Perseris: Package Insert / Prescribing Info
Package insert / product label
Generic name: risperidone
Dosage form: extended-release injectable suspension
Drug class: Atypical antipsychotics
J Code (medical billing code): J2798 (0.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Feb 23, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
PERSERIS (risperidone) for extended-release injectable suspension, for subcutaneous use.
Initial U.S. Approval: 1993
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
See full prescribing information for complete boxed warning.
- Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death.
- PERSERIS is not approved for use in patients with dementia-related psychosis. ( 5.1)
Recent Major Changes
| Warnings and Precautions (5.6) | 1/2025 |
Indications and Usage for Perseris
PERSERIS is an atypical antipsychotic indicated for the treatment of schizophrenia in adults. ( 1)
Perseris Dosage and Administration
- Establish tolerability with oral risperidone prior to initiating PERSERIS. ( 2.1)
- Administer monthly by subcutaneous injection in the abdomen or back of the upper arm by a healthcare provider. Do not administer by any other route. ( 2.1)
- PERSERIS may be initiated at a dose of 90 mg or 120 mg once monthly. Do not administer more than one dose per month. ( 2.1)
- Supplementation with oral risperidone is not recommended. ( 2.1)
- See Full Prescribing Information for important preparation and administration information. Failure to fully mix the medication could result in incorrect dosage. ( 2.4)
Dosage Forms and Strengths
For extended-release injectable suspension: 90 mg and 120 mg risperidone. ( 3)
Contraindications
Known hypersensitivity to risperidone, paliperidone, or other components of PERSERIS. ( 4)
Warnings and Precautions
- Cerebrovascular Adverse Reactions, in Elderly Patients with Dementia-Related Psychosis: Increased risk of cerebrovascular adverse reactions (e.g., stroke, transient ischemic attack). ( 5.2)
- Neuroleptic Malignant Syndrome (NMS): Manage with immediate discontinuation and close monitoring. ( 5.3)
- Tardive Dyskinesia: Discontinue treatment if clinically appropriate. ( 5.4)
- Metabolic Changes: Monitor for hyperglycemia, dyslipidemia, and weight gain. ( 5.5)
- Hyperprolactinemia: Prolactin elevations occur and persist during chronic administration. ( 5.6)
- Orthostatic Hypotension and Syncope: Monitor heart rate and blood pressure and warn patients with known cardiovascular disease or cerebrovascular disease, and risk of dehydration or syncope. ( 5.7)
- Leukopenia, Neutropenia, and Agranulocytosis: Perform complete blood counts (CBC) in patients with a history of a clinically significant low white blood cell count (WBC) or history of leukopenia or neutropenia. Consider discontinuing PERSERIS if a clinically significant decline in WBC occurs in absence of other causative factors. ( 5.9)
- Potential for Cognitive and Motor Impairment: Use caution when operating machinery. ( 5.10)
- Seizures: Use cautiously in patients with a history of seizures or with conditions that lower the seizure threshold. ( 5.11)
Adverse Reactions/Side Effects
The most common adverse reactions in clinical trials (≥ 5% and greater than twice placebo) were increased weight, sedation/somnolence, and musculoskeletal pain. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Indivior Inc. at 1-877-782-6966 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2025
Full Prescribing Information
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. PERSERIS®is not approved for the treatment of patients with dementia-related psychosis[see Warnings and Precautions ( 5.1)].
1. Indications and Usage for Perseris
PERSERIS is indicated for the treatment of schizophrenia in adults [see Clinical Studies ( 14)] .
2. Perseris Dosage and Administration
2.1 Recommended Dosage
Administer PERSERIS by a healthcare provider as a subcutaneous injection in the abdomen or back of the upper arm. Do not administer PERSERIS by any other route.
For detailed preparation and administration instructions, see Dosage and Administration ( 2.4) .
For patients who have never taken risperidone, establish tolerability with oral risperidone prior to initiating PERSERIS.
Initiate PERSERIS at a dose of 90 mg or 120 mg once monthly by subcutaneous injection. Do not administer more than one dose (90 mg or 120 mg total) per month.
For patients switching from oral risperidone:
- 3 mg of oral risperidone per day, administer a 90 mg PERSERIS dose one day after the last oral risperidone dose.
- 4 mg of oral risperidone per day, administer a 120 mg PERSERIS dose one day after the last oral risperidone dose.
Patients who are on stable oral risperidone doses lower than 3 mg per day or higher than 4 mg per day may not be candidates for PERSERIS [see Clinical Pharmacology ( 12.3) and Clinical Studies ( 14)] .
Neither a loading dose nor any supplemental oral risperidone is recommended. When a dose of PERSERIS is missed, administer the next PERSERIS injection as soon as possible.
2.2 Dosage Recommendations for Patients with Renal or Hepatic Impairment
Prior to initiating treatment with PERSERIS in patients with renal or hepatic impairment, titrate with oral risperidone up to at least 3 mg daily. Following oral titration, and based on clinical response and tolerability, the recommended dosage of PERSERIS is 90 mg once monthly [see Use in Specific Populations ( 8.6, 8.7) and Clinical Pharmacology ( 12.3)].
2.3 Dosage Recommendations for Concomitant Use with Strong CYP2D6 Inhibitors and Strong CYP3A4 Inducers
Co-administration with Strong CYP2D6 Inhibitors
Between 2 to 4 weeks prior to initiating a strong CYP2D6 inhibitor (such as fluoxetine or paroxetine), switch patients (if applicable) to the lowest PERSERIS dosage (90 mg once monthly) to adjust for the expected increase in plasma concentrations of risperidone [see Drug Interactions ( 7.1)] .
Co-administration with Strong CYP3A4 Inducers
With concomitant use of PERSERIS 90 mg and strong CYP3A4 inducers (such as carbamazepine), increase the PERSERIS dosage to 120 mg once monthly and consider starting additional oral risperidone therapy. In patients already receiving PERSERIS 120 mg once monthly, additional oral risperidone therapy may need to be considered.
Upon discontinuation of a strong CYP3A4 inducer in a patients receiving PERSERIS 120 mg once monthly, re-evaluate the dosage of PERSERIS or any additional oral risperidone therapy and, if necessary, decrease to adjust for the expected increase in plasma concentration of risperidone.
Upon discontinuation of a strong CYP3A4 inducer in a patient receiving PERSERIS 90 mg once monthly, continue treatment with the 90 mg dose unless clinical judgment necessitates interruption of PERSERIS treatment [see Drug Interactions ( 7.1)] .
2.4 Preparation and Administration Instructions
- Read the instructions for preparation and administration below and consider referring to the separate Healthcare Provider “Instructions for Use” for additional preparation and administration considerations.
- For subcutaneous injection only. Do not inject by any other route.
- Allow package to come to room temperature for at least 15 minutes prior to preparation. Prepare medication when you are ready to administer the dose.
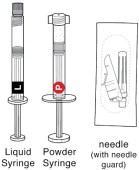
1 CHECK CONTENTS
Each carton of PERSERIS contains ( Figure 1):
-
One Liquid Syringe(
 ) prefilled with the delivery system. Inspect liquid solution for foreign particles. This is the syringe you will use to inject the patient.
) prefilled with the delivery system. Inspect liquid solution for foreign particles. This is the syringe you will use to inject the patient.
-
One Powder Syringe(
 ) prefilled with Risperidone powder. Inspect syringe for consistency of powder color and for foreign particles.
) prefilled with Risperidone powder. Inspect syringe for consistency of powder color and for foreign particles.
-
One sterile 18-gauge, 5/8-inch safety needle.
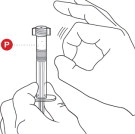
2 TAP POWDER SYRINGE
Hold the Powder Syringeupright and tap the barrel of the syringe to dislodge the packed powder ( Figure 2). Note: Powder can become packed during shipping.
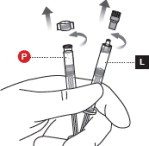
3 UNCAP LIQUID AND POWDER SYRINGES
Remove the cap from the Liquid Syringe, then remove the cap from the Powder Syringe( Figure 3).
Holding both syringes in your non-dominant hand can help with this step.
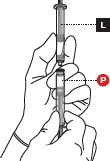
4 CONNECT THE SYRINGES
Place the Liquid Syringeon top of the Powder Syringe(to prevent powder spillage) and connect the syringes by twisting approximately ¾ turn ( Figure 4). Do not over tighten.
Keep your fingers off the plungers during this step to avoid spillage of the medication.
5 MIX THE PRODUCT
Failure to fully mix the medication could result in incorrect dosage.
Premixing
- Transfer the contents of the Liquid Syringeinto the Powder Syringe.
- Gently push the Powder Syringeplunger until you feel resistance (to wet powder and avoid compacting).
- Repeat this gentle back-and-forth process for 5 cycles.
Complete mixing
- Continue mixing the syringes for an additional 55 cycles.
- This mixing can be more vigorous than when premixing.
See Figure 5for an illustration of a correct full cycle.
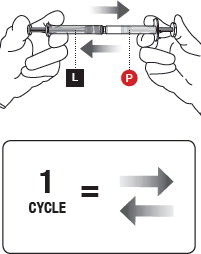
When fully mixed, the product should be a cloudy suspension that is uniform in color. It can vary from white to yellow-green in color. If you see any clear areas in the mixture, continue to mix until the distribution of the color is uniform. The product is designed to deliver risperidone 90 mg or 120 mg.
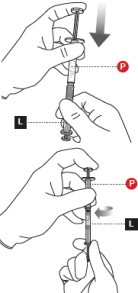
6 PREPARE INJECTION SYRINGE
Failure to aspirate the liquid from the Powder Syringemay result in incorrect dosage.
- First, transfer all contents into the Liquid Syringe( Figure 6).
- Next, perform the following actions
SIMULTANEOUSLY:
- maintain slight pressure on the Powder Syringeplunger and
- pull back gentlyon the Liquid Syringeplunger while twisting the syringes apart.
- Finally, attach the safety needle by twisting until finger tight.
- Check that medication is uniform in color and free from foreign particles.
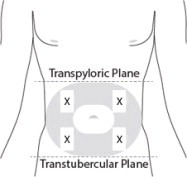
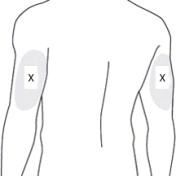
7 PREPARE THE SUBCUTANEOUS INJECTION SITE(S)
This medication is to be injected subcutaneously in the abdomen or back of the upper arm ( Figure 7).
Choose an injection site with adequate subcutaneous tissue that is free of skin conditions (e.g., nodules, lesions, excessive pigment).
Do notinject into an area where the skin is irritated, reddened, bruised, infected or scarred in any way.
Clean the injection site well with an alcohol pad.
To help minimize irritation, rotate injection sites following a pattern similar to the illustration. If you want to use the same injection site, make sure it is not the same spot on the injection site you used the last time.
8 REMOVE EXCESS AIR FROM SYRINGE
Hold the syringe upright for several seconds to allow air bubbles to rise. Remove needle cover and slowly depress the plunger to push out the excess air from the syringe ( Figure 8).
If medication is seen at the needle tip, pull back slightly on the plunger to prevent medication spillage.
Due to the viscous nature of the medication, bubbles will not rise as quickly as those in an aqueous solution.
Figure 8
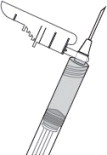
9 PINCH INJECTION SITE
Pinch the skin around the injection area. Be sure to pinch enough skin to accommodate the size of the needle ( Figure 9). Lift the adipose tissue from the underlying muscle to prevent accidental intramuscular injection.
Figure 9

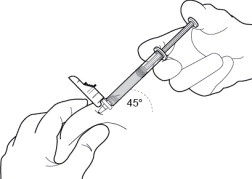
10 INJECT THE MEDICATION
Insert needle fully into the subcutaneous tissue. Inject the medication slow and steady ( Figure 10).
PERSERIS is for subcutaneous administration only. Do not inject by any other route. Actual angle of injection will depend on the amount of subcutaneous tissue.
Figure 10
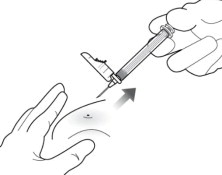
11 WITHDRAW NEEDLE
Withdraw the needle at the same angle used for insertion and release pinched skin ( Figure 11).
Do not rub the injection area after the injection. If there is bleeding, apply a gauze pad or bandage but use minimal pressure.
Figure 11
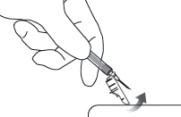
12 LOCK THE NEEDLE GUARD AND DISPOSE OF SYRINGE
Lock the needle guard into place by pushing it against a hard surface such as a table ( Figure 12).
13 INSTRUCT THE PATIENT
The patient may have a lump for several weeks that will decrease in size over time ( Figure 13). It is important that the patient not rub or massage the injection site and to be aware of the placement of any belts, waistbands, sleeves, cuffs or other parts of clothing.

3. Dosage Forms and Strengths
PERSERIS (risperidone) for extended-release injectable suspension for subcutaneous use is available in strengths of 90 mg and 120 mg.
Each strength is provided as a kit which includes: one pre-filled syringe containing a white to yellow risperidone powder in a sealed pouch, one pre-filled syringe containing a colorless to yellow delivery system in a sealed pouch, and one 18-gauge, 5/8-inch needle.
4. Contraindications
PERSERIS is contraindicated in patients with a known hypersensitivity to risperidone, its metabolite, paliperidone, or to any of its components. Hypersensitivity reactions, including anaphylactic reactions and angioedema, have been reported in patients treated with risperidone or paliperidone.
5. Warnings and Precautions
5.1 Increased Mortality in Elderly Patients with Dementia-Related Psychosis
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of 17 placebo-controlled trials (modal duration of 10-weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients between 1.6- to 1.7-times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. Observational studies suggest that, similar to atypical antipsychotic drugs, treatment with conventional antipsychotic drugs may increase mortality. The extent to which the findings of increased mortality in observational studies may be attributed to the antipsychotic drug as opposed to some characteristic(s) of the patients is not clear.
PERSERIS is not approved for the treatment of patients with dementia-related psychosis [see Warnings and Precautions ( 5.2)] .
5.2 Cerebrovascular Adverse Reactions, Including Stroke, in Elderly Patients with Dementia-Related Psychosis
Cerebrovascular adverse reactions (e.g., stroke, transient ischemic attack), including fatalities, were reported in patients (mean age 85-years; range 73 to 97) in trials of oral risperidone in elderly patients with dementia-related psychosis. In placebo-controlled trials, there was a significantly higher incidence of cerebrovascular adverse reactions in patients treated with oral risperidone compared to patients treated with placebo. PERSERIS is not approved for the treatment of patients with dementia-related psychosis [see Warnings and Precautions ( 5.1)] .
5.3 Neuroleptic Malignant Syndrome (NMS)
NMS, a potentially fatal symptom complex, has been reported in association with antipsychotic drugs. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status including delirium, and autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmia). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure.
If NMS is suspected, immediately discontinue PERSERIS and provide symptomatic treatment and monitoring.
5.4 Tardive Dyskinesia
Tardive dyskinesia, a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements, may develop in patients treated with antipsychotic drugs. Although the prevalence of the syndrome appears to be highest among the elderly, especially elderly women, it is impossible to rely upon prevalence estimates to predict which patients will develop the syndrome. Whether antipsychotic drug products differ in their potential to cause tardive dyskinesia is unknown.
The risk of developing tardive dyskinesia and the likelihood that it will become irreversible are believed to increase with the duration of treatment and the total cumulative dose. The syndrome can develop after relatively brief treatment periods, even at low doses. It may also occur after discontinuation of treatment.
Tardive dyskinesia may remit, partially or completely, if antipsychotic treatment is discontinued. Antipsychotic treatment, itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome and thereby may possibly mask the underlying process. The effect that symptomatic suppression has upon the long-term course of the syndrome is unknown.
Given these considerations, PERSERIS should be prescribed in a manner that is most likely to minimize the occurrence of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients: 1) who suffer from a chronic illness that is known to respond to antipsychotic drugs, and (2) for whom alternative, equally effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment producing a satisfactory clinical response. Periodically reassess the need for continued treatment.
If signs and symptoms of tardive dyskinesia appear in a patient treated with PERSERIS, drug discontinuation should be considered. However, some patients may require treatment with PERSERIS despite the presence of the syndrome.
5.5 Metabolic Changes
Atypical antipsychotic drugs have been associated with metabolic changes that may increase cardiovascular/cerebrovascular risk. These metabolic changes include hyperglycemia, dyslipidemia, and body weight gain. While all of the drugs in the class have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia and Diabetes Mellitus
Hyperglycemia and diabetes mellitus, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, have been reported in patients treated with atypical antipsychotics including risperidone. Assessment of the relationship between atypical antipsychotic use and glucose abnormalities is complicated by the possibility of an increased background risk of diabetes mellitus in patients with schizophrenia and the increasing incidence of diabetes mellitus in the general population. Given these confounders, the relationship between atypical antipsychotic use and hyperglycemia-related adverse events is not completely understood. However, epidemiological studies suggest an increased risk of hyperglycemia-related events in patients treated with the atypical antipsychotics. Precise risk estimates for hyperglycemia-related adverse events in patients treated with atypical antipsychotics are not available.
Patients with an established diagnosis of diabetes mellitus who are started on atypical antipsychotics, including PERSERIS, should be monitored regularly for worsening of glucose control. Patients with risk factors for diabetes mellitus (e.g., obesity, family history of diabetes) who are starting treatment with atypical antipsychotics, including PERSERIS, should undergo fasting blood glucose testing at the beginning of treatment and periodically during treatment. Any patient treated with atypical antipsychotics, including PERSERIS, should be monitored for symptoms of hyperglycemia including polydipsia, polyuria, polyphagia, and weakness. Patients who develop symptoms of hyperglycemia during treatment with atypical antipsychotics, including PERSERIS, should undergo fasting blood glucose testing. In some cases, hyperglycemia has resolved when the atypical antipsychotic, including risperidone, was discontinued; however, some patients required continuation of anti-diabetic treatment despite discontinuation of risperidone.
Data from an 8-week double-blind, placebo-controlled study with PERSERIS in adult patients with schizophrenia are presented in Table 1.
|
†The “n”s in the Serum Glucose mean row are the number of patients with data at baseline and EOS visits. |
|||
|
‡Data shown as number of patients with at least one postbaseline value as denominator and number of patients satisfying the predefined criterion as numerator. |
|||
| PERSERIS
90 mg | PERSERIS
120 mg | Placebo | |
| n = 98 | n = 106 | n = 96 | |
| Serum Glucose, mg/dL, mean† | |||
| Mean Change from Baseline to EOS | 5.7 | 6.3 | -0.9 |
| Glucose, > 126 mg/dL | |||
| Proportion of Patients with Postbaseline Abnormal Values ‡ | 12/104 (11.5%) | 14/111 (12.6%) | 8/109 (7.3%) |
Similar changes from baseline in serum glucose were observed in patients receiving PERSERIS during an open-label, 12-month long-term safety study. Additionally, the mean HbA 1cincreased from 5.6 to 5.7% over the 12-months.
Dyslipidemia
Undesirable alterations in lipids have been observed in patients treated with atypical antipsychotics. Before or soon after initiation of antipsychotic medications, obtain a fasting lipid profile at baseline and monitor periodically during treatment.
Data from an 8-week double-blind, placebo-controlled study with PERSERIS in adult patients with schizophrenia are presented in Table 2.
|
†The “n”s in the Cholesterol mean row are the number of patients with data at baseline and EOS visits. |
|||
|
‡Data shown as number of patients with at least one postbaseline value as denominator and number of patients satisfying the predefined criterion as numerator. |
|||
| PERSERIS
90 mg | PERSERIS
120 mg | Placebo | |
| Cholesterol, mg/dL, mean† | n = 98 | n = 106 | n = 96 |
| Mean Change from Baseline to EOS | -0.5 | -0.5 | 1.1 |
| Cholesterol, ≥ 300 mg/dL | |||
| Proportion of Patients with Postbaseline Abnormal Values ‡ | 2/104 (1.9%) | 2/111 (1.8%) | 2/109 (1.8%) |
Weight Gain
Weight gain has been observed with atypical antipsychotic use. Clinical monitoring of weight is recommended.
Data from an 8-week double-blind, placebo-controlled study with PERSERIS in adult patients with schizophrenia are presented in Table 3.
|
†The “n”s in the Weight Change mean row are the number of patients with data at baseline and end of study visits. |
|||
|
‡ Data shown as number of patients with at least one postbaseline value as denominator and number of patients satisfying the predefined criterion as numerator. |
|||
| PERSERIS
90 mg | PERSERIS
120 mg | Placebo | |
| Weight† | n = 105 | n = 112 | n = 107 |
| Mean Change from Baseline to EOS, kg | 4.4 | 5.3 | 2.6 |
| Weight Gain | |||
| ≥ 7% Increase from Baseline ‡ | 35/107 (32.7%) | 48/114 (42.1%) | 20/111 (18.0%) |
In an open-label, 12-month long-term safety study, for all patients receiving PERSERIS, mean weight increased approximately 2 kg from baseline to Day 85, then remained stable for the remainder of the study.
5.6 Hyperprolactinemia
As with other drugs that antagonize dopamine D 2receptors, risperidone elevates prolactin levels and the elevation persists during chronic administration. Risperidone is associated with higher levels of prolactin elevation than other antipsychotic agents.
Hyperprolactinemia may suppress hypothalamic GnRH, resulting in reduced pituitary gonadotropin secretion. This, in turn, may inhibit reproductive function by impairing gonadal steroidogenesis in both female and male patients [see Use in Specific Populations ( 8.3)] . Galactorrhea, amenorrhea, gynecomastia, and impotence have been reported in patients receiving prolactin-elevating compounds. Long-standing hyperprolactinemia when associated with hypogonadism may lead to decreased bone density in both female and male patients.
Tissue culture experiments indicate that approximately one-third of human breast cancers are prolactin dependent in vitro, a factor of potential importance if the prescription of these drugs is contemplated in a patient with previously detected breast cancer. An increase in pituitary gland, mammary gland, and pancreatic islet cell neoplasia (mammary adenocarcinomas, pituitary and pancreatic adenomas) was observed in the risperidone carcinogenicity studies conducted in mice and rats [see Nonclinical Toxicology ( 13.1)] . Published epidemiologic studies have shown inconsistent results when exploring the potential association between hyperprolactinemia and breast cancer.
5.7 Orthostatic Hypotension and Syncope
Risperidone may induce orthostatic hypotension associated with dizziness, tachycardia, and in some patients, syncope, particularly at the time of initiating treatment, re-initiating treatment, or increasing the dose, probably reflecting its alpha-adrenergic antagonistic properties.
PERSERIS should be used with particular caution in (1) patients with known cardiovascular disease (history of myocardial infarction or ischemia, heart failure, or conduction abnormalities), cerebrovascular disease, and conditions which would predispose patients to hypotension, e.g., dehydration and hypovolemia, and (2) in the elderly and patients with renal or hepatic impairment. Monitoring of orthostatic vital signs should be considered in all such patients, and a dose reduction should be considered if hypotension occurs. Clinically significant hypotension has been observed with concomitant use of oral risperidone and antihypertensive medication.
5.8 Falls
Somnolence, postural hypotension, motor instability, and sensory instability have been reported with the use of antipsychotics, including PERSERIS, which may lead to falls and, consequently, fractures or other fall-related injuries. For patients, particularly the elderly, with diseases, conditions, or medications that could exacerbate these effects, assess the risk of falls when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic therapy.
5.9 Leukopenia, Neutropenia, and Agranulocytosis
In clinical trial and/or postmarketing experience, events of leukopenia/neutropenia have been reported temporally related to antipsychotic agents, including risperidone. Agranulocytosis has also been reported.
Possible risk factors for leukopenia and neutropenia include pre-existing low white blood cell count (WBC) or absolute neutrophil count (ANC) and a history of drug-induced leukopenia or neutropenia. In patients with a pre-existing history of a clinically significant low WBC or ANC or a history of drug-induced leukopenia or neutropenia, perform a complete blood count (CBC) frequently during the first few months of therapy. In such patients, consider discontinuation of PERSERIS at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Monitor patients with clinically significant neutropenia for fever or other symptoms or signs of infection and treat promptly if such symptoms or signs occur. Discontinue PERSERIS in patients with absolute neutrophil count <1000/mm 3and follow their WBC until recovery.
5.10 Potential for Cognitive and Motor Impairment
PERSERIS, like other antipsychotics, may cause somnolence and has the potential to impair judgement, thinking, and motor skills.
In an 8-week, double-blind, placebo-controlled study, somnolence/sedation was reported by 7.0% and 7.7% of patients treated with PERSERIS 90 mg and 120 mg, respectively.
Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that treatment with PERSERIS does not affect them adversely.
5.11 Seizures
Seizures were observed during premarketing studies of risperidone in adult patients with schizophrenia. PERSERIS should be used cautiously in patients with a history of seizures or other conditions that potentially lower the seizure threshold.
5.12 Dysphagia
Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Aspiration pneumonia is a common cause of morbidity and mortality in patients with advanced Alzheimer's dementia. Antipsychotic drugs, including PERSERIS, should be used cautiously in patients at risk for aspiration [see Warnings and Precautions ( 5.1)] .
5.13 Priapism
Priapism has been reported during postmarketing surveillance for other risperidone products. Severe priapism may require surgical intervention.
5.14 Body Temperature Regulation
Atypical antipsychotics may disrupt the body's ability to reduce core body temperature. Both hyperthermia and hypothermia have been reported in association with oral risperidone use. Strenuous exercise, exposure to extreme heat, dehydration, and anticholinergic medications may contribute to an elevation in core body temperature; use PERSERIS with caution in patients who may experience these conditions.
6. Adverse Reactions/Side Effects
The following are discussed in more detail in previous sections of the labeling:
- Increased Mortality in Elderly Patients with Dementia-Related Psychosis [see Boxed Warningand Warnings and Precautions ( 5.1)]
- Cerebrovascular Adverse Reactions, Including Stroke, in Elderly Patients with Dementia-Related Psychosis [see Warnings and Precautions ( 5.2)]
- Neuroleptic Malignant Syndrome (NMS) [see Warnings and Precautions ( 5.3)]
- Tardive Dyskinesia [see Warnings and Precautions ( 5.4)]
- Metabolic Changes [see Warnings and Precautions ( 5.5)]
- Hyperprolactinemia [see Warnings and Precautions ( 5.6)]
- Orthostatic Hypotension and Syncope [see Warnings and Precautions ( 5.7)]
- Falls [see Warnings and Precautions ( 5.8)]
- Leukopenia, Neutropenia and Agranulocytosis [see Warnings and Precautions ( 5.9)]
- Potential for Cognitive and Motor Impairment [see Warnings and Precautions ( 5.10)]
- Seizures [see Warnings and Precautions ( 5.11)]
- Dysphagia [see Warnings and Precautions ( 5.12)]
- Priapism [see Warnings and Precautions ( 5.13)]
- Body Temperature Regulation [see Warnings and Precautions ( 5.14)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of PERSERIS was evaluated in a total of 837 adult patients with schizophrenia who received at least 1 dose of PERSERIS during the clinical development program. A total of 322 patients were exposed to PERSERIS for at least 6 months, of which 234 patients were exposed to PERSERIS for at least 12 months; 281 and 176 of these, respectively, received the 120 mg dose.
Adverse drug reactions in adult patients with schizophrenia (≥ 5% in any PERSERIS-treated group and greater than placebo) during the 8-week double-blind, placebo-controlled study) were weight increased, constipation, sedation/somnolence, pain in extremity, back pain, akathisia, anxiety, and musculoskeletal pain. In addition, the frequency of reported injection site reactions was similar across treatment groups with both PERSERIS and placebo; the most common (≥ 5%) of which were injection site pain, and erythema. The systemic safety profile for PERSERIS was consistent with the known safety profile of oral risperidone.
Commonly-Observed Adverse Drug Reactions in Double-Blind, Placebo-Controlled Clinical Studies – Schizophrenia
Adverse Reactions with an incidence of 2% or more and greater than placebo are shown in Table 4.
|
* Sedation includes sedation and somnolence |
|||
| System Organ Class
Preferred Term | PERSERIS
90 mg (n = 115) | PERSERIS
120 mg (n = 117) | Placebo
(n = 118) |
| Percentage of Patients Reporting ADR | |||
| Gastrointestinal disorders | |||
| Constipation | 7.0 | 7.7 | 5.1 |
| Abdominal discomfort | 2.6 | 2.6 | 1.7 |
| Dry mouth | 1.7 | 2.6 | 1.7 |
| Investigations | |||
| Weight increased | 13.0 | 12.8 | 3.4 |
| Metabolism and nutrition disorders | |||
| Increased appetite | 1.7 | 3.4 | 1.7 |
| Musculoskeletal and connective tissue disorders | |||
| Back pain | 3.5 | 6.8 | 4.2 |
| Pain in extremity | 0.9 | 7.7 | 5.1 |
| Musculoskeletal pain | 5.2 | 5.1 | 2.5 |
| Musculoskeletal stiffness | 2.6 | 0.9 | 1.7 |
| Muscle spasms | 0 | 2.6 | 0 |
| Nervous system disorders | |||
| Sedation* | 7.0 | 7.7 | 0 |
| Akathisia | 2.6 | 6.8 | 4.2 |
| Extrapyramidal disorder | 4.3 | 1.7 | 0.8 |
| Psychiatric disorders | |||
| Anxiety | 2.6 | 6.8 | 5.1 |
Other Adverse Drug Reactions Observed During the Clinical Trial Evaluation of PERSERIS
The following list does not include reactions: 1) already listed in previous tables or elsewhere in labeling, 2) which are part of the disease state, 3) for which a drug cause was remote, 4) which were so general as to be uninformative, or 5) which were not considered to have significant clinical implications.
Blood and Lymphatic System Disorders:neutropenia
Ear and Labyrinth Disorders:vertigo
Endocrine Disorders:hyperprolactinemia
Eye Disorders:blepharospasm
Gastrointestinal Disorders:nausea, dyspepsia, vomiting, diarrhea, abdominal pain upper, salivary hypersecretion, hypoesthesia oral, tongue movement disturbance
General Disorders and Administration Site Conditions:injection site reaction (including injection site pain, induration, pruritus, bruising, erythema, inflammation, swelling and irritation) fatigue, edema peripheral, asthenia, chest discomfort
Investigations:blood prolactin increased, blood glucose increased, glycosylated hemoglobin increased, electrocardiogram abnormal, electrocardiogram QT prolonged, blood creatine phosphokinase increased
Metabolism and Nutrition Disorders:diabetes mellitus, decreased appetite
Musculoskeletal, Connective Tissue, and Bone Disorders:arthralgia, muscle twitching, joint stiffness, trismus
Nervous System Disorders:headache, dizziness, tremor, drooling, dyskinesia, lethargy, dystonia, hypoesthesia, oromandibular dystonia, tardive dyskinesia, cogwheel rigidity, dysarthria, balance disorder, parkinsonian rest tremor, parkinsonism, slow speech
Psychiatric Disorders:insomnia, libido decreased, bruxism, restlessness, anorgasmia, loss of libido
Reproductive System and Breast Disorders:erectile dysfunction, galactorrhea, breast tenderness, breast pain, amenorrhea, breast engorgement, ejaculation delayed, ejaculation disorder, gynecomastia, hypomenorrhea, breast discharge, breast enlargement, ejaculation failure, menstruation delayed, menstruation irregular, polymenorrhea
Skin and Subcutaneous Tissue Disorders:night sweats
Vascular Disorders:hypertension, hypotension, orthostatic hypotension
Other Adverse Reactions Observed During the Clinical Trial Evaluations of Oral Risperidone
The following is a list of additional ADRs that have been reported during the clinical trial evaluation of oral risperidone, regardless of frequency of occurrence:
Blood and Lymphatic System Disorders:anemia, granulocytopenia
Cardiac Disorders:tachycardia, sinus bradycardia, sinus tachycardia, atrioventricular block first degree, bundle branch block left, bundle branch block right, atrioventricular block
Ear and Labyrinth Disorders:ear pain, tinnitus
Eye Disorders:vision blurred, oculogyration, ocular hyperemia, eye discharge, conjunctivitis, eye rolling, eyelid edema, eye swelling, eyelid margin crusting, dry eye, lacrimation increased, photophobia, glaucoma, visual acuity reduced
Gastrointestinal Disorders:dysphagia, fecaloma, fecal incontinence, gastritis, lip swelling, cheilitis, aptyalism
General Disorders:thirst, gait disturbance, chest pain, influenza-like illness, pitting edema, edema, chills, sluggishness, malaise, face edema, discomfort, generalized edema, drug withdrawal syndrome, peripheral coldness, feeling abnormal
Immune System Disorders:drug hypersensitivity
Infections and Infestations:nasopharyngitis, upper respiratory tract infection, sinusitis, urinary tract infection, pneumonia, influenza, ear infection, viral infection, pharyngitis, tonsillitis, bronchitis, eye infection, localized infection, cystitis, cellulitis, otitis media, onychomycosis, acarodermatitis, bronchopneumonia, respiratory tract infection, tracheobronchitis, otitis media chronic
Investigations:body temperature increased, alanine aminotransferase increased, heart rate increased, eosinophil count increased, white blood cell count decreased, hemoglobin decreased, blood creatine phosphokinase increased, hematocrit decreased, body temperature decreased, blood pressure decreased, transaminases increased
Metabolism and Nutrition Disorders:polydipsia, anorexia
Musculoskeletal, Connective Tissue, and Bone Disorders:joint swelling, musculoskeletal chest pain, posture abnormal, myalgia, neck pain, muscular weakness, muscle rigidity, muscle contracture, rhabdomyolysis
Nervous System Disorders:dizziness postural, disturbance in attention, unresponsive to stimuli, depressed level of consciousness, movement disorder, hypokinesia, bradykinesia, transient ischemic attack, coordination abnormal, cerebrovascular accident, masked facies, speech disorder, syncope, loss of consciousness, muscle contractions involuntary, Parkinson's disease, tongue paralysis, akinesia, cerebral ischemia, cerebrovascular disorder, neuroleptic malignant syndrome, diabetic coma, head titubation
Psychiatric Disorders:agitation, blunted affect, confusional state, middle insomnia, nervousness, sleep disorder, listlessness
Renal and Urinary Disorders:enuresis, dysuria, pollakiuria, urinary incontinence
Reproductive System and Breast Disorders:vaginal discharge, menstrual disorder, retrograde ejaculation, sexual dysfunction
Respiratory, Thoracic, and Mediastinal Disorders:nasal congestion, dyspnea, epistaxis, wheezing, pneumonia aspiration, sinus congestion, dysphonia, productive cough, pulmonary congestion, respiratory tract congestion, rales, respiratory disorder, hyperventilation, nasal edema
Skin and Subcutaneous Tissue Disorders:rash, dry skin, erythema, skin discoloration, skin lesion, pruritus, skin disorder, rash erythematous, rash papular, acne, hyperkeratosis, seborrheic dermatitis, rash generalized, rash maculopapular
Vascular Disorders:flushing
Discontinuations Due to Adverse Drug Reactions (ADRs)
There was no single adverse reaction leading to discontinuation that occurred at a rate of ≥ 2% in PERSERIS-treated patients and greater than placebo.
Changes in Body Weight
Data from the double-blind placebo-controlled study indicated there was a dose-dependent increase in mean changes in weight from baseline to postdose assessments in the PERSERIS 90 mg and 120 mg groups compared with the placebo group [see Warnings and Precautions ( 5.5), Adverse Reactions (6.1, Table 4)] .
Increased Prolactin
In the 8-week double-blind, placebo-controlled study, there was a typical increase in mean prolactin levels in fasting blood samples from baseline to the EOS assessments in both the PERSERIS 90 mg and 120 mg groups, while mean prolactin for the placebo group remained stable during the study. Changes in mean prolactin were dose-dependent and more pronounced in female patients than male patients.
Extrapyramidal Symptoms (EPS)
Several methods were used to measure EPS, including: (1) the Barnes Akathisia Rating Scale (BARS) global clinical rating score which evaluates akathisia, (2) the Abnormal Involuntary Movement Scale (AIMS) scores which evaluates dyskinesia, (3) the Simpson-Angus Scale (SAS) global score which broadly evaluates parkinsonism, and (4) the incidence of spontaneous reports of EPS-related adverse reactions.
In the 8-week double-blind, placebo-controlled study, the mean changes from baseline in BARS, AIMS, and SAS total scores were comparable between PERSERIS- and placebo-treated patients. At all postbaseline assessments, mean changes from baseline were between -0.1 and 0.2 (inclusive) for the BARS, between 0 and 0.2 (inclusive) for the AIMS and between -0.1 and 0.2 (inclusive) for the SAS.
The rates of ADRs associated with EPS were similar across treatment groups, including placebo. There was a higher incidence of akathisia in the PERSERIS 120 mg (6.8%) group compared with the PERSERIS 90 mg (2.6%) and placebo group (4.2%); reports of extrapyramidal disorders were higher in the PERSERIS 90 mg group (4.3%) compared with the PERSERIS 120 mg (1.7%) and placebo group (0.8%). In contrast, there was a higher incidence of dystonia in the placebo group (2.5%) compared with the PERSERIS groups (0 and 0.9%, respectively).
Dystonia
Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. Although these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first generation antipsychotic drugs. An elevated risk of acute dystonia has been observed in males and younger age groups.
Changes in ECG
In the 8-week double-blind, placebo-controlled study, there were no clinically relevant differences in mean changes from baseline to EOS in ECG parameters, including QT cF (Fridericia's corrected QT interval), QRS and PR intervals, and heart rate, in patients in either PERSERIS treatment group (90 mg and 120 mg) compared with placebo. Similarly, in the 12-month, long-term safety study, there were no clinically relevant changes in mean ECG interval values from baseline to postdose assessments.
Pain Assessment and Local Injection Site Reactions
Local injection site pain was assessed using patient-reported VAS scales (0 = no pain to 100 = unbearably painful). In the 8-week, double-blind placebo-controlled study, the mean patient-reported injection site pain VAS scores were similar for all treatment groups following both injections. Pain scores decreased from a mean of 27 (VAS score) 1 minute after the first dose to a range of 3 to 7 (VAS score) 30 to 60 minutes postdose. In the 12-month, long-term safety study, the 1-minute postdose injection site pain VAS scores were highest on Day 1 (mean of 25) and decreased over time with subsequent injections (14 to 16 following last injection).
The local injection site was assessed by appropriately trained personnel. Throughout the clinical development program, the maximum reported intensity at any time point for each injection site assessment (pain, tenderness, inflammation/swelling and erythema) was none or mild for most patients receiving PERSERIS.
Most patients (≥ 79%) reported no tenderness and most who had tenderness reported mild severity. Less than 1% of patients had moderate tenderness at any time point and 1 patient at Injections 1, 2, and 5 had severe tenderness. At each time point, most patients (≥ 75%) reported no pain on injection. Of patients who did have pain on injection, almost all of these were mild at each time point; only 1 or 2 patients at Injections 1, 2, 7, and 12 had moderate pain on injection. At least 92% of patients reported no erythema on each injection. All reports of erythema were of mild severity except for 2 cases of moderate erythema on Injection 1. Inflammation/swelling had a similar profile, with at least 88% of patients reporting no inflammation/swelling and only mild symptoms except for 1 case of moderate severity on Injection 1.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of oral risperidone. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These adverse reactions include: alopecia, anaphylactic reaction, angioedema, atrial fibrillation, cardiopulmonary arrest, catatonia, diabetic ketoacidosis in patients with impaired glucose metabolism, dysgeusia, hypoglycemia, hypothermia, ileus, inappropriate antidiuretic hormone secretion, intestinal obstruction, jaundice, mania, pancreatitis, pituitary adenoma, precocious puberty, pulmonary embolism, QT prolongation, sleep apnea syndrome, somnambulism, Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN), sudden death, thrombocytopenia, thrombotic thrombocytopenic purpura, urinary retention, and water intoxication.
Postmarketing cases of extrapyramidal symptoms (dystonia and dyskinesia) have been reported in patients concomitantly taking methylphenidate and risperidone when there was an increase or decrease in dosage, initiation, or discontinuation of either or both medications.
Related/similar drugs
7. Drug Interactions
The interactions of PERSERIS with co-administration of other drugs have not been studied. The drug interaction data provided in this section is based on studies with oral risperidone.
7.1 Drugs Having Clinically Important Interactions with PERSERIS
Table 5includes clinically significant drug interactions with PERSERIS.
| Strong CYP2D6 Inhibitors | |
| Clinical Impact: | Concomitant use of PERSERIS with strong CYP2D6 inhibitors may increase the plasma exposure of risperidone and lower the plasma exposure of a major active metabolite, 9-hydroxyrisperidone [see Clinical Pharmacology ( 12.3)] . |
| Intervention: | When initiation of strong CYP2D6 inhibitors is considered, patients may be placed on the lowest dose (90 mg) of PERSERIS between 2 to 4 weeks before the planned start of strong CYP2D6 inhibitors to adjust for the expected increase in plasma concentrations of risperidone. When strong CYP2D6 inhibitors is initiated in patients receiving PERSERIS 90 mg, it is recommended to continue treatment with 90 mg unless clinical judgment necessitates interruption of PERSERIS treatment. The effects of discontinuation of strong CYP2D6 inhibitors on the pharmacokinetics of risperidone and 9-hydroxyrisperidone have not been studied [see Clinical Pharmacology ( 12.3)] . |
| Strong CYP3A4 Inducers | |
| Clinical Impact: | Concomitant use of PERSERIS and a strong CYP3A4 inducer may cause decreases in the combined plasma concentrations of risperidone and 9-hydroxyrisperidone which could lead to decreased efficacy of PERSERIS [see Clinical Pharmacology ( 12.3)] . |
| Intervention: | Changes in efficacy and safety should be carefully monitored with any dose adjustment of PERSERIS. At the initiation of therapy with a strong CYP3A4 inducer, patients should be closely monitored during the first 4 to 8 weeks. In patients receiving PERSERIS 90 mg, consider increasing the dose to 120 mg. In patients receiving PERSERIS 120 mg, additional oral risperidone therapy may need to be considered. On discontinuation of a strong CYP3A4 inducer, the dosage of PERSERIS or any additional oral risperidone therapy should be re-evaluated and, if necessary, decreased to adjust for the expected increase in plasma concentration of risperidone and 9-hydroxyrisperidone. For patients treated with PERSERIS 90 mg and discontinuing from a strong CYP3A4 inducer, it is recommended to continue treatment with the 90 mg dose unless clinical judgment necessitates interruption of PERSERIS treatment [see Dosage and Administration ( 2.3)] . |
| Centrally-Acting Drugs and Alcohol | |
| Clinical Impact: | Due to additive pharmacologic effects, the concomitant use of centrally-acting drugs, including alcohol, may increase nervous system disorders. |
| Intervention: | Caution should be used when PERSERIS is administered in combination with other centrally-acting drugs or alcohol. |
| Hypotensive Agents | |
| Clinical Impact: | Because of its potential for inducing hypotension, PERSERIS may enhance the hypotensive effects of other therapeutic agents with this potential. |
| Intervention: | Caution should be used when PERSERIS is administered in combination with other therapeutic agents with hypotensive effects. |
| Dopamine Agonists | |
| Clinical Impact: | Agents with central antidopaminergic activity such as PERSERIS may antagonize the pharmacologic effects of dopamine agonists. |
| Intervention: | Caution should be used when PERSERIS is administered in combination with levodopa and dopamine agonists. |
| Methylphenidate | |
| Clinical Impact: | Concomitant use with methylphenidate, when there is change in dosage of either medication, may increase the risk of extrapyramidal symptoms (EPS) [see Adverse Reactions ( 6.2)] . |
| Intervention: | Monitor for symptoms of EPS with concomitant use of PERSERIS and methylphenidate. |
7.2 Drugs Having No Clinically Important Interactions with PERSERIS
Based on pharmacokinetic studies with oral risperidone, no dosage adjustment of PERSERIS is required when administered concomitantly with amitriptyline, cimetidine, ranitidine, clozapine, topiramate and moderate CYP3A4 inhibitors (erythromycin). Additionally, no dosage adjustment is necessary for lithium, valproate, topiramate, digoxin and CYP2D6 substrates (donepezil and galantamine) when co-administered with PERSERIS [see Clinical Pharmacology ( 12.3)] .
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to atypical antipsychotics, including PERSERIS, during pregnancy. Healthcare providers are encouraged to register patients by contacting the National Pregnancy Registry for Atypical Antipsychotics at 1-866-961-2388 or online at http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/.
Risk Summary
Neonates exposed to antipsychotic drugs during the third trimester of pregnancy are at risk for extrapyramidal and/or withdrawal symptoms following delivery (see Clinical Considerations) . Overall available data from published epidemiologic studies of pregnant women exposed to risperidone have not established a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes (see Data). There are risks to the mother associated with untreated schizophrenia and with exposure to antipsychotics, including PERSERIS, during pregnancy (see Clinical Considerations) .
Oral administration of risperidone to pregnant mice caused cleft palate at doses 3 to 4 times the oral maximum recommended human dose (MRHD) of 16 mg/day with maternal toxicity observed at 4 times the MRHD based on mg/m 2body surface area. Risperidone was not teratogenic in rats or rabbits at doses up to 6 times the oral MRHD based on mg/m 2body surface area. Increased stillbirths and decreased birth weight occurred after oral risperidone administration to pregnant rats at 1.5 times the oral MRHD based on mg/m 2body surface area. Learning was impaired in offspring of rats when the dams were dosed at 0.6 times the oral MRHD and offspring mortality increased at doses 0.1 to 3 times the oral MRHD based on mg/m 2body surface area.
Subcutaneous administration of the delivery system to pregnant rats and rabbits during the period of organogenesis caused developmental toxicity that included post-implantation loss, decreased number of live fetuses, decreased fetal weight and fetal malformations (external, skeletal, and visceral), at doses that are 52 (rat) and 43 (rabbit) times the delivery system amount present in 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area. These effects could be attributed to N-methyl-2-pyrrolidone (NMP) an excipient in the delivery system based on information in the published literature (see Data) . Subcutaneous administration of the delivery system to pregnant and lactating rats had no effect on embryofetal and postnatal development at doses up to 17 times the delivery system amount present in 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area.
The estimated background risks of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
There is a risk to the mother from untreated schizophrenia, including increased risk of relapse, hospitalization, and suicide. Schizophrenia is associated with increased adverse perinatal outcomes, including preterm birth. It is not known if this is a direct result of the illness or other comorbid factors.
Fetal/neonatal adverse reactions
Extrapyramidal and/or withdrawal symptoms, including agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress, and feeding disorder have been reported in neonates who were exposed to antipsychotic drugs, including risperidone, during the third trimester of pregnancy. These symptoms have varied in severity. Monitor neonates for extrapyramidal and/or withdrawal symptoms and manage symptoms appropriately. Some neonates recovered within hours or days without specific treatment; others required prolonged hospitalization.
Data
Human Data
Published data from observational studies, birth registries, and case reports on the use of atypical antipsychotics during pregnancy do not report a clear association with antipsychotics and major birth defects. A prospective observational study including 6 women treated with risperidone demonstrated placental passage of risperidone. A retrospective cohort study from a Medicaid database of 9258 women exposed to antipsychotics during pregnancy did not indicate an overall increased risk for major birth defects. There was a small increase in the risk of major birth defects (RR = 1.26, 95% CI 1.02 to 1.56) and of cardiac malformations (RR = 1.26, 95% CI 0.88 to 1.81) in a subgroup of 1566 women exposed to risperidone during the first trimester of pregnancy; however, there is no mechanism of action to explain the difference in malformation rates.
Animal data
No developmental toxicity studies were conducted with subcutaneous risperidone suspension.
Oral administration of risperidone to pregnant mice during organogenesis caused cleft palate at 10 mg/kg/day which is 3 times the oral MRHD of 16 mg/day based on mg/m 2body surface area; maternal toxicity occurred at 4 times the oral MRHD. Risperidone was not teratogenic when administered orally to rats at 0.6 to 10 mg/kg/day and rabbits at 0.3 to 5 mg/kg/day, which are up to 6 times the oral MRHD of 16 mg/day risperidone based on mg/m 2body surface area. Learning was impaired in offspring of rats dosed orally throughout pregnancy at 1 mg/kg/day which is 0.6 times the oral MRHD and neuronal cell death increased in fetal brains of offspring of rats dosed during pregnancy at 1 and 2 mg/kg/day which are 0.6 and 1.2 times the oral MRHD based on mg/m 2body surface area; postnatal development and growth of the offspring were also delayed.
Rat offspring mortality increased during the first 4 days of lactation when pregnant rats were dosed throughout gestation at 0.16 to 5 mg/kg/day which are 0.1 to 3 times the oral MRHD of 16 mg/day based on mg/m 2body surface area. It is not known whether these deaths were due to a direct effect on the fetuses or pups or to effects on the dams; a no-effect dose could not be determined. The rate of stillbirths was increased at 2.5 mg/kg or 1.5 times the oral MRHD based on mg/m 2body surface area. In a rat cross-fostering study the number of live offspring was decreased, the number of stillbirths increased, and the birth weight was decreased in offspring of drug-treated pregnant rats. In addition, the number of deaths increased by Day 1 among offspring of drug-treated pregnant rats, regardless of whether or not the offspring were cross-fostered. Risperidone also appeared to impair maternal behavior in that offspring body weight gain and survival (from Day 1 to 4 of lactation) were reduced in offspring born to control but reared by drug-treated dams. All of these effects occurred at 5 mg/kg which is 3 times the oral MRHD based on mg/m 2 and the only dose tested in the study.
Subcutaneous administration of the delivery system to pregnant rats and rabbits during the period of organogenesis caused maternal toxicity (decreased body weight, weight gain and food intake), post-implantation loss, decrease in number of live fetuses and decrease in fetal weight at doses that are 52 (rat), and 43 (rabbit) times the delivery system amount present in monthly 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area. Developmental toxicity in both rat and rabbit included skeletal and visceral malformations at doses 35 (rat), and 43 (rabbit) times the delivery system amount present in monthly 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area. The NOAEL dose for these effects in both species is 17 times the delivery system amount present in monthly 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area. These effects could be related to N-methyl-2-pyrrolidone (NMP), an excipient present in the delivery system. In published animal developmental toxicity studies, NMP administered orally daily to pregnant rats during organogenesis produced developmental toxicity below maternally toxic levels and resulted in dose-dependent decrease in fetal body weights, increased incidence of post-implantation loss, incomplete ossification and increased incidence of external, visceral and skeletal malformations. These toxicities occurred at doses that are ~3 to 12 times the NMP amount present in monthly 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area.
8.2 Lactation
Risk Summary
Limited data from published literature reports the presence of risperidone and its metabolite, 9-hydroxyrisperidone, in human breast milk at relative infant dose ranging between 2.3 and 4.7% of the maternal weight-adjusted dosage. There are reports of sedation, failure to thrive, jitteriness, and extrapyramidal symptoms (tremors and abnormal muscle movements) in breastfed infants exposed to risperidone (see Clinical Considerations).There is no information on the effects of risperidone on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PERSERIS and any potential adverse effects on the breastfed child from PERSERIS or from the mother's underlying condition.
8.3 Females and Males of Reproductive Potential
Females
Based on the pharmacologic action of risperidone (D 2receptor antagonism), treatment with PERSERIS may result in an increase in serum prolactin levels, which may lead to a reversible reduction in fertility in females of reproductive potential [see Warnings and Precautions ( 5.6) and Nonclinical Toxicology ( 13.1)].
8.4 Pediatric Use
Safety and effectiveness of PERSERIS have not been established in pediatric patients.
8.5 Geriatric Use
Clinical studies of PERSERIS in the treatment of schizophrenia did not include patients aged 65 and older to determine whether or not they respond differently from younger patients.
In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Elderly patients with dementia-related psychosis treated with PERSERIS are at an increased risk of death compared to placebo. PERSERIS is not approved for the treatment of patients with dementia related psychosis [see Boxed Warningand Warnings and Precautions ( 5.1, 5.2)] .
8.6 Renal Impairment
In patients with renal impairment, carefully titrate with oral risperidone (up to at least 3 mg) before initiating treatment with PERSERIS [see Dosage and Administration ( 2.2) and Clinical Pharmacology ( 12.3)] .
PERSERIS was not studied in patients with renal impairment, however, such effect has been investigated with oral risperidone.
8.7 Hepatic Impairment
In patients with hepatic impairment, carefully titrate with oral risperidone (up to at least 3 mg) before initiating treatment with PERSERIS [see Dosage and Administration ( 2.2) and Clinical Pharmacology ( 12.3)] .
PERSERIS was not studied in patients with hepatic impairment, however, such effect has been investigated with oral risperidone.
8.8 Patients with Parkinson's Disease or Dementia with Lewy Bodies
Patients with Parkinson's disease or dementia with Lewy bodies can experience increased sensitivity to risperidone. Manifestations can include confusion, obtundation, postural instability with frequent falls, extrapyramidal symptoms, and clinical features consistent with neuroleptic malignant syndrome.
10. Overdosage
10.1 Human Experience
No cases of overdose were reported in premarketing studies with PERSERIS. Because PERSERIS is to be administered by healthcare providers, the potential for overdosage by patients is low.
10.2 Management of Overdosage
In case of overdosage, consult a Poison Control Center at 1-800-222-1222.
In case of acute overdosage, establish and maintain an airway and ensure adequate oxygenation and ventilation. Cardiovascular monitoring should commence immediately and should include continuous electrocardiographic monitoring to detect possible arrhythmias. If antiarrhythmic therapy is administered, disopyramide, procainamide, and quinidine carry a theoretical hazard of QT prolonging effects that might be additive to those of risperidone. Similarly, it is reasonable to expect that the alpha-blocking properties of bretylium might be additive to those of risperidone, resulting in problematic hypotension.
There is no specific antidote to risperidone. Appropriate supportive measures should be instituted. Hypotension and circulatory collapse should be treated with appropriate measures, such as intravenous fluids and/or sympathomimetic agents (epinephrine and dopamine should not be used, since beta stimulation may worsen hypotension in the setting of risperidone-induced alpha blockade). In cases of severe extrapyramidal symptoms, anticholinergic medication should be administered. Close medical supervision and monitoring should continue until the patient recovers.
Consider the long-acting nature of PERSERIS when assessing treatment needs and recovery.
11. Perseris Description
PERSERIS contains risperidone, an atypical antipsychotic. Risperidone belongs to the chemical class of benzisoxazole derivatives. The chemical designation 3-[2-[4-(6-fluoro-1,2-benzoxazol-3-yl) piperidin-1-yl] ethyl]-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a] pyrimidin-4-one. Its molecular formula is C 23H 27FN 4O 2and its molecular weight is 410.5 g/mol.
The structural formula is:
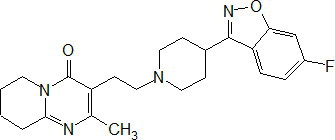
Risperidone is a white to off-white powder. It is practically insoluble in water and soluble in methanol and 0.1 N HCl.
PERSERIS is available as a sterile two-syringe mixing system: a liquid syringe prefilled with the delivery system (colorless to yellow solution), and a powder syringe prefilled with risperidone (white to yellow).
PERSERIS for extended release injectable suspension, for subcutaneous use, is available in 90 mg and 120 mg risperidone strengths. The quantitative composition is provided below ( Table 6).
|
*PLGH poly D,L(lactide co-glycolide); 80:20 molar ratio of lactide to glycolide |
||
| Component | PERSERIS 90 mg | PERSERIS 120 mg |
| Risperidone | 90 mg | 120 mg |
| PLGH* | 228 mg | 304 mg |
| N-methyl-pyrrolidine | 282 mg | 376 mg |
| Total mass | 600 mg | 800 mg |
| Total volume | 0.6 mL | 0.8 mL |
12. Perseris - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of risperidone, in schizophrenia, is unclear. The drug's therapeutic activity in schizophrenia could be mediated through a combination of dopamine Type 2 (D 2) and serotonin Type 2 (5HT 2) receptor antagonism. The clinical effect from risperidone results from the combined concentrations of risperidone and its major metabolite, 9-hydroxyrisperidone (paliperidone) [see Clinical Pharmacology ( 12.3)] . Antagonism at receptors other than D 2and 5HT 2may explain some of the other effects of risperidone.
12.2 Pharmacodynamics
Risperidone is a monoaminergic antagonist with high affinity (Ki of 0.12 to 7.3 nM) for the serotonin Type 2 (5HT 2), dopamine Type 2 (D 2), α 1and α 2 adrenergic, and H 1histaminergic receptors. Risperidone showed low to moderate affinity (Ki of 47 to 253 nM) for the serotonin 5HT 1C, 5HT 1D, and 5HT 1Areceptors, weak affinity (Ki of 620 to 800 nM) for the dopamine D 1and haloperidol-sensitive sigma site, and no affinity (when tested at concentrations > 10 -5M) for cholinergic muscarinic or β 1and β 2adrenergic receptors.
12.3 Pharmacokinetics
The pharmacokinetics of risperidone and total active moiety following subcutaneous injection of PERSERIS was evaluated in patients with clinically stable schizophrenia after single doses (60 mg, 90 mg, and 120 mg) (n = 101) and repeated doses [60 mg, 90 mg, 120 mg, 180 mg (1.5 times the maximum recommended dosage of PERSERIS)] (n = 68) separated by 28 days for up to 4 doses following oral risperidone.
Plasma concentrations of risperidone, 9-hydroxyrisperidone and total active moiety approached steady-state levels after the first dose of PERSERIS. Mean accumulation ratios for risperidone ranged from 1.2 to 1.7 based on mean area under the curve (AUC), and from 0.9 to 1.3 based on overall mean peak plasma concentrations (overall C max), indicating no to modest accumulation. For 9-hydroxyrisperidone, accumulation ratios ranged from 1.2 to 1.6 (AUC) and 0.99 to 1.3 (overall C max). For total active moiety, accumulation ratios ranged from 1.2 to 1.6 (AUC tau) and 0.97 to 1.3 (overall C max).
Following multiple doses of PERSERIS, plasma exposure (AUC tauand C max) of risperidone, 9-hydroxyrisperidone, and total active moiety increased in an approximately dose proportional manner over the dose range of 60 to 120 mg. At steady-state, a 2-fold increase in dose resulted in a 1.7-fold increase in C max(6.33 to 10.9 ng/mL) and AUC tau(2262 to 3891 ng*hr/mL) for risperidone. For 9-hydroxyrisperidone, a 2-fold increase in dose resulted in a 2.1-fold increase in C max(13.7 to 28.9 ng/mL) and 2-fold increase in AUC tau(5706 to 11658 ng*hr/mL). For total active moiety, a 2-fold increase in dose resulted in a 2.0-fold increase in C max(19.6 to 38.5 ng/mL) and a 1.9-fold increase in AUC tau(8102 to 15370 ng*hr/mL).
Plasma exposures at steady-state were compared between oral risperidone and PERSERIS. The average plasma concentrations (C avg) of total active moiety were 18.3 ng/mL and 18.1 ng/mL for 3 mg oral risperidone and 90 mg PERSERIS, respectively. The C avgof total active moiety were 25.2 ng/mL and 22.9 ng/mL for 4 mg oral risperidone and 120 mg PERSERIS, respectively.
Absorption
PERSERIS contains risperidone in a liquid delivery system. Following subcutaneous injection, it forms a depot that provides sustained plasma levels of risperidone over the monthly dosing interval.
After single subcutaneous injection, PERSERIS shows two absorption peaks for risperidone in plasma. The first peak of risperidone occurs with a T maxof 4 to 6 hours and is due to an initial release of the drug during the depot formation process. A second peak of risperidone is observed at 10 to 14 days post-dose and is associated with the slow release of risperidone from the subcutaneous depot. The first and second peaks of risperidone are of similar magnitude. For both 9-hydroxyrisperidone and total active moiety, the median T maxof the first peak ranges from 4 to 48 hours and the second peak ranges from 7 to 11 days.
Distribution
Following a subcutaneous injection of PERSERIS, the apparent volume of distribution is large. The extensively large values are because PERSERIS is administered as a depot injection. Risperidone is bound to albumin and α1-acid glycoprotein. The plasma protein binding of risperidone is approximately 90%, and that of its major metabolite, 9-hydroxyrisperidone, is 77%. Neither risperidone nor 9-hydroxyrisperidone displaces each other from plasma binding sites.
Elimination
Metabolism
Risperidone is extensively metabolized in the liver. The main metabolic pathway is through hydroxylation of risperidone to 9-hydroxyrisperidone by the enzyme cytochrome CYP2D6 with minor contribution by CYP3A4. A minor metabolic pathway is through N-dealkylation. The main metabolite, 9-hydroxyrisperidone, has similar pharmacological activity as risperidone. Consequently, the clinical effect of the drug results from the combined concentrations of risperidone plus 9-hydroxyrisperidone).
CYP2D6, is the enzyme responsible for metabolism of many neuroleptics, antidepressants, antiarrhythmics, and other drugs. CYP2D6 is subject to genetic polymorphism (about 6 to 8% of Caucasians, and a very low percentage of Asians, have little or no activity and are “poor metabolizers”) and to inhibition by a variety of substrates and some non-substrates, notably quinidine. Extensive CYP2D6 metabolizers convert risperidone rapidly into 9-hydroxyrisperidone, whereas poor CYP2D6 metabolizers convert it much more slowly. Plasma exposure to total active moiety was similar in CYP2D6 extensive, intermediate and poor metabolizers following subcutaneous injection with PERSERIS, supporting no need for dose adjustment based on genotype of CYP2D6.
Excretion
Risperidone and its metabolites are eliminated via the urine and, to a much lesser extent, via the feces. As illustrated by a mass balance study of a single 1 mg oral dose of 14C-risperidone administered as solution to 3 healthy male volunteers, total recovery of radioactivity at 1 week was 84%, including 70% in the urine and 14% in the feces.
Following a single subcutaneous injection of PERSERIS, the apparent terminal half-life of risperidone ranges between 9 and 11 days on average. This half-life is related to the slow release of risperidone from the subcutaneous depot and subsequent absorption of risperidone into the systemic circulation. The mean apparent terminal half-life ranges between 8 to 9 days for both 9-hydroxyrisperidone and total active moiety.
Drug Interaction Studies
No specific drug interaction studies have been performed with PERSERIS. The drug interaction data provided in this section is based on studies with oral risperidone. Effects of other drugs on the exposures of risperidone, 9-hydroxyrisperidone and total active moiety as well as the effects of risperidone on the exposures of other drugs is summarized below.
Effects of Other Drugs on Risperidone, 9-hydroxyrisperidone and Total Active Moiety Pharmacokinetics
Strong CYP2D6 Inhibitors (Fluoxetine and Paroxetine)
Fluoxetine (20 mg once daily) and paroxetine (20 mg once daily), potent CYP2D6 inhibitors, have been shown to increase the plasma concentration of risperidone by 2.5 to 2.8 folds and 3 to 9 folds, respectively. Fluoxetine did not affect the plasma concentration of 9-hydroxyrisperidone. Paroxetine lowered the concentration of 9-hydroxyrisperidone by about 10%. The effects of discontinuation of concomitant fluoxetine or paroxetine therapy on the pharmacokinetics of risperidone and 9-hydroxyrisperidone have not been studied.
Moderate CYP3A4 Inhibitor (Erythromycin)
There were no significant interactions between oral risperidone and erythromycin, a moderate CYP3A4 inhibitor.
Strong CYP3A4 Inducer (Carbamazepine)
Carbamazepine co-administration with oral risperidone decreased the steady-state plasma concentrations of risperidone and 9-hydroxyrisperidone by about 50%. Plasma concentrations of carbamazepine did not appear to be affected. Co-administration of other known CYP3A4 enzyme inducers (e.g., phenytoin, rifampin, and phenobarbital) with risperidone may cause similar decreases in the combined plasma concentrations of risperidone and 9-hydroxyrisperidone, which could lead to decreased efficacy of PERSERIS.
Amitriptyline, Cimetidine, Ranitidine, Clozapine, Topiramate
Clinically meaningful pharmacokinetic interaction between PERSERIS and other drugs, such as amitriptyline, cimetidine, ranitidine and clozapine, is not expected.
- Amitriptyline did not affect the pharmacokinetics of risperidone or of risperidone and 9-hydroxyrisperidone combined following concomitant administration with oral risperidone.
- Cimetidine and ranitidine increased the bioavailability of oral risperidone by 64% and 26%, respectively. However, cimetidine did not affect the AUC of risperidone and 9-hydroxyrisperidone combined, whereas ranitidine increased the AUC of risperidone and 9-hydroxyrisperidone combined by 20%.
- Chronic administration of clozapine with oral risperidone have shown to affect the clearance of risperidone, however, clinical relevance is unknown.
- There was no clinically relevant effect of oral risperidone (1 to 6 mg/day) on the pharmacokinetics of topiramate 400 mg/day.
Effects of Oral Risperidone on Pharmacokinetics of Other Drugs
Lithium
Repeated doses of oral risperidone (3 mg twice daily) did not affect the exposure (AUC) or peak plasma concentrations (C max) of lithium (n = 13).
Valproate
Repeated doses of oral risperidone (4 mg once daily) did not affect the pre-dose or average plasma concentrations and exposure (AUC) of valproate (1000 mg/day in three divided doses) compared to placebo (n = 21). However, there was a 20% increase in valproate peak plasma concentration (C max) after concomitant administration of oral risperidone.
Topiramate
Oral risperidone administered at doses from 1 to 6 mg/day concomitantly with topiramate 400 mg/day resulted in a 23% decrease in risperidone C maxand a 33% decrease in risperidone AUC 0-12hour at steady state. Minimal reductions in the exposure to risperidone and 9-hydroxyrisperidone combined, and no change for 9-hydroxyrisperidone were observed. This interaction is unlikely to be of clinical significance. There was no clinically relevant effect of oral risperidone on the pharmacokinetics of topiramate.
Digoxin
Oral risperidone (0.25 mg twice daily) did not show a clinically relevant effect on the pharmacokinetics of digoxin.
CYP2D6 Substrates (Donepezil and Galantamine)
In vitrostudies indicate that risperidone is a relatively weak inhibitor of CYP2D6. Therefore, PERSERIS is not expected to substantially inhibit the clearance of drugs that are metabolized by this enzymatic pathway. In drug interaction studies, oral risperidone did not significantly affect the pharmacokinetics of donepezil and galantamine, which are metabolized by CYP2D6.
Specific Populations
Based on population pharmacokinetic analyses, age, sex and race do not have a clinically meaningful effect on the pharmacokinetics of PERSERIS.
Renal Impairment
PERSERIS was not studied in patients with renal impairment, however, such effect has been investigated with oral risperidone. In patients with moderate to severe renal disease treated with oral risperidone, the apparent clearance (CL/F) of total active moiety was decreased by 60% in patients with moderate to severe renal disease compared with young healthy subjects [see Use in Specific Populations ( 8.6)].
Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of PERSERIS has not been studied.
The effect of hepatic impairment on the pharmacokinetics of oral risperidone has been evaluated in a dedicated phase I study. While the pharmacokinetics of risperidone in subjects with liver disease were comparable to those in young healthy subjects, the mean free fraction of risperidone in plasma was increased by about 35% because of the diminished concentration of both albumin and α1-acid glycoprotein [see Use in Specific Populations ( 8.7)] .
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No carcinogenicity studies were conducted with subcutaneous risperidone suspension. Carcinogenicity studies were conducted with oral risperidone in mice and rats. Risperidone was administered in the diet at doses of 0.63, 2.5, and 10 mg/kg for 18 months to mice and for 25 months to rats. These doses are equivalent to approximately 0.2, 0.75, and 3 times (mice) and 0.4, 1.5, and 6 times (rats) the oral MHRD of 16 mg/day, based on a mg/m 2body surface area. A maximum tolerated dose was not achieved in male mice. There were statistically significant increases in pituitary gland adenomas, endocrine pancreas adenomas, and mammary gland adenocarcinomas. The table below summarizes the multiples of the human oral dose on a mg/m 2 (mg/kg) basis at which these tumors occurred.
| Multiples of Maximum Human Oral Dose in mg/m2(mg/kg) | ||||
| Tumor Type | Species | Sex | Lowest Effect Level | Highest No-Effect Level |
| Pituitary adenomas | mouse | Female | 0.75 (9.4) | 0.2 (2.4) |
| Endocrine pancreas adenomas | rat | Male | 1.5 (9.4) | 0.4 (2.4) |
| Mammary gland adenocarcinomas | mouse | Female | 0.2 (2.4) | None |
| rat | Female | 0.4 (2.4) | None | |
| rat | Male | 6 (37.5) | 1.5 (9.4) | |
| Mammary gland neoplasm, Total | rat | Male | 1.5 (9.4) | 0.4 (2.4) |
Antipsychotic drugs have been shown to chronically elevate prolactin levels in rodents. Serum prolactin levels were not measured during the risperidone carcinogenicity studies; however, measurements during subchronic toxicity studies showed that risperidone elevated serum prolactin levels 5- to 6-fold in mice and rats at the same doses used in the carcinogenicity studies. An increase in mammary, pituitary, and endocrine pancreas neoplasms has been found in rodents after chronic administration of other antipsychotic drugs and is considered to be prolactin-mediated. The relevance for human risk of the findings of prolactin-mediated endocrine tumors in rodents is unclear [see Warnings and Precautions ( 5.6)] .
Mutagenesis
No evidence of mutagenic or clastogenic potential for risperidone was found in the in vitrotests of Ames gene mutation, the mouse lymphoma assay, rat hepatocyte DNA-repair assay, the chromosomal aberration test in human lymphocytes, Chinese hamster ovary cells, or in the in vivooral micronucleus test in mice and the sex-linked recessive lethal test in Drosophila.
No evidence of mutagenic potential was observed with risperidone subcutaneous injectable suspension or its delivery system alone at doses of 150 mg/kg risperidone or 943 mg/kg delivery system in an in vivomicronucleus test in rats. The safety margins of risperidone were 12 to 19 times the maximum monthly plasma risperidone concentration observed for humans at the monthly MRHD of 120 mg risperidone based on plasma exposure, and 13 times the delivery system amount present in monthly 120 mg risperidone.
Impairment of Fertility
No mating and fertility studies were conducted with subcutaneous risperidone suspension. Oral risperidone (0.16 to 5 mg/kg) impaired mating, but not fertility, in rat reproductive studies at doses 0.1 to 3 times the oral maximum recommended human dose (MRHD), of 16 mg/day based on mg/m 2body surface area. The effect appeared to be in females, since impaired mating behavior was not noted in the male fertility study. In a subchronic study in Beagle dogs in which risperidone was administered orally at doses of 0.31 to 5 mg/kg, sperm motility and concentration were decreased at doses 0.6 to 10 times the oral MRHD based on mg/m 2body surface area. Dose-related decreases were also noted in serum testosterone at the same doses. Serum testosterone and sperm parameters partially recovered, but remained decreased after treatment was discontinued. A no-effect dose could not be determined in either rat or dog.
Subcutaneous administration of the delivery system to rats had no effect on fertility parameters in either sex up to a dose that is 17 (delivery system), and 23 ( N-methyl-2-pyrrolidone) times the amount present in monthly 120 mg risperidone subcutaneous injectable suspension based on mg/m 2body surface area, respectively.
14. Clinical Studies
Efficacy for PERSERIS was demonstrated in an 8-week, randomized, double-blind, placebo-controlled study (Study 1, NCT #02109562). The study evaluated the efficacy, safety and tolerability of PERSERIS (90 and 120 mg subcutaneous every 4-weeks) compared with placebo in adults (age 18- to 55-years, inclusive) experiencing acute exacerbations of schizophrenia. Patients were required to have a Positive and Negative Syndrome Scale (PANSS) total score of 80- to 120-inclusive (moderate to severely ill) at the screening visit, occurring 3- to 8-days before the start of double-blind treatment, without an improvement in the PANSS total score of ≥ 20% between screening and the first dosing day.
At the screening visit, all patients received two doses of 0.25 mg oral risperidone 24-hours apart to establish tolerability. Patients were then placed in an inpatient setting, if not already hospitalized, and tapered off their current oral antipsychotic medication (if they were taking one) over a period of 3- to 8-days. Patients were randomized to receive 2 doses of subcutaneous PERSERIS (90 mg or 120 mg) or placebo 28-days apart (on Day 1 and Day 29). No supplemental oral risperidone was permitted during the study.
The primary endpoint was the change in PANSS total score from baseline to end of study (Day 57). Both PERSERIS 90 and 120 mg doses demonstrated a statistically significant improvement compared with placebo based on the primary endpoint ( Table 8). The results at each scheduled visit are displayed in Figure 14.
Characteristics of the patient population were balanced across the treatment groups. The mean baseline PANSS total score ranged from 94 to 96 across the groups. Most patients were male (74 to 83% per group), and the mean ages were 40 to 43 in each group. Most patients in this study were black or African American (71 to 75% per group). Of the 354 patients randomized to treatment, 337 were included in the intent-to-treat (ITT) population, and 259 (73%) completed the study.
Subgroup analyses by gender, age, and race did not suggest any clear evidence of differential responsiveness to PERSERIS.
|
ITT: intent-to-treat; SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: unadjusted confidence interval |
||||
|
ªDifference (drug minus placebo) in least-squares mean change from baseline |
||||
|
*Doses that are statistically significantly superior to placebo |
||||
| Primary Efficacy Measure: PANSS | ||||
| Treatment Group
| N
(# ITT patients) | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted
Differencea(95% CI) |
| PERSERIS 90 mg* | 111 | 95.5 (9.23) | -19.86 (1.56) | -6.50 (-10.87, -2.13)*
|
| PERSERIS 120 mg* | 114 | 94.9 (8.09) | -23.61 (1.58) | -10.24 (-14.64, -5.85)*
|
| Placebo | 112 | 94.1 (8.89) | -13.37 (1.58) | -- |
Figure 14. Least Square Mean Change from Baseline (+/- Standard Error) in PANSS Total Scores by Days
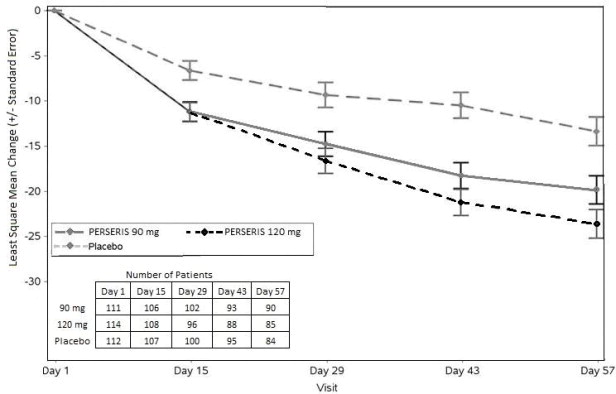
The secondary efficacy endpoint was defined as the CGI-S score at Day 57. Both PERSERIS treatment groups demonstrated statistically significantly better CGI-S scores versus placebo.
16. How is Perseris supplied
PERSERIS (risperidone) for extended-release injectable suspension, for subcutaneous use is, when fully mixed a viscous suspension that, varies from white to yellow-green and is available in dosage strengths of 90 mg and 120 mg.
PERSERIS 90 mg is supplied in a single-dose kit, packaged in a carton (NDC 12496-0090-1), containing the following:
- One pouch with a sterile syringe (labelled ‘P’) prefilled with risperidone powder
- One pouch with a sterile syringe (labelled ‘L’) prefilled with the delivery system, and desiccant.
- One 18-gauge, 5/8-inch sterile safety needle.
PERSERIS 120 mg is supplied in a single-dose kit, packaged in a carton (NDC 12496-0120-1), containing the following:
- One pouch with a sterile syringe (labelled ‘P') prefilled with risperidone powder.
- One pouch with a sterile syringe (labelled ‘L') prefilled with the delivery system, and desiccant.
- One 18-gauge, 5/8-inch sterile safety needle.
Storage and Handling
Store in refrigerator at 2°C to 8°C (36°F to 46°F). Allow PERSERIS kit to come to room temperature, 20°C to 25°C (68°F to 77°F), for at least 15 minutes prior to mixing.
PERSERIS may be stored in its unopened original packaging at room temperature, 20°C to 25°C (68°F to 77°F), for up to 30 days prior to administration. After removal from the refrigerator, use PERSERIS within 30 days or discard.
17. Patient Counseling Information
General Instructions
Advise patients not to rub or massage the injection site and to be aware of the placement of any belts or clothing waistbands, sleeves, cuffs, or other parts of clothing [see Dosage and Administration ( 2.4)] .
Neuroleptic Malignant Syndrome
Counsel patients about a potentially fatal adverse reaction, Neuroleptic Malignant Syndrome (NMS), that has been reported in association with administration of antipsychotic drugs. Advise patients, family members, or caregivers to contact the healthcare provider or report to the emergency room if they experience signs and symptoms of NMS [see Warnings and Precautions ( 5.3)] .
Tardive Dyskinesia
Counsel patients on the signs and symptoms of tardive dyskinesia and to contact their healthcare provider if these abnormal movements occur [see Warnings and Precautions ( 5.4)] .
Metabolic Changes
Educate patients about the risk of metabolic changes, how to recognize symptoms of hyperglycemia and diabetes mellitus, and the need for specific monitoring, including blood glucose, lipids, and weight [see Warnings and Precautions ( 5.5)] .
Hyperprolactinemia
Counsel patients on signs and symptoms of hyperprolactinemia that may be associated with chronic use of PERSERIS. Advise them to seek medical attention if they experience any of the following: amenorrhea or galactorrhea in females, erectile dysfunction or gynecomastia in males [see Warnings and Precautions ( 5.6)] .
Orthostatic Hypotension and Syncope
Educate patients of the risk of orthostatic hypotension and syncope, particularly at the time of initiating treatment, re-initiating treatment, or increasing the dose [see Warnings and Precautions ( 5.7)].
Leukopenia/Neutropenia
Advise patients with a pre-existing low WBC or a history of drug induced leukopenia/neutropenia they should have their CBC monitored while taking PERSERIS [see Warnings and Precautions ( 5.9)] .
Potential for Cognitive and Motor Impairment
Inform patients that PERSERIS has the potential to impair judgment, thinking, and motor skills. Caution patients about performing activities requiring mental alertness, such as operating hazardous machinery or operating a motor vehicle, until they are reasonably certain that PERSERIS does not affect them adversely [see Warnings and Precautions ( 5.10)] .
Priapism
Advise patients of the possibility of painful or prolonged penile erections (priapism). Instruct the patient to seek immediate medical attention in the event of priapism [Warnings and Precautions ( 5.13)] .
Heat Exposure and Dehydration
Educate patients regarding appropriate care in avoiding overheating and dehydration [see Warnings and Precautions ( 5.14)].
Concomitant Medication
Advise patients to inform their physicians if they are taking, or plan to take, any prescription or over-the-counter drugs, since there is a potential for interaction [see Drug Interactions ( 7)] .
Alcohol
Advise patients to avoid alcohol during treatment with PERSERIS [see Drug Interactions ( 7.1)] .
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with PERSERIS. Advise patients that PERSERIS may cause extrapyramidal and/or withdrawal symptoms (agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress, and feeding disorder) in a neonate. Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to PERSERIS during pregnancy [see Use in Specific Populations ( 8.1)] .
Lactation
Advise breastfeeding women using PERSERIS to monitor infants for somnolence, failure to thrive, jitteriness, and extrapyramidal symptoms (tremors and abnormal muscle movements) and to seek medical care if they notice these signs [see Use in Specific Populations ( 8.2)] .
Infertility
Advise females of reproductive potential that PERSERIS may impair fertility due to an increase in serum prolactin levels. The effects on fertility are reversible [see Use in Specific Populations ( 8.3)].
Manufactured for:
Indivior Inc., North Chesterfield, VA 23235.
© 2025, Indivior UK Limited. All Rights Reserved.
PERSERIS is a registered trademark of Indivior UK Limited.
Powder syringe manufactured by Patheon Manufacturing Services, Greenville, NC 27834.
Liquid syringe manufactured by Curia, Burlington, MA 01803.
 ) pre-filled with the delivery system – Inspect liquid solution for foreign particles. This is the syringe you will use to inject the patient.
) pre-filled with the delivery system – Inspect liquid solution for foreign particles. This is the syringe you will use to inject the patient.
 ) prefilled with Risperidone powder – Inspect syringe for consistency of powder color and for foreign particles.
) prefilled with Risperidone powder – Inspect syringe for consistency of powder color and for foreign particles.
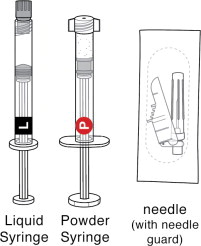
 Powder can become packed during shipping.
Powder can become packed during shipping.
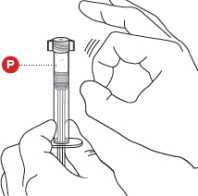
 Holding both syringes in your non-dominant hand can help with this step.
Holding both syringes in your non-dominant hand can help with this step.
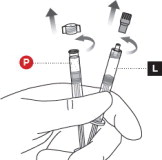
 Keep your fingers off the plungers during this step to avoid spillage of the medication.
Keep your fingers off the plungers during this step to avoid spillage of the medication.
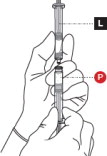
 Failure to fully mix the medication could result in incorrect dosage.
Failure to fully mix the medication could result in incorrect dosage.
 Graphic to the right illustrates a correct full cycle.
Graphic to the right illustrates a correct full cycle.
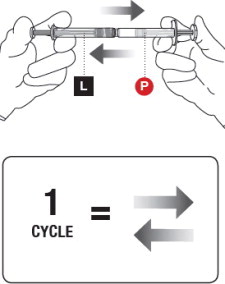
 Failure to aspirate the liquid from the
Failure to aspirate the liquid from the
 Check that medication is uniform in color and free from foreign particles.
Check that medication is uniform in color and free from foreign particles.

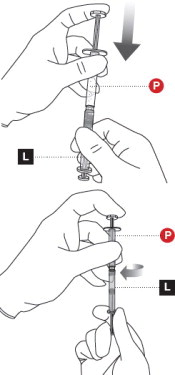
 To help minimize irritation, rotate injection sites following a pattern similar to the illustration. If you want to use the same injection site, make sure it is not the same spot on the injection site you used the last time.
To help minimize irritation, rotate injection sites following a pattern similar to the illustration. If you want to use the same injection site, make sure it is not the same spot on the injection site you used the last time.
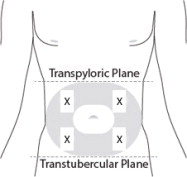
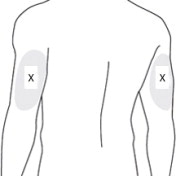
 Due to the viscous nature of the medication, bubbles will not rise as quickly as those in an aqueous solution.
Due to the viscous nature of the medication, bubbles will not rise as quickly as those in an aqueous solution.
 PERSERIS is for subcutaneous administration only. Do not inject by any other route.
PERSERIS is for subcutaneous administration only. Do not inject by any other route.
 Actual angle of injection will depend on the amount of subcutaneous tissue.
Actual angle of injection will depend on the amount of subcutaneous tissue.
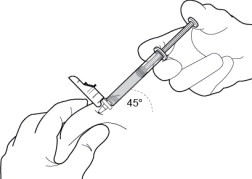
 Do not rub the injection area after the injection. If there is bleeding, apply a gauze pad or bandage but use minimal pressure.
Do not rub the injection area after the injection. If there is bleeding, apply a gauze pad or bandage but use minimal pressure.
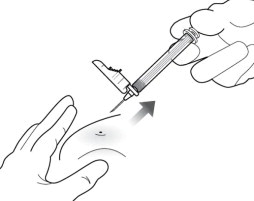
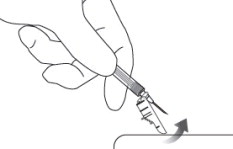
|
This Instructions for Use has been approved by the U.S. Food and Drug Administration. |
Revised 12/2022 |
| FREQUENTLY ASKED QUESTIONS
Q What is the mixture supposed to look like? AWhen fully mixed, the product should be a cloudy suspension that is uniform in color. It can vary from white to yellow-green in color. If you see any clear areas in the mixture, continue to mix until the distribution of the color is uniform. Q Why is it important to put the liquid syringe on top? AThe liquid is viscous and will not fall out when placing on top. However, powder may be lost if the powder syringe is inverted, which could affect the ultimate dosage. Q What do I do if there is residual left in the powder syringe? AA small ring of residual powder may be observed in the end of the powder syringe barrel after mixing. This is normal. If you have followed the instructions carefully and the rest of the mixture is a cloudy suspension that is uniform in color, proceed with the injection. Q What do I do if a foreign particle is found in either of the syringes? ADo not use if you suspect foreign particles in either of the syringes. Q How soon must I inject after mixing PERSERIS®? AThis product should be used immediately following preparation. Q Can I inject into the leg? AThis injection is only approved for injection into the subcutaneous tissue of abdomen and back of the upper arm. Q How do I get rid of large air gaps? ASmall bubbles, also known as champagne bubbles, are not a problem and common with this medication. Large air gaps, however, can be minimized by pulling back on the plunger rod to pop air bubbles prior to expelling the air very slowly. Air should be expelled very carefully to avoid loss of medication. Q Should I massage or put my finger on the injection site following the injection? AIt is not advisable to palpate or massage the area following the injection. Q Will the deposit be palpable? ADepending on the patient's subcutaneous tissue, the deposit may be more or less palpable. Patients should be advised that a bump may be palpable (decreasing in size) for several weeks. |
|
| Manufactured for: Indivior Inc., North Chesterfield, VA 23235.
© 2022, Indivior UK Limited. All Rights Reserved. PERSERIS is a registered trademark of Indivior UK Limited. Powder syringe manufactured by Patheon Manufacturing Services, Greenville, NC 27834. Liquid syringe manufactured by Curia, Burlington, MA 01803. |
|
Principal Display Panel - Perseris Kit 90 mg Carton Label
NDC 12496-0090-1
once-monthly
PERSERIS®
(risperiDONE)
for extended-release
injectable suspension
90 mg
Liquid
Syringe
+
Mix
Powder
Syringe
1 Injection
Rx only
Sterile
Single-dose only
For subcutaneous injection only. Please read complete instructions prior to use.

Principal Display Panel - Perseris Kit 120 mg Carton Label
NDC 12496-0120-1
once-monthly
PERSERIS®
(risperiDONE)
for extended-release
injectable suspension
120 mg
Liquid
Syringe
+
Mix
Powder
Syringe
1 Injection
Rx only
Sterile
Single-dose only
For subcutaneous injection only. Please read complete instructions prior to use.

| PERSERIS
risperidone kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| PERSERIS
risperidone kit |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Indivior Inc. (797408549) |
| Registrant - Indivior Inc. (797408549) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Curia Massachusetts, Inc. | 556991748 | analysis(12496-0090, 12496-0120) , manufacture(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Curia Global, Inc. | 080046430 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Assia Chemical Industries Ltd (Teva Tech site) | 649323474 | api manufacture(12496-0090, 12496-0120) , analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Eurofins Lancaster Laboratories, Inc | 069777290 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Indivior Inc. | 080660711 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Isomedix Operations Inc | 004303926 | sterilize(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Nelson Laboratories, LLC | 151663234 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Manufacturing Services LLC | 079415560 | analysis(12496-0090, 12496-0120) , manufacture(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SGS North America Inc | 049859261 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sharp Packaging Services, LLC | 143696495 | pack(12496-0090, 12496-0120) , label(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Spectral Data Services, Inc. | 144318722 | analysis(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SYNERGY HEALTH AST, LLC | 044622190 | sterilize(12496-0090, 12496-0120) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Teva API India Private Ltd. | 677604388 | analysis(12496-0090, 12496-0120) | |
Frequently asked questions
- What is the difference between Perseris and Risperdal Consta?
- What are the strongest sleeping pills?
- What drugs cause tardive dyskinesia?
More about Perseris (risperidone)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: atypical antipsychotics
- Breastfeeding
- En español
Patient resources
Professional resources
Other brands
Risperdal, Uzedy, Risperdal Consta, Risvan, Rykindo