Sivextro: Package Insert / Prescribing Info
Package insert / product label
Generic name: tedizolid phosphate
Dosage forms: tablet, film coated, lyophilized powder for injection
Drug class: Oxazolidinone antibiotics
J Code (medical billing code): J3090 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Apr 22, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
SIVEXTRO (tedizolid phosphate) for injection, for intravenous use
SIVEXTRO (tedizolid phosphate) tablet, for oral use
Initial U.S. Approval: 2014
Recent Major Changes
Indications and Usage for Sivextro
SIVEXTRO is an oxazolidinone antibacterial indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by designated susceptible microorganisms in adult and pediatric patients (at least 26 weeks gestational age and weighing at least 1 kg) (1.1)
Usage to Reduce Development of Drug-Resistant Bacteria
To reduce the development of drug-resistant bacteria and maintain the effectiveness of SIVEXTRO and other antibacterial drugs, SIVEXTRO should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria.
Sivextro Dosage and Administration
- Do not administer SIVEXTRO Tablets to pediatric patients weighing less than 35 kg. (2.1)
- Adult Patients Intravenous and Oral Dosage: 200 mg administered once daily orally or as an intravenous (IV) infusion over 1 hour for 6 days as specified in Table 1 in the full prescribing information. (2.2)
- Pediatric Patients Intravenous Dosage (at least 26 weeks gestational age and weighing at least 1 kg): Weight-based dosing as an intravenous infusion as specified in Table 2 in the full prescribing information. (2.3)
- Pediatric Patients Oral Dosage (weighing greater than or equal to 35 kg): Weight-based dosing as an oral tablet administered once daily as specified in Table 3 in the full prescribing information. (2.3)
Dosage Forms and Strengths
Contraindications
None (4)
Warnings and Precautions
- Patients with neutropenia: The safety and efficacy of SIVEXTRO in patients with neutropenia (neutrophil counts <1000 cells/mm3) have not been adequately evaluated. In an animal model of infection, the antibacterial activity of SIVEXTRO was reduced in the absence of granulocytes. Consider alternative therapies in neutropenic patients. (5.1)
- Clostridioides difficile-associated diarrhea: Evaluate if diarrhea occurs. (5.2)
Adverse Reactions/Side Effects
- The most common adverse reactions (≥2%) in adult patients are nausea, headache, diarrhea, infusion- or injection-related adverse reactions, vomiting, and dizziness. (6.1)
- The most common adverse reactions (>2%) in pediatric patients (12 years to less than 18 years of age) are phlebitis and increased hepatic transaminases. (6.1)
- The most common adverse reactions (>2%) in pediatric patients (less than 12 years of age) are infusion- or injection-related adverse reactions and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme LLC at 1-877-888-4231 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch .
Drug Interactions
Use In Specific Populations
Pregnancy: Based on animal data, SIVEXTRO may cause fetal harm. Advise pregnant women of the potential risks to a fetus. (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
Full Prescribing Information
1. Indications and Usage for Sivextro
1.1 Acute Bacterial Skin and Skin Structure Infections
SIVEXTRO® is indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of the following gram-positive microorganisms: Staphylococcus aureus (including methicillin-resistant [MRSA] and methicillin-susceptible [MSSA] isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus anginosus Group (including Streptococcus anginosus, Streptococcus intermedius, and Streptococcus constellatus), and Enterococcus faecalis, in adult and pediatric patients (at least 26 weeks gestational age and weighing at least 1 kg).
1.2 Usage to Reduce Development of Drug-Resistant Bacteria
To reduce the development of drug-resistant bacteria and maintain the effectiveness of SIVEXTRO and other antibacterial drugs, SIVEXTRO should be used only to treat infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2. Sivextro Dosage and Administration
2.1 Important Administration Instructions for Pediatric Patients Weighing Less than 35 kg
Do not administer SIVEXTRO Tablets to pediatric patients weighing less than 35 kg [see Dosage and Administration 2.3].
2.2 Recommended Dosage for Adult Patients
The recommended dosage of SIVEXTRO is 200 mg administered once daily for six (6) days either as an oral tablet (with or without food) or as an intravenous (IV) infusion in adult patients.
The recommended dosage and administration of SIVEXTRO in adult patients are described in Table 1.
| Infection | Route | Dose | Frequency | Infusion Time | Duration of Treatment |
|---|---|---|---|---|---|
| Acute Bacterial Skin and Skin Structure Infections (ABSSSI) | Intravenous | 200 mg | Once daily | 1 hour | 6 days |
| Oral | 200 mg | Once daily | Not Applicable |
No dose adjustment is necessary when changing from intravenous to oral SIVEXTRO.
2.3 Recommended Dosage for Pediatric Patients
Recommended Dosage of SIVEXTRO for Injection for Pediatric Patients: Intravenous Dosage
The recommended intravenous dosage of SIVEXTRO for Injection for pediatric patients (at least 26 weeks gestational age and weighing at least 1 kg) is presented in Table 2.
| Weight Band (kg) | Dose | Frequency | Infusion Time | Duration of Treatment |
|---|---|---|---|---|
|
||||
| Pediatric Patients Weighing Less than 2 kg | ||||
| 1 to less than 2* | 3 mg/kg | Twice daily | 1 hour | 6 days |
| Pediatric Patients Weighing at Least 2 kg | ||||
| 2 to less than 3 | 6 mg | Twice daily | 1 hour | 6 days |
| 3 to less than 6 | 12 mg | |||
| 6 to less than 10 | 20 mg | |||
| 10 to less than 14 | 30 mg | |||
| 14 to less than 20 | 40 mg | |||
| 20 to less than 35 | 60 mg | |||
| Pediatric Patients Weighing at Least 35 kg | ||||
| Greater than or equal to 35 | 200 mg | Once daily | 1 hour | 6 days |
Recommended Dosage of SIVEXTRO Tablets for Pediatric Patients: Oral Dosage
The recommended oral dosage of SIVEXTRO Tablets for pediatric patients is presented in Table 3. SIVEXTRO Tablets can be administered with or without food. Do not administer SIVEXTRO Tablets to pediatric patients weighing less than 35 kg.
| Weight Band (kg) | Dose | Frequency | Duration of Treatment |
|---|---|---|---|
| Greater than or equal to 35 | 200 mg | Once daily | 6 days |
No dose adjustment is necessary when changing from intravenous to oral SIVEXTRO.
2.4 Recommendations Regarding Missed Doses(s)
For Once Daily Oral Dosing of SIVEXTRO Tablets
If patients miss a dose, administer the dose as soon as possible anytime up to 8 hours prior to their next scheduled dose. If less than 8 hours remain before the next dose, instruct the patients to wait until their next scheduled dose.
2.5 Preparation and Administration of Intravenous Solution
SIVEXTRO is supplied as a sterile, lyophilized powder for injection in single-dose vials of 200 mg. Each 200 mg vial must be reconstituted with Sterile Water for Injection and subsequently diluted with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP.
SIVEXTRO vials contain no antimicrobial preservatives and are intended for single dose only. Discard any unused portion.
Preparation of SIVEXTRO for Injection
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
The contents of the vial should be reconstituted using aseptic technique as follows:
Note: To minimize foaming, AVOID vigorous agitation or shaking of the vial during or after reconstitution.
For Adults and Pediatric Patients Weighing at Least 35 kg:
- Reconstitute the SIVEXTRO vial with 4 mL of Sterile Water for Injection to provide a concentration of 50 mg/mL in each vial.
-
Gently swirl the contents and let the vial stand until the cake has completely dissolved and any foam disperses.
Inspect the vial to ensure the solution contains no particulate matter and no cake or powder remains attached to the sides of the vial. If necessary, invert the vial to dissolve any remaining powder and swirl gently to prevent foaming. The reconstituted solution is clear and colorless to pale-yellow in color; the total time from reconstitution to the end of administration should not exceed 24 hours at either room temperature or under refrigeration at 2°C to 8°C (36°F to 46°F).
Tilt the upright vial and insert a syringe with appropriately sized needle into the bottom corner of the vial and remove 4 mL of the reconstituted solution for the 200 mg dose. Do not invert the vial during extraction. - The reconstituted solution must be further diluted in 250 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. Slowly inject the required volume of reconstituted solution as determined in Step 2 into a 250 mL bag of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. Invert the bag gently to mix. Do NOT shake the bag as this may cause foaming.
For Pediatric Patients Weighing Less than 35 kg:
- Reconstitute the SIVEXTRO vial with 4 mL of Sterile Water for Injection to provide a concentration of 50 mg/mL in each vial.
- Gently swirl the contents and let the vial stand until the cake has completely dissolved and any foam disperses.
-
Inspect the vial to ensure the solution contains no particulate matter and no cake or powder remains attached to the sides of the vial. If necessary, invert the vial to dissolve any remaining powder and swirl gently to prevent foaming. The reconstituted solution is clear and colorless to pale-yellow in color; the total time from reconstitution to the end of administration should not exceed 24 hours at either room temperature or under refrigeration at 2°C to 8°C (36°F to 46°F).
Prepare a stock solution (100 mL of 0.8 mg/mL tedizolid phosphate): Tilt the upright vial and insert a syringe with appropriately sized needle into the bottom corner of the vial and remove 1.6 mL of the reconstituted solution and add it to an infusion bag containing 98.4 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP. Do not invert the vial during extraction. - Prepare the required volume of stock solution for infusion: Refer to Table 4 to convert the dose in mg to the appropriate volume of stock solution to be administered. Transfer this volume of stock solution to an adequately sized infusion bag or infusion syringe. It may be necessary to round to the nearest graduation mark of an appropriately sized syringe for smaller volumes. The total time from reconstitution to the end of administration should not exceed 24 hours at either room temperature or under refrigeration at 2°C to 8°C (36°F to 46°F).
| Body Weight (kg) | Amount of SIVEXTRO per dose (given twice daily) | Volume of stock solution (0.8 mg/mL) to be transferred to an adequately sized infusion bag or infusion syringe |
|---|---|---|
| Pediatric Patients Weighing Less than 2 kg | ||
| 1 to less than 2 | 3 mg/kg | Volume (mL) = Weight (kg) x 3.75 mL/kg |
| Pediatric Patients Weighing at Least 2 kg | ||
| 2 to less than 3 | 6 mg | 7.5 mL |
| 3 to less than 6 | 12 mg | 15 mL |
| 6 to less than 10 | 20 mg | 25 mL |
| 10 to less than 14 | 30 mg | 37.5 mL |
| 14 to less than 20 | 40 mg | 50 mL |
| 20 to less than 35 | 60 mg | 75 mL |
Administration of SIVEXTRO for Injection
Administer SIVEXTRO for Injection as an intravenous infusion only.
Do not administer as an intravenous push or bolus. Do not mix SIVEXTRO for Injection with other drugs when administering. It is not intended for intra-arterial, intramuscular, intrathecal, intraperitoneal, or subcutaneous administration.
The intravenous bag containing the reconstituted and diluted intravenous solution should be inspected visually for particulate matter prior to administration. Discard if visible particles are observed. The resulting solution is clear and colorless to pale-yellow in color.
After reconstitution and dilution, SIVEXTRO for Injection is to be administered via intravenous infusion using a total time of 1 hour.
The total time from reconstitution to the end of administration of SIVEXTRO for Injection should not exceed 24 hours at either room temperature or under refrigeration at 2°C to 8°C (36°F to 46°F).
Discard unused portion.
2.6 Compatible Intravenous Solutions
SIVEXTRO is compatible with 0.9% Sodium Chloride Injection, USP and 5% Dextrose Injection, USP.
Limited data are available on the compatibility of SIVEXTRO for Injection with other intravenous substances, additives or other medications and they should not be added to SIVEXTRO single-dose vials or infused simultaneously. If the same intravenous line is used for sequential infusion of several different drugs, the line should be flushed before and after infusion of SIVEXTRO with 0.9% Sodium Chloride Injection, USP.
3. Dosage Forms and Strengths
SIVEXTRO 200 mg tablet is a yellow film-coated oval tablet; each tablet is debossed with "TZD" on one side and "200" on the other side.
SIVEXTRO for Injection is a sterile, white to off-white lyophilized powder for injection in single-dose vials of 200 mg. Each 200 mg vial must be reconstituted with Sterile Water for Injection and subsequently diluted with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP.
5. Warnings and Precautions
5.1 Patients with Neutropenia
The safety and efficacy of SIVEXTRO in patients with neutropenia (neutrophil counts <1000 cells/mm3) have not been adequately evaluated. In an animal model of infection, the antibacterial activity of SIVEXTRO was reduced in the absence of granulocytes [see Clinical Pharmacology (12.2)]. Alternative therapies should be considered when treating patients with neutropenia and ABSSSI.
5.2 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents including SIVEXTRO, with severity ranging from mild diarrhea to fatal colitis. Treatment with antibacterial agents can alter the normal flora of the colon and may permit overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antibacterial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary because CDAD has been reported to occur more than two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, antibacterial use not directed against C. difficile should be discontinued, if possible. Appropriate measures such as fluid and electrolyte management, protein supplementation, antibacterial treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be compared directly to rates from clinical trials of another drug and may not reflect rates observed in practice.
Clinical Trials Experience in Adult Patients
Adverse reactions were evaluated for 1425 adult patients treated with SIVEXTRO in two Phase 2 and four Phase 3 clinical trials (three Phase 3 trials for 6 days of therapy and one Phase 3 trial for 7-21 days of therapy). The median age of adult patients treated with SIVEXTRO in the Phase 2 and Phase 3 trials was 44 years, ranging between 17 and 94 years old. The majority of adult patients treated with SIVEXTRO were male (66%) and White (67%).
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation in Adults
Serious adverse reactions occurred in 37/1425 (2.6%) of adult patients treated with SIVEXTRO and in 25/1000 (2.5%) of adult patients treated with the comparator. SIVEXTRO was discontinued due to an adverse reaction in 14/1425 (1%) of adult patients and the comparator was discontinued due to an adverse reaction in 13/1000 (1.3%) of adult patients.
Most Common Adverse Reactions in Adults
The most common adverse reactions in adult patients treated with SIVEXTRO were nausea (7.1%), headache (4.5%), diarrhea (3.6%), vomiting (2.7%), and dizziness (1.6%). The median time of onset of adverse reactions was 5 days for both SIVEXTRO and linezolid with 12% occurring on the second day of treatment in both treatment groups.
Table 5 lists selected adverse reactions occurring in at least 2% of adult patients treated with SIVEXTRO in clinical trials.
| Adverse Reactions | Pooled Phase 3 ABSSSI Clinical Trials | |
|---|---|---|
| SIVEXTRO (200 mg oral/intravenous once daily for 6 days) (N=1037) | Linezolid (600 mg oral/intravenous twice daily for 10 days) (N=1000) |
|
|
||
| Gastrointestinal Disorders | ||
| Nausea | 7% | 10% |
| Diarrhea | 4% | 5% |
| Vomiting | 3% | 5% |
| Nervous System Disorder | ||
| Headache | 5% | 5% |
| Dizziness | 2% | 2% |
| Infusion- or Injection-Related Adverse Reactions* | ||
| 4% | 2% | |
The following selected adverse reactions were reported in SIVEXTRO-treated adult patients at a rate of less than 2% in these clinical trials:
Blood and Lymphatic System Disorders: anemia
Cardiovascular: palpitations, tachycardia
Eye Disorders: asthenopia, vision blurred, visual impairment, vitreous floaters
Immune System Disorders: drug hypersensitivity
Infections and Infestations: Clostridioides difficile colitis, oral candidiasis, vulvovaginal mycotic infection
Investigations: hepatic transaminases increased (ALT increased, AST increased), gamma-glutamyltransferase (GGT) increased, white blood cell count decreased
Nervous System Disorders: hypoesthesia, paresthesia, VIIth nerve paralysis
Psychiatric Disorders: insomnia
Skin and Subcutaneous Tissue Disorders: pruritus, urticaria, dermatitis
Vascular Disorders: flushing, hypertension
Laboratory Parameters
Hematology laboratory abnormalities that were determined to be potentially clinically significant in the pooled Phase 3 ABSSSI clinical trials are provided in Table 6.
| Laboratory Assay | Potentially Clinically Significant Values*,† | |
|---|---|---|
| SIVEXTRO (200 mg oral/intravenous once daily for 6 days) (N)‡ | Linezolid (600 mg oral/intravenous twice daily for 10 days) (N)‡ |
|
| M = male; F = female | ||
|
||
| Hemoglobin (<10.1 g/dL [M]) (<9 g/dL [F]) | (994) 3.4% | (957) 3.4% |
| Platelet count (<112 × 103/mm3) | (989) 2.1% | (950) 3.8% |
| Absolute neutrophil count (<0.8 × 103/mm3) | (980) 0.4% | (941) 0.6% |
Myelosuppression
Phase 1 studies conducted in healthy adults exposed to SIVEXTRO for 21 days showed a possible dose and duration effect on hematologic parameters beyond 6 days of treatment. In the Phase 3 trials, clinically significant changes in these parameters were generally similar for both treatment arms (see Table 6). In postmarketing experience, thrombocytopenia has been reported in patients treated with SIVEXTRO. In one postmarketing report, patients who experienced thrombocytopenia were treated with tedizolid for a median duration of 26.5 days. A duration of treatment beyond 6 days is not approved.
Peripheral and Optic Neuropathy
Peripheral and optic neuropathy have been described in patients treated with another member of the oxazolidinone class for longer than 28 days. In Phase 3 trials in adults, reported adverse reactions for peripheral neuropathy and optic nerve disorders were similar between both treatment arms (peripheral neuropathy 1.2% vs. 0.7% for tedizolid phosphate and linezolid, respectively; optic nerve disorders 0.3% vs. 0.1%, respectively).
Clinical Trials Experience in Pediatric Patients
SIVEXTRO was evaluated in 166 pediatric patients with ABSSSI in two randomized, active-controlled clinical trials, including one in patients aged 12 years to less than 18 years and one in patients aged 4 months to less than 12 years (Pediatric Trial 1 and Pediatric Trial 2, respectively). Additionally, SIVEXTRO was evaluated in 47 pediatric patients less than 2 years of age with a suspected or confirmed gram-positive bacterial infection in an open-label clinical trial (pediatric Trial 3).
Pediatric Trial 1
Adverse Reactions
Pediatric Trial 1 was a randomized, active-controlled, single-blind clinical trial that enrolled pediatric patients aged 12 to less than 18 years with ABSSSI. A total of 91 pediatric patients were treated with IV and/or oral SIVEXTRO 200 mg for 6 days and 29 patients were treated with a comparator agent for 10 days. The majority of pediatric patients treated with SIVEXTRO were male (64%) and white (88%).
Serious adverse reactions occurred in 1/91 (1%) of pediatric patients treated with SIVEXTRO and in none of the 29 patients treated with the comparator. Adverse reactions leading to discontinuation occurred in 1 (1%) pediatric patient in the SIVEXTRO arm and in none in the comparator arm.
The most common adverse reactions occurring in those receiving SIVEXTRO in Pediatric Trial 1 were phlebitis (3%), increased hepatic transaminases (alanine aminotransferase, aspartate aminotransferase) (3%), anemia (1%), and vomiting (1%).
Laboratory Parameters
| Laboratory Assay | Potentially Clinically Significant Values*,† | |
|---|---|---|
| SIVEXTRO (200 mg oral/intravenous once daily for 6 days) (N)‡ | Comparators§
(for 10 days) (N)‡ |
|
| M = male; F = female | ||
|
||
| Hemoglobin (<10.1 g/dL [M]) (<9 g/dL [F]) | (85) 2.4% | (26) 0.0% |
| Platelet count (<112 × 103/mm3) | (82) 1.2% | (26) 0.0% |
| Absolute neutrophil count (<0.8 × 103/mm3) | (85) 0.0% | (26) 0.0% |
Pediatric Trial 2
Pediatric Trial 2 was a randomized, active-controlled, single-blind clinical trial that enrolled pediatric patients aged 4 months to less than 12 years with ABSSSI. A total of 75 patients were treated with IV and/or oral SIVEXTRO for 6 to 10 days and 25 were treated with a comparator agent for 10 to 14 days. The oral suspension formulation (currently not an approved formulation) was used in the clinical trial for those receiving oral therapy [see Clinical Studies (14.1)]. The majority of patients treated with SIVEXTRO were male (53%) and white (77%), with a median age of 7 years and range of 0.3 to 11 years. The most common adverse reactions occurring in >2% of patients receiving SIVEXTRO in Pediatric Trial 2 were infusion- or injection-related adverse reactions (including catheter site pain, catheter occlusion, infusion site extravasation, phlebitis; 5%), and vomiting (4%).
Potentially clinically significant laboratory abnormalities in Pediatric Trial 2 included one patient treated with SIVEXTRO who developed thrombocytopenia with platelet count less than 100 × 103/mm3.
Pediatric Trial 3
Pediatric Trial 3 was an open-label, pharmacokinetic and safety trial enrolling 47 pediatric patients less than 2 years of age as follows: ages 28 days to less than 2 years (N=14), term neonates from birth to less than 28 days (N=16), and pre-term neonates (gestational age ≥ 26 weeks) from birth to less than 28 days (N=17). In this single-arm trial, 39 patients aged less than 2 years with a suspected or confirmed gram-positive bacterial infection received a single-dose of IV or oral SIVEXTRO and 8 patients aged less than 28 days with a suspected or confirmed gram-positive bacterial infection received multiple doses of IV SIVEXTRO for 3 days. The majority of patients were male (62%) and white (55%), with a median age of 16 days (range 1 day to 608 days).
The safety profile observed in this pediatric population was similar to that observed in Pediatric Trials 1 and 2.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of SIVEXTRO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders: thrombocytopenia
Related/similar drugs
7. Drug Interactions
Orally administered SIVEXTRO inhibits Breast Cancer Resistance Protein (BCRP) in the intestine, which can increase the plasma concentrations of orally administered BCRP substrates, and the potential for adverse reactions. If possible, an interruption in the treatment of the co-administered BCRP substrate medicinal product should be considered during treatment with SIVEXTRO, especially for BCRP substrates with a narrow therapeutic index (e.g., methotrexate or topotecan). If coadministration cannot be avoided, monitor for adverse reactions related to the concomitantly administered BCRP substrates, including rosuvastatin. [see Clinical Pharmacology (12.3).]
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on animal reproduction studies, SIVEXTRO may cause fetal harm when administered to pregnant women. The available data on the use of SIVEXTRO in pregnant women are insufficient to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Advise pregnant women of the potential risks to a fetus. Fetal developmental toxicities were observed in mice and rats treated with SIVEXTRO. In embryo-fetal studies in mice and rats, tedizolid phosphate was shown to produce fetal developmental toxicities in mice and maternal toxicity and fetal developmental toxicities in rats. Tedizolid phosphate administered orally during organogenesis to pregnant animals was associated with reduced fetal weights and an increased incidence of costal cartilage anomalies in the absence of maternal toxicity in mice; and maternal toxicity, decreased fetal weights, and increased skeletal variations in rats at plasma exposures approximately 4 and 6 times respectively, the human plasma exposure at the maximum recommended human dose (MRHD) of 200 mg/day. In female rats administered tedizolid phosphate during organogenesis through lactation, there was no evidence of fetal toxicity, developmental delays, or impaired reproduction in the offspring at plasma exposures approximately equivalent to the human plasma exposure at the MRHD. (see Data)
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study, tedizolid phosphate administered orally to pregnant mice at doses of 1, 5, and 25 mg/kg/day during organogenesis (Gestational Day [GD] 6 to GD15) was associated with fetal developmental effects occurring in the absence of maternal toxicity, including reduced fetal weights and an increased incidence of costal cartilage anomalies at the high dose (approximately 4-times the human plasma exposure at the MRHD based on plasma AUC comparison). Tedizolid phosphate administered orally at doses of 2.5, 5, and 15 mg/kg/day to pregnant rats during organogenesis (GD6 through GD17) was associated with maternal toxicity (reduced maternal body weights), decreased fetal weights, and increased skeletal variations including reduced ossification of the sternebrae, vertebrae, and skull at the high dose of 15 mg/kg/day (approximately 6-times the human plasma exposure at the MRHD based on plasma AUC comparison). The doses not associated with fetal toxicity in mice and maternal and fetal toxicity in rats were 5 and 2.5 mg/kg/day respectively (for both species approximately equivalent to the human plasma exposure at the MRHD based on plasma AUC comparison).
In a pre-postnatal study, oral tedizolid phosphate administered to female rats at doses of 1.25, 2.5, and 3.75 mg/kg/day during gestation and lactation (GD6 through Lactational Day 20) was not associated with maternal toxicity, fetal toxicity, developmental delays, or impaired reproduction at doses up to the high dose of 3.75 mg/kg/day (approximately equivalent to the human plasma exposure at the MRHD based on plasma AUC comparison).
8.2 Lactation
Risk Summary
There is no information on the presence of tedizolid in human milk. Tedizolid is present in rat milk. When a drug is present in animal milk, it is likely that the drug will be present in human milk. There are no data on the effects of SIVEXTRO on the breastfed child or on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for SIVEXTRO and any potential adverse effects on the breastfed child from SIVEXTRO or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of SIVEXTRO for the treatment of ABSSSI have been established in pediatric patients at least 26 weeks gestational age and weighing at least 1 kg. Use of SIVEXTRO for the treatment of ABSSSI is supported by evidence from adequate and well-controlled studies in adults with additional pharmacokinetic and safety data in pediatric patients from birth (includes neonates at least 26 weeks gestational age) to less than 18 years of age [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.1)].
The safety and effectiveness of SIVEXTRO in pediatric patients less than 26 weeks gestational age and weighing less than 1 kg have not been established.
10. Overdosage
In the event of overdosage, SIVEXTRO should be discontinued and general supportive treatment given. Hemodialysis does not result in meaningful removal of tedizolid from systemic circulation.
11. Sivextro Description
SIVEXTRO (tedizolid phosphate), a phosphate prodrug, is converted to tedizolid in the presence of phosphatases.
Tedizolid phosphate has the chemical name [(5R)-(3-{3-Fluoro-4-[6-(2-methyl-2H-tetrazol- 5-yl) pyridin-3-yl]phenyl}-2-oxooxazolidin- 5-yl]methyl hydrogen phosphate.
Its empirical formula is C17H16FN6O6P and its molecular weight is 450.32. Its structural formula is:
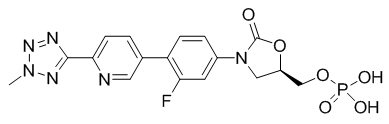
Tedizolid phosphate is a white to yellow solid and is administered orally or by intravenous infusion.
The pharmacologically active moiety, tedizolid, is an antibacterial agent of the oxazolidinone class.
SIVEXTRO Tablets contain 200 mg of tedizolid phosphate, and the following inactive ingredients: crospovidone, magnesium stearate, mannitol, microcrystalline cellulose, and povidone. In addition, the film coating contains the following inactive ingredients: polyethylene glycol/macrogol, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide.
SIVEXTRO for Injection is a sterile, white to off-white sterile lyophilized powder supplied in a clear glass single-dose vial. Each vial contains 200 mg of tedizolid phosphate and the inactive ingredient, mannitol (105 mg). Sodium hydroxide and hydrochloric acid are used as needed for pH adjustment. When reconstituted as directed with 4 mL of Sterile Water for Injection, each mL contains 50 mg of tedizolid phosphate. The pH of the reconstituted solution is 7.4 to 8.1.
12. Sivextro - Clinical Pharmacology
12.2 Pharmacodynamics
The AUC/minimum inhibitory concentration (MIC) was shown to best correlate with tedizolid activity in animal infection models.
In the mouse thigh infection model of S. aureus, antistaphylococcal killing activity was impacted by the presence of granulocytes. In granulocytopenic mice (neutrophil count <100 cells/mL), bacterial stasis was achieved at a human-equivalent dose of approximately 2000 mg/day; whereas, in non-granulocytopenic animals, stasis was achieved at a human-equivalent dose of approximately 100 mg/day. The safety and efficacy of SIVEXTRO for the treatment of neutropenic patients (neutrophil counts <1000 cells/mm3) have not been evaluated.
Cardiac Electrophysiology
In a randomized, positive- and placebo-controlled crossover thorough QTc study, 48 enrolled subjects were administered a single oral dose of SIVEXTRO at a therapeutic dose of 200 mg, SIVEXTRO at a supratherapeutic dose of 1200 mg, placebo, and a positive control; no significant effects of SIVEXTRO on heart rate, electrocardiogram morphology, PR, QRS, or QT interval were detected. Therefore, SIVEXTRO does not affect cardiac repolarization.
12.3 Pharmacokinetics
Tedizolid phosphate is a prodrug that is converted by phosphatases to tedizolid, the microbiologically active moiety, following oral and intravenous administration. Only the pharmacokinetic profile of tedizolid is discussed further due to negligible systemic exposure of tedizolid phosphate following oral and intravenous administration. Following multiple once-daily oral or intravenous administration, steady-state concentrations are achieved within approximately three days with tedizolid accumulation of approximately 30% (tedizolid half-life of approximately 12 hours). Pharmacokinetic (PK) parameters of tedizolid following oral and intravenous administration of 200 mg once daily tedizolid phosphate in adults are shown in Table 8.
| Pharmacokinetic Parameters of Tedizolid* | Oral | Intravenous | ||
|---|---|---|---|---|
| Single Dose | Steady State | Single Dose | Steady State | |
|
||||
| Cmax (mcg/mL) | 2.0 (0.7) | 2.2 (0.6) | 2.3 (0.6) | 3.0 (0.7) |
| Tmax (hr)† | 2.5 (1.0 - 8.0) | 3.5 (1.0 - 6.0) | 1.1 (0.9 - 1.5) | 1.2 (0.9 - 1.5) |
| AUC (mcg∙hr/mL)‡ | 23.8 (6.8) | 25.6 (8.5) | 26.6 (5.2) | 29.2 (6.2) |
| CL or CL/F (L/hr) | 7.5 (2.3) | 6.9 (1.7) | 6.4 (1.2) | 5.9 (1.4) |
Absorption
Peak plasma tedizolid concentrations are achieved within approximately 3 hours following oral administration of the oral tablet under fasting conditions or at the end of the 1 hour intravenous infusion of tedizolid phosphate. The absolute bioavailability of the oral tablet is approximately 91% and no dosage adjustment is necessary between intravenous and oral administration.
Tedizolid phosphate (oral tablet) may be administered with or without food as total systemic exposure (AUC0-∞) is unchanged between fasted and fed (high-fat, high-calorie) conditions.
Distribution
Protein binding of tedizolid to human plasma proteins is approximately 70 to 90%. The mean steady state volume of distribution of tedizolid in healthy adults following a single intravenous dose of tedizolid phosphate 200 mg ranged from 67 to 80 L (approximately twice total body water). Tedizolid penetrates into the interstitial space fluid of adipose and skeletal muscle tissue with exposure similar to free drug exposure in plasma.
Elimination
Metabolism
Other than tedizolid, which accounts for approximately 95% of the total radiocarbon AUC in plasma, there are no other significant circulating metabolites in humans.
There was no degradation of tedizolid in human liver microsomes indicating tedizolid is unlikely to be a substrate for hepatic CYP450 enzymes.
In vitro studies showed that conjugation of tedizolid is mediated via multiple sulfotransferase (SULT) isoforms (SULT1A1, SULT1A2, and SULT2A1).
Excretion
Following single oral administration of 14C-labeled tedizolid phosphate under fasted conditions in adult healthy volunteers, the majority of elimination occurred via the liver, with 82% of the radioactive dose recovered in feces and 18% in urine, primarily as a non-circulating and microbiologically inactive sulfate conjugate. Most of the elimination of tedizolid (>85%) occurs within 96 hours. Less than 3% of the tedizolid phosphate-administered dose is excreted in feces and urine as unchanged tedizolid.
Specific Populations
Based on the population pharmacokinetic analysis, there are no clinically relevant demographic or clinical patient factors (including age, gender, race, ethnicity, weight, body mass index, and measures of renal or liver function) that impact the pharmacokinetics of tedizolid.
Patients with Hepatic Impairment
Following administration of a single 200 mg oral dose of SIVEXTRO, no clinically meaningful changes in mean tedizolid Cmax and AUC0-∞ were observed in adult patients with moderate (n=8) or severe (n=8) hepatic impairment (Child-Pugh Class B and C) compared to 8 matched healthy control subjects. No dose adjustment is necessary for patients with hepatic impairment.
Patients with Renal Impairment
Following administration of a single 200 mg intravenous dose of SIVEXTRO to 8 adult subjects with severe renal impairment defined as eGFR <30 mL/min/1.73 m2, the Cmax was essentially unchanged and AUC0-∞ was decreased by less than 10% compared to 8 matched healthy control adult subjects. Hemodialysis does not result in meaningful removal of tedizolid from systemic circulation, as assessed in subjects with end-stage renal disease (eGFR <15 mL/min/1.73 m2). No dosage adjustment is necessary in patients with renal impairment or patients on hemodialysis.
Geriatric Patients
The pharmacokinetics of tedizolid were evaluated in a Phase 1 study conducted in elderly healthy volunteers (age 65 years and older, with at least 5 subjects at least 75 years old; n=14) compared to younger control subjects (25 to 45 years old; n=14) following administration of a single oral dose of SIVEXTRO 200 mg. There were no clinically meaningful differences in tedizolid Cmax and AUC0-∞ between elderly subjects and younger control subjects. No dosage adjustment of SIVEXTRO is necessary in elderly patients.
Male and Female Patients
The impact of gender on the pharmacokinetics of SIVEXTRO was evaluated in clinical trials of adult healthy males and females and in a population pharmacokinetics analysis. The pharmacokinetics of tedizolid were similar in males and females. No dosage adjustment of SIVEXTRO is necessary based on gender.
Pediatric Patients
The pharmacokinetics of tedizolid was evaluated in clinical studies in pediatric patients from birth (includes neonates who are at least 26 weeks gestational age and weighing at least 1 kg) to less than 18 years of age (N=249).
Compared to adult patients, tedizolid exposures are higher in pediatric patients (12 to <18 years of age, mean weight: 59.7 kg, weight range: 28 kg to 126 kg in Pediatric Trial 1) following multiple dose administration of a once daily dose of 200 mg IV or oral SIVEXTRO (geometric mean Cmax 3.1 vs. 2.0 mcg/mL, AUC24h 28.6 vs. 21.0 mcg*h/mL); however, this increase in exposure is not considered clinically significant.
The predicted steady state mean pharmacokinetic parameters of tedizolid by the pediatric weight-based dosage in Table 2 and Table 3 are shown in Table 9.
| Weight (kg) | Dosage Regimen | Total Daily Dose | Route | Steady-State AUC24h (mcg·h/mL)† | Steady-State Cmax (mcg/mL) |
|---|---|---|---|---|---|
| AUC, area under the concentration-time curve; Cmax, maximum concentration; %CV, coefficient of variation. | |||||
| 1 to less than 2 | 3 mg/kg Twice daily | 6 mg to less than12 mg | IV | 33.1 (29.8) | 2.3 (17.2) |
| 2 to less than 3 | 6 mg Twice daily | 12 mg | IV | 20.8 (25.6) | 1.8 (15.9) |
| 3 to less than 6 | 12 mg Twice daily | 24 mg | IV | 26.4 (34.4) | 2.4 (20.7) |
| 6 to less than 10 | 20 mg Twice daily | 40 mg | IV | 22.5 (43.9) | 2.2 (20.4) |
| 10 to less than 14 | 30 mg Twice daily | 60 mg | IV | 27.6 (32.1) | 2.7 (19.6) |
| 14 to less than 20 | 40 mg Twice daily | 80 mg | IV | 28.4 (22.1) | 2.7 (13.0) |
| 20 to less than 35 | 60 mg Twice daily | 120 mg | IV | 31.4 (29.9) | 2.6 (21.6) |
| At least 35 | 200 mg Once daily | 200 mg | IV | 30.6 (29.7) | 3.8 (23.9) |
| Oral (tablet) | 29.3 (29.7) | 2.5 (24.4) | |||
Drug Interaction Studies
Drug Metabolizing Enzymes
Transformation via Phase 1 hepatic oxidative metabolism is not a significant pathway for elimination of SIVEXTRO.
Neither SIVEXTRO nor tedizolid detectably inhibited or induced the metabolism of selected CYP enzyme substrates, suggesting that drug-drug interactions based on oxidative metabolism are unlikely.
Membrane Transporters
The potential for tedizolid or tedizolid phosphate to inhibit transport of probe substrates of important drug uptake (OAT1, OAT3, OATP1B1, OATP1B3, OCT1, and OCT2) and efflux transporters (P-gp and BCRP) was tested in vitro. No clinically relevant interactions are expected to occur with these transporters except BCRP.
Coadministration of multiple oral doses of SIVEXTRO (200 mg once daily) increased the Cmax and AUC of rosuvastatin (10 mg single oral dose), a known BCRP substrate, by approximately 55% and 70%, respectively, in healthy adult subjects [see Drug Interactions (7)].
Monoamine Oxidase Inhibition
Tedizolid is a reversible inhibitor of monoamine oxidase (MAO) in vitro. The interaction with MAO inhibitors could not be evaluated in Phase 2 and 3 trials, as subjects taking such medications were excluded from the trials.
Adrenergic Agents
Two placebo-controlled crossover studies were conducted to assess the potential of 200 mg oral SIVEXTRO at steady state to enhance pressor responses to pseudoephedrine and tyramine in healthy adults. No meaningful changes in blood pressure or heart rate were seen with pseudoephedrine. The median tyramine dose required to cause an increase in systolic blood pressure of ≥30 mmHg from pre-dose baseline was 325 mg with SIVEXTRO compared to 425 mg with placebo. Palpitations were reported in 21/29 (72.4%) adult subjects exposed to SIVEXTRO compared to 13/28 (46.4%) exposed to placebo in the tyramine challenge study.
Serotonergic Agents
Serotonergic effects at doses of tedizolid phosphate up to 30-fold above the human equivalent dose did not differ from vehicle control in a mouse model that predicts serotonergic activity. In Phase 3 trials, subjects taking serotonergic agents including antidepressants such as selective serotonin reuptake inhibitors (SSRIs), tricyclic antidepressants, and serotonin 5-hydroxytryptamine (5-HT1) receptor agonists (triptans), meperidine, or buspirone were excluded.
12.4 Microbiology
Mechanism of Action
The antibacterial activity of tedizolid is mediated by binding to the 50S subunit of the bacterial ribosome resulting in inhibition of protein synthesis. Tedizolid inhibits bacterial protein synthesis through a mechanism of action different from that of other non-oxazolidinone class antibacterial drugs; therefore, cross-resistance between tedizolid and other classes of antibacterial drugs is unlikely. The results of in vitro time-kill studies show that tedizolid is bacteriostatic against enterococci, staphylococci, and streptococci.
Resistance
Organisms resistant to oxazolidinones via mutations in chromosomal genes encoding 23S rRNA or ribosomal proteins (L3 and L4) are generally cross-resistant to tedizolid. In the limited number of Staphylococcus aureus strains tested, the presence of the chloramphenicol-florfenicol resistance (cfr) gene did not result in resistance to tedizolid in the absence of chromosomal mutations. Mutations in 23SrRNA (G2576T mutation) have been associated with tedizolid resistance in S. aureus isolates.
Spontaneous mutations conferring reduced susceptibility to tedizolid occur in vitro at a frequency rate of approximately 10-10.
Interaction with Other Antimicrobial Drugs
In vitro drug combination studies with tedizolid and aztreonam, ceftriaxone, ceftazidime, imipenem, rifampin, trimethoprim/sulfamethoxazole, minocycline, clindamycin, ciprofloxacin, daptomycin, vancomycin, gentamicin, amphotericin B, ketoconazole, and terbinafine demonstrate neither synergy nor antagonism.
Antimicrobial Activity
Tedizolid has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections, as described in Indications and Usage (1).
Aerobic bacteria
Gram-positive bacteria
- Staphylococcus aureus (including methicillin-resistant [MRSA] and methicillin-susceptible [MSSA] isolates)
- Streptococcus pyogenes
- Streptococcus agalactiae
- Streptococcus anginosus Group (including S. anginosus, S. intermedius, and S. constellatus)
- Enterococcus faecalis
The following in vitro data are available, but their clinical significance is unknown. At least 90% of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for tedizolid against isolates of similar genus or organism group. However, the efficacy of SIVEXTRO in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Mutagenesis
Tedizolid phosphate was negative for genotoxicity in all in vitro assays (bacterial reverse mutation (Ames), Chinese hamster lung (CHL) cell chromosomal aberration) and in all in vivo tests (mouse bone marrow micronucleus, rat liver unscheduled DNA synthesis). Tedizolid, generated from tedizolid phosphate after metabolic activation (in vitro and in vivo), was also tested for genotoxicity. Tedizolid was positive in an in vitro CHL cell chromosomal aberration assay, but negative for genotoxicity in other in vitro assays (Ames, mouse lymphoma mutagenicity) and in vivo in a mouse bone marrow micronucleus assay.
Impairment of Fertility
In a fertility study, oral tedizolid phosphate administered in doses of 5, 15, and 50 mg/kg/day for 28 days before mating and during mating to male rats had no adverse effects on the fertility or reproductive performance, including spermatogenesis, at the maximum tested dose (50 mg/kg/day) with a plasma tedizolid AUC approximately 5-fold greater than the plasma AUC value in humans at the maximum recommended human dose (MRHD). Tedizolid phosphate administered in doses of 2.5, 5, and 15 mg/kg/day for 14 days before mating, during mating, and until Gestation Day (GD)7 to female rats also had no adverse effects on the fertility or reproductive performance at doses up to the maximum tested dose of 15 mg/kg/day (approximately 4-fold higher than exposures in humans at the MRHD based on plasma AUC comparison).
13.2 Animal Toxicology and/or Pharmacology
Repeated-oral and intravenous dosing of tedizolid phosphate in rats in 1 month and 3 month toxicology studies produced dose- and time-dependent bone marrow hypocellularity (myeloid, erythroid, and megakaryocyte), with associated reduction in circulating RBCs, WBCs, and platelets. These effects showed evidence of reversibility and occurred at plasma tedizolid exposure levels (AUC) ≥6-fold greater than the plasma exposure associated with the human therapeutic dose. In a 1-month immunotoxicology study in rats, repeated oral dosing of tedizolid phosphate was shown to significantly reduce splenic B cells and T cells and reduce plasma IgG titers. These effects occurred at plasma tedizolid exposure levels (AUC) ≥3-fold greater than the expected human plasma exposure associated with the therapeutic dose.
14. Clinical Studies
14.1 Acute Bacterial Skin and Skin Structure Infections
Adults
A total of 1333 adults with acute bacterial skin and skin structure infections (ABSSSI) were randomized in two multicenter, multinational, double-blind, non-inferiority trials. Both trials compared SIVEXTRO 200 mg once daily for 6 days versus linezolid 600 mg every 12 hours for 10 days. In Trial 1, patients were treated with oral therapy, while in Trial 2, patients could receive oral therapy after a minimum of one day of intravenous therapy. Patients with cellulitis/erysipelas, major cutaneous abscess, or wound infection were enrolled in the trials. Patients with wound infections could have received aztreonam and/or metronidazole as adjunctive therapy for gram-negative bacterial coverage, if needed. The intent-to-treat (ITT) patient population included all randomized patients.
In Trial 1, 332 patients with ABSSSI were randomized to SIVEXTRO and 335 patients were randomized to linezolid. The majority (91%) of patients treated with SIVEXTRO in Trial 1 were less than 65 years old with a median age of 43 years (range: 18 to 86 years). Patients treated with SIVEXTRO were predominantly male (61%) and White (84%); 13% had BMI ≥35 kg/m2, 8% had diabetes mellitus, 35% were current or recent intravenous drug users, and 2% had moderate to severe renal impairment. The overall median surface area of infection was 188 cm2. The types of ABSSSI included were cellulitis/erysipelas (41%), wound infection (29%), and major cutaneous abscess (30%). In addition to local signs and symptoms of infection, patients were also required to have at least one regional or systemic sign of infection at baseline, defined as lymphadenopathy (87% of patients), temperature 38°C or higher (16% of patients), white blood cell count greater than 10,000 cells/mm3 or less than 4000 cells/mm3 (42%), or 10% or more band forms on white blood cell differential (4%).
The primary endpoint in Trial 1 was early clinical response defined as no increase from baseline lesion area at 48-72 hours after the first dose and oral temperature of ≤37.6°C, confirmed by a second temperature measurement within 24 hours in the ITT population.
In Trial 2, 332 patients with ABSSSI were randomized to SIVEXTRO and 334 patients were randomized to linezolid. The majority (87%) of patients treated with SIVEXTRO in Trial 2 were less than 65 years old with a median age of 46 years (range: 17 to 86 years). Patients treated with SIVEXTRO were predominantly male (68%) and White (86%); 16% had BMI ≥35 kg/m2, 10% had diabetes mellitus, 20% were current or recent intravenous drug users, and 4% had moderate to severe renal impairment. The overall median surface area of infection was 231 cm2. The types of ABSSSI included were cellulitis/erysipelas (50%), wound infection (30%), and major cutaneous abscess (20%). In addition to local signs and symptoms of infection, patients were also required to have at least one regional or systemic sign of infection at baseline, defined as lymphadenopathy (71% of patients), temperature 38°C or higher (31% of patients), white blood cell count greater than 10,000 cells/mm3 or less than 4000 cells/mm3 (53%), or 10% or more band forms on white blood cell differential (16%).
The primary endpoint in Trial 2 was early clinical response defined as at least a 20% decrease from baseline lesion area at 48-72 hours after the first dose in the ITT population (Table 10).
| SIVEXTRO (200 mg) | Linezolid (1200 mg) | Treatment Difference (2-sided 95% CI) |
|
|---|---|---|---|
| CI=confidence interval | |||
| No increase in lesion surface area from baseline and oral temperature of ≤37.6°C, confirmed by a second temperature measurement within 24 hours at 48-72 hours* | |||
| Trial 1, N | 332 | 335 | |
| Responder, n (%) | 264 (79.5) | 266 (79.4) | 0.1 (-6.1, 6.2) |
| Trial 2, N | 332 | 334 | |
| Responder, n (%) | 286 (86.1) | 281 (84.1) | 2.0 (-3.5, 7.3) |
| At least a 20% decrease from baseline in lesion area at 48-72 hours† | |||
| Trial 1, N | 332 | 335 | |
| Responder, n (%) | 259 (78.0) | 255 (76.1) | 1.9 (-4.5, 8.3) |
| Trial 2, N | 332 | 334 | |
| Responder, n (%) | 283 (85.2) | 276 (82.6) | 2.6 (-3.0, 8.2) |
An investigator assessment of clinical response was made at the post-therapy evaluation (PTE) (7 - 14 days after the end of therapy) in the ITT and CE (Clinically Evaluable) populations. Clinical success was defined as resolution or near resolution of most disease-specific signs and symptoms, absence or near resolution of systemic signs of infection if present at baseline (lymphadenopathy, fever, >10% immature neutrophils, abnormal WBC count), and no new signs, symptoms, or complications attributable to the ABSSSI requiring further treatment of the primary lesion (Table 11).
| SIVEXTRO (200 mg) n/N (%) | Linezolid (1200 mg) n/N (%) | Treatment Difference (2-sided 95% CI) |
|
|---|---|---|---|
| CI=confidence interval; ITT=intent-to-treat; CE=clinically evaluable | |||
| Trial 1 | |||
| ITT | 284/332 (85.5) | 288/335 (86.0) | -0.5 (-5.8, 4.9) |
| CE | 264/279 (94.6) | 267/280 (95.4) | -0.8 (-4.6, 3.0) |
| Trial 2 | |||
| ITT | 292/332 (88.0) | 293/334 (87.7) | 0.3 (-4.8, 5.3) |
| CE | 268/290 (92.4) | 269/280 (96.1) | -3.7 (-7.7, 0.2) |
Clinical success by baseline pathogens from the primary infection site or blood cultures for the microbiological intent-to-treat (MITT) patient population for two integrated Phase 3 ABSSSI studies are presented in Table 12 and Table 13.
| Pathogen | No increase in lesion surface area from baseline and oral temperature of ≤37.6°C* | At least a 20% decrease from baseline in lesion area† | ||
|---|---|---|---|---|
| SIVEXTRO (200 mg) n/N (%) | Linezolid (1200 mg) n/N (%) | SIVEXTRO (200 mg) n/N (%) | Linezolid (1200 mg) n/N (%) |
|
| Pooled analysis; n=number of patients in the specific category; N=Number of patients with the specific pathogen isolated from the ABSSSI | ||||
| Staphylococcus aureus | 276/329 (83.9) | 278/342 (81.3) | 280/329 (85.1) | 276/342 (80.7) |
| Methicillin-resistant S. aureus | 112/141 (79.4) | 113/146 (77.4) | 114/141 (80.9) | 111/146 (76.0) |
| Methicillin-susceptible S. aureus | 164/188 (87.2) | 167/198 (84.3) | 166/188 (88.3) | 167/198 (84.3) |
| Streptococcus pyogenes | 27/33 (81.8) | 18/20 (90.0) | 25/33 (75.8) | 16/20 (80.0) |
| Streptococcus anginosus Group | 22/30 (73.3) | 26/28 (92.9) | 22/30 (73.3) | 25/28 (89.3) |
| Streptococcus agalactiae | 6/9 (66.7) | 8/10 (80.0) | 6/9 (66.7) | 7/10 (70.0) |
| Enterococcus faecalis | 7/10 (70.0) | 3/4 (75.0) | 6/10 (60.0) | 1/4 (25.0) |
Baseline bacteremia in the tedizolid arm with relevant pathogens included two subjects with MRSA, four subjects with MSSA, two subjects with S. pyogenes, one subject with S. agalactiae, and one subject with S. constellatus. All of these subjects were Responders at the 48-72 hour evaluation. At the Post-therapy Evaluation (PTE), 8 of 10 subjects were considered clinical successes.
| Pathogen | Clinical Response at PTE | |
|---|---|---|
| SIVEXTRO (200 mg) n/N (%) | Linezolid (1200 mg) n/N (%) |
|
| Pooled analysis; n=number of patients in the specific category; N=Number of patients with the specific pathogen isolated from the ABSSSI | ||
| Staphylococcus aureus | 291/329 (88.5) | 303/342 (88.6) |
| Methicillin-resistant S. aureus | 118/141 (83.7) | 119/146 (81.5) |
| Methicillin-susceptible S. aureus | 173/188 (92.0) | 186/198 (93.9) |
| Streptococcus pyogenes | 30/33 (90.9) | 19/20 (95.0) |
| Streptococcus anginosus Group | 21/30 (70.0) | 25/28 (89.3) |
| Streptococcus agalactiae | 8/9 (88.9) | 8/10 (80.0) |
| Enterococcus faecalis | 7/10 (70.0) | 4/4 (100.0) |
Baseline bacteremia in the tedizolid arm with relevant pathogens included two subjects with MRSA, four subjects with MSSA, two subjects with S. pyogenes, one subject with S. agalactiae, and one subject with S. constellatus. All of these subjects were Responders at the 48-72 hour evaluation. At the Post-therapy Evaluation (PTE) 8 of 10 subjects were considered clinical successes.
Pediatric Patients
The safety and efficacy of SIVEXTRO were investigated in Pediatric Trials 1 and 2. Pediatric Trial 1 included pediatric patients 12 to < 18 years of age who were investigated in a randomized, single blind, active-controlled trial of 120 patients with clinically documented ABSSSI (91 receiving tedizolid, 29 receiving comparator). Patients were randomized in a 3:1 ratio with stratification by geographic region to receive SIVEXTRO IV and/or oral therapy, dosed 200 mg once daily for 6 days, or comparator IV and/or oral therapy, dosed over 10 days. Comparator therapy was selected by the investigator from a list of 5 IV and 4 oral comparators per local standard of care. The most frequently used comparators were cefazolin (11 patients) and vancomycin (8 patients).
Additionally, Pediatric Trial 2 (NCT03176134) evaluated the safety and efficacy of SIVEXTRO in pediatric patients 4 months to <12 years of age in a randomized, single blind, active-controlled trial of 100 patients with clinically documented ABSSSI (75 receiving tedizolid, 25 receiving comparator). Patients were randomized in a 3:1 ratio to receive SIVEXTRO for 6 to 10 days or comparator for 10 to 14 days. SIVEXTRO (IV and/or oral) was dosed as a weight-based dose of 2 to 2.5 mg/kg every 12 hours or as 200 mg given once daily. Patients who received oral therapy received an oral suspension formulation that is not currently approved for use. Comparator therapy was selected from a pre-specified list by the investigator and dosed per local standard of care.
The primary objective of Pediatric Trials 1 and 2 was to evaluate the safety and tolerability of SIVEXTRO. The trials were not powered for comparative inferential efficacy analysis. Clinical response at the test of cure visit (Day 18-25) was assessed by a blinded investigator in the ITT population (all randomized patients). Clinical successes were required to have resolution or near resolution of all related signs and symptoms such that no further antibacterial therapy was needed. Early clinical response, defined as at least a 20% reduction in lesion size at 48-72 hours after start of treatment, was also assessed in the ITT population.
In Pediatric Trial 1, clinical success at test of cure was 96.7% (88/91) in the tedizolid group and 93.1% (27/29) in the comparator group (difference: 3.6%, 95% CI: -6.3, 13.5). Early clinical response at 48-72 hours was 92.3% (84/91) in the tedizolid group and 96.6% (28/29) in the comparator group (difference: -4.2%, 95% CI: -12.9, 4.4).
In Pediatric Trial 2, clinical success at test of cure was 93.3% (70/75) in the tedizolid group and 92.0% (23/25) in the comparator group (difference: 1.3%, 95% CI: -10.7, 13.4). Early clinical response was not adequately assessed in Pediatric Trial 2 because an in-person outpatient visit at this timepoint was not required during the COVID-19 pandemic.
16. How is Sivextro supplied
16.1 Tablets
SIVEXTRO Tablets are yellow film-coated oval tablets containing 200 mg of tedizolid phosphate; each tablet is debossed with "TZD" on one side and "200" on the other side.
They are supplied as follows:
HDPE bottles of 30 tablets with child-resistant closure (NDC 67919-041-04)
Unit dose blister packs of 6 tablets (NDC 67919-041-05)
16.2 For Injection
SIVEXTRO is supplied as a sterile, white to off-white lyophilized powder for injection in single-dose vials of 200 mg. Each 200 mg vial must be reconstituted with Sterile Water for Injection and subsequently diluted with 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP.
They are supplied as follows:
Package of ten 200 mg single-dose vials (NDC 67919-040-02)
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Administration with Food
Patients should be informed that SIVEXTRO Tablets may be taken with or without food and without any dietary restrictions [see Dosage and Administration (2.2, 2.3) and Clinical Pharmacology (12.3)].
Antibacterial Resistance
Patients should be counseled that antibacterial drugs including SIVEXTRO should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When SIVEXTRO is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by SIVEXTRO or other antibacterial drugs in the future [see Indications and Usage (1.2)].
Missed Doses
Patients should be informed that if they miss a SIVEXTRO oral dose, they should take the dose as soon as possible anytime up to 8 hours prior to their next scheduled dose. If less than 8 hours remains before the next dose, then they should wait until their next scheduled dose. Patients should take the prescribed number of doses [see Dosage and Administration (2.2, 2.3)].
Potentially Serious Adverse Reactions
Patients should be advised that diarrhea is a common problem caused by antibacterial drugs including SIVEXTRO and usually resolves when the drug is discontinued. Sometimes after starting treatment with antibacterial drugs, patients can develop frequent watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibacterial drug and may be a sign of a more serious intestinal infection [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)]. If this occurs, patients should contact their healthcare provider as soon as possible.
Embryo-Fetal Toxicity
Based on animal data, advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Manuf. for: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
Sivextro Tablets
Manufactured by: Patheon Inc.
Whitby, Ontario, L1N 5Z5 Canada
Sivextro for Injection
Manufactured by: Patheon Italia S.p.A.
03013, Ferentino, FR Italy
For patent information: www.msd.com/research/patent
uspi-mk1986-mf-2504r012
| Patient Information SIVEXTRO®(sih-vex-tro) (tedizolid phosphate) tablets |
What you need to know about SIVEXTRO
|
| What is SIVEXTRO? |
SIVEXTRO tablet is for adults and for children weighing at least 77 pounds (35 kg) who have a skin infection or an infection in the tissue below the skin. SIVEXTRO is an antibiotic that works by stopping the growth of certain bacteria.
| What should I tell my doctor before taking SIVEXTRO? |
Medical Conditions
Tell your doctor if you:
- have diarrhea or have ever had diarrhea while taking antibiotics. Tell your doctor, even if you had diarrhea that occurred up to 2 months after you took the antibiotic.
- are allergic to tedizolid phosphate or any of the ingredients in SIVEXTRO. See the end of this Patient Information for a complete list of ingredients in SIVEXTRO.
- are pregnant or plan to get pregnant, tell your doctor if you become pregnant while taking SIVEXTRO. It is not known if SIVEXTRO will harm your baby while you are pregnant. You and your doctor should decide together if you will take SIVEXTRO.
- are breastfeeding or plan to breastfeed, tell your doctor before you take SIVEXTRO. It is not known if SIVEXTRO passes into your breast milk. You and your doctor should decide together if you will take SIVEXTRO or breastfeed.
| Are you taking other medicines? |
- SIVEXTRO can affect the way other medicines work, and other medicines can affect how SIVEXTRO works. Some medicines cannot be taken with SIVEXTRO at all. Your doctor will tell you if it is safe to take SIVEXTRO with other medicines.
- It is especially important to tell your doctor if you take any of the following medicines:
- Methotrexate (for cancer or rheumatoid arthritis)
- Topotecan (for cancer)
- Rosuvastatin (for cholesterol)
- Tell your doctor about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal and dietary supplements.
- Know the medicines you take. Keep a list of them and show the list to your doctor and pharmacist when you get a new medicine.
| How do I take SIVEXTRO? |
- Do not give SIVEXTRO tablets to children weighing less than 77 pounds (35 kg).
- Take SIVEXTRO exactly how your doctor tells you to take it.
- Take 1 SIVEXTRO tablet 1 time each day.
- Take SIVEXTRO for 6 days, at the same time every day.
- Take SIVEXTRO by mouth, with or without food.
| What if I forget to take SIVEXTRO? |
- If you miss a dose, take the missed dose as soon as you remember.
If it is less than 8 hours until your next dose, skip the missed dose and take the next tablet at the time you usually take it. - Do not take 2 doses of SIVEXTRO at the same time to make up for a missed dose.
- If you are not sure how to take SIVEXTRO, call your doctor or pharmacist.
- Take all 6 tablets to finish your SIVEXTRO, even if you have missed a dose.
If you do not finish your medicine, SIVEXTRO may not work.
You may get sick again and the remaining bacteria may be harder to treat.
| What are the possible side effects of SIVEXTRO? |
SIVEXTRO may cause serious side effects, including diarrhea from C-diff (Clostridioides difficile) infection. Call your healthcare provider right away if you get stomach cramps, fever, watery diarrhea, diarrhea that does not go away, or bloody stools. C-diff infection can happen 2 or more months after you have finished your antibacterial medicine.
C-diff is an infection of your intestines (bowels) that can happen with many antibiotics like SIVEXTRO and may cause mild diarrhea to life-threatening swelling of your intestines (colitis).
Common side effects of SIVEXTRO include:
|
|
Some less common side effects are:
Problems with your skin
- itching, red or itchy rash, hives, acne
- hot flushes or feeling like you are blushing or your face, neck or chest is red
- not able to feel something as well
- a tingling or prickling sensation
Problems with your sleep
- hard time sleeping
Problems with your body
- numbness
- facial paralysis
Problems with infections
- vagina that is infected, inflamed, or itchy
- fungal infections of skin, mouth
Problems with your eyes
- eye strain
- blurred or impaired vision
- seeing dots or spots in your eyes
Problems with your heart
- Your heartbeat does not feel normal. It could feel like your heart is beating too fast or pumping harder than usual.
Problems with your vascular system
- high blood pressure
Problems with your blood work
Your doctor may tell you that you have the following while taking SIVEXTRO:
- a low white blood cell count
- anemia (low red blood cells)
- low platelet count, the small cells involved in clotting your blood
Side effects where the frequency is not known:
- bleeding or bruising easily
If you have any side effect that bothers you or does not go away, tell your doctor.
These are not all the possible side effects of SIVEXTRO. For information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
| How should I store SIVEXTRO? |
- Store SIVEXTRO at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep SIVEXTRO and all medicines out of the reach of children.
| General information about SIVEXTRO. |
- Medicines are sometimes prescribed for purposes that are not mentioned here.
- Do not use SIVEXTRO for a condition for which it was not prescribed.
| What if I have questions? |
- Call your doctor.
- Call the company that makes SIVEXTRO at 1-800-444-2080.
- Go to the website – www.SIVEXTRO.com.
- You can ask your doctor or pharmacist for information about SIVEXTRO that is written for health professionals.
| What are the ingredients in SIVEXTRO? |
- The active ingredient: tedizolid phosphate.
- The inactive ingredients: crospovidone, magnesium stearate, mannitol, microcrystalline cellulose, and povidone. Film coating: polyethylene glycol/macrogol, polyvinyl alcohol, talc, titanium dioxide, and yellow iron oxide.
Manuf. for: Merck Sharp & Dohme LLC
Rahway, NJ 07065, USA
Sivextro tablets Manufactured by: Patheon Inc., Whitby, Ontario, L1N 5Z5 Canada
Sivextro for injection Manufactured by: Patheon Italia S.p.A., 03013, Ferentino, FR Italy
For patent information: www.msd.com/research/patent. The trademarks depicted herein are owned by their respective companies.
Copyright © 2017-2025 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved.
usppi-mk1986-mf-2504r005
This Patient Information has been approved by the U.S. Food and Drug Administration Revised: 4/2025
PRINCIPAL DISPLAY PANEL - 200 mg Tablet Bottle Label
NDC 67919-041-04
30 tablets
Sivextro®
(tedizolid phosphate) tablets
200 mg per tablet
Rx only


| SIVEXTRO
tedizolid phosphate tablet, film coated |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| SIVEXTRO
tedizolid phosphate injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Merck Sharp & Dohme LLC (118446553) |
More about Sivextro (tedizolid)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Imprints, shape & color data
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: oxazolidinone antibiotics
- Breastfeeding
- En español
Patient resources
- Sivextro drug information
- Sivextro (Tedizolid Intravenous) (Advanced Reading)
- Sivextro (Tedizolid Oral) (Advanced Reading)
Professional resources
Related treatment guides
Copyright © 2015-2025 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates.
All rights reserved.