prilocaine cream, 2.5%/2.5% has an ototoxic effect when instilled into the middle ear. In these same studies, animals exposed to lidocaine and prilocaine cream, 2.5%/2.5% in the external auditory canal only, showed no abnormality. Lidocaine and prilocaine cream, 2.5%/2.5% should not be used in any clinical situation when its penetration or migration beyond the tympanic membrane into the middle ear is possible.
Methemoglobinemia: Lidocaine and prilocaine cream, 2.5%/2.5% should not be used in those rare patients with congenital or idiopathic methemoglobinemia and in infants under the age of twelve months who are receiving treatment with methemoglobin-inducing agents.
Very young patients or patients with glucose-6-phosphate dehydrogenase deficiencies are more susceptible to methemoglobinemia.
Patients taking drugs associated with drug-induced methemoglobinemia such as sulfonamides, acetaminophen, acetanilid, aniline dyes, benzocaine, chloroquine, dapsone, naphthalene, nitrates and nitrites, nitrofurantoin, nitroglycerin, nitroprusside, pamaquine, para-aminosalicylic acid, phenacetin, phenobarbital, phenytoin, primaquine, quinine, are also at greater risk for developing methemoglobinemia.
There have been reports of significant methemoglobinemia (20 to 30%) in infants and children following excessive applications of lidocaine and prilocaine cream, 2.5%/2.5%. These cases involved the use of large doses, larger than recommended areas of application, or infants under the age of 3 months who did not have fully mature enzyme systems. In addition, a few of these cases involved the concomitant administration of methemoglobin-inducing agents. Most patients recovered spontaneously after removal of the cream. Treatment with IV methylene blue may be effective if required.
Physicians are cautioned to make sure that parents or other caregivers understand the need for careful application of lidocaine and prilocaine cream, 2.5%/2.5%, to ensure that the doses and areas of application recommended in Table 2 are not exceeded (especially in children under the age of 3 months) and to limit the period of application to the minimum required to achieve the desired anesthesia.
Neonates and infants up to 3 months of age should be monitored for Met-Hb levels before, during, and after the application of lidocaine and prilocaine cream, 2.5%/2.5%, provided the test results can be obtained quickly.
Precautions
General: Repeated doses of lidocaine and prilocaine cream, 2.5%/2.5% may increase blood levels of lidocaine and prilocaine. Lidocaine and prilocaine cream, 2.5%/2.5% should be used with caution in patients who may be more sensitive to the systemic effects of lidocaine and prilocaine including acutely ill, debilitated, or elderly patients.
Lidocaine and prilocaine cream, 2.5%/2.5% should not be applied to open wounds. Care should be taken not to allow lidocaine and prilocaine cream, 2.5%/2.5% to come in contact with the eye because animal studies have demonstrated severe eye irritation. Also the loss of protective reflexes can permit corneal irritation and potential abrasion. Absorption of lidocaine and prilocaine cream, 2.5%/2.5% in conjunctival tissues has not been determined. If eye contact occurs, immediately wash out the eye with water or saline and protect the eye until sensation returns.
Patients allergic to para-aminobenzoic acid derivatives (procaine, tetracaine, benzocaine, etc.) have not shown cross sensitivity to lidocaine and/or prilocaine, however, lidocaine and prilocaine cream, 2.5%/2.5% should be used with caution in patients with a history of drug sensitivities, especially if the etiologic agent is uncertain.
Patients with severe hepatic disease, because of their inability to metabolize local anesthetics normally, are at greater risk of developing toxic plasma concentrations of lidocaine and prilocaine.
Lidocaine and prilocaine have been shown to inhibit viral and bacterial growth. The effect of lidocaine and prilocaine cream, 2.5%/2.5% on intradermal injections of live vaccines has not been determined.
Information for Patients: When lidocaine and prilocaine cream, 2.5%/2.5% is used, the patient should be aware that the production of dermal analgesia may be accompanied by the block of all sensations in the treated skin. For this reason, the patient should avoid inadvertent trauma to the treated area by scratching, rubbing, or exposure to extreme hot or cold temperatures until complete sensation has returned. Lidocaine and prilocaine cream, 2.5%/2.5% should not be applied near the eyes or on open wounds,
Drug Interactions: Lidocaine and prilocaine cream, 2.5%/2.5% should be used with caution in patients receiving Class I antiarrhythmic drugs (such as tocainide and mexiletine) since the toxic effects are additive and potentially synergistic.
Prilocaine may contribute to the formation of methemoglobin in patients treated with other drugs known to cause this condition (see Methemoglobinemia subsection of WARNINGS).
Specific interaction studies with lidocaine/prilocaine and class III anti-arrhythmic drugs (eg, amiodarone, bretylium, sotalol, dofetilide) have not been performed, but caution is advised (see WARNINGS).
Should lidocaine and prilocaine cream, 2.5%/2.5% be used concomitantly with other products containing lidocaine and/or prilocaine, cumulative doses from all formulations must be considered.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis- Long-term studies in animals designed to evaluate the carcinogenic potential of lidocaine and prilocaine have not been conducted.
Metabolites of prilocaine have been shown to be carcinogenic in laboratory animals. In the animal studies reported below, doses or blood levels are compared to the Single Dermal Administration (SDA) of 60 g of lidocaine and prilocaine cream, 2.5%/2.5% to 400 cm2 for 3 hours to a small person (50 kg). The typical application of lidocaine and prilocaine cream, 2.5%/2.5% for one or two treatments for venipuncture sites (2.5 or 5 g) would be 1/24 or 1/12 of that dose in an adult or about the same mg/kg dose in an infant.
Chronic oral toxicity studies of ortho-toluidine, a metabolite of prilocaine, in mice (450 to 7200 mg/m2; 60 to 960 times SDA) and rats (900 to 4,800 mg/m2; 60 to 320 times SDA) have shown that ortho-toluidine is a carcinogen in both species. The tumors included hepatocarcinomas/adenomas in female mice, multiple occurrences of hemangiosarcomas/hemangiomas in both sexes of mice, sarcomas of multiple organs, transitional-cell carcinomas/papillomas of urinary bladder in both sexes of rats, subcutaneous fibromas/fibrosarcomas and mesotheliomas in male rats, and mammary gland fibroadenomas/adenomas in female rats. The lowest dose tested (450 mg/m2 in mice; 900 mg/m2 in rats, 60 times SDA) was carcinogenic in both species. Thus the no-effect dose must be less than 60 times SDA. The animal studies were conducted at 150 to 2,400 mg/kg in mice and at 150 to 800 mg/kg in rats. The dosages have been converted to mg/m2 for the SDA calculations above.
Mutagenesis- The mutagenic potential of lidocaine HCl has been tested in a bacterial reverse (Ames) assay in Salmonella, an in vitro chromosomal aberration assay using human lymphocytes, an in vivo micronucleus test in mice. There was no indication of mutagenicity or structural damage to chromosomes in these tests.
Ortho-toluidine, a metabolite of prilocaine, at a concentration of 0.5 mcg/mL, was genotoxic in Escherichia coli DNA repair and phage-induction assays. Urine concentrates from rats treated with ortho-toluidine (300 mg/kg orally; 300 times SDA) were mutagenic when examined in Salmonella typhimuriumin the presence of metabolic activation. Several other tests on ortho-toluidine, including reverse mutations in five different Salmonella typhimurium strains in the presence or absence of metabolic activation and a study to detect single strand breaks in DNA of V79 Chinese hamster cells, were negative.
Use in Pregnancy: Teratogenic Effects: Pregnancy Category B.
Reproduction studies with lidocaine have been performed in rats and have revealed no evidence of harm to the fetus (30 mg/kg subcutaneously; 22 times SDA). Reproduction studies with prilocaine have been performed in rats and have revealed no evidence of impaired fertility or harm to the fetus (300 mg/kg intramuscularly; 188 times SDA). There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, lidocaine and prilocaine cream, 2.5%/2.5% should be used during pregnancy only if clearly needed.
Reproduction studies have been performed in rats receiving subcutaneous administration of an aqueous mixture containing lidocaine HCl and prilocaine HCl at 1:1 (w/w). At 40 mg/kg each, a dose equivalent to 29 times SDA lidocaine and 25 times SDA prilocaine, no teratogenic, embryotoxic or fetotoxic effects were observed.
Labor and Delivery: Neither lidocaine nor prilocaine are contraindicated in labor and delivery. Should lidocaine and prilocaine cream, 2.5%/2.5% be used concomitantly with other products containing lidocaine and/or prilocaine, cumulative doses from all formulations must be considered.
Nursing Mothers: Lidocaine, and probably prilocaine, are excreted in human milk. Therefore, caution should be exercised when lidocaine and prilocaine cream, 2.5%/2.5% is administered to a nursing mother since the milk:plasma ratio of lidocaine is 0.4 and is not determined for prilocaine.
Pediatric Use: Controlled studies of lidocaine and prilocaine cream, 2.5%/2.5% in children under the age of seven years have shown less overall benefit than in older children or adults. These results illustrate the importance of emotional and psychological support of younger children undergoing medical or surgical procedures.
Lidocaine and prilocaine cream, 2.5%/2.5% should be used with care in patients with conditions or therapy associated with methemoglobinemia (see Methemoglobinemia subsection of WARNINGS).
When using lidocaine and prilocaine cream, 2.5%/2.5% in young children, especially infants under the age of 3 months, care must be taken to insure that the caregiver understands the need to limit the dose and area of application, and to prevent accidental ingestion (see DOSAGE AND ADMINISTRATION and Methemoglobinemia).
In neonates (minimum gestation age: 37 weeks) and children weighing less than 20 kg, the area and duration of application should be limited (see TABLE 2 in Individualization of Dose). Studies have not demonstrated the efficacy of lidocaine and prilocaine cream, 2.5%/2.5% for heel lancing in neonates.
Geriatric Use
Of the total number of patients in clinical studies of lidocaine and prilocaine cream, 2.5%/2.5%, 180 were age 65 to 74 and 138 were 75 and over. No overall differences in safety or efficacy were observed between these patients and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Plasma levels of lidocaine and prilocaine in geriatric and non-geriatric patients following application of a thick layer of lidocaine and prilocaine cream, 2.5%/2.5% are very low and well below potentially toxic levels. However, there are no sufficient data to evaluate quantitative differences in systemic plasma levels of lidocaine and prilocaine between geriatric and non-geriatric patients following application of lidocaine and prilocaine cream, 2.5%/2.5%. Consideration should be given for those elderly patients who have enhanced sensitivity to systemic absorption. (See PRECAUTION).
After intravenous dosing, the elimination half-life of lidocaine is significantly longer in elderly patients (2.5 hours) than in younger patients (1.5 hours). (See CLINICAL PHARMACOLOGY).
Adverse Reactions/Side Effects
Localized Reactions: During or immediately after treatment with lidocaine and prilocaine cream, 2.5%/2.5% on intact skin, the skin at the site of treatment may develop erythema or edema or may be the locus of abnormal sensation. Rare cases of discrete purpuric or petechial reactions at the application site have been reported. Rare cases of hyperpigmentation following the use of lidocaine and prilocaine cream, 2.5%/2.5% have been reported. The relationship to lidocaine and prilocaine cream, 2.5%/2.5% or the underlying procedure has not been established. In clinical studies on intact skin involving over 1,300 lidocaine and prilocaine cream, 2.5%/2.5%-treated subjects, one or more such local reactions were noted in 56% of patients, and were generally mild and transient, resolving spontaneously within 1 or 2 hours. There were no serious reactions which were ascribed to lidocaine and prilocaine cream, 2.5%/2.5%.
Two recent reports describe blistering on the foreskin in neonates about to undergo circumcision. Both neonates received 1 g of lidocaine and prilocaine cream, 2.5%/2.5%.
In patients treated with lidocaine and prilocaine cream, 2.5%/2.5% on intact skin, local effects observed in the trials included: paleness (pallor or blanching) 37%, redness (erythema) 30%, alterations in temperature sensations 7%, edema 6%, itching 2% and rash, less than 1%.
In clinical studies on genital mucous membranes involving 378 lidocaine and prilocaine cream, 2.5%/2.5%-treated patients, one or more application site reactions, usually mild and transient, were noted in 41% of patients. The most common application site reactions were redness (21%), burning sensation (17%) and edema (10%).
Allergic Reactions: Allergic and anaphylactoid reactions associated with lidocaine or prilocaine can occur. They are characterized by urticaria, angioedema, bronchospasm, and shock. If they occur they should be managed by conventional means. The detection of sensitivity by skin testing is of doubtful value.
Systemic (Dose Related) Reactions: Systemic adverse reactions following appropriate use of lidocaine and prilocaine cream, 2.5%/2.5% are unlikely due to the small dose absorbed (see Pharmacokinetics subsection of CLINICAL PHARMACOLOGY). Systemic adverse effects of lidocaine and/or prilocaine are similar in nature to those observed with other amide local anesthetic agents including CNS excitation and/or depression (light-headedness, nervousness, apprehension, euphoria, confusion, dizziness, drowsiness, tinnitus, blurred or double vision, vomiting, sensations of heat, cold or numbness, twitching, tremors, convulsions, unconsciousness, respiratory depression and arrest). Excitatory CNS reactions may be brief or not occur at all, in which case the first manifestation may be drowsiness merging into unconsciousness. Cardiovascular manifestations may include bradycardia, hypotension and cardiovascular collapse leading to arrest.
Related/similar drugs
Overdosage
Peak blood levels following a 60 g application to 400 cm2 of intact skin for 3 hours are 0.05 to 0.16 mcg/mL for lidocaine and 0.02 to 0.10 mcg/mL for prilocaine. Toxic levels of lidocaine (>5 mcg/mL) and/or prilocaine (>6 mcg/mL) cause decreases in cardiac output, total peripheral resistance and mean arterial pressure. These changes may be attributable to direct depressant effects of these local anesthetic agents on the cardiovascular system. In the absence of massive topical overdose or oral ingestion, evaluation should include evaluation of other etiologies for the clinical effects or overdosage from other sources of lidocaine, prilocaine or other local anesthetics. Consult the package inserts for parenteral Xylocaine (lidocaine HCl) or Citanest (prilocaine HCl) for further information for the management of overdose.
DermacinRx Cinlone-II CPI Dosage and Administration
Adult Patients-Intact Skin
A thick layer of lidocaine and prilocaine cream, 2.5%/2.5% is applied to intact skin and covered with an occlusive dressing.
Minor Dermal Procedures: For minor procedures such as intravenous cannulation and venipuncture, apply 2.5 grams (1/2 the 5 g tube) of lidocaine and prilocaine cream, 2.5%/2.5% over 20 to 25 cm2 of skin surface for at least 1 hour. In controlled clinical trials using lidocaine and prilocaine cream, 2.5%/2.5%, two sites were usually prepared in case there was a technical problem with cannulation or venipuncture at the first site.
Major Dermal Procedures: For more painful dermatological procedures involving a larger skin area such as split thickness skin graft harvesting, apply 2 grams of lidocaine and prilocaine cream, 2.5%/2.5% per 10 cm2 of skin and allow to remain in contact with the skin for at least 2 hours.
Adult Male Genital Skin: As an adjunct prior to local anesthetic infiltration, apply a thick layer of lidocaine and prilocaine cream, 2.5%/2.5% (1 g/10 cm2) to the skin surface for 15 minutes. Local anesthetic infiltration should be performed immediately after removal of lidocaine and prilocaine cream, 2.5%/2.5%.
Dermal analgesia can be expected to increase for up to 3 hours under occlusive dressing and persist for 1 to 2 hours after removal of the cream. The amount of lidocaine and prilocaine absorbed during the period of application can be estimated from the information in Table 2, ** footnote, in Individualization of Dose.
Adult Female Patients-Genital Mucous Membranes
For minor procedures on the female external genitalia, such as removal of condylomata acuminata, as well as for use as pretreatment for anesthetic infiltration, apply a thick layer (5-10 grams) of lidocaine and prilocaine cream, 2.5%/2.5% for 5 to 10 minutes.
Occlusion is not necessary for absorption, but may be helpful to keep the cream in place. Patients should be lying down during the lidocaine and prilocaine cream, 2.5%/2.5% application, especially if no occlusion is used. The procedure or the local anesthetic infiltration should be performed immediately after the removal of lidocaine and prilocaine cream, 2.5%/2.5%.
Pediatric Patients-Intact Skin
The following are the maximum recommended doses, application areas and application times for lidocaine and prilocaine cream, 2.5%/2.5% based on a child's age and weight:
Practitionersshould carefully instruct caregivers to avoid application of excessive amounts of lidocaine and prilocaine cream, 2.5%/2.5% (see PRECAUTIONS).
|
Age and Body Weight Requirements |
Maximum total Dose of |
Maximum |
Maximum |
|
0 up to 3 months or <5 kg |
1g |
10 cm2 |
1 hour |
|
3 up to 12 months and >5 kg |
2g |
20 cm2 |
4 hours |
|
1 to 6 years and >10 kg |
10g |
100 cm2 |
4 hours |
|
7 to 12 years and >20 kg |
20g |
200 cm2 |
4 hours |
Please note: If a patient greater than 3 months old does not meet the minimum weight requirement, the maximum total dose of lidocaine and prilocaine cream, 2.5%/2.5% should be restricted to that which corresponds to the patient's weight.
When applying lidocaine and prilocaine cream, 2.5%/2.5% to the skin of young children, care must be taken to maintain careful observation of the child to prevent accidental ingestion of lidocaine and prilocaine cream, 2.5%/2.5% or the occlusive dressing. A secondary protective covering to prevent inadvertent disruption of the application site may be useful.
Lidocaine and prilocaine cream, 2.5%/2.5% should not be used in neonates with a gestational age less than 37 weeks nor in infants under the age of 12 months who are receiving treatment with methemoglobin-inducing agents (see Methemoglobinemia subsection of WARNINGS).
When lidocaine and prilocaine cream, 2.5%/2.5% is used concomitantly with other products containing local anesthetic agents, the amount absorbed from all formulations must be considered (see Individualization of Dose). The amount absorbed in the case of lidocaine and prilocaine cream, 2.5%/2.5% is determined by the area over which it is applied and the duration of application under occlusion (see Table 2, ** footnote, in Individualization of Dose).
Although the incidence of systemic adverse reactions with lidocaine and prilocaine cream, 2.5%/2.5% is very low, caution should be exercised, particularly when applying it over large areas and leaving it on for longer than 2 hours. The incidence of systemic adverse reactions can be expected to be directly proportional to the area and time of exposure (see Individualization of Dose).
How is DermacinRx Cinlone-II CPI supplied
Lidocaine and Prilocaine Cream, 2.5%/2.5% is available as follows:
NDC 0168-0357-05 5 gram tube
NOT FOR OPHTHALMIC USE.
KEEP CONTAINER TIGHTLY CLOSED AT ALL TIMES WHEN NOT IN USE.
WARNING: Keep out of reach of children.
Store at 20°-25°C (68°-77°F) (See USP Controlled Room Temperature).
INSTRUCTIONS FOR APPLICATION
Lidocaine and Prilocaine Cream, 2.5%/2.5%
NOT FOR OPHTHALMIC USE.
FOR EXTERNAL USE ONLY.
Rx only
1.In adults, apply 2.5 g of cream per 20 to 25 cm2(approx. 2 in. by 2 in.) of skin in a thick layer at the site of the procedure. For pediatric patients, apply ONLY as prescribed by your physician. If your child is below the age of 3 months or small for their age, please inform your doctor before applying lidocaine and prilocaine cream, 2.5%/2.5%, which can be harmful, if applied over too much skin at one time in young children. If your child becomes very dizzy, excessively sleepy, or develops duskiness of the face or lips after applying lidocaine and prilocaine cream, 2.5%/2.5%, remove the cream and contact your physician at once.
2.Take the dressing (may be supplied in this package) or other occlusive dressing and remove the center cut-out piece.
3.Peel the paper liner from the dressing. (Instructions continued on reverse side.)
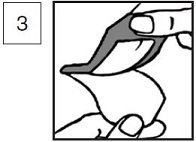
4.Cover the lidocaine and prilocaine cream, 2.5%/2.5% so that you get a thick layer underneath. Do not spread out the cream. Smooth down the dressing edges carefully and ensure it is secure to avoid leakage. (This is especially important when the patient is a child.)
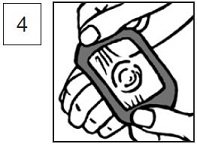
5.The time of application can be marked directly on the occlusive dressing. Lidocaine and prilocaine cream, 2.5%/2.5% must be applied at least 1 hour before the start of a routine procedure and for 2 hours before the start of a painful procedure.
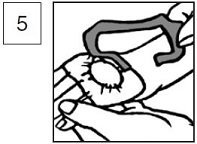
6.Remove the occlusive dressing, wipe off the lidocaine and prilocaine cream, 2.5%/2.5%, clean the entire area with an antiseptic solution and prepare the patient for the procedure. The duration of effective skin anesthesia will be at least 1 hour after removal of the occlusive dressing.
PRECAUTIONS
1. Do not apply near eyes or on open wounds.
2. Keep out of reach of children.
NOT FOR USE IN NEONATES
CONTAINS BENZYL ALCOHOL
For Intramuscular or Intra-articular Use Only
NOT FOR INTRAVENOUS, INTRADERMAL, INTRAOCULAR, EPIDURAL, OR INTRATHECAL USE
DermacinRx Cinlone-II CPI Description
Kenalog®-40 Injection (triamcinolone acetonide injectable suspension, USP) is a synthetic glucocorticoid corticosteroid with anti-inflammatory action. THIS FORMULATION IS SUITABLE FOR INTRAMUSCULAR AND INTRA-ARTICULAR USE ONLY. THIS FORMULATION IS NOT FOR INTRADERMAL INJECTION.
Each mL of the sterile aqueous suspension provides 40 mg triamcinolone acetonide, with 0.66% sodium chloride for isotonicity, 0.99% (w/v) benzyl alcohol as a preservative, 0.63% carboxymethylcellulose sodium, and 0.04% polysorbate 80. Sodium hydroxide or hydrochloric acid may be present to adjust pH to 5.0 to 7.5. At the time of manufacture, the air in the container is replaced by nitrogen.
The chemical name for triamcinolone acetonide is 9-Fluoro-11β,16α,17,21-tetrahydroxypregna-1,4-diene-3,20-dione cyclic 16,17-acetal with acetone. Its structural formula is:
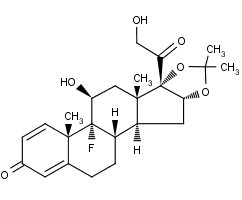
MW 434.50
Triamcinolone acetonide occurs as a white to cream-colored, crystalline powder having not more than a slight odor and is practically insoluble in water and very soluble in alcohol.
DermacinRx Cinlone-II CPI - Clinical Pharmacology
Glucocorticoids, naturally occurring and synthetic, are adrenocortical steroids that are readily absorbed from the gastrointestinal tract.
Naturally occurring glucocorticoids (hydrocortisone and cortisone), which also have salt-retaining properties, are used as replacement therapy in adrenocortical deficiency states. Synthetic analogs such as triamcinolone are primarily used for their anti-inflammatory effects in disorders of many organ systems.
Kenalog-40 Injection has an extended duration of effect which may be sustained over a period of several weeks. Studies indicate that following a single intramuscular dose of 60 mg to 100 mg of triamcinolone acetonide, adrenal suppression occurs within 24 to 48 hours and then gradually returns to normal, usually in 30 to 40 days. This finding correlates closely with the extended duration of therapeutic action achieved with the drug.
Indications and Usage for DermacinRx Cinlone-II CPI
Where oral therapy is not feasible, injectable corticosteroid therapy, including Kenalog-40 Injection (triamcinolone acetonide injectable suspension, USP) is indicated for intramuscular use as follows:
Allergic states: Control of severe or incapacitating allergic conditions intractable to adequate trials of conventional treatment in asthma, atopic dermatitis, contact dermatitis, drug hypersensitivity reactions, perennial or seasonal allergic rhinitis, serum sickness, transfusion reactions.
Dermatologic diseases: Bullous dermatitis herpetiformis, exfoliative erythroderma, mycosis fungoides, pemphigus, severe erythema multiforme (Stevens-Johnson syndrome).
Endocrine disorders: Primary or secondary adrenocortical insufficiency (hydrocortisone or cortisone is the drug of choice; synthetic analogs may be used in conjunction with mineralocorticoids where applicable; in infancy, mineralocorticoid supplementation is of particular importance), congenital adrenal hyperplasia, hypercalcemia associated with cancer, nonsuppurative thyroiditis.
Gastrointestinal diseases: To tide the patient over a critical period of the disease in regional enteritis and ulcerative colitis.
Hematologic disorders: Acquired (autoimmune) hemolytic anemia, Diamond-Blackfan anemia, pure red cell aplasia, selected cases of secondary thrombocytopenia.
Miscellaneous: Trichinosis with neurologic or myocardial involvement, tuberculous meningitis with subarachnoid block or impending block when used with appropriate antituberculous chemotherapy.
Neoplastic diseases: For the palliative management of leukemias and lymphomas.
Nervous system: Acute exacerbations of multiple sclerosis; cerebral edema associated with primary or metastatic brain tumor or craniotomy.
Ophthalmic diseases: Sympathetic ophthalmia, temporal arteritis, uveitis, and ocular inflammatory conditions unresponsive to topical corticosteroids.
Renal diseases: To induce diuresis or remission of proteinuria in idiopathic nephrotic syndrome or that due to lupus erythematosus.
Respiratory diseases: Berylliosis, fulminating or disseminated pulmonary tuberculosis when used concurrently with appropriate antituberculous chemotherapy, idiopathic eosinophilic pneumonias, symptomatic sarcoidosis.
Rheumatic disorders: As adjunctive therapy for short-term administration (to tide the patient over an acute episode or exacerbation) in acute gouty arthritis; acute rheumatic carditis; ankylosing spondylitis; psoriatic arthritis; rheumatoid arthritis, including juvenile rheumatoid arthritis (selected cases may require low-dose maintenance therapy). For the treatment of dermatomyositis, polymyositis, and systemic lupus erythematosus.
Intra-Articular
The intra-articular or soft tissue administration of Kenalog-40 Injection is indicated as adjunctive therapy for short-term administration (to tide the patient over an acute episode or exacerbation) in acute gouty arthritis, acute and subacute bursitis, acute nonspecific tenosynovitis, epicondylitis, rheumatoid arthritis, synovitis, or osteoarthritis.
Contraindications
Kenalog-40 Injection is contraindicated in patients who are hypersensitive to any components of this product (see WARNINGS: General).
Intramuscular corticosteroid preparations are contraindicated for idiopathic thrombocytopenic purpura.
Warnings
Serious Neurologic Adverse Reactions with Epidural Administration
Serious neurologic events, some resulting in death, have been reported with epidural injection of corticosteroids (see WARNINGS: Neurologic). Specific events reported include, but are not limited to, spinal cord infarction, paraplegia, quadriplegia, cortical blindness, and stroke. These serious neurologic events have been reported with and without use of fluoroscopy. The safety and effectiveness of epidural administration of corticosteroids have not been established, and corticosteroids are not approved for this use.
General
Exposure to excessive amounts of benzyl alcohol has been associated with toxicity (hypotension, metabolic acidosis), particularly in neonates, and an increased incidence of kernicterus, particularly in small preterm infants. There have been rare reports of deaths, primarily in preterm infants, associated with exposure to excessive amounts of benzyl alcohol. The amount of benzyl alcohol from medications is usually considered negligible compared to that received in flush solutions containing benzyl alcohol. Administration of high dosages of medications containing this preservative must take into account the total amount of benzyl alcohol administered. The amount of benzyl alcohol at which toxicity may occur is not known. If the patient requires more than the recommended dosages or other medications containing this preservative, the practitioner must consider the daily metabolic load of benzyl alcohol from these combined sources (see PRECAUTIONS: Pediatric Use).
Rare instances of anaphylaxis have occurred in patients receiving corticosteroid therapy (see ADVERSE REACTIONS). Cases of serious anaphylaxis, including death, have been reported in individuals receiving triamcinolone acetonide injection, regardless of the route of administration.
Because Kenalog-40 Injection (triamcinolone acetonide injectable suspension, USP) is a suspension, it should not be administered intravenously.
Unless a deep intramuscular injection is given, local atrophy is likely to occur. (For recommendations on injection techniques, see DOSAGE AND ADMINISTRATION.) Due to the significantly higher incidence of local atrophy when the material is injected into the deltoid area, this injection site should be avoided in favor of the gluteal area.
Increased dosage of rapidly acting corticosteroids is indicated in patients on corticosteroid therapy subjected to any unusual stress before, during, and after the stressful situation. Kenalog-40 Injection is a long-acting preparation, and is not suitable for use in acute stress situations. To avoid drug-induced adrenal insufficiency, supportive dosage may be required in times of stress (such as trauma, surgery, or severe illness) both during treatment with Kenalog-40 Injection and for a year afterwards.
Results from one multicenter, randomized, placebo-controlled study with methylprednisolone hemisuccinate, an intravenous corticosteroid, showed an increase in early (at 2 weeks) and late (at 6 months) mortality in patients with cranial trauma who were determined not to have other clear indications for corticosteroid treatment. High doses of systemic corticosteroids, including Kenalog-40 Injection, should not be used for the treatment of traumatic brain injury.
Cardio-Renal
Average and large doses of corticosteroids can cause elevation of blood pressure, salt and water retention, and increased excretion of potassium. These effects are less likely to occur with the synthetic derivatives except when they are used in large doses. Dietary salt restriction and potassium supplementation may be necessary (see PRECAUTIONS). All corticosteroids increase calcium excretion.
Literature reports suggest an apparent association between use of corticosteroids and left ventricular free wall rupture after a recent myocardial infarction; therefore, therapy with corticosteroids should be used with great caution in these patients.
Endocrine
Corticosteroids can produce reversible hypothalamic-pituitary-adrenal (HPA) axis suppression with the potential for glucocorticosteroid insufficiency after withdrawal of treatment.
Metabolic clearance of corticosteroids is decreased in hypothyroid patients and increased in hyperthyroid patients. Changes in thyroid status of the patient may necessitate adjustment in dosage.
Infections
General
Patients who are on corticosteroids are more susceptible to infections than are healthy individuals. There may be decreased resistance and inability to localize infection when corticosteroids are used. Infection with any pathogen (viral, bacterial, fungal, protozoan, or helminthic) in any location of the body may be associated with the use of corticosteroids alone or in combination with other immunosuppressive agents. These infections may be mild to severe. With increasing doses of corticosteroids, the rate of occurrence of infectious complications increases. Corticosteroids may also mask some signs of current infection.
Fungal Infections
Corticosteroids may exacerbate systemic fungal infections and therefore should not be used in the presence of such infections unless they are needed to control drug reactions. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure (see PRECAUTIONS: Drug Interactions:Amphotericin B injection and potassium-depleting agents).
Special Pathogens
Latent disease may be activated or there may be an exacerbation of intercurrent infections due to pathogens, including those caused by Amoeba, Candida, Cryptococcus, Mycobacterium, Nocardia, Pneumocystis, or Toxoplasma.
It is recommended that latent amebiasis or active amebiasis be ruled out before initiating corticosteroid therapy in any patient who has spent time in the tropics or in any patient with unexplained diarrhea.
Similarly, corticosteroids should be used with great care in patients with known or suspected Strongyloides (threadworm) infestation. In such patients, corticosteroid-induced immunosuppression may lead to Strongyloides hyperinfection and dissemination with widespread larval migration, often accompanied by severe enterocolitis and potentially fatal gram-negative septicemia.
Corticosteroids should not be used in cerebral malaria.
Tuberculosis
The use of corticosteroids in patients with active tuberculosis should be restricted to those cases of fulminating or disseminated tuberculosis in which the corticosteroid is used for the management of the disease in conjunction with an appropriate anti-tuberculosis regimen. If corticosteroids are indicated in patients with latent tuberculosis or tuberculin reactivity, close observation is necessary as reactivation of the disease may occur. During prolonged corticosteroid therapy, these patients should receive chemoprophylaxis.
Vaccination
Administration of live or live, attenuated vaccines is contraindicated in patients receiving immunosuppressive doses of corticosteroids. Killed or inactivated vaccines may be administered. However, the response to such vaccines cannot be predicted. Immunization procedures may be undertaken in patients who are receiving corticosteroids as replacement therapy, eg, for Addison’s disease.
Viral Infections
Chicken pox and measles can have a more serious or even fatal course in pediatric and adult patients on corticosteroids. In pediatric and adult patients who have not had these diseases, particular care should be taken to avoid exposure. The contribution of the underlying disease and/or prior corticosteroid treatment to the risk is also not known. If exposed to chicken pox, prophylaxis with varicella zoster immune globulin (VZIG) may be indicated. If exposed to measles, prophylaxis with immunoglobulin (IG) may be indicated. (See the respective package inserts for complete VZIG and IG prescribing information.) If chicken pox develops, treatment with antiviral agents should be considered.
Neurologic
Epidural and intrathecal administration of this product is not recommended. Reports of serious medical events, including death, have been associated with epidural and intrathecal routes of corticosteroid administration (see ADVERSE REACTIONS:Gastrointestinal and Neurologic/Psychiatric).
Ophthalmic
Use of corticosteroids may produce posterior subcapsular cataracts, glaucoma with possible damage to the optic nerves, and may enhance the establishment of secondary ocular infections due to bacteria, fungi, or viruses. The use of oral corticosteroids is not recommended in the treatment of optic neuritis and may lead to an increase in the risk of new episodes. Corticosteroids should not be used in active ocular herpes simplex.
Adequate studies to demonstrate the safety of Kenalog Injection use by intraturbinal, subconjunctival, sub-Tenons, retrobulbar, and intraocular (intravitreal) injections have not been performed. Endophthalmitis, eye inflammation, increased intraocular pressure, and visual disturbances including vision loss have been reported with intravitreal administration. Administration of Kenalog Injection intraocularly or into the nasal turbinates is not recommended.
Intraocular injection of corticosteroid formulations containing benzyl alcohol, such as Kenalog Injection, is not recommended because of potential toxicity from the benzyl alcohol.
Precautions
General
This product, like many other steroid formulations, is sensitive to heat. Therefore, it should not be autoclaved when it is desirable to sterilize the exterior of the vial.
The lowest possible dose of corticosteroid should be used to control the condition under treatment. When reduction in dosage is possible, the reduction should be gradual.
Since complications of treatment with glucocorticoids are dependent on the size of the dose and the duration of treatment, a risk/benefit decision must be made in each individual case as to dose and duration of treatment and as to whether daily or intermittent therapy should be used.
Kaposi’s sarcoma has been reported to occur in patients receiving corticosteroid therapy, most often for chronic conditions. Discontinuation of corticosteroids may result in clinical improvement.
Cardio-Renal
As sodium retention with resultant edema and potassium loss may occur in patients receiving corticosteroids, these agents should be used with caution in patients with congestive heart failure, hypertension, or renal insufficiency.
Endocrine
Drug-induced secondary adrenocortical insufficiency may be minimized by gradual reduction of dosage. This type of relative insufficiency may persist for months after discontinuation of therapy; therefore, in any situation of stress occurring during that period, hormone therapy should be reinstituted. Since mineralocorticoid secretion may be impaired, salt and/or a mineralocorticoid should be administered concurrently.
Gastrointestinal
Steroids should be used with caution in active or latent peptic ulcers, diverticulitis, fresh intestinal anastomoses, and nonspecific ulcerative colitis, since they may increase the risk of a perforation.
Signs of peritoneal irritation following gastrointestinal perforation in patients receiving corticosteroids may be minimal or absent.
There is an enhanced effect of corticosteroids in patients with cirrhosis.
Intra-Articular and Soft Tissue Administration
Intra-articularly injected corticosteroids may be systemically absorbed.
Appropriate examination of any joint fluid present is necessary to exclude a septic process.
A marked increase in pain accompanied by local swelling, further restriction of joint motion, fever, and malaise are suggestive of septic arthritis. If this complication occurs and the diagnosis of sepsis is confirmed, appropriate antimicrobial therapy should be instituted.
Injection of a steroid into an infected site is to be avoided. Local injection of a steroid into a previously infected joint is not usually recommended.
Corticosteroid injection into unstable joints is generally not recommended.
Intra-articular injection may result in damage to joint tissues (see ADVERSE REACTIONS:Musculoskeletal).
Musculoskeletal
Corticosteroids decrease bone formation and increase bone resorption both through their effect on calcium regulation (ie, decreasing absorption and increasing excretion) and inhibition of osteoblast function. This, together with a decrease in the protein matrix of the bone secondary to an increase in protein catabolism, and reduced sex hormone production, may lead to inhibition of bone growth in pediatric patients and the development of osteoporosis at any age. Special consideration should be given to patients at increased risk of osteoporosis (ie, postmenopausal women) before initiating corticosteroid therapy.
Neuro-Psychiatric
Although controlled clinical trials have shown corticosteroids to be effective in speeding the resolution of acute exacerbations of multiple sclerosis, they do not show that they affect the ultimate outcome or natural history of the disease. The studies do show that relatively high doses of corticosteroids are necessary to demonstrate a significant effect. (See DOSAGE AND ADMINISTRATION.)
An acute myopathy has been observed with the use of high doses of corticosteroids, most often occurring in patients with disorders of neuromuscular transmission (eg, myasthenia gravis), or in patients receiving concomitant therapy with neuromuscular blocking drugs (eg, pancuronium). This acute myopathy is generalized, may involve ocular and respiratory muscles, and may result in quadriparesis. Elevation of creatinine kinase may occur. Clinical improvement or recovery after stopping corticosteroids may require weeks to years.
Psychiatric derangements may appear when corticosteroids are used, ranging from euphoria, insomnia, mood swings, personality changes, and severe depression to frank psychotic manifestations. Also, existing emotional instability or psychotic tendencies may be aggravated by corticosteroids.
Ophthalmic
Intraocular pressure may become elevated in some individuals. If steroid therapy is continued for more than 6 weeks, intraocular pressure should be monitored.
Information for Patients
Patients should be warned not to discontinue the use of corticosteroids abruptly or without medical supervision, to advise any medical attendants that they are taking corticosteroids, and to seek medical advice at once should they develop fever or other signs of infection.
Persons who are on corticosteroids should be warned to avoid exposure to chicken pox or measles. Patients should also be advised that if they are exposed, medical advice should be sought without delay.
Drug Interactions
Aminoglutethimide: Aminoglutethimide may lead to a loss of corticosteroid-induced adrenal suppression.
Amphotericin B injection and potassium-depleting agents: When corticosteroids are administered concomitantly with potassium-depleting agents (ie, amphotericin B, diuretics), patients should be observed closely for development of hypokalemia. There have been cases reported in which concomitant use of amphotericin B and hydrocortisone was followed by cardiac enlargement and congestive heart failure.
Antibiotics: Macrolide antibiotics have been reported to cause a significant decrease in corticosteroid clearance.
Anticholinesterases: Concomitant use of anticholinesterase agents and corticosteroids may produce severe weakness in patients with myasthenia gravis. If possible, anticholinesterase agents should be withdrawn at least 24 hours before initiating corticosteroid therapy.
Anticoagulants, oral: Coadministration of corticosteroids and warfarin usually results in inhibition of response to warfarin, although there have been some conflicting reports. Therefore, coagulation indices should be monitored frequently to maintain the desired anticoagulant effect.
Antidiabetics: Because corticosteroids may increase blood glucose concentrations, dosage adjustments of antidiabetic agents may be required.
Antitubercular drugs: Serum concentrations of isoniazid may be decreased.
Cholestyramine: Cholestyramine may increase the clearance of corticosteroids.
Cyclosporine: Increased activity of both cyclosporine and corticosteroids may occur when the two are used concurrently. Convulsions have been reported with this concurrent use.
Digitalis glycosides: Patients on digitalis glycosides may be at increased risk of arrhythmias due to hypokalemia.
Estrogens, including oral contraceptives: Estrogens may decrease the hepatic metabolism of certain corticosteroids, thereby increasing their effect.
Hepatic enzyme inducers (eg, barbiturates, phenytoin, carbamazepine, rifampin): Drugs which induce hepatic microsomal drug metabolizing enzyme activity may enhance the metabolism of corticosteroids and require that the dosage of the corticosteroid be increased.
Ketoconazole: Ketoconazole has been reported to decrease the metabolism of certain corticosteroids by up to 60%, leading to an increased risk of corticosteroid side effects.
Nonsteroidal anti-inflammatory drugs (NSAIDs): Concomitant use of aspirin (or other nonsteroidal anti-inflammatory drugs) and corticosteroids increases the risk of gastrointestinal side effects. Aspirin should be used cautiously in conjunction with corticosteroids in hypoprothrombinemia. The clearance of salicylates may be increased with concurrent use of corticosteroids.
Skin tests: Corticosteroids may suppress reactions to skin tests.
Vaccines: Patients on prolonged corticosteroid therapy may exhibit a diminished response to toxoids and live or inactivated vaccines due to inhibition of antibody response. Corticosteroids may also potentiate the replication of some organisms contained in live attenuated vaccines. Routine administration of vaccines or toxoids should be deferred until corticosteroid therapy is discontinued if possible (see WARNINGS: Infections: Vaccination).
Carcinogenesis, Mutagenesis, Impairment of Fertility
No adequate studies have been conducted in animals to determine whether corticosteroids have a potential for carcinogenesis or mutagenesis.
Steroids may increase or decrease motility and number of spermatozoa in some patients.
Pregnancy
Teratogenic Effects: Pregnancy Category C
Corticosteroids have been shown to be teratogenic in many species when given in doses equivalent to the human dose. Animal studies in which corticosteroids have been given to pregnant mice, rats, and rabbits have yielded an increased incidence of cleft palate in the offspring. There are no adequate and well-controlled studies in pregnant women. Corticosteroids should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. Infants born to mothers who have received corticosteroids during pregnancy should be carefully observed for signs of hypoadrenalism.
Nursing Mothers
Systemically administered corticosteroids appear in human milk and could suppress growth, interfere with endogenous corticosteroid production, or cause other untoward effects. Caution should be exercised when corticosteroids are administered to a nursing woman.
Pediatric Use
This product contains benzyl alcohol as a preservative. Benzyl alcohol, a component of this product, has been associated with serious adverse events and death, particularly in pediatric patients. The “gasping syndrome” (characterized by central nervous system depression, metabolic acidosis, gasping respirations, and high levels of benzyl alcohol and its metabolites found in the blood and urine) has been associated with benzyl alcohol dosages >99 mg/kg/day in neonates and low-birth-weight neonates. Additional symptoms may include gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Although normal therapeutic doses of this product deliver amounts of benzyl alcohol that are substantially lower than those reported in association with the “gasping syndrome,” the minimum amount of benzyl alcohol at which toxicity may occur is not known. Premature and low-birth-weight infants, as well as patients receiving high dosages, may be more likely to develop toxicity. Practitioners administering this and other medications containing benzyl alcohol should consider the combined daily metabolic load of benzyl alcohol from all sources.
The efficacy and safety of corticosteroids in the pediatric population are based on the well-established course of effect of corticosteroids which is similar in pediatric and adult populations. Published studies provide evidence of efficacy and safety in pediatric patients for the treatment of nephrotic syndrome (>2 years of age), and aggressive lymphomas and leukemias (>1 month of age). Other indications for pediatric use of corticosteroids, eg, severe asthma and wheezing, are based on adequate and well-controlled trials conducted in adults, on the premises that the course of the diseases and their pathophysiology are considered to be substantially similar in both populations.
The adverse effects of corticosteroids in pediatric patients are similar to those in adults (see ADVERSE REACTIONS). Like adults, pediatric patients should be carefully observed with frequent measurements of blood pressure, weight, height, intraocular pressure, and clinical evaluation for the presence of infection, psychosocial disturbances, thromboembolism, peptic ulcers, cataracts, and osteoporosis. Pediatric patients who are treated with corticosteroids by any route, including systemically administered corticosteroids, may experience a decrease in their growth velocity. This negative impact of corticosteroids on growth has been observed at low systemic doses and in the absence of laboratory evidence of HPA axis suppression (ie, cosyntropin stimulation and basal cortisol plasma levels). Growth velocity may therefore be a more sensitive indicator of systemic corticosteroid exposure in pediatric patients than some commonly used tests of HPA axis function. The linear growth of pediatric patients treated with corticosteroids should be monitored, and the potential growth effects of prolonged treatment should be weighed against clinical benefits obtained and the availability of treatment alternatives. In order to minimize the potential growth effects of corticosteroids, pediatric patients should be titrated to the lowest effective dose.
Geriatric Use
No overall differences in safety or effectiveness were observed between elderly subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Adverse Reactions/Side Effects
(listed alphabetically under each subsection)
The following adverse reactions may be associated with corticosteroid therapy:
Allergic reactions: Anaphylaxis including death, angioedema.
Cardiovascular: Bradycardia, cardiac arrest, cardiac arrhythmias, cardiac enlargement, circulatory collapse, congestive heart failure, fat embolism, hypertension, hypertrophic cardiomyopathy in premature infants, myocardial rupture following recent myocardial infarction (see WARNINGS), pulmonary edema, syncope, tachycardia, thromboembolism, thrombophlebitis, vasculitis.
Dermatologic: Acne, allergic dermatitis, cutaneous and subcutaneous atrophy, dry scaly skin, ecchymoses and petechiae, edema, erythema, hyperpigmentation, hypopigmentation, impaired wound healing, increased sweating, lupus erythematosus-like lesions, purpura, rash, sterile abscess, striae, suppressed reactions to skin tests, thin fragile skin, thinning scalp hair, urticaria.
Endocrine: Decreased carbohydrate and glucose tolerance, development of cushingoid state, glycosuria, hirsutism, hypertrichosis, increased requirements for insulin or oral hypoglycemic agents in diabetes, manifestations of latent diabetes mellitus, menstrual irregularities, secondary adrenocortical and pituitary unresponsiveness (particularly in times of stress, as in trauma, surgery, or illness), suppression of growth in pediatric patients.
Fluid and electrolyte disturbances: Congestive heart failure in susceptible patients, fluid retention, hypokalemic alkalosis, potassium loss, sodium retention.
Gastrointestinal: Abdominal distention, bowel/bladder dysfunction (after intrathecal administration [see WARNINGS: Neurologic]), elevation in serum liver enzyme levels (usually reversible upon discontinuation), hepatomegaly, increased appetite, nausea, pancreatitis, peptic ulcer with possible perforation and hemorrhage, perforation of the small and large intestine (particularly in patients with inflammatory bowel disease), ulcerative esophagitis.
Metabolic: Negative nitrogen balance due to protein catabolism.
Musculoskeletal: Aseptic necrosis of femoral and humeral heads, calcinosis (following intra-articular or intralesional use), Charcot-like arthropathy, loss of muscle mass, muscle weakness, osteoporosis, pathologic fracture of long bones, post injection flare (following intra-articular use), steroid myopathy, tendon rupture, vertebral compression fractures.
Neurologic/Psychiatric: Convulsions, depression, emotional instability, euphoria, headache, increased intracranial pressure with papilledema (pseudotumor cerebri) usually following discontinuation of treatment, insomnia, mood swings, neuritis, neuropathy, paresthesia, personality changes, psychiatric disorders, vertigo. Arachnoiditis, meningitis, paraparesis/paraplegia, and sensory disturbances have occurred after intrathecal administration. Spinal cord infarction, paraplegia, quadriplegia, cortical blindness, and stroke (including brainstem) have been reported after epidural administration of corticosteroids (see WARNINGS: Serious Neurologic Adverse Reactions with Epidural Administration and WARNINGS: Neurologic).
Ophthalmic: Exophthalmos, glaucoma, increased intraocular pressure, posterior subcapsular cataracts, rare instances of blindness associated with periocular injections.
Other: Abnormal fat deposits, decreased resistance to infection, hiccups, increased or decreased motility and number of spermatozoa, malaise, moon face, weight gain.
Overdosage
Treatment of acute overdosage is by supportive and symptomatic therapy. For chronic overdosage in the face of severe disease requiring continuous steroid therapy, the dosage of the corticosteroid may be reduced only temporarily, or alternate day treatment may be introduced.
DermacinRx Cinlone-II CPI Dosage and Administration
General
NOTE: CONTAINS BENZYL ALCOHOL(see PRECAUTIONS).
The initial dose of Kenalog-40 Injection may vary from 2.5 mg to 100 mg per day depending on the specific disease entity being treated (see DOSAGE section below). However, in certain overwhelming, acute, life-threatening situations, administration in dosages exceeding the usual dosages may be justified and may be in multiples of the oral dosages.
IT SHOULD BE EMPHASIZED THAT DOSAGE REQUIREMENTS ARE VARIABLE AND MUST BE INDIVIDUALIZED ON THE BASIS OF THE DISEASE UNDER TREATMENT AND THE RESPONSE OF THE PATIENT. After a favorable response is noted, the proper maintenance dosage should be determined by decreasing the initial drug dosage in small decrements at appropriate time intervals until the lowest dosage which will maintain an adequate clinical response is reached. Situations which may make dosage adjustments necessary are changes in clinical status secondary to remissions or exacerbations in the disease process, the patients individual drug responsiveness, and the effect of patient exposure to stressful situations not directly related to the disease entity under treatment. In this latter situation it may be necessary to increase the dosage of the corticosteroid for a period of time consistent with the patients condition. If after long-term therapy the drug is to be stopped, it is recommended that it be withdrawn gradually rather than abruptly.
Dosage
SYSTEMIC
The suggested initial dose is 60 mg, injected deeply into the gluteal muscle. Atrophy of subcutaneous fat may occur if the injection is not properly given. Dosage is usually adjusted within the range of 40 mg to 80 mg, depending upon patient response and duration of relief. However, some patients may be well controlled on doses as low as 20 mg or less.
Hay fever or pollen asthma: Patients with hay fever or pollen asthma who are not responding to pollen administration and other conventional therapy may obtain a remission of symptoms lasting throughout the pollen season after a single injection of 40 mg to 100 mg.
In the treatment of acute exacerbations of multiple sclerosis, daily doses of 160 mg of triamcinolone for a week followed by 64 mg every other day for one month are recommended (see PRECAUTIONS: NEURO-PSYCHIATRIC).
In pediatric patients, the initial dose of triamcinolone may vary depending on the specific disease entity being treated. The range of initial doses is 0.11 to 1.6 mg/kg/day in 3 or 4 divided doses (3.2 to 48 mg/m2bsa/day).
For the purpose of comparison, the following is the equivalent milligram dosage of the various glucocorticoids:
|
Cortisone, 25 |
Triamcinolone, 4 |
|
Hydrocortisone, 20 |
Paramethasone, 2 |
|
Prednisolone, 5 |
Betamethasone, 0.75 |
|
Prednisone, 5 |
Dexamethasone, 0.75 |
|
Methylprednisolone, 4 |
These dose relationships apply only to oral or intravenous administration of these compounds. When these substances or their derivatives are injected intramuscularly or into joint spaces, their relative properties may be greatly altered.
LOCAL
Intra-articular administration: A single local injection of triamcinolone acetonide is frequently sufficient, but several injections may be needed for adequate relief of symptoms.
Initial dose: 2.5 mg to 5 mg for smaller joints and from 5 mg to 15 mg for larger joints, depending on the specific disease entity being treated. For adults, doses up to 10 mg for smaller areas and up to 40 mg for larger areas have usually been sufficient. Single injections into several joints, up to a total of 80 mg, have been given.
Administration
GENERAL
STRICT ASEPTIC TECHNIQUE IS MANDATORY. The vial should be shaken before use to ensure a uniform suspension. Prior to withdrawal, the suspension should be inspected for clumping or granular appearance (agglomeration). An agglomerated product results from exposure to freezing temperatures and should not be used. After withdrawal, Kenalog-40 Injection should be injected without delay to prevent settling in the syringe. Careful technique should be employed to avoid the possibility of entering a blood vessel or introducing infection.
SYSTEMIC
For systemic therapy, injection should be made deeply into the gluteal muscle (see WARNINGS). For adults, a minimum needle length of 1½ inches is recommended. In obese patients, a longer needle may be required. Use alternative sites for subsequent injections.
LOCAL
For treatment of joints, the usual intra-articular injection technique should be followed. If an excessive amount of synovial fluid is present in the joint, some, but not all, should be aspirated to aid in the relief of pain and to prevent undue dilution of the steroid.
With intra-articular administration, prior use of a local anesthetic may often be desirable. Care should be taken with this kind of injection, particularly in the deltoid region, to avoid injecting the suspension into the tissues surrounding the site, since this may lead to tissue atrophy.
In treating acute nonspecific tenosynovitis, care should be taken to ensure that the injection of the corticosteroid is made into the tendon sheath rather than the tendon substance. Epicondylitis may be treated by infiltrating the preparation into the area of greatest tenderness.
How is DermacinRx Cinlone-II CPI supplied
Kenalog®-40 Injection (triamcinolone acetonide injectable suspension, USP) is supplied in vials providing 40 mg triamcinolone acetonide per mL.
|
40 mg/mL, 1 mL vial |
NDC 0003-0293-05 |
Storage
Store at controlled room temperature, 20°–25°C (68°–77°F), avoid freezing and protect from light. Do not refrigerate.
DermacinRx® Cinlone-II™ CPI(NDC 59088-352-00)RX Only
Cinlone-II™ contains:
(1) Lidocaine and Prilocaine Cream, 2.5% / 2.5%, 5 g
(2) Kenalog-40 (Triamcinolone Acetonide Injectable Suspension, USP), 40 mg / mL, 1 mL†
(1) Povidone-Iodine Swabsticks, 3 swabs
(4) Isopropyl Alcohol Prep Pad
(1) Gauze Pad, 4" x 4"
†For full prescribing information of Kenalog-40, please see insert enclosed in the Kenalog-40 carton.
Packaged in the USA by:
Puretek Corporation
San Fernando, CA 91340
For questions or informationcall toll-free: 877-921-7873
Visit: dermacinrx.com
Rev. 37806 05/16
| DERMACINRX CINLONE-II CPI
lidocaine and prilocaine, triamcinolone acetonide kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - PureTek Corporation (785961046) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| PureTek Corporation | 785961046 | pack(59088-352) | |
More about lidocaine / prilocaine topical
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (30)
- Side effects
- Dosage information
- During pregnancy
- Drug class: topical anesthetics
- En español
Patient resources
Professional resources
Other brands
Emla, Dolotranz, Lidotor, Oraqix, ... +23 more