Caplyta: Package Insert / Prescribing Info
Package insert / product label
Generic name: lumateperone
Dosage form: capsule
Drug class: Atypical antipsychotics
Medically reviewed by Drugs.com. Last updated on Jul 24, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
CAPLYTA® (lumateperone) capsules, for oral use
Initial U.S. Approval: 2019
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS; and SUICIDAL THOUGHTS AND BEHAVIORS
See full prescribing information for complete boxed warning.
- Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. CAPLYTA is not approved for the treatment of patients with dementia-related psychosis. (5.1)
- Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adult patients. Closely monitor all antidepressant-treated patients for worsening and emergence of suicidal thoughts and behaviors. Safety and effectiveness of CAPLYTA have not been established in pediatric patients. (5.2, 8.4)
Indications and Usage for Caplyta
Caplyta Dosage and Administration
Dosage Forms and Strengths
Capsules: 42 mg, 21 mg, 10.5 mg (3)
Contraindications
Known hypersensitivity to lumateperone or any components of CAPLYTA. (4)
Warnings and Precautions
- Cerebrovascular Adverse Reactions in Elderly Patients with Dementia-Related Psychosis: Increased incidence of cerebrovascular adverse reactions (e.g., stroke and transient ischemic attack). (5.3)
- Neuroleptic Malignant Syndrome: Manage with immediate discontinuation and close monitoring. (5.4)
- Tardive Dyskinesia: Discontinue treatment if clinically appropriate. (5.5)
- Metabolic Changes: Monitor for hyperglycemia/diabetes mellitus, dyslipidemia, and weight gain. (5.6)
- Leukopenia, Neutropenia, and Agranulocytosis: Perform complete blood counts (CBC) in patients with pre-existing low white blood cell count (WBC) or history of leukopenia or neutropenia. Consider discontinuing CAPLYTA if clinically significant decline in WBC occurs in absence of other causative factors. (5.7)
- Orthostatic Hypotension and Syncope: Monitor heart rate and blood pressure and warn patients with known cardiovascular or cerebrovascular disease, and risk of dehydration or syncope. (5.8)
- Seizures: Use cautiously in patients with a history of seizure or with conditions that lower seizure threshold. (5.10)
- Potential for Cognitive and Motor Impairment: Use caution when operating machinery. (5.11)
Adverse Reactions/Side Effects
Most common adverse reactions in clinical trials (incidence > 5% and greater than twice placebo) were (6.1):
- Schizophrenia: somnolence/sedation and dry mouth.
- Bipolar depression: somnolence/sedation, dizziness, nausea, dry mouth.
To report SUSPECTED ADVERSE REACTIONS, contact Intra-Cellular Therapies, Inc. at 1-888-611-4824 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
- Pregnancy: May cause extrapyramidal and/or withdrawal symptoms in neonates with third trimester exposure. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2023
Full Prescribing Information
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS; and SUICIDAL THOUGHTS AND BEHAVIORS
Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. CAPLYTA is not approved for the treatment of patients with dementia-related psychosis [see Warnings and Precautions (5.1)].
Suicidal Thoughts and Behaviors
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric and young adults in short-term studies. Closely monitor all antidepressant-treated patients for clinical worsening, and for emergence of suicidal thoughts and behaviors [see Warnings and Precautions (5.2)]. The safety and effectiveness of CAPLYTA have not been established in pediatric patients [see Use in Specific Populations (8.4)].
1. Indications and Usage for Caplyta
CAPLYTA is indicated for the treatment of:
- Schizophrenia in adults [see Clinical Studies (14.1)].
- Depressive episodes associated with bipolar I or II disorder (bipolar depression) in adults, as monotherapy and as adjunctive therapy with lithium or valproate [see Clinical Studies (14.2)].
2. Caplyta Dosage and Administration
2.1 Recommended Dosage
The recommended dosage of CAPLYTA is 42 mg administered orally once daily with or without food. Dose titration is not required.
2.2 Dosage Recommendations for Concomitant Use with Moderate or Strong CYP3A4 Inhibitors
Coadministration with Strong CYP3A4 Inhibitors
The recommended dosage for patients receiving strong CYP3A4 inhibitors is CAPLYTA 10.5 mg once daily [see Drug Interactions (7.1)].
Coadministration with Moderate CYP3A4 Inhibitors
The recommended dosage for patients receiving moderate CYP3A4 inhibitors is CAPLYTA 21 mg once daily [see Drug Interactions (7.1)].
2.3 Dosage Recommendations for Patients with Hepatic Impairment
For patients with moderate or severe hepatic impairment (Child-Pugh class B or C), the recommended dosage of CAPLYTA is 21 mg once daily [see Use in Specific Populations (8.6)].
3. Dosage Forms and Strengths
CAPLYTA capsules are available in three strengths:
- 42 mg: Blue cap and opaque white body imprinted with “ITI-007 42 mg”
- 21 mg: Opaque white cap and body imprinted with “ITI-007 21 mg”
- 10.5 mg: Opaque light pink cap and body imprinted with “ITI-007 10.5 mg”
4. Contraindications
CAPLYTA is contraindicated in patients with history of hypersensitivity reaction to lumateperone. Reactions have included pruritus, rash (e.g. allergic dermatitis, papular rash, and generalized rash), and urticaria.
5. Warnings and Precautions
5.1 Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of 17 placebo-controlled trials (modal duration of 10 weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in the drug-treated patients of between 1.6 to 1.7 times that in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in placebo-treated patients. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. CAPLYTA is not approved for the treatment of patients with dementia-related psychosis [see Boxed Warning, Warnings and Precautions (5.3)].
5.2 Suicidal Thoughts and Behaviors in Children, Adolescents and Young Adults
In pooled analyses of placebo-controlled trials of antidepressant drugs (SSRIs and other antidepressant classes) that included approximately 77,000 adult patients and 4,500 pediatric patients, the incidence of suicidal thoughts and behaviors in antidepressant-treated patients age 24 years and younger was greater than in placebo-treated patients. There was considerable variation in risk of suicidal thoughts and behaviors among drugs, but there was an increased risk identified in young patients for most drugs studied. There were differences in absolute risk of suicidal thoughts and behaviors across the different indications, with the highest incidence in patients with MDD. The drug-placebo differences in the number of cases of suicidal thoughts and behaviors per 1000 patients treated are provided in Table 1.
| Age Range |
Drug-Placebo Difference in Number of Patients of Suicidal Thoughts or Behaviors per 1000 Patients Treated |
| Increases Compared to Placebo | |
| <18 years old | 14 additional patients |
| 18-24 years old | 5 additional patients |
| Decreases Compared to Placebo | |
| 25-64 years old | 1 fewer patient |
| >65 years old | 6 fewer patients |
*CAPLYTA is not approved for use in pediatric patients.
It is unknown whether the risk of suicidal thoughts and behaviors in children, adolescents, and young adults extends to longer-term use, i.e., beyond four months. However, there is substantial evidence from placebo-controlled maintenance trials in adults with MDD that antidepressants delay the recurrence of depression and that depression itself is a risk factor for suicidal thoughts and behaviors.
Monitor all antidepressant-treated patients for any indication for clinical worsening and emergence of suicidal thoughts and behaviors, especially during the initial few months of drug therapy, and at times of dosage changes. Counsel family members or caregivers of patients to monitor for changes in behavior and to alert the healthcare provider. Consider changing the therapeutic regimen, including possibly discontinuing CAPLYTA, in patients whose depression is persistently worse, or who are experiencing suicidal thoughts or behaviors.
5.3 Cerebrovascular Adverse Reactions, Including Stroke, in Elderly Patients with Dementia-Related Psychosis
In placebo-controlled trials in elderly subjects with dementia, patients randomized to risperidone, aripiprazole, and olanzapine had a higher incidence of stroke and transient ischemic attack, including fatal stroke. CAPLYTA is not approved for the treatment of patients with dementia-related psychosis [see Warnings and Precautions (5.1)].
5.4 Neuroleptic Malignant Syndrome
Neuroleptic Malignant Syndrome (NMS), a potentially fatal symptom complex, has been reported in association with administration of antipsychotic drugs. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, delirium, and autonomic instability. Additional signs may include elevated creatinine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. If NMS is suspected, immediately discontinue CAPLYTA and provide intensive symptomatic treatment and monitoring.
5.5 Tardive Dyskinesia
Tardive dyskinesia, a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements, may develop in patients treated with antipsychotic drugs. The risk appears to be highest among the elderly, especially elderly women, but it is not possible to predict which patients are likely to develop the syndrome. Whether antipsychotic drug products differ in their potential to cause tardive dyskinesia is unknown.
The risk of tardive dyskinesia and the likelihood that it will become irreversible increase with the duration of treatment and the cumulative dose. The syndrome can develop after a relatively brief treatment period, even at low doses. It may also occur after discontinuation of treatment.
Tardive dyskinesia may remit, partially or completely, if antipsychotic treatment is discontinued. Antipsychotic treatment itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome, possibly masking the underlying process. The effect that symptomatic suppression has upon the long-term course of tardive dyskinesia is unknown.
Given these considerations, CAPLYTA should be prescribed in a manner most likely to reduce the risk of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients: 1) who suffer from a chronic illness that is known to respond to antipsychotic drugs; and 2) for whom alternative, effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment producing a satisfactory clinical response. Periodically reassess the need for continued treatment.
If signs and symptoms of tardive dyskinesia appear in a patient on CAPLYTA, drug discontinuation should be considered. However, some patients may require treatment with CAPLYTA despite the presence of the syndrome.
5.6 Metabolic Changes
Antipsychotic drugs have caused metabolic changes, including hyperglycemia, diabetes mellitus, dyslipidemia, and weight gain. Although all of the drugs in the class have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia and Diabetes Mellitus
Hyperglycemia, in some cases extreme and associated with ketoacidosis, hyperosmolar coma or death, has been reported in patients treated with antipsychotics. There have been reports of hyperglycemia in patients treated with CAPLYTA. Assess fasting plasma glucose before or soon after initiation of antipsychotic medication and monitor periodically during long-term treatment.
Schizophrenia
In pooled data from short-term (4- to 6-week), placebo-controlled trials of adult patients with schizophrenia, mean changes from baseline and the proportion of patients with shifts from normal to greater than normal levels of fasting glucose in patients treated with CAPLYTA were similar to those in patients treated with placebo.
In an uncontrolled open-label trial of CAPLYTA for up to 1 year in patients with stable schizophrenia, the percentages of patients with shifts in fasting glucose and insulin values from normal to high were 8% and 12%, respectively. 4.7% of patients with normal hemoglobin A1c (<6.5%) at baseline developed elevated levels (≥6.5%) post-baseline.
Bipolar Depression
In data from short-term (6-week), placebo-controlled monotherapy and adjunctive therapy bipolar depression trials, mean changes from baseline and the proportion of patients with shifts from normal to greater than normal levels of fasting glucose and insulin in patients treated with CAPLYTA were similar to those in patients treated with placebo.
Dyslipidemia
Antipsychotics have caused adverse alterations in lipids. Before or soon after initiation of antipsychotic medications, obtain a fasting lipid profile at baseline and monitor periodically during treatment.
Schizophrenia
In pooled data from short-term (4- to 6-week), placebo-controlled trials of adult patients with schizophrenia, mean changes from baseline and the proportion of patients with shifts to higher levels of fasting total cholesterol and triglycerides were similar in patients treated with CAPLYTA and placebo.
In an uncontrolled open-label trial of CAPLYTA for up to 1 year in patients with stable schizophrenia, the percentages of patients with a shift from normal to high were 8%, 5%, and 4% for total cholesterol, triglycerides, and LDL cholesterol, respectively.
Bipolar Depression
In data from short-term (6-week), placebo-controlled monotherapy and adjunctive therapy bipolar depression trials, mean changes from baseline and the proportion of patients with shifts to higher levels of fasting total cholesterol and triglycerides were similar in patients treated with CAPLYTA and placebo.
In an uncontrolled open-label trial of CAPLYTA for up to 6 months in patients with bipolar depression, the proportion of patients with a shift from normal to high were 10%, 5%, and 2% for total cholesterol, triglycerides, and LDL cholesterol, respectively.
Weight Gain
Weight gain has been observed with use of antipsychotics. Monitor weight at baseline and frequently thereafter.
Schizophrenia
In pooled data from placebo-controlled trials of adult patients with schizophrenia, mean changes from baseline and the proportion of patients with an increase in weight ≥7% from baseline to end of study was similar in patients treated with CAPLYTA and placebo.
In an uncontrolled open-label trial of CAPLYTA for up to 1 year in patients with stable schizophrenia, the mean change in body weight was approximately -2 kg (SD 5.6) at Day 175 and approximately - 3.2 kg (SD 7.4) at Day 350.
Bipolar Depression
In data from short-term (6-week), placebo-controlled monotherapy and adjunctive therapy bipolar depression trials, mean changes from baseline and the proportion of patients with an increase in weight ≥7% from baseline to end of study were similar in patients treated with CAPLYTA and placebo.
In an uncontrolled open-label trial of CAPLYTA for up to 6 months in patients with bipolar depression, the mean change in body weight was -0.01 kg (SD 3.1) at Day 175.
5.7 Leukopenia, Neutropenia, and Agranulocytosis
Leukopenia and neutropenia have been reported during treatment with antipsychotic agents, including CAPLYTA. Agranulocytosis (including fatal cases) has been reported with other agents in the class.
Possible risk factors for leukopenia and neutropenia include pre-existing low white blood cell count (WBC) or absolute neutrophil count (ANC) and history of drug-induced leukopenia or neutropenia. In patients with a pre-existing low WBC or ANC or a history of drug-induced leukopenia or neutropenia, perform a complete blood count (CBC) frequently during the first few months of therapy. In such patients, consider discontinuation of CAPLYTA at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Monitor patients with clinically significant neutropenia for fever or other symptoms or signs of infection and treat promptly if such symptoms or signs occur. Discontinue CAPLYTA in patients with absolute neutrophil count < 1000/mm3 and follow their WBC until recovery.
5.8 Orthostatic Hypotension and Syncope
Atypical antipsychotics cause orthostatic hypotension and syncope. Generally, the risk is greatest during initial dose administration. Orthostatic vital signs should be monitored in patients who are vulnerable to hypotension (e.g., elderly patients, patients with dehydration, hypovolemia, and concomitant treatment with antihypertensive medications), patients with known cardiovascular disease (history of myocardial infarction, ischemic heart disease, heart failure, or conduction abnormalities), and patients with cerebrovascular disease. CAPLYTA has not been evaluated in patients with a recent history of myocardial infarction or unstable cardiovascular disease. Such patients were excluded from pre-marketing clinical trials.
Schizophrenia
In pooled data from short-term (4- to 6-week), placebo-controlled schizophrenia trials, the frequencies of orthostatic hypotension for CAPLYTA and placebo were 0.7% and 0%, respectively. The rates of syncope for CAPLYTA and placebo were 0.2% and 0.2%.
Bipolar Depression
In data from short-term (6-week), placebo-controlled monotherapy and adjunctive therapy bipolar depression trials, the frequencies of orthostatic hypotension for CAPLYTA and placebo were both 0%. The rates of syncope for CAPLYTA and placebo were 0.3% and 0.5%, respectively in the monotherapy trials, and there were no reports for CAPLYTA or placebo in the adjunctive therapy trial.
5.9 Falls
Antipsychotics, including CAPLYTA, may cause somnolence, postural hypotension, and motor and sensory instability, which may lead to falls and, consequently, fractures and other injuries. For patients with diseases, conditions or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and periodically during long-term treatment.
5.10 Seizures
Like other antipsychotic drugs, CAPLYTA may cause seizures. The risk is greatest in patients with a history of seizures or with conditions that lower the seizure threshold. Conditions that lower the seizure threshold may be more prevalent in older patients.
5.11 Potential for Cognitive and Motor Impairment
CAPLYTA, like other antipsychotics, may cause somnolence and has the potential to impair judgment, thinking, and motor skills. Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that therapy with CAPLYTA does not affect them adversely.
Schizophrenia
In short-term (i.e., 4- to 6-week), placebo-controlled clinical trials of patients with schizophrenia, somnolence and sedation were reported in 24% of CAPLYTA-treated patients, compared to 10% of placebo-treated patients.
Bipolar Depression
In short term (6-week), placebo-controlled monotherapy and adjunctive therapy bipolar depression clinical trials, somnolence and sedation were reported in 13% of CAPLYTA-treated patients, compared to 3% of placebo-treated patients.
5.12 Body Temperature Dysregulation
Atypical antipsychotics may disrupt the body’s ability to reduce core body temperature. Strenuous exercise, exposure to extreme heat, dehydration, and anticholinergic medications may contribute to an elevation in core body temperature; use CAPLYTA with caution in patients who may experience these conditions.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in detail in other sections of the labeling:
- Increased Mortality in Elderly Patients with Dementia-Related Psychosis [see Boxed Warning, Warnings and Precautions (5.1)]
- Suicidal Thoughts and Behaviors [see Boxed Warning, Warnings and Precautions (5.2)]
- Cerebrovascular Adverse Reactions, Including Stroke, in Elderly Patients with Dementia-related Psychosis [see Warnings and Precautions (5.3)]
- Neuroleptic Malignant Syndrome [see Warnings and Precautions (5.4)]
- Tardive Dyskinesia [see Warnings and Precautions (5.5)]
- Metabolic Changes [see Warnings and Precautions (5.6)]
- Leukopenia, Neutropenia, and Agranulocytosis [see Warnings and Precautions (5.7)]
- Orthostatic Hypotension and Syncope [see Warnings and Precautions (5.8)]
- Falls [see Warnings and Precautions (5.9)]
- Seizures [see Warnings and Precautions (5.10)]
- Potential for Cognitive and Motor Impairment [see Warnings and Precautions (5.11)]
- Body Temperature Dysregulation [see Warnings and Precautions (5.12)]
- Dysphagia [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of CAPLYTA has been evaluated in placebo-controlled clinical trials in 2664 adult patients with schizophrenia and bipolar depression exposed to one or more doses. A total of 402 CAPLYTA-exposed patients had at least 6 months of exposure and 108 had at least 1 year of exposure to the 42-mg dose of CAPLYTA.
Schizophrenia
The following findings are based on the pooled short-term (4- to 6-week), placebo-controlled studies in adult patients with schizophrenia in which CAPLYTA was administered at a daily dose of 42 mg (N=406).
There was no single adverse reaction leading to discontinuation that occurred at a rate of >2% in CAPLYTA-treated patients.
The most common adverse reactions (incidence of at least 5% of patients exposed to CAPLYTA and greater than twice the rate of placebo) are somnolence/sedation and dry mouth.
Adverse reactions associated with CAPLYTA (incidence of at least 2% in patients exposed to CAPLYTA and greater than placebo) are shown in Table 2.
| CAPLYTA 42 mg (N=406) | Placebo (N=412) |
|
| Somnolence/Sedation | 24% | 10% |
| Nausea | 9% | 5% |
| Dry Mouth | 6% | 2% |
| Dizziness1 | 5% | 3% |
| Creatine Phosphokinase Increased | 4% | 1% |
| Fatigue | 3% | 1% |
| Vomiting | 3% | 2% |
| Hepatic Transaminases Increased2 | 2% | 1% |
| Decreased Appetite | 2% | 1% |
1 Dizziness, dizziness postural
2 ALT, AST, “hepatic enzymes” increased, or liver function test abnormal
Bipolar Depression - Monotherapy
The following findings are based on the pooled short-term (6-week), placebo-controlled monotherapy bipolar depression studies in adult patients treated with CAPLYTA administered at a daily dose of 42 mg (N=372).
There was no single adverse reaction leading to discontinuation that occurred at a rate of >2% in CAPLYTA-treated patients.
The most common adverse reactions (incidence of at least 5% of patients exposed to CAPLYTA and greater than twice the rate of placebo) are somnolence/sedation, dizziness, nausea, and dry mouth.
Adverse reactions associated with CAPLYTA (incidence of at least 2% in patients exposed to CAPLYTA and greater than placebo) are shown in Table 3.
| CAPLYTA 42 mg (N=372) | Placebo (N=374) |
|
| Headache | 14% | 8% |
| Somnolence/Sedation | 13% | 3% |
| Dizziness1 | 8% | 4% |
| Nausea | 8% | 3% |
| Dry mouth | 5% | 1% |
| Diarrhea | 4% | 2% |
| Vomiting | 4% | 0% |
| Abdominal pain2 | 2% | 1% |
| Upper respiratory tract infection | 2% | 1% |
1 Dizziness, dizziness postural
2 Abdominal discomfort, abdominal pain, abdominal pain upper and lower
Bipolar Depression - Adjunctive Therapy with Lithium or Valproate
The following findings are based on a 6-week, placebo-controlled adjunctive therapy bipolar depression study in adult patients treated with CAPLYTA administered at a daily dose of 42 mg (N=177).
There was no single adverse reaction leading to discontinuation that occurred at a rate of >2% in CAPLYTA-treated patients.
The most common adverse reactions (incidence of at least 5% of patients exposed to CAPLYTA and greater than twice the rate of placebo) are somnolence/sedation, dizziness, nausea, and dry mouth.
Adverse reactions associated with CAPLYTA (incidence of at least 2% in patients exposed to CAPLYTA and greater than placebo) are shown in Table 4.
|
CAPLYTA 42 mg (N=177) |
Placebo (N=175) |
|
| Somnolence/Sedation | 13% | 3% |
| Dizziness1 | 11% | 2% |
| Nausea | 9% | 4% |
| Dry mouth | 5% | 1% |
| Vomiting | 4% | 0% |
| Diarrhea | 3% | 2% |
| Upper respiratory tract infection | 3% | 1% |
| Blurred vision | 3% | 1% |
| Increased blood prolactin | 2% | 0% |
1 Dizziness, dizziness postural
Dystonia
Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. Although these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and higher doses of first-generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups.
Extrapyramidal Symptoms (EPS)
In the short-term, placebo-controlled schizophrenia and bipolar depression studies, data was objectively collected on the Simpson-Angus Scale (SAS) for EPS (total score ranges from 0 to 40), the Barnes Akathisia Rating Scale (BARS) for akathisia (total score ranges from 0 to 14) and the Abnormal Involuntary Movement Scale (AIMS) for dyskinesia (total score ranges from 0 to 28).
Schizophrenia
In the 4- to 6-week, placebo-controlled schizophrenia trials, the frequency of reported events related to extrapyramidal symptoms (EPS), including akathisia, extrapyramidal disorder, muscle spasms, restlessness, musculoskeletal stiffness, dyskinesia, dystonia, muscle twitching, tardive dyskinesia, tremor, drooling, and involuntary muscle contractions was 6.7% for CAPLYTA and 6.3% for placebo.
In the 4- to 6-week schizophrenia trials, the mean changes from baseline for CAPLYTA-treated patients and placebo-treated patients were 0.1 and 0 for the SAS, -0.1 and 0 for the BARS, and 0.1 and 0 for the AIMS, respectively.
Bipolar Depression
In the 6-week, monotherapy bipolar depression trials, the frequency of reported reactions related to EPS, including muscle spasms, dyskinesia, extrapyramidal disorder, movement disorder, tremor, restlessness, and akathisia was 1.3% for CAPLYTA and 1.1% for placebo.
In a 6-week, adjunctive therapy bipolar depression trial, the frequency of reported reactions related to EPS, including tremor, muscle spasms, akathisia, extrapyramidal disorder, gait disturbance, and restlessness was 4.0% for CAPLYTA and 2.3% for placebo.
In the 6-week, monotherapy bipolar depression trials, the mean changes from baseline for CAPLYTA-treated patients and placebo-treated patients were 0 and 0 for the SAS, -0.1 and -0.1 for the BARS, and 0 and 0 for the AIMS, respectively. In the 6-week adjunctive therapy bipolar depression trial, the mean changes from baseline for CAPLYTA-treated patients and placebo-treated patients were 0 and 0 for the SAS, 0 and -0.1 for the BARS, and 0and 0 for the AIMS, respectively.
6.2 Postmarketing Experience
The following adverse reaction has been identified during post-approval use of CAPLYTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or establish a causal relationship to drug exposure.
Central and Peripheral Nervous System Disorders: burning sensation, including skin burning sensation
Related/similar drugs
7. Drug Interactions
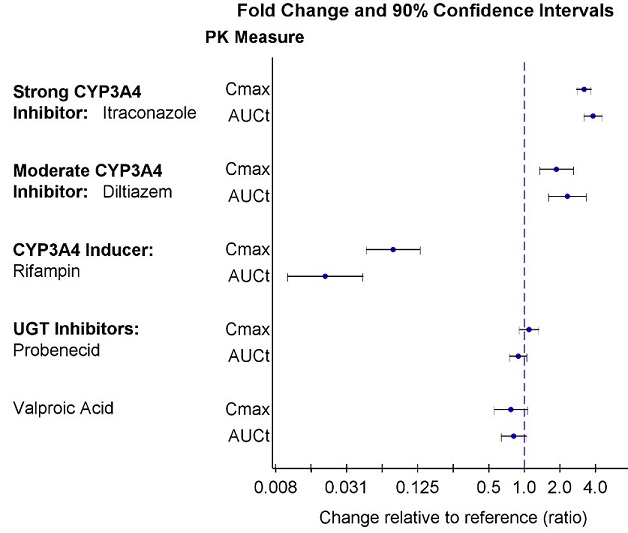
7.1 Drugs Having Clinically Important Interactions with CAPLYTA
| CYP3A4 Inducers | ||
| Clinical Impact | Concomitant use of CAPLYTA with CYP3A4 inducers decreases the exposure of lumateperone [see Clinical Pharmacology (12.3)]. | |
| Intervention | Avoid concomitant use of CAPLYTA with CYP3A4 inducers. | |
| Moderate or Strong CYP3A4 inhibitors | ||
| Clinical Impact | Concomitant use of CAPLYTA with moderate or strong CYP3A4 inhibitors increases lumateperone exposure [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reactions. | |
| Intervention |
Reduce CAPLYTA dose when used concomitantly with moderate or strong CYP3A4 inhibitors [see Dosage and Administration (2.2)]. |
|
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to atypical antipsychotics, including CAPLYTA, during pregnancy. Healthcare providers are encouraged to register patients by contacting the National Pregnancy Registry for Atypical Antipsychotics at 1-866-961-2388 or online at http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/.
Risk Summary
Neonates exposed to antipsychotic drugs during the third trimester are at risk for extrapyramidal and/or withdrawal symptoms following delivery (see Clinical Considerations). Available data from case reports on CAPLYTA use in pregnant women are insufficient to establish any drug associated risks for birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks to the mother associated with untreated schizophrenia and with exposure to antipsychotics, including CAPLYTA, during pregnancy (see Clinical Considerations). In animal reproduction studies, no malformations were observed with oral administration of lumateperone to pregnant rats and rabbits during organogenesis at doses up to 2.4 and 9.7 times, respectively, the maximum recommended human dose (MRHD) of 42 mg/day on a mg/m2 basis. When pregnant rats were administered lumateperone during the period of organogenesis through lactation, the number of perinatal deaths of pups was increased at 4.9 times the MRHD, with no adverse effects on pups at 2.4 times the MRHD (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease associated maternal and/or embryo/fetal risk
There is risk to the mother from untreated schizophrenia, including increased risk of relapse, hospitalization, and suicide. Schizophrenia is associated with increased adverse perinatal outcomes, including preterm birth. It is not known if this is a direct result of the illness or other comorbid factors.
Fetal/neonatal adverse reactions
Extrapyramidal and/or withdrawal symptoms, including agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress, and feeding disorder have been reported in neonates who were exposed to antipsychotic drugs during the third trimester of pregnancy. These symptoms have varied in severity. Monitor neonates for extrapyramidal and/or withdrawal symptoms and manage symptoms appropriately. Some neonates recovered within hours or days without specific treatment; others required prolonged hospitalization.
Data
Animal Data
Pregnant rats were treated with oral doses of 3.5, 10.5, 21, and 63 mg/kg/day lumateperone (0.8, 2.4, 4.9, and 14.6 times the MRHD on a mg/m2 basis) during the period of organogenesis. No malformations were observed with lumateperone at doses up to 2.4 times the MRHD. Findings of decreased body weight were observed in fetuses at 4.9 and 14.6 times the MRHD. Findings of incomplete ossification and increased incidences of visceral and skeletal variations were recorded in fetuses at 14.6 times the MRHD, a dose that induced maternal toxicity.
Pregnant rabbits were treated with oral doses of 2.1, 7, and 21 mg/kg/day lumateperone (1.0, 3.2, and 9.7 times the MRHD on a mg/m2 basis) during the period of organogenesis. Lumateperone did not cause adverse developmental effects at doses up to 9.7 times the MRHD.
In a study in which pregnant rats were administered oral doses of 3.5, 10.5, and 21 mg/kg/day lumateperone (0.8, 2.4, and 4.9 times the MRHD on a mg/m2 basis) during the period of organogenesis and through lactation, the number of live-born pups was decreased at 2.4 and 4.9 times the MRHD, and early postnatal deaths increased at a dose 4.9 times the MRHD. Impaired nursing and decreased body weight gain in pups were observed at 4.9 times, but not at 2.4 times, the MRHD.
Pregnant rats were treated with a human metabolite of lumateperone (reduced ketone metabolite) at oral doses of 15, 60, and 100 mg/kg/day (1.2, 19, and 27 times the exposure to this metabolite at the MRHD of lumateperone based on AUC plasma exposure) during the period of organogenesis. This metabolite did not cause adverse developmental effects at a dose 1.2 times the exposure at the MRHD of lumateperone; however, it caused an increase in visceral malformations (cleft palate) at 27 times and skeletal malformations at 19 times the exposure at the MRHD of lumateperone, a dose that induced maternal toxicity.
8.2 Lactation
Risk Summary
Lumateperone and its metabolites are present in human breast milk in low amounts. In a clinical lactation study, lumateperone was detected in human milk at an estimated daily infant dose 0.0004 mg/kg, with a relative infant dose (RID) of 0.06% the maternal weight-adjusted dosage. Several major circulating metabolites were similarly detected in breast milk in low amounts; however, aniline metabolites were not present in milk or maternal plasma at quantifiable levels (see Data). There are no data on the effects of lumateperone on the breastfed infant or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for CAPLYTA and any potential adverse effects on the breastfed child from CAPLYTA or from the underlying maternal condition.
Data
A lactation study in 17 lactating women evaluated the concentrations of lumateperone and its metabolites in plasma and mature breast milk following a single dose of 42 mg CAPLYTA. The estimated daily infant dose of lumateperone in human milk was 0.0004 mg/kg/day (with assumed average milk consumption of 200 ml/kg/day). The mean relative infant dose (RID) (with assumed mean milk consumption of 200 mL/kg/day and average maternal weight of 71 kg) was 0.06% of the maternal weight-adjusted dosage. Several major circulating metabolites were also present in breast milk at estimated daily infant dose of 0.0004 mg/kg/day. Aniline metabolites were not present in milk or maternal plasma at quantifiable levels.
8.3 Females and Males of Reproductive Potential
Infertility
Based on findings from animal studies, lumateperone may impair male and female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of CAPLYTA have not been established in pediatric patients.
Antidepressants increased the risk of suicidal thoughts and behaviors in pediatric patients [see Boxed Warning, Warnings and Precautions (5.2)].
8.5 Geriatric Use
Controlled clinical studies of CAPLYTA in the treatment of schizophrenia did not include any patients aged 65 or older to determine whether or not they respond differently from younger patients. Controlled clinical studies of CAPLYTA in the treatment of bipolar depression included patients aged 65 or older; the number of patients was not sufficient to determine whether or not they respond differently from younger patients.
Antipsychotic drugs increase the risk of death in elderly patients with dementia-related psychosis. CAPLYTA is not approved for the treatment of patients with dementia-related psychosis [see Boxed Warning, Warnings and Precautions (5.1) and (5.3)].
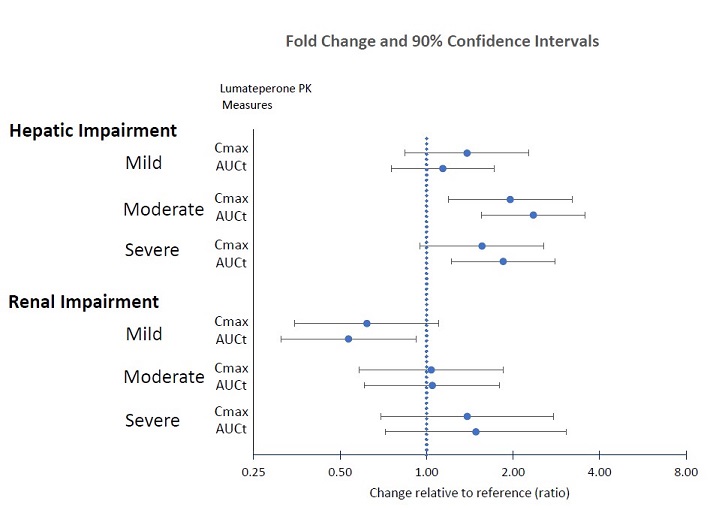
8.6 Hepatic Impairment
Patients with moderate (Child-Pugh class B) and severe (Child-Pugh class C) hepatic impairment generally had higher exposure to lumateperone than patients with normal hepatic function; therefore, a dosage reduction of CAPLYTA is recommended in patients with moderate or severe hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
No dosage adjustment is recommended for patients with mild hepatic impairment (Child-Pugh class A).
10. Overdosage
No specific antidotes for CAPLYTA are known. In managing overdose, provide supportive care, including close medical supervision and monitoring and consider the possibility of multiple drug involvement. In case of overdose, consult a Certified Poison Control Center (1-800-222-1222 or www.poison.org).
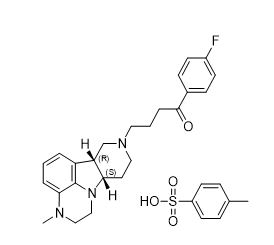
11. Caplyta Description
CAPLYTA capsules contains lumateperone, an atypical antipsychotic, present as lumateperone tosylate salt with the chemical name 4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H,7H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8-yl)-1-(4-fluoro-phenyl)-butan-1-one 4-methylbenzenesulfonate. Its molecular formula is C31H36FN3O4S, and its molecular weight is 565.71 g/mol with the following structure:
CAPLYTA capsules are intended for oral administration. Each CAPLYTA capsule contains lumateperone and are available in 42 mg (equivalent to 60 mg of lumateperone tosylate), 21 mg (equivalent to 30 mg of lumateperone tosylate), and 10.5 mg (equivalent to 15 mg of lumateperone tosylate) strengths. Capsules include the following inactive ingredients: croscarmellose sodium, gelatin, magnesium stearate, mannitol, and talc. Colorants include FD&C blue #1 and red #3 (42 mg), FDA/E172 black iron oxide, FDA/E172 red iron oxide and FD&C red #3 (10.5 mg), and titanium dioxide (42 mg, 21 mg and 10.5 mg).
12. Caplyta - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of lumateperone in the treatment of schizophrenia and depressive episodes associated with bipolar I or II disorder is unknown. However, the efficacy of lumateperone could be mediated through a combination of antagonist activity at central serotonin 5-HT2A receptors and postsynaptic antagonist activity at central dopamine D2 receptors.
12.2 Pharmacodynamics
Lumateperone has high binding affinity for serotonin 5-HT2A receptors (Ki = 0.54 nM) and moderate binding affinity for dopamine D2 (Ki = 32 nM) receptors. Lumateperone has moderate binding affinity for serotonin transporters (Ki = 33 nM). Lumateperone also has moderate binding affinity for dopamine D1 (41 nM) and D4 and adrenergic alpha1A and alpha1B receptors (Ki projected at < 100 nM) but has low binding affinity (less than 50% inhibition at 100 nM) for muscarinic and histaminergic receptors.
Cardiac Electrophysiology
QTcF interval was evaluated in a randomized, placebo- and active- (moxifloxacin 400 mg) controlled, four-arm crossover study utilizing concentration-QTc effect modeling in 33 patients with schizophrenia. The placebo-corrected change from baseline QTcF (90% two-sided upper confidence interval) values of 4.9 (8.9) and 15.8 (19.8) ms for the 42 mg and the supratherapeutic dose of 126 mg (three times the recommended daily dosage) CAPLYTA, respectively, administered orally once daily for 5 days.
12.3 Pharmacokinetics
Following once daily oral administration of CAPLYTA, lumateperone steady state is reached in about 5 days. Lumateperone steady-state exposure is approximately dose-proportional in the range of 3.5 mg to 56 mg (0.08 to 1.3 times the approved recommended daily dosage). A large inter-subject variability in lumateperone PK parameters was observed, with coefficients of variation for Cmax (peak plasma concentration) and AUC (area under the concentration vs time curve) ranging from 68% to 97% at steady state.
Absorption
The absolute bioavailability of lumateperone capsules is about 4.4%. Cmax of lumateperone is reached approximately 1-2 hours after CAPLYTA dosing.
Effect of Food
Ingestion of a high-fat meal with CAPLYTA lowers lumateperone mean Cmax by 33% and increases mean AUC by 9%. Median Tmax was delayed about 1 hour (from 1 hour at fasted state to 2 hours in the presence of food).
Distribution
Protein binding of lumateperone is 97.4% at 5 µM (about 70-fold higher than therapeutic concentrations) in human plasma. The volume of distribution of lumateperone following intravenous administration is about 4.1 L/kg.
Elimination
The clearance of lumateperone is approximately 27.9 L/hour and the terminal half-life is about 18 hours after intravenous administration.
Metabolism
Lumateperone is extensively metabolized with more than twenty metabolites identified in vivo. After a single 14C-labeled oral dose, lumateperone and glucuronidated metabolites represent about 2.8% and 51% of the total plasma radioactivity, respectively. In vitro studies show that multiple enzymes, including but not limited to uridine 5'-diphospho-glucuronosyltransferases (UDP-glucuronosyltransferase, UGT) 1A1, 1A4, and 2B15, aldoketoreductase (AKR)1C1, 1B10, and 1C4, and cytochrome P450 (CYP) 3A4, 2C8, and 1A2, are involved in the metabolism of lumateperone.
Excretion
In a human mass‑balance study, 58% and 29% of the radioactive dose was recovered in the urine and feces, respectively. Less than 1% of the dose was excreted as unchanged lumateperone in the urine.
Specific Populations
Effects of hepatic or renal impairment on lumateperone exposure are presented in Figure 1. No clinically significant differences in the pharmacokinetics of lumateperone were observed based on age, sex, or race.
Figure 1: Effects of Intrinsic Factors on Lumateperone Pharmacokinetics
Drug Interaction Studies
Clinical Studies
The effects of other drugs on the exposures of lumateperone are presented in Figure 2.
Figure 2: Effects of Other Drugs on Lumateperone Pharmacokinetics
CYP3A4 substrates: No clinically significant differences in the pharmacokinetics of midazolam (CYP3A4 substrate) or its metabolite 1-hydroxymidazolam were observed when used concomitantly with single or multiple doses of lumateperone in patients with schizophrenia.
In Vitro Studies
Lumateperone showed little to no inhibition of CYP1A2, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5. It showed no induction of CYP1A2, CYP2B6, or CYP3A4.
Lumateperone did not appear to be a P-gp or BCRP substrate. It showed little to no inhibition of OCT2, OAT1, OAT3, OATP1B3, or OATP1B1.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies were conducted in rats and mice, and results showed no carcinogenic potential in either species.
In Sprague-Dawley rats, males were administered lumateperone (free base) at oral doses of 3.5, 7 or 14 mg/kg/day and females were administered lumateperone at oral doses of 3.5, 10.5, or 21 mg/kg/day for the first 385 days, then doses were reduced for the two higher dose groups so that the females were administered 3.5, 7 or 14 mg/kg/day, respectively, for the duration of the study. In this study the no adverse effect level for neoplastic lesions was determined to be 14 mg/kg/day (84 mg/m2/day) for males and 10.5/7 mg/kg/day (42 mg/m2/day) for females, which are 1.6 times (females) to 3.2 times (males) the MRHD on a mg/m2 basis.
Male and female CD-1 mice were administered lumateperone at oral doses of 3.5, 10.5 or 21 mg/kg/day for the first 35 days, then doses were reduced to 1.4, 4.9, and 14 mg/kg/day, respectively, for the duration of the study. In this study, the no adverse effect level for neoplastic lesions was determined to be 10.5/4.9 mg/kg/day (15 mg/m2/day) for each sex which is 0.6 times the MRHD on a mg/m2 basis.
Mutagenesis
No evidence of mutagenic potential was found in the in vitro bacterial reverse mutation assay (Ames test) and the mouse lymphoma test without metabolic activation. Lumateperone was positive in the Ames test only in the presence of metabolic activation and only in the TA1537 strain and was positive in the mouse lymphoma test only in the presence of metabolic activation and only at high concentrations that inhibited cell growth; together these results were thought to be related to solubility limits and/or nonspecific effects on cellular function. Lumateperone was negative for clastogenic activity in the in vivo micronucleus assay in rats and was not genotoxic in the in vivo Comet assay in rats.
Impairment of Fertility
Female rats were treated with oral doses of 3.5, 10.5, 21 or 42 mg/kg/day lumateperone (free base) (0.8, 2.4, 4.9, and 9.7 times the MRHD on a mg/m2 basis) prior to mating and continuing through conception and implantation. Estrus cycle irregularities were observed at doses ≥10.5 mg/kg/day. Decreases in the median number of corpora lutea and implantation sites, and increases in the number of non-gravid uteruses, were recorded at 42 mg/kg/day. Decreased gestation body weight and body weight gain, and increases in time to mating, were observed at 21 and 42 mg/kg/day.
Male rats were treated with oral doses of 3.5, 10.5, 21 or 42 mg/kg/day lumateperone (0.8, 2.4, 4.9, and 9.7 times the MRHD on a mg/m2 basis) for 9 weeks prior to mating and throughout 14 days of mating. Decreased sperm motility, changes in sperm morphology, reduced epididymal counts, and adverse histopathology changes in testes and epididymides were observed at 21 and 42 mg/kg/day.
13.2 Animal Toxicology and/or Pharmacology
Oral administration of lumateperone caused systemic intracytoplasmic accumulation of pigmented material in dogs, rats, and mice at clinically relevant exposures (AUC). Intracytoplasmic pigmentation appeared to be localized in lysosomes. Accumulation of pigmented material persisted without reversal at the end of 1- to 2-month drug-free periods. Pigmented material was observed in the brain and spinal cord of all three species, and in the heart and eye of rats. Although the composition of the pigmented material was not established, the material is likely polymers or protein adducts formed from aniline metabolites of lumateperone.
In the dog, accumulation of pigmented material in the brain and spinal cord was associated with neuronal degeneration and necrosis, followed by axonal degeneration and histiocytic inflammation after oral administration of lumateperone for up to 9 months. In the rat, accumulation of pigmented material was associated with degenerative changes and signs of an inflammatory response in the spinal cord, peripheral nervous system, eye, and heart after oral administration of lumateperone for up to 2 years. Although overt degenerative changes were not observed in the rat brain, the presence of pigment-containing infiltrating macrophages is consistent with an inflammatory response.
The role of intracytoplasmic pigmented material in causing these lesions was not definitively established; however, the colocalization of pigmented material in tissues with degenerative changes and signs of inflammation is supportive. Alternatively, the aniline metabolites of lumateperone may undergo metabolic activation forming reactive metabolites that contribute to the observed toxicities. The role of intracellular accumulation of lumateperone or its non-aniline metabolites in these toxicities could not be ruled out.
The aniline metabolites thought to be responsible for these toxicities were detected in dogs and rats but were not present in humans at quantifiable levels. Based on all the available evidence, these toxicities do not appear to be relevant to humans.
14. Clinical Studies
14.1 Schizophrenia
CAPLYTA was evaluated for the treatment of schizophrenia in two placebo-controlled trials.
Study 1 (NCT01499563) was a four-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients with a diagnosis of schizophrenia according to the DSM-IV-TR criteria. The primary efficacy measure was change from baseline in the Positive and Negative Syndrome Scale (PANSS) total score at Week 4. The PANSS is a 30-item scale used to measure symptoms of schizophrenia. Each item is rated by a clinician on a seven-point scale. A score of 1 indicates the absence of symptoms, and a score of 7 indicates extremely severe symptoms. The PANSS total score may range from 30 to 210 with higher scores reflecting greater overall symptom severity.
A total of 335 patients were randomized to receive CAPLYTA 42 mg, CAPLYTA 84 mg (two times the recommended daily dose), an active comparator, or placebo. The study was not designed to allow for efficacy comparison of CAPLYTA and the active comparator. Demographic and baseline disease characteristics were similar for the CAPLYTA, active comparator, and placebo groups. Median age was 42 years (range 20 to 55 years). 17% were female, 19% were Caucasian, and 78% were African American.
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant reduction from baseline to Day 28 in the PANSS total score. The treatment effect in the CAPLYTA 84 mg group (vs. placebo) was not statistically significant. The results of Study 1 are shown in Table 6.
Study 2 (NCT02282761) was a four-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients with a diagnosis of schizophrenia according to the DSM-5 criteria. The primary efficacy measure was change from baseline in the PANSS total score at Week 4.
A total of 450 patients were randomized to receive CAPLYTA 28 mg (two-thirds the recommended daily dose), CAPLYTA 42 mg, or placebo. Demographic and baseline disease characteristics were similar for the CAPLYTA and placebo groups. Median age was 44 years (range 19 to 60 years); 23% were female, 26% were Caucasian and 66% were African American.
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant reduction from baseline to Day 28 in the PANSS total score. The treatment effect in the CAPLYTA 28 mg group (vs. placebo) was not statistically significant. The results of Study 2 are shown in Table 6.
Studies 1 and 2 did not include any patients aged 65 or older. Examination of subgroups by sex and race did not suggest differences in response in either study.
| Primary Efficacy Endpoint: PANSS Total Score | |||||
|
Study Number | Treatment Group | N | Mean Baseline Score (SD) |
LS Mean Change from Baseline (SE) |
Placebo-subtracted Difference (95% CI) |
| 1 | CAPLYTA (42 mg)* | 84 | 88.1 (11.0) | -13.2 (1.7) | -5.8 (-10.5, -1.1)a |
| Placebo | 85 | 86.3 (13.1) | -7.4 (1.7) | -- | |
| 2 | CAPLYTA (42 mg)* | 150 | 90.0 (9.6) | -14.5 (1.3) | -4.2 (-7.8, -0.6) |
| Placebo | 150 | 89.0 (10.3) | -10.3 (1.3) | -- | |
The PANSS total score may range from 30 to 210; higher scores reflect greater symptom severity.
SD: standard deviation; SE: standard error; LS Mean: least squares mean; CI: unadjusted confidence interval.
aDifference (drug minus placebo) in LS mean change from baseline not adjusted for sample size increase after unblinded interim analysis.
*Statistically significantly superior to placebo.
Figure 3: Change from Baseline in PANSS Total Score by Time (Weeks) in Patients with Schizophrenia in Study 2.
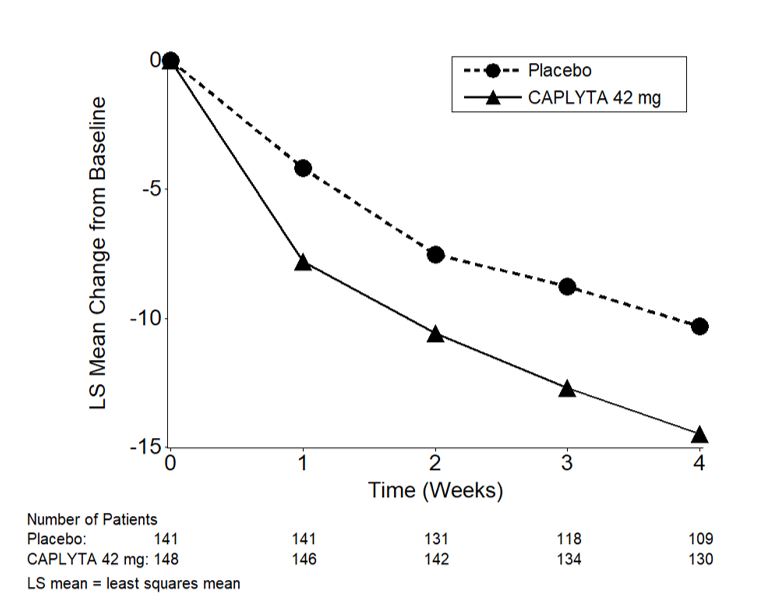
14.2 Depressive Episodes Associated with Bipolar I or II Disorder (Bipolar Depression)
Monotherapy
The efficacy of CAPLYTA, as monotherapy, was established in a 6-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients who met DSM-5 criteria for depressive episodes associated with bipolar I or bipolar II disorder (Study 3; NCT03249376). The primary efficacy measure was the change from baseline in Montgomery-Asberg Depression Rating Scale (MADRS) total score at Week 6. The MADRS is a 10-item clinician-rated scale with total scores ranging from 0 (no depressive features) to 60 (maximum score). The secondary endpoint was the change from baseline in Clinical Global Impression-Bipolar-Severity of Illness scale (CGI-BP-S) total score at Week 6. The CGI-BP-S total score is a clinician-rated scale that measures the patient’s current illness state on a 21-point scale that assesses depression, mania, and overall illness, where a higher score is associated with greater illness severity.
A total of 381 patients were randomized to receive CAPLYTA 42 mg or placebo. Demographic and baseline characteristics were similar for the CAPLYTA and placebo groups. Median age was 45 (range 18 to 72). 58% were female, 91% were Caucasian, and 8% were African-American.
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant improvement from baseline to Day 43 in the MADRS total score and CGI-BP-S total score. The results of Study 3 are shown in Table 7.
Examination of subgroups by age, sex, and race did not suggest differences in response in the study.
Adjunctive Therapy with Lithium or Valproate
The efficacy of CAPLYTA, as adjunctive therapy with lithium or valproate, was established in a 6-week, randomized, double-blind, placebo-controlled, multi-center study in adult patients who met DSM-5 criteria for depressive episodes associated with bipolar I or bipolar II disorder (Study 4; NCT02600507). The primary efficacy measure was the change from baseline in MADRS total score at Week 6. The secondary endpoint was the change from baseline in CGI-BP-S depression score at Week 6. The CGI-BP-S depression score is a clinician-rated scale that measures the patient’s current illness state on a 7-point scale, where a higher score is associated with greater illness severity.
A total of 529 patients were randomized to receive CAPLYTA 28 mg (two-thirds the recommended daily dose), CAPLYTA 42 mg, or placebo. Demographic and baseline characteristics were similar for the CAPLYTA and placebo groups. Median age was 46 (range 18 to 74). 58% were female, 88% were Caucasian, and 11% were African-American.
Compared to the placebo group, patients randomized to CAPLYTA 42 mg showed a statistically significant improvement from baseline to Day 43 in the MADRS total score and CGI-BP-S depression score. The treatment effect in the CAPLYTA 28 mg group (vs. placebo) was not statistically significant. The results of Study 4 are shown in Table 7.
Examination of subgroups by age, sex, and race did not suggest differences in response in the study.
| Primary Efficacy Endpoint: MADRS Total Score | ||||||
|
Study Number | Treatment Group | N | Mean Baseline Score (SD) |
LS Mean Change from Baseline (SE) |
Placebo-subtracted Differencea (95% CI) |
|
| Monotherapy | ||||||
| 3 | CAPLYTA (42 mg)* | 188 | 30.8 (4.9) | -16.7 (0.7) | -4.6 (-6.3, -2.8) | |
| Placebo | 188 | 30.3 (4.6) | -12.1 (0.7) | -- | ||
| Adjunctive Therapy | ||||||
| 4 | CAPLYTA (42 mg)* + lithium or valproate | 174 | 32.2 (5.0) | -16.9 (0.8) | -2.4 (-4.4, -0.4) | |
| Placebo + lithium or valproate | 174 | 32.1 (5.2) | -14.5 (0.8) |
|
||
The MADRS total score ranges from 0 to 60; higher scores reflect greater symptom severity
SD: standard deviation; SE: standard error; LS Mean: least squares mean; CI: confidence interval
a Difference (drug minus placebo) in LS mean change from baseline
*Statistically significantly superior to placebo.
Figure 4. Change from Baseline in MADRS Total Score by Visits (Study 3) in Patients With Depressive Episodes Associated with Bipolar I or II Disorder (Monotherapy)
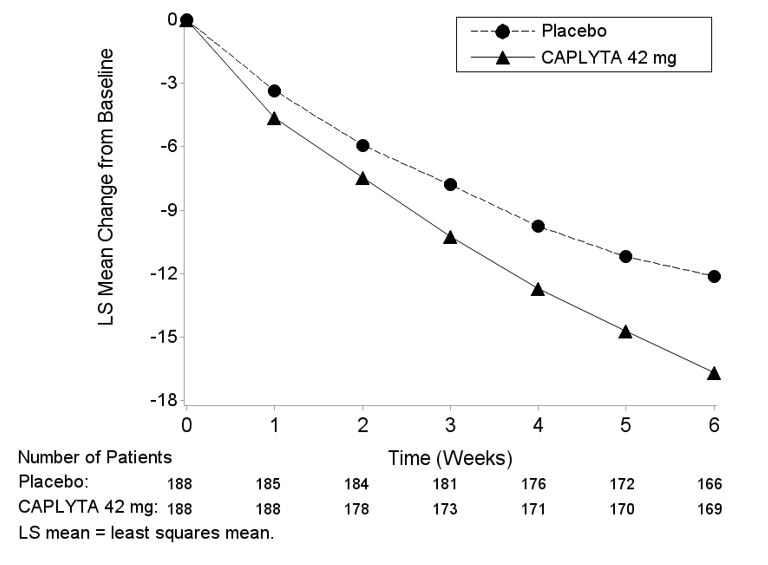
16. How is Caplyta supplied
CAPLYTA (lumateperone) capsules are supplied as follows:
| Capsule Strength | Capsule Color | Imprint Codes | Package Configuration | NDC Code | |
| 42 mg | Blue cap and opaque white body | ITI-007 42 mg | Box of 30 (3 Blister Packs of 10 capsules) | 72060-142-30 | |
| 42 mg | Blue cap and opaque white body | ITI-007 42 mg | Bottle of 30 | 72060-142-40 | |
| 21 mg | Opaque white cap and body | ITI-007 21 mg | Bottle of 30 | 72060-121-40 | |
| 10.5 mg | Opaque light pink cap and body | ITI-007 10.5 mg | Bottle of 30 | 72060-110-40 | |
Store at controlled room temperature 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide).
Suicidal Thoughts and Behavior
Advise patients and caregivers to look for the emergence of suicidality, especially early during treatment and instruct them to report such symptoms to their healthcare provider [see Boxed Warning, Warnings and Precautions (5.2)].
Neuroleptic Malignant Syndrome
Counsel patients about a potentially fatal adverse reaction, Neuroleptic Malignant Syndrome (NMS), that has been reported with administration of antipsychotic drugs. Advise patients, family members, or caregivers to contact the healthcare provider or to report to the emergency room if they experience signs and symptoms of NMS [see Warnings and Precautions (5.4)].
Tardive Dyskinesia
Counsel patients on the signs and symptoms of tardive dyskinesia and to contact their healthcare provider if these abnormal movements occur [see Warnings and Precautions (5.5)].
Metabolic Changes
Educate patients about the risk of metabolic changes, how to recognize symptoms of hyperglycemia and diabetes mellitus, and the need for specific monitoring, including blood glucose, lipids, and weight [see Warnings and Precautions (5.6)].
Leukopenia/Neutropenia
Advise patients with a pre-existing low WBC or a history of drug induced leukopenia/ neutropenia that they should have their CBC monitored while taking CAPLYTA [see Warnings and Precautions (5.7)].
Orthostatic Hypotension and Syncope
Educate patients about the risk of orthostatic hypotension and syncope, especially early in treatment, and also at times of re-initiating treatment [see Warnings and Precautions (5.8)].
Interference with Cognitive and Motor Performance
Caution patients about performing activities requiring mental alertness, such as operating hazardous machinery or operating a motor vehicle, until they are reasonably certain that CAPLYTA therapy does not affect them adversely [see Warnings and Precautions (5.11)].
Heat Exposure and Dehydration
Educate patients regarding appropriate care in avoiding overheating and dehydration [see Warnings and Precautions (5.12)].
Concomitant Medications
Advise patients to inform their health care providers of any changes to their current prescription or over-the-counter medications because there is a potential for interactions [see Drug Interactions (7.1)].
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with CAPLYTA. Advise patients that CAPLYTA used during the third trimester may cause extrapyramidal and/or withdrawal symptoms (agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress, and feeding disorder) in the neonate. Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to CAPLYTA during pregnancy [see Use in Specific Populations (8.1)].
Infertility
Advise males and females of reproductive potential that CAPLYTA may impair fertility [see Use in Specific Populations (8.3)].
Distributed by Intra-Cellular Therapies, Inc.
New York, NY 10016
CAPLYTA is a registered trademark of Intra-Cellular Therapies, Inc.
© 2023 Intra-Cellular Therapies, Inc. All rights reserved.
Medication Guide
| MEDICATION GUIDE CAPLYTA (kap-LITE-ah) (lumateperone) capsules |
|
|
What is the most important information I should know about CAPLYTA? CAPLYTA may cause serious side effects, including:
|
|
|
• thoughts about suicide or dying |
• attempts to commit suicide |
|
• new or worse depression |
• new or worse anxiety |
|
• feeling very agitated or restless |
• panic attacks |
|
• trouble sleeping (insomnia) |
• new or worse irritability |
|
• acting aggressive, being angry, or violent |
• acting on dangerous impulses |
|
• an extreme increase in activity and talking (mania) |
• other unusual changes in behavior or mood |
| What is CAPLYTA?
CAPLYTA is a prescription medicine used in adults:
It is not known if CAPLYTA is safe and effective in children. |
|
| Do not take CAPLYTA if you are allergic to lumateperone or any of the ingredients in CAPLYTA. See the end of this Medication Guide for a complete list of ingredients in CAPLYTA. | |
Before taking CAPLYTA, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. CAPLYTA and other medicines may affect each other causing possible serious side effects. CAPLYTA may affect the way other medicines work, and other medicines may affect how CAPLYTA works. Your healthcare provider can tell you if it is safe to take CAPLYTA with your other medicines. Do not start or stop any medicines during treatment with CAPLYTA without first talking to your healthcare provider. Know the medicines you take. Keep a list of your medicines to show your healthcare provider and pharmacist when you get a new medicine. |
|
How should I take CAPLYTA?
|
|
What should I avoid while taking CAPLYTA?
|
|
|
What are the possible side effects of CAPLYTA? CAPLYTA may cause serious side effects, including:
|
|
|
◦ high fever |
◦ confusion |
|
◦ changes in your breathing, heart rate, and blood pressure |
◦ stiff muscles |
|
◦ increased sweating | |
|
|
|
• feel very thirsty |
• need to urinate more than usual |
|
• feel very hungry |
• feel weak or tired |
|
• feel sick to your stomach |
• feel confused, or your breath smells fruity |
|
|
The most common side effects of CAPLYTA include sleepiness, dizziness, nausea, and dry mouth. CAPLYTA may cause fertility problems in females and males. Talk to your healthcare provider if this is a concern for you. These are not all the possible side effects of CAPLYTA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
| How should I store CAPLYTA?
• Store CAPLYTA at room temperature between 68°F to 77°F (20°C to 25°C). Keep CAPLYTA and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of CAPLYTA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use CAPLYTA for a condition for which it was not prescribed. Do not give CAPLYTA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about CAPLYTA that is written for healthcare professionals. |
|
| What are the ingredients in CAPLYTA?
Active ingredient: lumateperone Inactive ingredients: croscarmellose sodium, gelatin, magnesium stearate, mannitol, and talc. Colorants include FD&C blue #1 and red #3 (42 mg), FDA/E172 black iron oxide, FDA/E172 red iron oxide and FD&C red #3 (10.5 mg), and titanium dioxide (42 mg, 21 mg and 10.5 mg). Distributed by: Intra-Cellular Therapies, Inc., New York, NY 10016 ©2023 Intra-Cellular Therapies, Inc. All rights reserved. For more information, go to www.CAPLYTA.com or call (888) 252-4824 |
|
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 6/2023



| CAPLYTA
lumateperone capsule |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| CAPLYTA
lumateperone capsule |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| CAPLYTA
lumateperone capsule |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Intra-Cellular Therapies, Inc (112765909) |
Frequently asked questions
- What is the mechanism of action for Caplyta?
- Caplyta savings card: Do I qualify and how much can I save?
- How long does it take Caplyta to work?
- Does Caplyta cause weight gain?
More about Caplyta (lumateperone)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (192)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: atypical antipsychotics
- Breastfeeding
- En español