Baxdela: Package Insert / Prescribing Info
Package insert / product label
Generic name: delafloxacin meglumine
Dosage form: tablet
Drug class: Quinolones and fluoroquinolones
Medically reviewed by Drugs.com. Last updated on Aug 12, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
BAXDELA (delafloxacin) tablets, for oral use
BAXDELA (delafloxacin) for injection, for intravenous use
Initial U.S. Approval: 2017
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS, and EXACERBATION OF MYASTHENIA GRAVIS
See full prescribing information for complete boxed warning.
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together (5.1), including:
- Tendinitis and tendon rupture (5.2)
- Peripheral neuropathy (5.3)
- Central nervous system effects (5.4)
Discontinue BAXDELA immediately and avoid the use of fluoroquinolones, including BAXDELA, in patients who experience any of these serious adverse reactions. (5.1)
- Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid BAXDELA in patients with known history of myasthenia gravis. (5.5)
Indications and Usage for Baxdela
BAXDELA is a fluoroquinolone antibacterial indicated for the treatment of adults with the following infections caused by designated susceptible bacteria:
- Acute Bacterial Skin and Skin Structure Infections (ABSSSI) (1.1)
- Community-Acquired Bacterial Pneumonia (CABP) (1.2)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of BAXDELA and other antibacterial drugs, BAXDELA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria. (1.3)
Baxdela Dosage and Administration
- For ABSSSI and CABP: Administer BAXDELA for injection 300 mg by intravenous infusion over 60 minutes, every 12 hours, or a 450 mg BAXDELA tablet orally every 12 hours. (2.1, 2.2)
- Recommended duration of treatment: (2.2)
- ABSSSI: 5 to 14 days
- CABP: 5 to 10 days
- Dosage for patients with renal impairment is based on the estimated glomerular filtration rate (eGFR) (2.3)
| Estimated Glomerular Filtration Rate (eGFR)(mL/min/1.73m2)* | Recommended Dosage Regimen for BAXDELA† | |
|---|---|---|
|
||
| Oral | Intravenous‡ | |
| 30-89 | No dosage adjustment | No dosage adjustment |
| 15-29 | No dosage adjustment | 200 mg every 12 hours |
| End Stage Renal Disease (ESRD) (< 15 including hemodialysis) | Not Recommended§ | |
Dosage Forms and Strengths
Warnings and Precautions
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥ 2%) are nausea, diarrhea, headache, transaminase elevations, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Melinta Therapeutics at 1-844-633-6568 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Use In Specific Populations
Renal Impairment: Closely monitor serum creatinine levels in patients with severe renal impairment (eGFR 15-29 mL/min/1.73 m2) receiving intravenous delafloxacin. If serum creatinine level increases occur, consider changing to oral delafloxacin. Discontinue BAXDELA if eGFR decreases to < 15 mL/min/1.73 m2 (8.6).
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2021
Full Prescribing Information
WARNING: SERIOUS ADVERSE REACTIONS INCLUDING TENDINITIS, TENDON RUPTURE, PERIPHERAL NEUROPATHY, CENTRAL NERVOUS SYSTEM EFFECTS and EXACERBATION OF MYASTHENIA GRAVIS
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions that have occurred together (5.1), including:
- Tendinitis and tendon rupture (5.2)
- Peripheral neuropathy (5.3)
- Central nervous system effects (5.4)
Discontinue BAXDELA immediately and avoid the use of fluoroquinolones, including BAXDELA, in patients who experience any of these serious adverse reactions (5.1)
Fluoroquinolones may exacerbate muscle weakness in patients with myasthenia gravis. Avoid BAXDELA in patients with known history of myasthenia gravis. (5.5)
1. Indications and Usage for Baxdela
1.1 Acute Bacterial Skin and Skin Structure Infections
BAXDELA is indicated in adults for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by the following susceptible microorganisms: Staphylococcus aureus (including methicillin-resistant [MRSA] and methicillin-susceptible [MSSA] isolates), Staphylococcus haemolyticus, Staphylococcus lugdunensis, Streptococcus agalactiae, Streptococcus anginosus Group (including Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus), Streptococcus pyogenes, Enterococcus faecalis, Escherichia coli, Enterobacter cloacae, Klebsiella pneumoniae, and Pseudomonas aeruginosa.
1.2 Community-Acquired Bacterial Pneumonia
BAXDELA is indicated in adults for the treatment of community-acquired bacterial pneumonia (CABP) caused by the following susceptible microorganisms: Streptococcus pneumoniae, Staphylococcus aureus (methicillin-susceptible [MSSA] isolates only), Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, Haemophilus influenzae, Haemophilus parainfluenzae, Chlamydia pneumoniae, Legionella pneumophila, and Mycoplasma pneumoniae.
1.3 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of BAXDELA and other antibacterial drugs, BAXDELA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2. Baxdela Dosage and Administration
2.1 Important Administration Instructions
BAXDELA Tablets
Administer BAXDELA at least 2 hours before or 6 hours after antacids containing magnesium, or aluminum, with sucralfate, with metal cations such as iron, or with multivitamin preparations containing zinc or iron, or with didanosine buffered tablets for oral suspension or the pediatric powder for oral solution [see Drug Interactions (7.1)].
BAXDELA Tablets can be taken with or without food [see Clinical Pharmacology (12.3)].
If patients miss a dose, they should take it as soon as possible anytime up to 8 hours prior to their next scheduled dose. If less than 8 hours remain before the next dose, wait until their next scheduled dose.
BAXDELA for Injection
Do NOT administer BAXDELA for Injection with any solution containing multivalent cations, e.g., calcium and magnesium, through the same intravenous line [see Drug Interactions (7.1)]. Do NOT co-infuse BAXDELA for Injection with other medications [see Dosage and Administration (2.4)].
2.2 Recommended Dosage Regimen
For treatment of adults with ABSSSI or CABP, the recommended dosage regimen of BAXDELA is described in Table 1 below.
| Infection | Dosage and Route of Administration | Total Duration (days) |
|---|---|---|
| ABSSSI |
| 5 to 14 |
| CABP | 5 to 10 |
2.3 Dosage in Patients with Renal Impairment
Table 2 below describes the dosage modification based on the estimated glomerular filtration rate (eGFR) that is recommended in patients with renal impairment. Dosage adjustment is required for patients with severe renal impairment (eGFR 15-29 mL/min/1.73m2).
In patients with severe renal impairment receiving BAXDELA intravenously, closely monitor serum creatinine levels and eGFR [see Use in Specific Populations (8.7)]. If serum creatinine level increases, consider switching to BAXDELA Tablets. Discontinue BAXDELA if eGFR decreases to < 15 mL/min/1.73 m2.
| Estimated Glomerular Filtration Rate (eGFR) (mL/min/1.73 m2)* | Recommended Dosage Regimen† | |
|---|---|---|
| BAXDELA Tablets | BAXDELA for Injection‡ | |
|
||
| 30-89 | No dosage adjustment | No dosage adjustment |
| 15-29 | No dosage adjustment | 200 mg every 12 hours Or 200 mg every 12 hours, then switch to a 450 mg BAXDELA tablet orally every 12 hours at the discretion of the physician |
| End Stage Renal Disease (ESRD) (< 15), including patients on hemodialysis (HD) | Not Recommended§ | |
2.4 Preparation and Administration of BAXDELA for Injection Intravenous Solution
Reconstitution and Dilution
- BAXDELA must be reconstituted and then further diluted under aseptic conditions. Reconstitute the powder in the BAXDELA vial using 10.5 mL of 5% Dextrose Injection (D5W) or 0.9% Sodium Chloride Injection for each 300 mg vial. Shake the vial vigorously until contents are completely dissolved. The reconstituted vial contains 300 mg per 12 mL (25 mg/mL) of BAXDELA as a clear yellow to amber colored solution.
- The reconstituted solution must then be diluted to a total volume of 250 mL using either 0.9% Sodium Chloride or D5W to achieve a concentration of 1.2 mg/mL, prior to administration. Prepare the required dose for intravenous infusion by withdrawing the appropriate volume from the reconstituted vial per Table 3 below:
Table 3 Preparation of BAXDELA Doses BAXDELA for Injection Dose Volume of Reconstituted Solution to Withdraw 300 mg 12 mL 200 mg 8 mL - Aseptically transfer the required volume of BAXDELA reconstituted solution from the vial to an intravenous bag to achieve a 250 mL volume of infusion solution. Discard any unused portion of the reconstituted solution.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Storage of the Reconstituted and Diluted Solutions
Reconstituted vials, as described above, may be stored either refrigerated at 2°C to 8°C (36°F to 46°F), or at controlled room temperature 20°C to 25°C (68°F to 77°F) for up to 24 hours. Do not freeze.
Once diluted into the intravenous bag, as described above, BAXDELA may be stored either refrigerated at 2°C to 8°C (36°F to 46°F) or at a controlled room temperature of 20°C to 25°C (68°F to 77°F) for up to 24 hours. Do not freeze.
Administration
After reconstitution and dilution, administer BAXDELA by intravenous infusion, using a total infusion time of 60 minutes [see Dosage and Administration (2.1)].
The compatibility of reconstituted BAXDELA with intravenous medications, additives, or substances other than D5W or 0.9% Sodium Chloride Injection has not been established. If a common intravenous line is being used to administer other drugs in addition to BAXDELA the line should be flushed before and after each BAXDELA infusion with 0.9% Sodium Chloride Injection or D5W.
3. Dosage Forms and Strengths
BAXDELA for Injection: A sterile, lyophilized powder containing 300 mg delafloxacin (equivalent to 433 mg delafloxacin meglumine) in a single-dose vial, which must be reconstituted and further diluted prior to intravenous infusion. The lyophilized powder is a light yellow to tan cake, which may exhibit cracking and shrinkage and slight variation in texture and color.
4. Contraindications
BAXDELA is contraindicated in patients with known hypersensitivity to delafloxacin or any of the fluoroquinolone class of antibacterial drugs, or any of the components of BAXDELA [see Warnings and Precautions (5.6)].
5. Warnings and Precautions
5.1 Disabling and Potentially Irreversible Serious Adverse Reactions Including Tendinitis and Tendon Rupture, Peripheral Neuropathy and Central Nervous System Effects
Fluoroquinolones have been associated with disabling and potentially irreversible serious adverse reactions from different body systems that can occur together in the same patient. Commonly seen adverse reactions include tendinitis, tendon rupture, arthralgia, myalgia, peripheral neuropathy, and central nervous system effects (hallucinations, anxiety, depression, insomnia, severe headaches, and confusion). These reactions could occur within hours to weeks after starting a fluoroquinolone. Patients of any age or without pre-existing risk factors have experienced these adverse reactions [see Warnings and Precautions (5.2, 5.3 and 5.4)].
Discontinue BAXDELA immediately at the first signs or symptoms of any serious adverse reaction. In addition, avoid the use of fluoroquinolones, including BAXDELA, in patients who have experienced any of these serious adverse reactions associated with fluoroquinolones.
5.2 Tendinitis and Tendon Rupture
Fluoroquinolones have been associated with an increased risk of tendinitis and tendon rupture in all ages. This adverse reaction most frequently involves the Achilles tendon, and has also been reported with the rotator cuff (the shoulder), the hand, the biceps, the thumb, and other tendons. Tendinitis or tendon rupture can occur, within hours or days of starting a fluoroquinolone, or as long as several months after completion of fluoroquinolone therapy. Tendinitis and tendon rupture can occur bilaterally.
This risk of developing fluoroquinolone-associated tendinitis and tendon rupture is increased in patients over age 60 years of age, in patients taking corticosteroid drugs, and, in patients with kidney, heart, and lung transplant. Other factors that may independently increase the risk of tendon rupture include strenuous physical activity, renal failure, and previous tendon disorders such as rheumatoid arthritis. Tendinitis and tendon rupture have also occurred in patients taking fluoroquinolones who do not have the above risk factors.
Discontinue BAXDELA immediately if the patient experiences pain, swelling, inflammation or rupture of a tendon. Advise patients, at the first sign of tendon pain, swelling, or inflammation, to stop taking BAXDELA, to avoid exercise and use of the affected area, and to promptly contact their healthcare provider about changing to a non-quinolone antimicrobial drug. Avoid BAXDELA in patients who have a history of tendon disorders or have experienced tendinitis or tendon rupture.
5.3 Peripheral Neuropathy
Fluoroquinolones have been associated with an increased risk of peripheral neuropathy. Cases of sensory or sensorimotor axonal polyneuropathy affecting small and/or large axons resulting in paresthesias, hypoesthesias, dysesthesias, and weakness have been reported in patients receiving fluoroquinolones, including BAXDELA. Symptoms may occur soon after initiation of fluoroquinolones and may be irreversible in some patients [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
Discontinue BAXDELA immediately if the patient experiences symptoms of peripheral neuropathy including pain, burning, tingling, numbness, and/or weakness or other alterations of sensation including light touch, pain, temperature, position sense, and vibratory sensation and/or motor strength in order to minimize the development of an irreversible condition. Avoid fluoroquinolones, including BAXDELA in patients who have previously experienced peripheral neuropathy [see Adverse Reactions (6.1)].
5.4 Central Nervous System Effects
Psychiatric Adverse Reactions
Fluoroquinolones, including BAXDELA, have been associated with an increased risk of psychiatric adverse reactions, including: toxic psychosis; hallucinations, or paranoia; depression, or suicidal thoughts or acts; delirium, disorientation, confusion, or disturbances in attention; anxiety, agitation, or nervousness; insomnia or nightmares; memory impairment. These adverse reactions may occur following the first dose. If these reactions occur in patients receiving BAXDELA, discontinue BAXDELA immediately and institute appropriate measures.
Central Nervous System Adverse Reactions
Fluoroquinolones have been associated with an increased risk of seizures (convulsions), increased intracranial pressure (including pseudotumor cerebri), dizziness, and tremors. As with all fluoroquinolones, use BAXDELA when the benefits of treatment exceed the risks in patients with known or suspected CNS disorders (e.g., severe cerebral arteriosclerosis, epilepsy) or in the presence of other risk factors that may predispose to seizures or lower the seizure threshold. If these reactions occur in patients receiving BAXDELA, discontinue BAXDELA immediately and institute appropriate measures.
5.5 Exacerbation of Myasthenia Gravis
Fluoroquinolones have neuromuscular blocking activity and may exacerbate muscle weakness in persons with myasthenia gravis. Post-marketing serious adverse reactions, including death and requirement for ventilator support, have been associated with fluoroquinolone use in persons with myasthenia gravis. Avoid BAXDELA in patients with known history of myasthenia gravis [see Patient Counseling Information (17)].
5.6 Hypersensitivity Reactions
Serious and occasionally fatal hypersensitivity (anaphylactic) reactions, some following the first dose, have been reported in patients receiving fluoroquinolone therapy. Some reactions were accompanied by cardiovascular collapse, loss of consciousness, tingling, pharyngeal or facial edema, dyspnea, urticaria, and itching. Hypersensitivity reactions have been reported in patients receiving BAXDELA. These reactions may occur after first or subsequent doses of BAXDELA [see Adverse Reactions (6.1)]. Discontinue BAXDELA at the first appearance of a skin rash or any other sign of hypersensitivity.
5.7 Clostridium difficile-Associated Diarrhea
Clostridium difficile-associated diarrhea (CDAD) has been reported in users of nearly all systemic antibacterial drugs, including BAXDELA, with severity ranging from mild diarrhea to fatal colitis. Treatment with antibacterial agents can alter the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B, which contribute to the development of CDAD. Hypertoxin-producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antibacterial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial use. Careful medical history is necessary because CDAD has been reported to occur more than 2 months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial use not directed against C. difficile should be discontinued, if possible. Appropriate measures such as fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.8 Risk of Aortic Aneurysm and Dissection
Epidemiologic studies report an increased risk of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients. The cause for the increased risk has not been identified. In patients with a known aortic aneurysm or patients who are at greater risk for aortic aneurysms, reserve BAXDELA for use only when there are no alternative antibacterial treatments available.
5.9 Development of Drug-Resistant Bacteria
Prescribing BAXDELA in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
5.10 Blood Glucose Disturbances
Fluoroquinolones have been associated with disturbances of blood glucose, including symptomatic hyperglycemia and hypoglycemia, usually in diabetic patients receiving concomitant treatment with an oral hypoglycemic agent (e.g., glyburide) or with insulin. In these patients, careful monitoring of blood glucose is recommended. Severe cases of hypoglycemia resulting in coma or death have been reported with other fluoroquinolones. If a hypoglycemic reaction occurs in a patient being treated with BAXDELA, discontinue BAXDELA and initiate appropriate therapy immediately [see Adverse Reactions (6.1)].
6. Adverse Reactions/Side Effects
The following serious and otherwise important adverse reactions are discussed in greater detail in other sections of labeling:
- Disabling and Potentially Irreversible Serious Adverse Reactions [see Warnings and Precautions (5.1)]
- Tendinitis and Tendon Rupture [see Warnings and Precautions (5.2)]
- Peripheral Neuropathy [see Warnings and Precautions (5.3)]
- Central Nervous System Effects [see Warnings and Precautions (5.4)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.6)]
- Clostridium difficile-Associated Diarrhea [see Warnings and Precautions (5.7)]
- Blood Glucose Disturbances [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of BAXDELA cannot be directly compared to rates in the clinical trials of another drug and may not reflect rates observed in practice.
Overview of the Safety Evaluation of BAXDELA
BAXDELA was evaluated in three Phase 3 multicenter, multinational, randomized, double-blind clinical trials. These trials included two trials in ABSSSI patients (Trial 1 and Trial 2) and one trial in CABP (Trial 3). A total of 1170 patients were treated with BAXDELA across all Phase 3 trials (741 patients in the two ABSSSI trials and 429 patients in the CABP trial).
Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
BAXDELA was evaluated in two multicenter, multinational, randomized, double-blind, double-dummy, non-inferiority trials (Trial 1 and Trial 2) in adults with ABSSSI. In Trial 1 patients received BAXDELA 300 mg by intravenous infusion every 12 hours and in Trial 2 the patients received BAXDELA 300 mg by intravenous infusion every 12 hours for 6 doses then were switched to BAXDELA 450 mg tablets every 12 hours. The total treatment duration was 5 to 14 days. Adverse reactions were evaluated for 741 patients treated with BAXDELA and 751 patients treated with comparator antibacterial drugs. The median age of patients treated with BAXDELA was 49 years, ranging between 18 and 94 years old; 15% were age 65 years and older. Patients treated with BAXDELA were predominantly male (62%) and Caucasian (86%). The BAXDELA treated population included 44% obese patients (BMI ≥ 30 kg/m2), 11% with diabetes, and 16% with baseline renal impairment (calculated creatinine clearance less than 90 mL/min).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
Serious adverse reactions occurred in 3/741 (0.4%) of patients treated with BAXDELA and in 6/751 (0.8%) of patients treated with the comparator.
BAXDELA was discontinued due to an adverse reaction in 7/741 (0.9%) patients and the comparator was discontinued due to an adverse reaction in 21/751 (2.8%) patients. The most commonly reported adverse reactions leading to study discontinuation in the BAXDELA arm included urticaria (2/741; 0.3%) and hypersensitivity (2/741; 0.3%); whereas, the most commonly reported adverse reactions leading to study discontinuation in the comparator arm included urticaria (5/751; 0.7%), rash (4/751; 0.5%), hypersensitivity and infusion site extravasation (2/751; 0.3%).
Most Common Adverse Reactions
The most common adverse reactions in patients treated with BAXDELA were nausea (8%), diarrhea (8%), headache (3%), transaminase elevations (3%), and vomiting (2%). Table 4 lists selected adverse reactions occurring in ≥ 2% of patients receiving BAXDELA in the pooled adult Phase 3 clinical trials.
Community-Acquired Bacterial Pneumonia
BAXDELA was evaluated in one multicenter, multinational, randomized, double-blind trial in adults with CABP (Trial 3). Patients received BAXDELA 300 mg over 60 minutes every 12 hours for a minimum of 6 doses with an option to switch to oral BAXDELA tablet 450 mg every 12 hours for the remaining doses (total of 10 to 20 doses of intravenous infusion and oral combined). Adverse reactions were evaluated for 429 patients treated with BAXDELA and 427 patients treated with moxifloxacin. The median age of patients treated with BAXDELA was 63 years, ranging between 18 and 89 years; 47.1% were 65 years of age and older and 19.6% were 75 years of age and older. Patients treated with BAXDELA were predominantly male (58.3%) and white (92.3%). The BAXDELA-treated population included patients with obesity (BMI greater than or equal to 30) (24.0%), COPD/asthma (14.2%), cardiac disease (24.2%), diabetes (16.3%), and baseline renal impairment including 36.4% with moderate renal impairment (CrCl less than 30-59 mL/min), and 4.0% with severe renal impairment (CrCl less than 29 mL/min). Overall, approximately 12.4% of patients were in PORT Risk Class II, 60.1% were in PORT Risk Class III, 26.6% were in PORT Risk Class IV, and 0.9% were in PORT Risk Class V.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
Serious adverse reactions occurred in 2/429 (0.5%) of patients treated with BAXDELA and in 1/427 (0.2%) of patients treated with moxifloxacin. Discontinuation due to an adverse reaction occurred in 9/429 (2.1%) patients treated with BAXDELA and in 4/427 (0.9%) treated with moxifloxacin. The most commonly reported adverse reactions leading to study drug discontinuation in the BAXDELA arm were transaminase elevations (2/429; 0.5%). The most commonly reported adverse reactions leading to study drug discontinuation in the comparator arm were infusion site reactions (1/427; 0.2%).
Most Common Adverse Reactions
The most common adverse reactions in patients treated with BAXDELA were diarrhea (5%) and transaminase elevations (5%). Table 5 lists selected adverse reactions occurring in ≥ 2% of patients receiving BAXDELA in the adult Phase 3 CABP clinical trial.
| Adverse Reactions | BAXDELA N = 429 | Moxifloxacin N = 427 |
|---|---|---|
|
||
| Diarrhea | 5% | 3% |
| Transaminase elevations* | 5% | 3% |
Adverse Reactions Occurring in Less Than 2% of Patients Receiving BAXDELA in the ABSSSI (Trials 1 and 2) and CABP (Trial 3) Clinical Trials
The following selected adverse reactions were reported in BAXDELA-treated patients at a rate of less than 2% in the ABSSSI (Trials 1 and 2) and CABP (Trial 3) clinical trials:
Blood and Lymphatic System Disorders: agranulocytosis, anemia, leukopenia, neutropenia, pancytopenia
Cardiac Disorders: sinus tachycardia, palpitations, bradycardia, ventricular extrasystoles
Ear and Labyrinth Disorders: tinnitus, vertigo, vestibular disorder
Eye Disorders: vision blurred
General disorders and administration site conditions: infusion related reactions
Gastrointestinal Disorders: abdominal pain, dyspepsia
Immune System Disorders: hypersensitivity
Infections and Infestations: Clostridium difficile infection, fungal infection, oral candidiasis, vulvovaginal candidiasis
Laboratory Investigations: blood alkaline phosphatase increased, blood creatinine increased, blood creatine phosphokinase increased
Metabolism and Nutrition Disorders: hyperglycemia, hypoglycemia
Musculoskeletal and Connective Tissue Disorders: myalgia
Nervous System Disorders: dizziness, hypoesthesia, paraesthesia, dysgeusia, presyncope, syncope
Psychiatric Disorders: agitation, anxiety, confusional state, insomnia, abnormal dreams
Renal and Urinary: renal impairment, renal failure
Skin and Subcutaneous Tissue Disorders: pruritus, urticaria, dermatitis, rash
Vascular Disorders: flushing, hypotension, hypertension
Related/similar drugs
7. Drug Interactions
7.1 Chelation Agents: Antacids, Sucralfate, Metal Cations, Multivitamins
Fluoroquinolones form chelates with alkaline earth and transition metal cations. Oral administration of BAXDELA with antacids containing aluminum or magnesium, with sucralfate, with metal cations such as iron, or with multivitamins containing iron or zinc, or with formulations containing divalent and trivalent cations such as didanosine buffered tablets for oral suspension or the pediatric powder for oral solution, may substantially interfere with the absorption of BAXDELA, resulting in systemic concentrations considerably lower than desired. Therefore, BAXDELA should be taken at least 2 hours before or 6 hours after these agents [see Dosage and Administration (2.1)].
There are no data concerning an interaction of intravenous BAXDELA with oral antacids, sucralfate, multivitamins, didanosine, or metal cations. However, BAXDELA should not be co-administered with any solution containing multivalent cations, e.g., magnesium, through the same intravenous line [see Dosage and Administration (2.1)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
The limited available data with BAXDELA use in pregnant women are insufficient to inform a drug-associated risk of major birth defects and miscarriages. When delafloxacin (as the N-methyl glucamine salt) was administered orally to rats during the period of organogenesis, no malformations or fetal death were observed at up to 7 times the estimated clinical exposure based on AUC. When rats were dosed intravenously in late pregnancy and through lactation, there were no adverse effects on offspring at exposures approximating the clinical intravenous (IV) exposure based on AUC [see Data].
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
In embryo-fetal studies, oral administration of delafloxacin to pregnant rats during the period of major organogenesis resulted in maternal toxicity and reduced fetal body weights at the highest dose (1600 mg/kg/day) and fetal ossification delays at all doses. No malformations were reported up to the highest dose tested (approximately 7 times the estimated human plasma exposure based on AUC). The lowest dose, 200 mg/kg/day (approximately 2.5 times the estimated human plasma exposure based on AUC), was still toxic to the fetus, based on ossification delays. In rabbits, a species known to be extremely sensitive to maternal toxicity of antibacterial drugs, no embryo-fetal developmental toxicity was observed up to the highest dose which induced maternal toxicity (1.6 mg/kg/day, or approximately 0.01 times the estimated human plasma exposure based on AUC). In a pre-postnatal study in rats of IV administered delafloxacin, dams at the highest dose tested (120 mg/kg/day) exhibited slightly lower body weights and slightly longer gestation length than control animals. Exposure at that dose was estimated to be approximately 5 times human plasma exposure based on AUC, as determined in a separate shorter term study at an earlier stage of pregnancy. Effects on pups at that dose included increased mortality during lactation, small stature, and lower body weights, but no changes in learning and memory, sensory function, locomotor activity, developmental landmarks, or reproductive performance were reported. The No Adverse Effect Level (NOAEL) for maternal toxicity pup development in that study was 60 mg/kg/day (approximately 580 mg/day IV for a 60 kg patient, or just below the clinical IV dose).
8.2 Lactation
Risk Summary
There are no data available on the presence of delafloxacin in human milk, the effects on the breast-fed infant, or the effects on milk production. Delafloxacin is excreted in the breast milk of rats [see Data]. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for BAXDELA and any potential adverse effects on the breast-fed child from BAXDELA or from the underlying maternal condition.
Data
After single oral dose of 20 mg/kg (approximately 194 mg for a 60 kg patient) 14C-labeled delafloxacin on post-natal day 11, the radioactivity was transferred into the milk of lactating rats. The mean milk/plasma radioactivity concentration ratios in dams at 4 and 8 hours after dosing were 8.5 and 4.0, respectively, and essentially background by 24 hours. The rate of elimination of radioactivity was similar in milk and plasma. Absorption of radioactive drug by rat pups following nursing was observed.
8.4 Pediatric Use
Use in patients under 18 years of age is not recommended. Safety and effectiveness in pediatric patients below the age of 18 years have not been established. Pediatric studies were not conducted because risk-benefit considerations do not support the use of BAXDELA for ABSSSI in this population. Fluoroquinolones cause arthropathy in juvenile animals.
8.5 Geriatric Use
Of the 754 adult ABSSSI patients treated with BAXDELA, in Trials 1 and 2, 111/754 (15%) were 65 years of age and older. The clinical response rates at 48-72 hours for the BAXDELA-treated and comparator-treated patients were 84/111 (75.7%) and 72/101 (71.3%), respectively in ABSSSI patients aged 65 years and older compared to patients aged less than 65 years of age 529/643 (82.3%) and 538/655 (82.1%), respectively. In the safety population, of the 741 adult patients treated with BAXDELA, 18/110 (16.4%) patients aged 65 years and older and 146/631 (23.1%) patients aged less than 65 years had at least one adverse drug reaction.
Of the 431 adult CABP patients treated with BAXDELA, in Trial 3, 203/431 (47.1%) were 65 years of age and older, while 85/431 (19.7%) were 75 and over. The clinical response rates at 72-120 hours for the BAXDELA-treated and moxifloxacin-treated patients were 177/203 (87.2%) and 161/179 (89.9%), respectively in the CABP patients aged 65 years and older compared to patients aged less than 65 years old (206/228 (90.4%) and 220/249 (88.4%), respectively). In the safety population, of the 429 adult patients treated with BAXDELA, 10/84 (11.9%) patients aged 75 and older, 27/202 (13.4%) patients aged 65 years and older and 38/227 (16.7%) patients aged less than 65 years old had at least one adverse drug reaction.
Geriatric patients are at increased risk for developing severe tendon disorders including tendon rupture when being treated with a fluoroquinolone. This risk is further increased in patients receiving concomitant corticosteroid therapy. Tendinitis or tendon rupture can involve the Achilles, hand, shoulder, or other tendon sites and can occur during or after completion of therapy; cases occurring up to several months after fluoroquinolone treatment have been reported. Caution should be used when prescribing BAXDELA to elderly patients especially those on corticosteroids. Patients should be informed of this potential adverse reaction and advised to discontinue BAXDELA and contact their healthcare provider if any symptoms of tendinitis or tendon rupture occur [see Warnings and Precautions (5.1)].
Epidemiologic studies report an increased rate of aortic aneurysm and dissection within two months following use of fluoroquinolones, particularly in elderly patients [see Warnings and Precautions (5.8)].
In elderly subjects (≥ 65 years), the mean Cmax and AUC∞ of delafloxacin were about 35% higher compared with young adults, which is not considered clinically significant [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dosage adjustment is necessary for BAXDELA in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment of BAXDELA is necessary in patients with mild (eGFR 60-89 mL/min/1.73 m2) or moderate (eGFR 30-59 mL/min/1.73 m2) renal impairment. The dose of BAXDELA intravenous IV infusion in patients with severe renal impairment (eGFR 15-29 mL/min/1.73 m2) should be decreased to 200 mg intravenously every 12 hours; the dose of oral BAXDELA in patients with severe renal impairment (eGFR 15-29 mL/min/1.73 m2) is 450 mg orally every 12 hours. BAXDELA is not recommended in patients with End Stage Renal Disease [ESRD] (eGFR of < 15 mL/min/1.73 m2) [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
In patients with severe renal impairment or ESRD (eGFR of < 15 mL/min/1.73 m2), accumulation of the intravenous vehicle, sulfobutylether-β-cyclodextrin (SBECD) occurs. Serum creatinine levels should be closely monitored in patients with severe renal impairment receiving intravenous BAXDELA. If serum creatinine level increases occur, consideration should be given to changing to oral BAXDELA. If eGFR decreases to < 15 mL/min/1.73 m2, BAXDELA should be discontinued.
10. Overdosage
Treatment of overdose with BAXDELA should consist of observation and general supportive measures. Hemodialysis removed about 19% of delafloxacin and 56% of SBECD (Sulfobutylether β cyclodextrin) after intravenous administration of BAXDELA [see Clinical Pharmacology (12.3)].
11. Baxdela Description
BAXDELA (delafloxacin) for Injection and BAXDELA (delafloxacin) Tablets contain meglumine salt of delafloxacin, a fluoroquinolone antibacterial. Delafloxacin meglumine is identified chemically as 1-Deoxy-1-(methylamino)-D-glucitol, 1-(6-amino-3,5-difluoropyridin-2-yl)-8-chloro-6-fluoro-7-(3-hydroxyazetidin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (salt), the chemical structure of which is shown below. The meglumine salt has a molecular weight of 635.97 g/mol, whereas the molecular weight of the delafloxacin free acid is 440.76 g/mol.
Figure 1 Chemical Structure
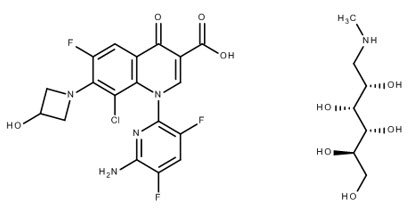
C18H12ClF3N4O4 ∙ C7H17NO5 M.W. 635.97
BAXDELA is intended for intravenous infusion or oral administration. BAXDELA is supplied as a sterile, lyophilized powder for injection and oral tablets as follows:
BAXDELA for Injection
Each vial of BAXDELA for Injection, 300 mg, is a sterile lyophilized powder that contains 300 mg delafloxacin (equivalent to 433 mg delafloxacin meglumine) and the following inactive ingredients: Edetate disodium (EDTA), (3.4 mg); meglumine (59 mg); sulfobutylether-β-cyclodextrin (2400 mg). Sodium hydroxide and/or hydrochloric acid may have been used to adjust the pH.
BAXDELA Tablets
Each BAXDELA tablet for oral use contains 450 mg delafloxacin (equivalent to 649 mg delafloxacin meglumine) and the following inactive ingredients: Citric acid anhydrous (5.5 mg); crospovidone (109 mg); magnesium stearate (10 mg); microcrystalline cellulose (417 mg); povidone (34 mg); sodium bicarbonate (140 mg); sodium phosphate monobasic monohydrate (5.5 mg).
12. Baxdela - Clinical Pharmacology
12.2 Pharmacodynamics
The antibacterial activity of delafloxacin appears to best correlate with the ratio of area under the concentration-time curve of free delafloxacin to minimal inhibitory concentration (fAUC/MIC) for Gram-positive organisms such as Staphylococcus aureus and Gram-negative organisms such as Escherichia coli based on animal models of infection.
Cardiac Electrophysiology
In a randomized, positive- and placebo-controlled, thorough QT/QTc study, 51 healthy subjects received BAXDELA 300 mg IV, BAXDELA 900 mg IV, oral moxifloxacin 400 mg, or placebo. Neither BAXDELA 300 mg nor BAXDELA 900 mg (three times the intravenous therapeutic dose) had any clinically relevant adverse effect on cardiac repolarization.
Photosensitivity Potential
A study of photosensitizing potential to ultraviolet (UVA and UVB) and visible radiation was conducted in 52 healthy volunteers (originally 13 subjects per treatment group). BAXDELA, at 200 mg/day and 400 mg/day (0.22 and 0.44 times the approved recommended daily oral dosage, respectively) for 7 days, and placebo did not demonstrate clinically significant phototoxic potential at any wavelengths tested (295 nm to 430 nm), including solar simulation. The active comparator (lomefloxacin) demonstrated a moderate degree of phototoxicity at UVA 335 nm and 365 nm and solar simulation wavelengths.
12.3 Pharmacokinetics
The pharmacokinetic parameters of delafloxacin following single- and multiple-dose (every 12 hours) oral (450 mg) and intravenous (300 mg) administration are shown in Table 6. Steady-state was achieved within approximately three days with accumulation of approximately 10% and 36% following IV and oral administration, respectively.
| Parameters | Tablet | Intravenous Injection | ||
|---|---|---|---|---|
| Single Dose 450 mg | Steady State 450 mg Q12h* | Single Dose 300 mg | Steady State 300 mg Q12h* |
|
| Cmax = maximum concentration; Tmax = time to reach Cmax; AUC = area under the concentration-time curve; CL = systemic clearance; CL/F = apparent oral clearance; Rac = accumulation ratio | ||||
| Tmax (h)† | 0.75 (0.5, 4.0) | 1.00 (0.50, 6.00) | 1.0 (1.0, 1.2) | 1.0 (1.0, 1.0) |
| Cmax (µg/mL) | 7.17 (2.01) | 7.45 (3.16) | 8.94 (2.54) | 9.29 (1.83) |
| AUC (µg∙h/mL)‡ | 22.7 (6.21) | 30.8 (11.4) | 21.8 (4.54) | 23.4 (6.90) |
| CL or CL/F(L/h)§ | 20.6 (6.07) | 16.8 (6.54) | 14.1 (2.81) | 13.8 (3.96) |
| CLr (L/h) | - | - | 5.89 (1.53) | 6.69 (2.19) |
| Rac | - | 1.36 | - | 1.1 |
Absorption
The absolute bioavailability for BAXDELA 450 mg oral tablet administered as a single dose was 58.8%. The AUC of delafloxacin following administration of a single 450 mg oral (tablet) dose was comparable to that following a single 300 mg intravenous dose. The Cmax of delafloxacin was achieved within about 1 hour after oral administration under fasting condition. Food (kcal: 917, Fat: 58.5%, Protein: 15.4%, Carbohydrate: 26.2%). did not affect the bioavailability of delafloxacin [see Dosage and Administration (2.1)].
Distribution
The steady state volume of distribution of delafloxacin is 30–48 L which approximates total body water. The plasma protein binding of delafloxacin is approximately 84%; delafloxacin primarily binds to albumin. Plasma protein binding of delafloxacin is not significantly affected by renal impairment.
Following IV administration of 7 doses of 300 mg of BAXDELA to 30 healthy volunteers, the mean BAXDELA AUC0-12 (3.6 hr*mcg/mL) in alveolar macrophages was 80% of the free-plasma AUC0-12, and the mean BAXDELA AUC0-12 (2.8 hr*mcg/mL) in epithelial lining fluid was 70% of the free-plasma AUC0-12.
Elimination
In a mass balance study, the mean half-life for delafloxacin was 3.7 hours (SD 0.7 hour) after a single dose intravenous administration. The mean half-life values for delafloxacin ranged from 4.2 to 8.5 hours following multiple oral administrations. Following administration of a single 300 mg intravenous dose of BAXDELA, the mean clearance (CL) of delafloxacin was 16.3 L/h (SD 3.7 L/h), and the renal clearance (CLr) of delafloxacin accounts for 35-45% of the total clearance.
Metabolism
Glucuronidation of delafloxacin is the primary metabolic pathway with oxidative metabolism representing about 1% of an administered dose. The glucuronidation of delafloxacin is mediated mainly by UGT1A1, UGT1A3, and UGT2B15. Unchanged parent drug is the predominant component in plasma. There are no significant circulating metabolites in humans.
Excretion
After single intravenous dose of 14C-labeled delafloxacin, 65% of the radioactivity was excreted in urine as unchanged delafloxacin and glucuronide metabolites and 28% was excreted in feces as unchanged delafloxacin. Following a single oral dose of 14C-labeled delafloxacin, 50% of the radioactivity was excreted in urine as unchanged delafloxacin and glucuronide metabolites and 48% was excreted in feces as unchanged delafloxacin.
Specific Populations
No clinical significance in the pharmacokinetics of delafloxacin was observed based on age, sex, race, weight, body mass index, and disease state (ABSSSI and CABP).
Patients with Hepatic Impairment
No clinically meaningful changes in delafloxacin Cmax and AUC were observed, following administration of a single 300 mg intravenous dose of BAXDELA to patients with mild, moderate or severe hepatic impairment (Child-Pugh Class A, B, and C) compared to matched healthy control subjects.
Patients with Renal Impairment
Following a single intravenous (300 mg) administration of delafloxacin to subjects with mild (eGFR = 51-80 mL/min/1.73 m2), moderate (eGFR = 31–50 mL/min/1.73 m2), severe (eGFR = 15-29 mL/min/1.73 m2) renal impairment, and ESRD on hemodialysis receiving intravenous delafloxacin within 1 hour before and 1 hour after hemodialysis, mean total exposure (AUCt) of delafloxacin was 1.3, 1.6, 1.8, 2.1, and 2.6-fold higher, respectively than that for matched normal control subjects. The mean dialysate clearance (CLd) of delafloxacin was 4.21 L/h (SD 1.56 L/h). After about 4 hours of hemodialysis, the mean fraction of administered delafloxacin recovered in the dialysate was about 19% [see Use in Specific Populations (8.7)].
Following a single oral (400 mg) administration of delafloxacin to subjects with mild (eGFR = 51-80 mL/min/1.73 m2), moderate (eGFR = 31-50 mL/min/1.73 m2), or severe (eGFR = 15-29 mL/min/1.73 m2) renal impairment, the mean total exposure (AUCt) of delafloxacin was about 1.5-fold higher for subjects with moderate and severe renal impairment compared with healthy subjects, whereas total systemic exposures of delafloxacin in subjects with mild renal impairment were comparable with healthy subjects.
In patients with moderate (eGFR = 31–50 mL/min/1.73 m2), or severe (eGFR = 15–29 mL/min/1.73 m2) renal impairment or ESRD on hemodialysis, accumulation of the intravenous vehicle SBECD occurs. The mean systemic exposure (AUC) increased 2-fold, 5-fold, 7.5-fold, and 27-fold for patients with moderate impairment, severe impairment, ESRD on hemodialysis receiving intravenous delafloxacin within 1 hour before, and 1 hour after hemodialysis respectively, compared to the healthy control group. In subjects with ESRD undergoing hemodialysis, SBECD is dialyzed with a clearance of 4.74 L/h. When hemodialysis occurred 1 hour after the BAXDELA infusion in subjects with ESRD, the mean fraction of SBECD recovered in the dialysate was 56.1% over approximately 4 hours.
Geriatric Patients
Following single oral administration of 250 mg delafloxacin (approximately 0.6 times the approved recommended oral dose), the mean delafloxacin Cmax and AUC∞ values in elderly subjects (≥ 65 years) were about 35% higher compared to values obtained in young adults (18 to 40 years). This difference is not considered clinically relevant. A population pharmacokinetic analysis of patients with ABSSSI or CABP indicated that patients over the age of 65 years have slower clearance than younger patients. However, the overall impact on delafloxacin pharmacokinetics is not considered clinically significant and dose adjustment in elderly patients is not warranted.
Male and Female Patients
Following single oral administration of 250 mg delafloxacin (approximately 0.6 times the approved recommended oral dose), the mean delafloxacin Cmax and AUC∞ values in male subjects were comparable to female subjects. Results from a population pharmacokinetic analysis showed that females have a 24% lower AUC than males. This difference is not considered clinically relevant.
Drug Interaction Studies
Drug Metabolizing Enzymes
Delafloxacin at clinically relevant concentrations does not inhibit the cytochrome P450 isoforms CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4/5 in vitro in human liver microsomes. At a delafloxacin concentration (500 µM) well above clinically relevant exposures, the activity of CYP2E1was increased.
In human hepatocytes, delafloxacin showed no potential for in vitro induction of CYP1A2, 2B6, 2C19, or 2C8 but was a mild inducer of CYP2C9 at a concentration of 100 µM and CYP3A4 at a clinically relevant concentration. Administration of BAXDELA 450 mg every 12 hours for 5 days to healthy male and female subjects (n = 22) prior to and on Day 6 with a single oral 5 mg dose of midazolam (a sensitive CYP3A substrate), did not affect the Cmax and AUC values for midazolam or 1-hydroxy midazolam compared to administration of midazolam alone.
Transporters
Delafloxacin was not an inhibitor of the following hepatic and renal transporters in vitro at clinically relevant concentrations: MDR1, BCRP, OAT1, OAT3, OATP1B1, OATP1B3, BSEP, OCT1 and OCT2. Delafloxacin was not a substrate of OAT1, OAT3, OCT1, OCT2, OATP1B1 or OATP. Delafloxacin was shown to be a substrate of P-gp and BCRP in vitro. The clinical relevance of co-administration of delafloxacin and P-gp and/or BCRP inhibitors is unknown.
12.4 Microbiology
Mechanism of Action
Delafloxacin belongs to the fluoroquinolone class of antibacterial drugs and is anionic in nature. The antibacterial activity of delafloxacin is due to the inhibition of both bacterial topoisomerase IV and DNA gyrase (topoisomerase II) enzymes which are required for bacterial DNA replication, transcription, repair, and recombination. Delafloxacin exhibits a concentration-dependent bactericidal activity against gram-positive and gram-negative bacteria in vitro.
Resistance
Resistance to fluoroquinolones, including delafloxacin, can occur due to mutations in defined regions of the target bacterial enzymes topoisomerase IV and DNA gyrase referred to as Quinolone-Resistance Determining Regions (QRDRs), or through altered efflux.
Fluoroquinolones, including delafloxacin, have a different chemical structure and mechanism of action relative to other classes of antibacterial compounds (e.g., aminoglycosides, macrolides, β-lactams, glycopeptides, tetracyclines and oxazolidinones).
In vitro resistance to delafloxacin develops by multiple step mutations in the QRDRs of gram-positive and gram-negative bacteria. Delafloxacin-resistant mutants were selected in vitro at a frequency of < 10-9.
Although cross-resistance between delafloxacin and other fluoroquinolone-class antibacterial agents has been observed, some isolates resistant to other fluoroquinolone-class antibacterial agents may be susceptible to BAXDELA including some S. aureus isolates carrying mutations in the quinolone resistance determining region (gyrA, parC and parE).
Additionally, delafloxacin has activity against some isolates of beta-lactamase positive H. influenzae and H. parainfluenzae.
Interaction With Other Antimicrobials
In vitro drug combination studies with delafloxacin and amoxicillin/clavulanate, azithromycin, aztreonam, ceftaroline, ceftazidime, ceftriaxone, colistin, daptomycin, doxycycline, linezolid, meropenem, penicillin, rifampin, tigecycline, trimethoprim/sulfamethoxazole and vancomycin demonstrated neither synergy nor antagonism.
Antimicrobial Activity
BAXDELA has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections, [see Indications and Usage (1.1, 1.2)].
Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
Aerobic bacteria
- Gram-positive bacteria
- Staphylococcus aureus (including methicillin-resistant and methicillin-susceptible isolates)
- Staphylococcus haemolyticus
- Staphylococcus lugdunensis
- Streptococcus pyogenes
- Streptococcus agalactiae
- Streptococcus anginosus Group (including S. anginosus, S. intermedius, and S. constellatus)
- Enterococcus faecalis
- Gram-negative bacteria
- Escherichia coli
- Klebsiella pneumoniae
- Enterobacter cloacae
- Pseudomonas aeruginosa
Community-Acquired Bacterial Pneumonia (CABP)
Aerobic bacteria
- Gram-positive bacteria
- Streptococcus pneumoniae
- Staphylococcus aureus (methicillin-susceptible isolates only)
- Gram-negative bacteria
- Escherichia coli
- Haemophilus influenzae
- Haemophilus parainfluenzae
- Klebsiella pneumoniae
- Pseudomonas aeruginosa
- Other microorganisms
- Chlamydia pneumoniae
- Legionella pneumophila
- Mycoplasma pneumoniae
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint of delafloxacin against isolates of similar genus or organism group. However, the efficacy of BAXDELA in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Aerobic bacteria
- Gram-positive bacteria
- Streptococcus dysgalactiae
- Gram-negative bacteria
- Enterobacter aerogenes
- Klebsiella oxytoca
- Proteus mirabilis
- Moraxella catarrhalis
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies have not been conducted with BAXDELA.
Delafloxacin was not mutagenic in a bacterial reverse mutation (Ames) assay, and was not clastogenic in a mouse bone marrow micronucleus test at ≥ 15 times the estimated human plasma exposure based on AUC. In an in vitro clastogenicity assay using isolated human lymphocytes, delafloxacin was negative in short incubations (~3 hours) and, at high cytotoxic concentrations (> 1.0 mM), was positive in a long incubation (~19 hours).
Delafloxacin did not affect the fertility of male and female rats up to the highest intravenous dose tested (120 mg/kg/day); female rats were dosed 2 weeks prior to mating and through gestation day 7 and male rats were treated for 28 days prior to mating and beyond for a total of 58-59 days. AUC in male and female (non-pregnant and pregnant) rats at 120 mg/kg/day delafloxacin intravenous was estimated to be approximately 5 times the estimated human plasma exposure based on AUC in separate intravenous toxicology studies in rats, one of which was a 2-week study that used a different vehicle for delafloxacin than in the fertility study, and another was an 8-day study in nonpregnant and pregnant (gestation day 13) rats that used the same vehicle for delafloxacin as in the fertility study.
13.2 Animal Toxicology and/or Pharmacology
Fluoroquinolone antibacterials are associated with degenerative changes in articular cartilage and arthropathy in skeletally immature animals. In a toxicology study of the formulated tablet in dogs, the femoral head of one of three high dose (480 mg/kg/day) females had minimal focal degeneration of the superficial articular cartilage and a small focal cleft in the articular cartilage. No other joints were examined.
14. Clinical Studies
14.1 Acute Bacterial Skin and Skin Structure Infections
A total of 1510 adults with acute bacterial skin and skin structure infections (ABSSSI) were randomized in 2 multicenter, multinational, double-blind, double-dummy, non-inferiority trials. Trial 1 compared BAXDELA 300 mg via intravenous infusion every 12 hours to comparator. In Trial 2, patients received BAXDELA 300 mg via intravenous infusion every 12 hours for 6 doses then made a mandatory switch to oral BAXDELA 450 mg every 12 hours. In both studies, the comparator was the intravenous combination of vancomycin 15 mg/kg actual body weight and aztreonam. Aztreonam therapy was discontinued if no gram-negative pathogens were identified in the baseline cultures.
In Trial 1, 331 patients with ABSSSI were randomized to BAXDELA and 329 patients were randomized to vancomycin plus aztreonam. Patients in this trial had the following infections: cellulitis (39%), wound infection (35%), major cutaneous abscess (25%), and burn infection (1%). The overall mean surface area of the infected lesion as measured by digital planimetry was 307 cm2. The average age of patients was 46 years (range 18 to 94 years). Patients were predominately male (63%) and white (91%); 32% had BMI ≥ 30 kg/m2. The population studied in Trial 1 included a distribution of patients with associated comorbidities such as hypertension (21%), diabetes (9%), and renal impairment (16%; 0.2% with severe renal impairment or ESRD). Current or recent history of drug abuse, including IV drug abuse, was reported by 55% of patients. Bacteremia was documented at baseline in 2% of patients.
In Trial 2, 423 patients were randomized to BAXDELA and 427 patients were randomized to vancomycin plus aztreonam. Patients in this trial had the following infections: cellulitis (48%), wound infection (26%), major cutaneous abscess (25%), and burn infection (1%). The overall mean surface area of the infected lesion, as measured by digital planimetry, was 353 cm2. The average age of patients was 51 years (range 18 to 93 years). Patients were predominately male (63%) and white (83%); 50 % had a BMI ≥ 30 kg/m2. The population studied in Trial 2 included a distribution of patients with associated comorbidities such as hypertension (31%), diabetes (13%) and renal impairment (16%; 0.2% with severe renal impairment or ESRD). Current or recent history of drug abuse, including IV drug abuse, was reported by 30% of patients. Bacteremia was documented at baseline in 2% of patients.
In both trials, objective clinical response at 48 to 72 hours post initiation of treatment was defined as a 20% or greater decrease in lesion size as determined by digital planimetry of the leading edge of erythema. Table 7 summarizes the objective clinical response rates in both of these trials.
| CI = Confidence Interval; ITT = Intent To Treat and includes all randomized patients | |||
|
|||
| Trial | BAXDELA (300 mg IV) | Vancomycin 15 mg/kg + Aztreonam | Treatment Difference†
(2-sided 95% CI) |
| Trial 1 | |||
| Total N | 331 | 329 | |
| Responder, n (%) | 259 (78.2%) | 266 (80.9%) | -2.6 (-8.8, 3.6) |
| BAXDELA (300 mg IV and 450 mg oral) | Vancomycin 15 mg/kg + Aztreonam | ||
| Trial 2 | |||
| Total N | 423 | 427 | |
| Responder, n (%) | 354 (83.7%) | 344 (80.6%) | 3.1 (-2.0, 8.3) |
In both trials, an investigator assessment of response was made at Follow-up (Day 14 ± 1) in the ITT and CE populations. Success was defined as "cure + improved," where patients had complete or near resolution of signs and symptoms, with no further antibacterial needed. The success rates in the ITT and CE populations are shown in Table 8.
| CI = Confidence Interval; ITT = Intent To Treat and includes all randomized patients; CE = Clinically Evaluable consisted of all ITT patients who had a diagnosis of ABSSSI, received at least 80% of expected doses of study drug, did not have any protocol deviations that would affect the assessment of efficacy and had investigator assessment at the Follow-Up Visit. | |||
| Trial | BAXDELA (300 mg IV) | Vancomycin 15 mg/kg + Aztreonam | Treatment Difference*
(2-sided 95% CI) |
| Trial 1 | |||
| Success†, n/N (%) ITT | 270/331 (81.6%) | 274/329 (83.3%) | -1.7 (-7.6, 4.1) |
| Success†, n/N (%) CE | 232/240 (96.7%) | 238/244 (97.5%) | -0.9 (-4.3, 2.4) |
| BAXDELA (300 mg IV and 450 mg Oral) | Vancomycin 15 mg/kg + Aztreonam | ||
| Trial 2 | |||
| Success, n/N (%) ITT | 369/423 (87.2%) | 362/427 (84.8%) | 2.5 (-2.2, 7.2) |
| Success, n/N (%) CE | 339/353 (96.0%) | 319/329 (97.0%) | -0.9 (-3.9, 2.0) |
Six delafloxacin patients had baseline S. aureus bacteremia with ABSSSI. Five of these 6 patients (83.3%) were clinical responders at 48 to 72 hours and 5/6 (83.3%) were considered clinical success for ABSSSI at Day 14 ± 1. Two delafloxacin patients had baseline Gram-negative bacteremia (K. pneumoniae and P. aeruginosa), and both were clinical responders and successes.
The investigator assessments of clinical success rates were also similar between treatment groups at Late Follow-up (LFU, day 21-28).
Objective clinical response and investigator-assessed success by baseline pathogens from the primary infection site or blood cultures for the microbiological ITT (MITT) patient population pooled across Trial 1 and Trial 2 are presented in Table 9.
| Clinical Response† at 48–72 hours | Investigator-Assessed Success‡ at Follow-up | |||
|---|---|---|---|---|
| BAXDELA | Comparator | BAXDELA | Comparator | |
| Pathogen | n/N (%) | n/N (%) | n/N (%) | n/N (%) |
|
||||
| Staphylococcus aureus | 271/319 (85.0) | 269/324 (83.0) | 275/319 (86.2) | 269/324 (83.0) |
| Methicillin-susceptible§ | 149/177 (84.2) | 148/183 (80.9) | 154/177 (87.0) | 153/183 (83.6) |
| Methicillin-resistant§ | 125/144 (86.8) | 121/141 (85.8) | 122/144 (84.7) | 116/141 (82.3) |
| Streptococcus pyogenes | 17/23 (73.9) | 9/18 (50.0) | 21/23 (91.3) | 16/18 (88.9) |
| Staphylococcus haemolyticus | 11/15 (73.3) | 7/8 (87.5) | 13/15 (86.7) | 7/8 (87.5) |
| Streptococcus agalactiae | 10/14 (71.4) | 9/12 (75.0) | 12/14 (85.7) | 11/12 (91.7) |
| Streptococcus anginosus Group | 59/64 (92.2) | 55/61 (90.2) | 54/64 (84.4) | 47/61 (77.0) |
| Staphylococcus lugdunensis | 8/11 (72.7) | 6/9 (66.7) | 10/11 (90.9) | 8/9 (88.9) |
| Enterococcus faecalis | 11/11 (100.0) | 12/16 (75.0) | 9/11 (81.8) | 14/16 (87.5) |
| Escherichia coli | 12/14 (85.7) | 16/20 (80.0) | 12/14 (85.7) | 18/20 (90.0) |
| Enterobacter cloacae | 10/14 (71.4) | 8/11 (72.7) | 12/14 (85.7) | 10/11 (90.9) |
| Klebsiella pneumoniae | 19/22 (86.4) | 22/23 (95.7) | 20/22 (90.9) | 21/23 (91.3) |
| Pseudomonas aeruginosa | 9/11 (81.8) | 11/12 (91.7) | 11/11 (100.0) | 12/12 (100.0) |
14.2 Community-Acquired Bacterial Pneumonia
A total of 859 adults with CABP were randomized in a multicenter, multinational, double-blind, double-dummy, noninferiority trial comparing BAXDELA to moxifloxacin (Trial 3, NCT 02679573). In this trial, BAXDELA for injection 300 mg was administered intravenously (IV) every 12 hours with an option to switch to BAXDELA tablet 450 mg orally every 12 hours. Moxifloxacin 400 mg was administered IV every 24 hours with an option to switch to moxifloxacin tablet 400 mg orally every 24 hours. Switch to oral treatment was allowed after a minimum of 3 days of IV dosing. Total treatment duration was 5 to 10 days. In the moxifloxacin arm, the investigator could switch patients to linezolid 600 mg every 12 hours if methicillin-resistant Staphylococcus aureus (MRSA) was confirmed.
A total of 431 patients were randomized to BAXDELA and 428 to moxifloxacin. Patient demographic and baseline characteristics were balanced between the treatment arms. In this trial, 12.9% of patients were in PORT Risk Class II, 60.3% were in PORT Risk Class III, 25.4% were in PORT Risk Class IV, and 1.4% were in PORT Risk Class V. The average age of patients was 60 years (range 18 to 93 years). Patients were predominantly male (58.7%) and white (91.5%); average BMI was 26.9 kg/m2. Associated comorbidities included pre-existing pulmonary disease (13.6%), cardiac disease (23.9%), diabetes (15.3%), and mild to severe renal impairment (76.9%). Bacteremia was documented at baseline in 1.5% of patients. The majority of sites were in Eastern Europe, which accounted for 82.8% of enrollment. One subject (0.2%) was enrolled in the BAXDELA arm and 5 (1.2%) in the moxifloxacin arm from the United States.
Early clinical response (ECR) at 72-120 hours after the first dose was defined as survival with improvement in at least two of four symptoms (cough, sputum production, chest pain, dyspnea) from baseline without deterioration in any of these symptoms, and without use of additional antimicrobial therapy for treatment of the current CABP infection due to lack of efficacy.
| Trial 3 | BAXDELA (300 mg IV and 450 mg oral) | Moxifloxacin (400 mg IV and 400 mg oral) | Treatment Difference†
(2-sided 95% CI) |
|---|---|---|---|
| CI = Confidence Interval; ITT = Intent To Treat includes all randomized patients | |||
|
|||
| Total N | 431 | 428 | |
| Responder n (%) | 383 (88.9) | 381 (89.0) | -0.2 (-4.4, 4.1) |
Clinical response was also assessed by the investigator at the test of cure (TOC) visit and defined as survival with resolution or near resolution of the symptoms of CABP present at study entry, and no use of additional antimicrobial therapy for the current CABP infection, and no new symptoms associated with the current CABP infection.
Clinical response rates at the TOC visit for the ITT and Clinically Evaluable (CE) populations are presented in Table 11.
| Trial 3 | BAXDELA (300 mg IV and 450 mg oral) | Moxifloxacin (400 mg IV and 400 mg oral) | Treatment Difference*
(2-sided 95% CI) |
|---|---|---|---|
| CI = Confidence Interval; ITT = Intent To Treat and includes all randomized patients; CE = Clinically Evaluable | |||
| Clinically Evaluable consisted of all ITT patients who had evidence of acute CABP, received at least 80% of expected doses of the correct study drug, did not receive any concomitant, systemic antibacterial therapy except for lack of efficacy, and did not have any protocol deviations that would affect the assessment of efficacy. | |||
|
|||
| Success†, n/N (%) ITT | 390/431 (90.5) | 384/428 (89.7) | 0.8 (-3.3, 4.8) |
| Success†, n/N (%) CE | 376/397 (94.7) | 373/394 (94.7) | 0.0 (-3.2, 3.3) |
Early clinical response and investigator-assessed clinical response at the TOC visit is presented in Table 12 by baseline pathogen for the Microbiological ITT (MITT) population which comprised all randomized patients who had a baseline pathogen identified that is known to cause CABP.
| Early Clinical Response† at 96 hours ± 24 hours | Investigator-Assessed Success‡ at Test-of Cure (TOC) | |||
|---|---|---|---|---|
| BAXDELA | Moxifloxacin | BAXDELA | Moxifloxacin | |
| Pathogen | n/N (%) | n/N (%) | n/N (%) | n/N (%) |
|
||||
| Staphylococcus aureus | 24/26 (92.3) | 25/28 (89.3) | 24/26 (92.3) | 26/28 (92.9) |
| Methicillin-susceptible | 22/24 (91.7) | 25/28 (89.3) | 22/24 (91.7) | 26/28 (92.9) |
| Streptococcus pneumoniae | 66/71 (93.0) | 51/62 (82.3) | 64/71 (90.1) | 54/62 (87.1) |
| Haemophilus influenzae | 25/26 (96.2) | 31/35 (88.6) | 24/26 (92.3) | 31/35 (88.6) |
| Haemophilus parainfluenzae | 30/32 (93.8) | 27/33 (81.8) | 30/32 (93.8) | 26/33 (78.8) |
| Escherichia coli | 15/16 (93.8) | 8/11 (72.7) | 15/16 (93.8) | 10/11 (90.9) |
| Klebsiella pneumoniae | 13/17 (76.5) | 15/16 (93.8) | 14/17 (82.4) | 16/16 (100.0) |
| Pseudomonas aeruginosa | 12/13 (92.3) | 10/11 (90.9) | 11/13 (84.6) | 11/11 (100.0) |
| Chlamydia pneumoniae | 24/25 (96.0) | 14/16 (87.5) | 25/25 (100.0) | 16/16 (100.0) |
| Legionella pneumophilia | 27/29 (93.1) | 28/33 (84.8) | 27/29 (93.1) | 32/33 (97.0) |
| Mycoplasma pneumoniae | 30/35 (85.7) | 29/30 (96.7) | 34/35 (97.1) | 30/30 (100.0) |
16. How is Baxdela supplied
16.1 BAXDELA for Injection
BAXDELA is supplied as a sterile, lyophilized powder in single-dose clear glass vials of 300 mg delafloxacin (equivalent to 433 mg delafloxacin meglumine). The lyophilized powder is a light yellow to tan cake, which may exhibit cracking and shrinkage and slight variation in texture and color.
They are supplied as follows: 300 mg single-dose vials (NDC 70842-102-01), packaged in cartons of 10 vials (NDC 70842-102-03).
16.2 BAXDELA Tablets
BAXDELA Tablets contain 450 mg delafloxacin (equivalent to 649 mg delafloxacin meglumine); each modified capsule-shaped tablet in beige to mottled beige color is debossed with RX3341 on one side. They are supplied as follows:
Bottles of 20 tablets with child-resistant closure (NDC 70842-101-01)
Unit dose blister packs which contain 20 tablets (2 blister cards of 10 tablets each)
(20 tablet blister pack: NDC 70842-101-02, 10 tablet blister card: NDC 70842-101-03)
16.3 Storage and Handling
BAXDELA Tablets and BAXDELA for Injection should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
The reconstituted powder may be stored for up to 24 hours under refrigerated or controlled room temperature and then further diluted for intravenous infusion. The reconstituted solution in the infusion bag may be stored under refrigerated or controlled room temperature conditions for up to 24 hours [see Dosage and Administration (2.4)]. Do not freeze.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Serious Adverse Reactions
Advise patients to stop taking BAXDELA if they experience an adverse reaction and to call their healthcare provider for advice on completing the full course of treatment with another antibacterial drug.
Inform patients of the following serious adverse reactions that have been associated with BAXDELA or other fluoroquinolone use:
- Disabling and Potentially Irreversible Serious Adverse Reactions that may occur together: Inform patients that disabling and potentially irreversible serious adverse reactions, including tendinitis and tendon rupture, peripheral neuropathies, and central nervous system effects, have been associated with use of fluoroquinolones and may occur together in the same patient. Inform patients to stop taking BAXDELA immediately if they experience an adverse reaction and to call their healthcare provider.
- Tendinitis and Tendon Rupture: Instruct patients to contact their healthcare provider if they experience pain, swelling, or inflammation of a tendon, or weakness or inability to use one of their joints; rest and refrain from exercise; and discontinue BAXDELA treatment. Symptoms may be irreversible. The risk of severe tendon disorder with fluoroquinolones is higher in older patients usually over 60 years of age, in patients taking corticosteroid drugs, and in patients with kidney, heart or lung transplants.
- Peripheral Neuropathy: Inform patients that peripheral neuropathies have been associated with BAXDELA use, symptoms may occur soon after initiation of therapy and may be irreversible. If symptoms of peripheral neuropathy including pain, burning, tingling, numbness and/or weakness develop, immediately discontinue BAXDELA and tell them to contact their physician.
- Central Nervous System Effects: (for example, convulsions, dizziness, lightheadedness, increased intracranial pressure): Inform patients that convulsions have been reported in patients receiving fluoroquinolones. Instruct patients to notify their physician before taking this drug if they have a history of convulsions. Inform patients that they should know how they react to BAXDELA before they operate an automobile or machinery or engage in other activities requiring mental alertness and coordination. Instruct patients to notify their physician if persistent headache with or without blurred vision occurs.
- Exacerbation of Myasthenia Gravis: Instruct patients to inform their physician of any history of myasthenia gravis. Instruct patients to notify their physician if they experience any symptoms of muscle weakness, including respiratory difficulties.
- Hypersensitivity Reactions: Inform patients that BAXDELA can cause hypersensitivity reactions, even following a single dose, and to discontinue BAXDELA at the first sign of a skin rash, hives or other skin reactions, a rapid heartbeat, difficulty in swallowing or breathing, any swelling suggesting angioedema (for example, swelling of the lips, tongue, face, tightness of the throat, hoarseness), or other symptoms of an allergic reaction.
- Diarrhea: Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, instruct patients to contact their physician as soon as possible.
- Aortic Aneurysm and Dissection: Inform patients to seek emergency medical care if they experience sudden chest, stomach, or back pain.
- Antibacterial Resistance: Patients should be counseled that antibacterial drugs including BAXDELA Tablets and Injection should only be used to treat bacterial infections. They do not treat viral infections (for example, the common cold). When BAXDELA Tablets and BAXDELA Injection are prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by BAXDELA Tablets and BAXDELA Injection or other antibacterial drugs in the future.
Administration with Food and Concomitant Medications
- Inform patients that BAXDELA Tablets may be taken with or without food and without any dietary restrictions [see Dosage and Administration (2.1) and Clinical Pharmacology (12.3)].
- Inform patients that BAXDELA Tablets should be taken at least 2 hours before or 6 hours after antacids containing magnesium, or aluminum, with sucralfate, with metal cations such as iron, or with multivitamin preparations containing zinc or iron, or with didanosine buffered tablets for oral suspension or the pediatric powder for oral solution.
Distributed by:
Melinta Therapeutics, LLC
Lincolnshire, Illinois 60069 USA
Trademarks depicted herein are the property of their respective owners.
©2021 Melinta Therapeutics, LLC. All rights reserved.
MEL001-R007
| This Medication Guide has been approved by the U.S. Food and Drug Administration MEL011-R007 | Revised: 6/2021 | ||
| MEDICATION GUIDE BAXDELA® (Bax-de'-lah) (delafloxacin) for injection |
|||
| BAXDELA® (Bax-de'-lah) (delafloxacin) tablets for oral use |
|||
| What is the most important information I should know about BAXDELA? | |||
| BAXDELA, a fluoroquinolone antibacterial medicine, can cause serious side effects. Some of these serious side effects can happen at the same time and could result in death. If you get any of the following serious side effects while you take BAXDELA, you should stop taking BAXDELA immediately and get medical help right away. | |||
|
|||
|
|
||
|
|||
|
|||
|
|
||
|
|||
| What is BAXDELA? | |||
BAXDELA is a fluoroquinolone antibacterial medicine used to treat certain types of infections caused by certain germs called bacteria in adults 18 years or older. These bacterial infections include:
|
|||
| It is not known if BAXDELA is safe and effective in people under 18 years of age, and use in people under 18 years of age is not recommended. Children younger than 18 years of age may have a higher chance of getting bone, joint, and tendon (musculoskeletal) problems while taking fluoroquinolone antibacterial medicines. | |||
| Sometimes infections are caused by viruses rather than by bacteria. Examples include viral infections in the sinuses and lungs, such as the common cold or flu. Antibacterial medicines, including BAXDELA, do not kill viruses. Call your healthcare provider if you think your condition is not getting better while you are taking BAXDELA. | |||
| Do not take BAXDELA if: | |||
| you have ever had a severe allergic reaction to an antibacterial known as a fluoroquinolone, or if you are allergic to any of the ingredients in BAXDELA. Ask your healthcare provider if you are not sure. See the list of ingredients in BAXDELA at the end of this Medication Guide. | |||
| What should I tell my healthcare provider before taking BAXDELA? | |||
| See "What is the most important information I should know about BAXDELA?" | |||
Before you take BAXDELA, tell your healthcare provider about all your medical conditions, including if you:
|
|||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal and dietary supplements. | |||
BAXDELA and other medicines can affect each other causing side effects. Especially tell your healthcare provider if you take:
|
|||
| Ask your healthcare provider if you are not sure if any of your medicines are listed above. Know the medicines you take. Keep a list of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine. | |||
How should I take BAXDELA?
|
|||
| What should I avoid while taking BAXDELA? | |||
| BAXDELA can make you feel dizzy and lightheaded. Do not drive, operate machinery, or do other activities that require mental alertness or coordination until you know how BAXDELA affects you. | |||
| What are the possible side effects of BAXDELA? | |||
BAXDELA may cause serious side effects, including:
|
|||
|
|
||
| Skin rash may happen in people taking fluoroquinolones even after only 1 dose. Stop taking BAXDELA at the first sign of a skin rash and call your healthcare provider. Skin rash may be a sign of a more serious reaction to BAXDELA. | |||
|
|||
| The most common side effects of BAXDELA include: | |||
|
|
||
| These are not all the possible side effects of BAXDELA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
| How should I store BAXDELA? | |||
BAXDELA Tablets:
|
|||
| Keep BAXDELA and all medicines out of the reach of children. | |||
| General information about the safe and effective use of BAXDELA. | |||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BAXDELA for a condition for which it was not prescribed. Do not give BAXDELA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about BAXDELA that is written for health professionals. | |||
What are the ingredients in BAXDELA?
|
|||
| Distributed by: Melinta Therapeutics, LLC 300 Tri-State International, Lincolnshire, Illinois, 60069 USA Trademarks depicted herein are the property of their respective owners. ©2021 Melinta Therapeutics, LLC All rights reserved For more information go to BAXDELA.com or call 1-844-633-6568. |
|||
PRINCIPAL DISPLAY PANEL - 450 mg Tablet Blister Pack Carton
NDC 70842-101-02
PHARMACIST: Please
dispense with medication
guide provided.
Baxdela®
(delafloxacin) tablets
450 mg per tablet
Contains 20 Tablets (2 blister cards of 10 tablets each)
Rx Only
Melinta
THERAPEUTICS
The
Antibiotics
Company
PRINCIPAL DISPLAY PANEL - 450 mg Tablet Bottle Carton
NDC 70842-101-01
Baxdela®
(delafloxacin) tablets
450 mg per tablet
Contains 20 Tablets
Rx Only
PHARMACIST: Please dispense
with medication guide provided.
melinta
therapeutics



| BAXDELA
delafloxacin meglumine tablet |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| BAXDELA
delafloxacin meglumine injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Melinta Therapeutics, LLC (034132850) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SGS North America Inc. | 808308303 | ANALYSIS(70842-101, 70842-102) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| ScinoPharm Taiwan, Ltd. | 657484726 | ANALYSIS(70842-101, 70842-102) , API MANUFACTURE(70842-101, 70842-102) , PACK(70842-101, 70842-102) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pharma Packaging Solutions, LLC dba Tjoapack, LLC | 928861723 | PACK(70842-101, 70842-102) , LABEL(70842-101, 70842-102) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Bostal LLC | 362785011 | ANALYSIS(70842-101, 70842-102) , MANUFACTURE(70842-101, 70842-102) | |
Frequently asked questions
More about Baxdela (delafloxacin)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: quinolones and fluoroquinolones
- Breastfeeding
- En español
Patient resources
- Baxdela drug information
- Baxdela (Delafloxacin Intravenous) (Advanced Reading)
- Baxdela (Delafloxacin Oral) (Advanced Reading)
