Natrecor: Package Insert / Prescribing Info
Package insert / product label
Generic name: nesiritide
Dosage form: injection, powder, lyophilized, for solution
Drug class: Vasodilators
Medically reviewed by Drugs.com. Last updated on May 19, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
NATRECOR (nesiritide) for injection, for intravenous use
Initial U.S. Approval: 2001
Indications and Usage for Natrecor
NATRECOR is a natriuretic peptide indicated for the treatment of patients with acutely decompensated heart failure who have dyspnea at rest or with minimal activity. In this population, the use of NATRECOR reduced pulmonary capillary wedge pressure and improved symptomatic dyspnea when measured at 3 hours of infusion. (1)
Natrecor Dosage and Administration
Monitor blood pressure closely during NATRECOR administration. (2)
The recommended dose of NATRECOR is an IV bolus of 2 mcg/kg followed by a continuous infusion of 0.01 mcg/kg/min. Do not initiate NATRECOR at a dose that is above the recommended dose. The bolus dose may not be appropriate for those with low SBP <110 mm Hg or those recently treated with afterload reducers such as nitroglycerin. (2.1)
Dosage Forms and Strengths
Single-use vial: 1.5 mg vial (3)
Contraindications
Warnings and Precautions
- Hypotension: Not recommended for patients for whom vasodilating agents are not appropriate, or other conditions in which cardiac output is dependent upon venous return, or for patients suspected to have low cardiac filling pressures. (5.1)
- Worsening renal impairment. Monitor serum creatinine both during and after NATRECOR administration. (5.2)
- Hypersensitivity: Serious hypersensitivity/allergic reactions have been reported. (5.3)
Adverse Reactions/Side Effects
The most common adverse reactions (≥3%) are hypotension, headache, nausea, and back pain. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Scios LLC at 1-877-462-8732 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2019
Full Prescribing Information
1. Indications and Usage for Natrecor
NATRECOR (nesiritide) is indicated for the treatment of patients with acutely decompensated heart failure who have dyspnea at rest or with minimal activity. In this population, the use of NATRECOR reduced pulmonary capillary wedge pressure and improved short term (3 hours) symptoms of dyspnea.
2. Natrecor Dosage and Administration
NATRECOR (nesiritide) is for intravenous (IV) use only. There is limited experience with administering NATRECOR for longer than 96 hours. Monitor blood pressure closely during NATRECOR administration.
2.1 Recommended Dosage
The recommended dose of NATRECOR is an IV bolus of 2 mcg/kg followed by a continuous infusion of 0.01 mcg/kg/min. Do not initiate NATRECOR at a dose that is above the recommended dose.
The loading dose may not be appropriate for those with low systolic blood pressure (SBP) <110 mm Hg or for patients recently treated with afterload reducers.
The administration of the recommended dose of NATRECOR is a two step process:
Step 1. Administration of the IV Bolus
After preparation of the infusion bag [see Dosage and Administration (2.3)], withdraw the bolus volume (see Table 1) from the NATRECOR infusion bag, and administer it over approximately 60 seconds through an IV port in the tubing.
Bolus Volume (mL) = Patient Weight (kg)/3
| Patient Weight (kg) | Volume of Bolus (mL=kg/3) |
|---|---|
| 60 | 20.0 |
| 70 | 23.3 |
| 80 | 26.7 |
| 90 | 30.0 |
| 100 | 33.3 |
| 110 | 36.7 |
Step 2. Administration of the Continuous Infusion
Immediately following the administration of the bolus, infuse NATRECOR at a flow rate of 0.1 mL/kg/hr. This will deliver a NATRECOR infusion dose of 0.01 mcg/kg/min.
To calculate the infusion flow rate to deliver a 0.01 mcg/kg/min dose, use the following formula (see Table 2):
Infusion Flow Rate (mL/hr) = Patient Weight (kg) × 0.1
| Patient Weight (kg) | Infusion Flow Rate (mL/hr) |
|---|---|
| 60 | 6 |
| 70 | 7 |
| 80 | 8 |
| 90 | 9 |
| 100 | 10 |
| 110 | 11 |
2.2 Dose Adjustments
The dose-limiting side effect of NATRECOR is hypotension. If hypotension occurs during the administration of NATRECOR, reduce the dose of or discontinue NATRECOR and initiate other measures to support blood pressure (IV fluids, changes in body position). When symptomatic hypotension occurs, discontinue NATRECOR. Because hypotension caused by NATRECOR may be prolonged (up to hours), a period of observation may be necessary before restarting the drug. NATRECOR may be subsequently restarted at a dose that is reduced by 30% (with no bolus administration) once the patient has stabilized.
Do not up-titrate NATRECOR more frequently than every 3 hours. Use central hemodynamic monitoring and do not exceed 0.03 mcg/kg/min.
2.3 Preparation and Administration Instructions
The NATRECOR bolus must be drawn from the prepared infusion bag. Prime the IV tubing with 5 mL of the solution for infusion prior to connecting to the patient's vascular access port and prior to administering the bolus or starting the infusion.
- Reconstitute one 1.5 mg vial of NATRECOR by adding 5 mL of diluent removed from a pre-filled 250 mL plastic IV bag containing the diluent of choice. After reconstitution of the vial, each mL contains 0.32 mg of nesiritide. The following preservative-free diluents are recommended for reconstitution: 5% Dextrose Injection (D5W), USP; 0.9% Sodium Chloride Injection, USP; 5% Dextrose and 0.45% Sodium Chloride Injection, USP, or 5% Dextrose and 0.2% Sodium Chloride Injection, USP.
- Do not shake the vial. Rock the vial gently so that all surfaces, including the stopper, are in contact with the diluent to ensure complete reconstitution. Use only a clear, essentially colorless solution.
- Withdraw the entire contents of the reconstituted NATRECOR vial and add to the 250 mL plastic IV bag. This will yield a solution with a concentration of NATRECOR of approximately 6 mcg/mL. Invert the IV bag several times to ensure complete mixing of the solution.
- Use the reconstituted solution within 24 hours, as NATRECOR contains no antimicrobial preservative. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Reconstituted vials of NATRECOR may be stored at 2 to 25°C (36 to 77°F) for up to 24 hours.
2.4 Chemical/Physical Interactions
NATRECOR is physically and/or chemically incompatible with injectable formulations of heparin, insulin, ethacrynate sodium, bumetanide, enalaprilat, hydralazine, and furosemide. Do not co-administer these drugs with NATRECOR through the same IV catheter. The preservative sodium metabisulfite is incompatible with NATRECOR. Do not administer injectable drugs that contain sodium metabisulfite in the same infusion line as NATRECOR. Flush the catheter between administration of NATRECOR and incompatible drugs.
NATRECOR binds to heparin and therefore could bind to the heparin lining of a heparin-coated catheter, decreasing the amount of NATRECOR delivered to the patient for some period of time. Therefore, do not administer NATRECOR through a central heparin-coated catheter. Concomitant administration of a heparin infusion through a separate catheter is acceptable.
3. Dosage Forms and Strengths
NATRECOR (nesiritide) is provided in a sterile, single-use vial. Each 1.5 mg vial contains a white- to off-white lyophilized powder for intravenous (IV) administration after reconstitution.
4. Contraindications
NATRECOR is contraindicated in patients with:
- Persistent systolic blood pressure <100 mm Hg prior to therapy because of an increased risk of symptomatic hypotension [see Warnings and Precautions (5.1)]
- Known hypersensitivity to any of its components [see Warnings and Precautions (5.3)]
- Cardiogenic shock
5. Warnings and Precautions
5.1 Hypotension
NATRECOR may cause hypotension. In the ASCEND-HF trial, the incidence of symptomatic hypotension was 7.1% in NATRECOR-treated patients compared to 4.0% in placebo-treated patients on a background of standard care. The risk of hypotension may be increased by the concomitant use of NATRECOR with drugs affecting the renin-angiotensin system (i.e., angiotensin receptor blockers and/or angiotensin-converting enzyme inhibitors) or other afterload reducers. In the VMAC trial, in patients given the recommended dose (2 mcg/kg bolus followed by a 0.01 mcg/kg/min infusion) or the adjustable dose, the incidence of symptomatic hypotension in the first 24 hours was similar for NATRECOR (4%) and IV nitroglycerin (5%). When hypotension occurred, however, the duration of symptomatic hypotension was longer with NATRECOR® (mean duration was 2.2 hours) than with nitroglycerin (mean duration was 0.7 hours).
Administer NATRECOR only in settings where blood pressure can be monitored closely and hypotension aggressively treated. Reduce the dose of or discontinue NATRECOR in patients who develop hypotension [see Dosage and Administration (2.2)].
Avoid administration of NATRECOR in patients suspected of having, or known to have, low cardiac filling pressures.
NATRECOR is not recommended for patients for whom vasodilating agents are not appropriate, such as patients with significant valvular stenosis, restrictive or obstructive cardiomyopathy, constrictive pericarditis, pericardial tamponade, or other conditions in which cardiac output is dependent upon venous return, or for patients suspected to have low cardiac filling pressures [see Contraindications (4)].
5.2 Worsening of Renal Function
NATRECOR may decrease renal function as judged by increases in serum creatinine. Monitor serum creatinine both during and after therapy has been completed. Monitor serum creatinine until values have stabilized. In patients with severe heart failure whose renal function may depend on the activity of the renin-angiotensin aldosterone system, treatment with NATRECOR may be associated with azotemia. When NATRECOR was initiated at doses higher than 0.01 mcg/kg/min (0.015 and 0.03 mcg/kg/min), there was an increased rate of elevated serum creatinine over baseline compared with standard therapies, although the rate of acute renal failure and need for dialysis was not increased.
5.3 Hypersensitivity
Serious hypersensitivity/allergic reactions following administration of NATRECOR have been reported.
These reactions are more likely to occur in individuals with a history of sensitivity to recombinant peptides. Before therapy with NATRECOR is instituted, careful inquiry should be made to determine whether the patient has had a previous hypersensitivity reaction to other recombinant peptides. If an allergic reaction to NATRECOR occurs, discontinue the drug. Some serious hypersensitivity/allergic reactions may require treatment with epinephrine, oxygen, IV fluids, antihistamines, corticosteroids, pressor amines and airway management, as clinically indicated.
6. Adverse Reactions/Side Effects
The following are discussed in more detail in other sections of the labeling:
- Hypotension [see Warnings and Precautions (5.1)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice. A causal relationship for NATRECOR cannot be reliably established in individual cases.
Adverse drug reactions that occurred at least ≥2% more frequently on NATRECOR® than on placebo during the first 24 hours of infusion (excluding the ASCEND-HF study) are shown in Table 3.
| System Organ Class Adverse Reaction | NATRECOR (N=331) 0.01 mcg/kg/min % (n)‡ | Placebo (N=188) % (n)§ |
|---|---|---|
|
||
| Vascular Disorders | ||
| Hypotension | 12 (41) | 4 (7) |
| GI Disorders | ||
| Nausea | 3 (11) | 1 (2) |
| Musculoskeletal Disorders | ||
| Back pain | 3 (11) | 1 (2) |
| Nervous System Disorders | ||
| Headache | 7 (24) | 6 (11) |
| Dizziness | 2 (8) | 2 (3) |
Laboratory adverse drug reactions that occurred in ≥2% of patients and collected during the first 14 days after the start of NATRECOR infusion included: hypoglycemia.
Worsening Renal Function
In the ASCEND-HF trial, through Day 30, the incidence of renal impairment as measured by a >25% decrease in glomerular filtration rate (calculated based on serum creatinine) was observed in 31.4% and 29.5% in the NATRECOR and placebo groups, respectively. Other metrics of decompensated renal function such as an increase in creatinine of > 0.5 mg/dl, a 50% increase in creatinine or a value of ≥ 2 or 100% increase in creatinine were more frequent in the NATRECOR group. At 30 days post enrollment, more subjects in the NATRECOR group had elevated levels of creatinine of 50% greater than baseline compared to placebo 4.6% versus 3.3%. In the ASCEND-HF study there were relatively few subjects requiring either hemofiltration or dialysis.
In the PRECEDENT trial, the incidence of elevations in serum creatinine to >0.5 mg/dL above baseline through Day 14 was higher in the NATRECOR 0.015 mcg/kg/min group (17%) and the NATRECOR 0.03 mcg/kg/min group (19%) than with standard therapy (11%). In the VMAC trial, through Day 30, the incidence of elevations in creatinine to >0.5 mg/dL above baseline was 28% and 21% in the NATRECOR (2 mcg/kg bolus followed by 0.01 mcg/kg/min) and nitroglycerin groups, respectively.
Neutral Effect on Mortality
A meta-analysis performed of seven clinical trials demonstrated NATRECOR did not increase mortality in patients with acute decompensated heart failure (ADHF) at Day 30 or Day 180 (see Figures 1 and 2). Data from seven studies in which 30-day data were collected are presented in Figure 1. The data depict hazard ratios (HR) and confidence intervals (CI) of mortality data for randomized and treated patients with NATRECOR relative to active or placebo controls through Day 30 for each of the seven individual studies along with the overall combined estimate (Studies 311, 325, 326, 329 [PRECEDENT], 339 [VMAC], 341 [PROACTION], and A093 [ASCEND-HF]).
Figure 1 (on logarithmic scale) also contains an estimate for the seven studies combined (n=8514). The results indicate that there is no increased mortality risk for NATRECOR at Day 30 (seven studies pooled: HR=0.99; 95% CI: 0.80, 1.22). The percentages are the Kaplan-Meier estimates.
Figure 1: 30-Day All-Cause Mortality Hazard Ratios
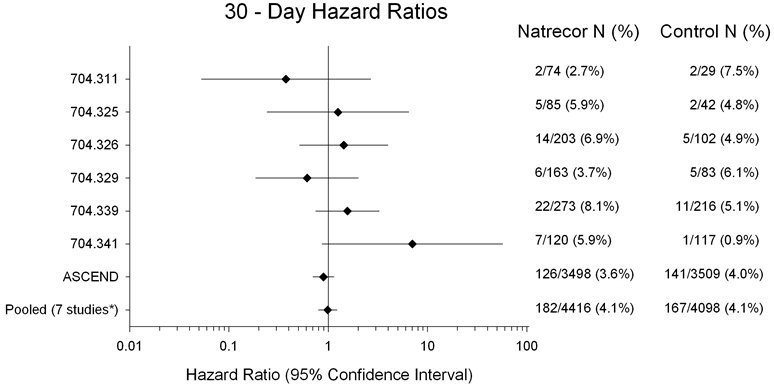
*Studies 704.311, 704.325, 704.326, 704.329, 704.339, 704.341 and ASCEND-HF
Figure 2 presents 180-day mortality hazard ratios from all six individual studies where 180-day data were collected (Studies 325, 326, 329, 339, 341 and A093 [ASCEND-HF]). The results indicate that, there is no increased mortality risk for NATRECOR at Day 180 (six studies pooled: HR=0.98; 95% CI: 0.88, 1.10).
Figure 2: 180-Day All-Cause Mortality Hazard Ratios
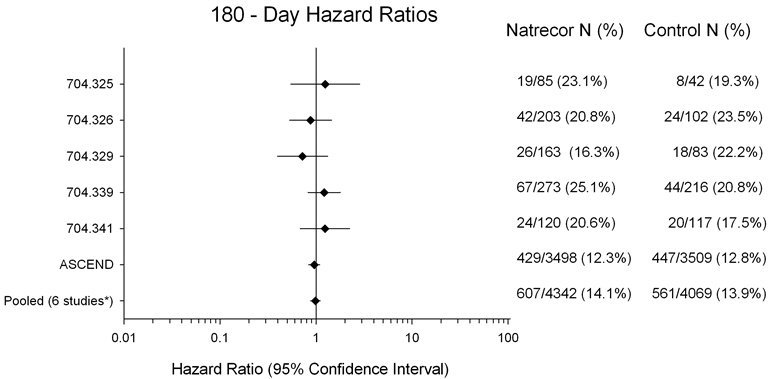
*Studies 704.325, 704.326, 704.329, 704.339, 704.341 and ASCEND-HF
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of NATRECOR. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to estimate their frequency reliably or to establish a causal relationship to drug exposure.
- Hypersensitivity reactions
- Infusion site extravasation
- Pruritus
- Rash
Related/similar drugs
7. Drug Interactions
No trials specifically examining potential drug interactions with NATRECOR were conducted, although many concomitant drugs (including IV nitroglycerin) were used in clinical trials [see Clinical Studies (14)]. No drug interactions were detected except for an increase in symptomatic hypotension in patients receiving afterload reducers or affecting the renin-angiotensin system (i.e., ARBs or ACE inhibitors).
The co-administration of NATRECOR with nitroprusside, milrinone, or IV ACE inhibitors has not been evaluated.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no data to assess a drug associated risk of major birth defects, miscarriage, or adverse maternal fetal outcomes with NATRECOR use in pregnant women. In animal reproduction studies, intravenous administration of NATRECOR to pregnant rabbits, during the period of organogenesis at 100 times the maximum recommended human dose (MRHD) did not result in any toxicities to the rabbits or their developing fetuses (see Data).
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15– 20%, respectively.
Data
Animal Data
A developmental reproductive toxicology study was conducted in pregnant rabbits. NATRECOR was infused at a constant rate of 1.0 µg/kg/min (equivalent to 1440 µg/kg/day) for 13 days from days 7 to 19 of gestation. During this time period, the pharmacokinetics properties were determined following initiation of infusion on these days (7 and 19). At the highest level of exposure (based on AUC, approximately at 100 times the maximum recommended human dose [MRHD]) no toxicities to the rabbits or developing fetuses up to the highest dose evaluated in the study were observed. Neither the NOAEL nor the lowest observed toxic effect levels were achieved in this study and no adverse effects on live births or fetal development were observed.
8.4 Pediatric Use
The safety and effectiveness of NATRECOR in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of patients in clinical trials treated with NATRECOR (n=4505), 52% were 65 years or older and 27% were 75 years or older. No overall differences in effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients. Some older individuals may be more sensitive to the effect of NATRECOR than younger individuals.
10. Overdosage
Overdose with NATRECOR therapy has been reported and is primarily the result of either a miscalculated NATRECOR dose or a mechanical error such as an infusion-pump malfunction or an infusion-pump programming error. The most frequently reported adverse event reported with NATRECOR overdose is hypotension, which may be symptomatic and may persist for several hours. Asymptomatic hypotensive events may resolve with drug stoppage. In some cases hypotension may persist for several hours beyond discontinuation. In the event of an overdose, discontinue NATRECOR and support blood pressure [see Warnings and Precautions (5.1)].
11. Natrecor Description
NATRECOR (nesiritide) is a sterile, purified preparation of human B-type natriuretic peptide (hBNP), and is manufactured from E. coli using recombinant DNA technology. Nesiritide has a molecular weight of 3464 g/mol and an empirical formula of C143H244N50O42S4. Nesiritide has the same 32 amino acid sequence as the endogenous peptide, which is produced by the ventricular myocardium.
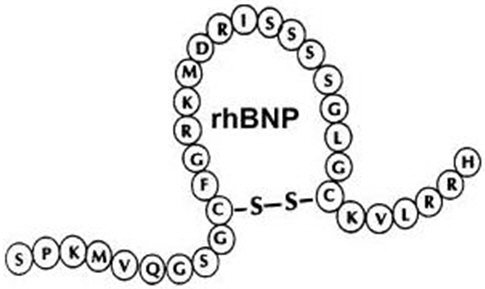
NATRECOR is formulated as the citrate salt of rhBNP, and is provided in a sterile, single-use vial. Each 1.5 mg vial contains a white- to off-white lyophilized powder for intravenous (IV) administration after reconstitution. The quantitative composition of the lyophilized drug per vial is: nesiritide 1.58 mg, citric acid monohydrate 2.1 mg, mannitol 20.0 mg, and sodium citrate dihydrate 2.94 mg.
12. Natrecor - Clinical Pharmacology
12.1 Mechanism of Action
Human BNP (hBNP) is secreted by the ventricular myocardium in response to stretch and exists in several isoforms in the human body. Elevated levels of BNP have been associated with advanced heart failure and are considered to be a compensatory mechanism in this disease. Human BNP binds to the particulate guanylate cyclase receptor of vascular smooth muscle and endothelial cells, leading to increased intracellular concentrations of guanosine 3'5'-cyclic monophosphate (cGMP) and smooth muscle cell relaxation. Cyclic GMP serves as a second messenger to dilate veins and arteries. Nesiritide has been shown to relax isolated human arterial and venous tissue preparations that were precontracted with either endothelin-1 or the alpha-adrenergic agonist, phenylephrine.
In animals, nesiritide had no effects on cardiac contractility or on measures of cardiac electrophysiology such as atrial and ventricular effective refractory times or atrioventricular node conduction.
12.2 Pharmacodynamics
With a dosing regimen of NATRECOR of 2 mcg/kg IV bolus followed by an intravenous infusion dose of 0.01 mcg/kg/min, Table 4 and Figure 3 summarize the changes in the VMAC trial in PCWP and other measures during the first 3 hours.
| Effects at 3 Hours | Placebo (n=62) | Nitroglycerin (n=60) | NATRECOR (n=124) |
|---|---|---|---|
| Pulmonary capillary wedge pressure (mm Hg) | -2.0 | -3.8 | -5.8* |
| Right atrial pressure (mm Hg) | 0.0 | -2.6 | -3.1* |
| Cardiac index (L/min/M2) | 0.0 | 0.2 | 0.1 |
| Mean pulmonary artery pressure (mm Hg) | -1.1 | -2.5 | -5.4* |
| Systemic vascular resistance (dynes∙sec∙cm-5) | -44 | -105 | -144 |
| Systolic blood pressure† (mm Hg) | -2.5 | -5.7* | -5.6* |
Figure 3: PCWP through 3 Hours in VMAC
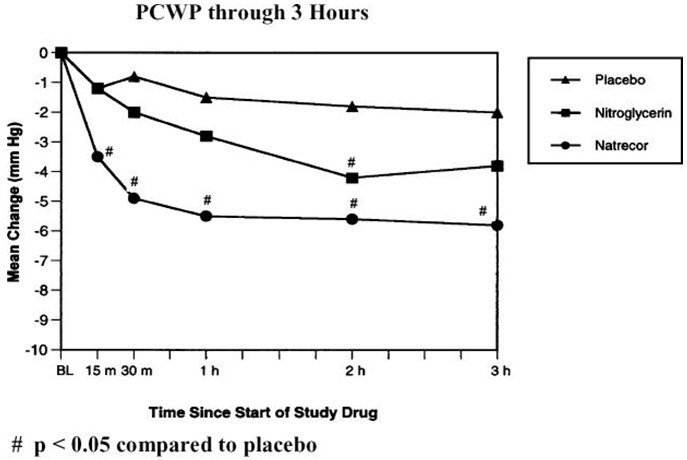
With this dosing regimen, 60% of the 3-hour effect on PCWP reduction is achieved within 15 minutes after the bolus, reaching 95% of the 3-hour effect within 1 hour. Approximately 70% of the 3-hour effect on SBP reduction is reached within 15 minutes. The pharmacodynamic (PD) half-life of the onset and offset of the hemodynamic effect of NATRECOR is longer than what the PK half-life of 18 minutes would predict. Longer infusions may exaggerate the discrepancy from onset and offset effects. For example, in patients who developed symptomatic hypotension in the VMAC (Vasodilation in the Management of Acute Congestive Heart Failure) trial, half of the recovery of SBP toward the baseline value after discontinuation or reduction of the dose of NATRECOR was observed in about 60 minutes. When higher doses of NATRECOR were infused, the duration of hypotension was sometimes several hours.
No rebound increase to levels above baseline state was observed. There was also no evidence of tachyphylaxis to the hemodynamic effects of NATRECOR in the clinical trials.
In the VMAC trial, in which the use of diuretics was not restricted, the mean change in volume status (output minus input) during the first 24 hours in the nitroglycerin and NATRECOR groups was similar: 1279 ± 1455 mL and 1257 ± 1657 mL, respectively.
12.3 Pharmacokinetics
Distribution
In patients with heart failure (HF), NATRECOR administered intravenously by infusion or bolus exhibits biphasic disposition from the plasma. The mean terminal elimination half-life (t1/2) of nesiritide is approximately 18 minutes and was associated with approximately 2/3 of the area-under-the-curve (AUC). The mean initial elimination phase was estimated to be approximately 2 minutes. In these patients, the mean volume of distribution of the central compartment (Vc) of nesiritide was estimated to be 0.073 L/kg, the mean steady-state volume of distribution (Vss) was 0.19 L/kg, and the mean clearance (CL) was approximately 9.2 mL/min/kg. At steady state, plasma BNP levels increase from baseline endogenous levels by approximately 3-fold to 6-fold with NATRECOR infusion doses ranging from 0.01 to 0.03 mcg/kg/min.
Metabolism and Excretion
The mechanism of elimination of nesiritide has not been studied specifically in humans.
Special Populations
Renal Impairment
Clinical data suggest that dose adjustment is not required in patients with renal impairment. The effects of nesiritide on PCWP, cardiac index (CI), and systolic blood pressure (SBP) were not significantly different in patients with chronic renal impairment (baseline serum creatinine ranging from 2 mg/dL to 4.3 mg/dL), and patients with normal renal function.
Body Weight
The population pharmacokinetic (PK) analyses carried out to determine the effects of demographics and clinical variables on PK parameters showed that clearance of nesiritide is proportional to body weight, supporting the administration of weight-adjusted dosing of nesiritide (i.e., administration on a mcg/kg/min basis).
Effects of Concomitant Medications
The co-administration of NATRECOR with enalapril did not have significant effects on the PK of NATRECOR. The PK effect of co-administration of NATRECOR with other IV vasodilators such as nitroglycerin, nitroprusside, milrinone, or IV ACE inhibitors has not been evaluated. During clinical studies, NATRECOR was administered concomitantly with other medications, including: diuretics, digoxin, oral ACE inhibitors, anticoagulants, oral nitrates, statins, class III antiarrhythmic agents, beta-blockers, dobutamine, calcium channel blockers, angiotensin II receptor antagonists, and dopamine. Although no PK interactions were specifically assessed, there did not appear to be evidence suggesting any clinically significant PK interaction.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Long-term studies in animals have not been performed to evaluate the carcinogenic potential or the effect on fertility of nesiritide. Nesiritide did not increase the frequency of mutations when used in an in vitro bacterial cell assay (Ames test). No other genotoxicity studies were performed.
14. Clinical Studies
NATRECOR has been studied in 11 clinical trials including 4505 patients with HF (NYHA class II–III 56%, NYHA class IV 27%; mean age 64 years, women 32%). There were six randomized, multi-center, placebo- or active-controlled studies (comparative agents included nitroglycerin, dobutamine, milrinone, nitroprusside, or dopamine) in which 4269 patients with decompensated HF received continuous infusions of NATRECOR at doses ranging from 0.01 to 0.03 mcg/kg/min. Of these patients, the majority (n=3358, 79%) received the NATRECOR infusion for at least 24 hours; 2182 (51%) received NATRECOR for 24 to 48 hours, and 1176 (28%) received NATRECOR for greater than 48 hours.
In the initial five of these six controlled trials, NATRECOR was used alone or in conjunction with other standard therapies, including diuretics (79%), digoxin (62%), oral ACE inhibitors (55%), anticoagulants (38%), oral nitrates (32%), statins (18%), class III antiarrhythmic agents (16%), beta-blockers (15%), dobutamine (15%), calcium channel blockers (11%), angiotensin II receptor antagonists (6%), and dopamine (4%).
In the ASCEND-HF trial (Acute Study of Clinical Effectiveness of Nesiritide in patients with Decompensated Heart Failure), NATRECOR was used alone or in conjunction with other standard therapies. Most patients (99.4%) received diuretic medications in conjunction with NATRECOR, with the most commonly used diuretic being furosemide (55%). The following standard therapies were used in ≥2% of patients: beta-blockers (72%), aspirin (64%); oral ACE inhibitors (60%), statins (50%), aldosterone antagonists (48%), digoxin/digitalis glycoside (39%), oral or topical nitrates (30%), oral anticoagulants (29%), clopidogrel/thienopyridine (21%), angiotensin receptor antagonists (19%), antiarrhythmic agents (16%), IV nitroglycerin (16%); calcium channel blockers (13%), hydralazine (11%), dobutamine (8%), dopamine (5%), alpha blockers (4%), IV opiates (5%), and NSAIDs (4%). The following standard therapies were used in <2% of patients: COX2 inhibitors, milrinone, epinephrine, levosimendan, nitroprusside, norepinephrine, phenylephrine, and vasopressin.
NATRECOR has been studied in a broad range of patients, including the elderly (53% >65 years of age), women (33%), minorities (17% black), and patients with a history of significant morbidities such as hypertension (71%), previous myocardial infarction (38%), diabetes (43%), atrial fibrillation/flutter (37%), ventricular tachycardia/fibrillation (10%), and preserved systolic function (20%). In trials other than the ASCEND-HF trial, NATRECOR was also studied in patients with nonsustained ventricular tachycardia (25%) and patients with acute coronary syndromes less than 7 days before the start of NATRECOR (4%).
The VMAC (Vasodilation in the Management of Acute Congestive Heart Failure) trial was a randomized, double-blind study of 489 patients (246 patients requiring a right heart catheter, 243 patients without a right heart catheter) who required hospitalization for management of shortness of breath at rest due to acutely decompensated HF. The study compared the effects of NATRECOR, placebo, and IV nitroglycerin when added to background therapy (IV and oral diuretics, non-IV cardiac medications, dobutamine, and dopamine). Patients with acute coronary syndrome, preserved systolic function, arrhythmia, and renal impairment were not excluded. The primary endpoints of the study were the change from baseline in PCWP and the change from baseline in patients' dyspnea, evaluated after three hours. Close attention was also paid to the occurrence and persistence of hypotension, given nesiritide's relatively long (compared to nitroglycerin) PK and PD half-life.
NATRECOR was administered as a 2 mcg/kg bolus over approximately 60 seconds, followed by a continuous fixed dose infusion of 0.01 mcg/kg/min. After the 3-hour placebo-controlled period, patients receiving placebo crossed over to double-blinded active therapy with either NATRECOR or nitroglycerin. The nitroglycerin dose was titrated at the physician's discretion. A subset of patients in the VMAC trial with central hemodynamic monitoring who were treated with NATRECOR (62 of 124 patients) were allowed dose increases of NATRECOR after the first 3 hours of treatment if the PCWP was ≥20 mm Hg and the SBP was ≥100 mm Hg. Dose increases of a 1 mcg/kg bolus followed by an increase of the infusion dose by 0.005 mcg/kg/min were allowed every 3 hours, up to a maximum dose of 0.03 mcg/kg/min. Overall, 23 patients in this subset had the dose of NATRECOR increased in the VMAC trial.
In the VMAC study, patients receiving NATRECOR reported greater improvement in their dyspnea at 3 hours than patients receiving placebo (p=0.034).
In the dose-response study, patients receiving both doses of NATRECOR reported greater improvement in dyspnea at 6 hours than patients receiving placebo.
NATRECOR was also studied in a randomized, double-blind, placebo-controlled, parallel-group, multicenter trial evaluating the efficacy and safety of NATRECOR compared with placebo in patients with ADHF, the ASCEND-HF Study. The study was divided into a screening phase, a double-blind treatment phase, and a follow-up phase, including a Day 30 visit and a telephone contact at Day 180. Patients who qualified for the study were ≥18 years of age, hospitalized for the management of ADHF or diagnosed with ADHF within 48 hours after being hospitalized for another reason. They were randomized to receive either NATRECOR as a continuous IV infusion at 0.010 mcg/kg/min with or without an initial 2 mcg/kg bolus (at the discretion of the physician) or a matching placebo bolus and infusion.
The primary objective of ASCEND-HF was to evaluate whether treatment with NATRECOR compared to placebo improved patient outcomes (as measured by a reduction in the composite of HF rehospitalization and all-cause mortality from randomization through Day 30) or HF symptoms (as measured by patient self-assessed Likert dyspnea scale which included Markedly Better, Moderately Better, Minimally Better, No Change, Minimally Worse, Moderately Worse and Markedly Worse at 6 hours and 24 hours after NATRECOR initiation).
A total of 7141 patients were randomized of which 7007 patients took at least one dose of study medication (modified intent-to-treat population) and received treatment for 24 to 168 hours (7 days), if the patient's clinical condition warranted continued treatment for dyspnea or pulmonary congestion, at the physician's discretion. The median treatment duration was 42.9 hours for the placebo group and 40.8 hours for the NATRECOR group. The patients' mean age was 65.5 years. The patient population was 65.8% male, 55.9% Caucasian, 24.7% Asian and 15.1% Black or African American.
The incidence rate for the composite of HF rehospitalization and all-cause mortality from randomization through Day 30 was 9.4% in the NATRECOR group compared with 10.1% in the placebo group. The difference was not statistically significant (p=0.313). The self-assessed dyspnea results did not meet the pre-specified criteria for statistical significance (p ≤0.005 for both or p ≤0.0025 for either) at either time point.
A total of 273 deaths were reported during the first 30 days after therapy and 876 (12.5%) deaths were reported from randomization through Day 180, 429 (12.3%) patients in the NATRECOR group and 447 (12.7%) patients in the placebo group. Approximately 65% of the deaths at 180 days were cardiovascular (mostly worsening heart failure). There was no statistically significant difference between treatment groups (p=0.5).
16. How is Natrecor supplied
NATRECOR (nesiritide) is provided as a sterile lyophilized powder in 1.5 mg, single-use vials. Each carton contains one vial and is available in the following package:
1 vial/carton (NDC 65847-205-25)
17. Patient Counseling Information
Advise patients of the potential benefits and risks of NATRECOR. Advise patients to notify their physician or healthcare professional if they have symptoms of hypotension [see Warnings and Precautions (5.1)].

| NATRECOR
nesiritide injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Scios LLC (015904436) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Hospira, Inc. | 030606222 | MANUFACTURE(65847-205) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sandoz GmbH, BiochemiestraB 10 | 300220969 | MANUFACTURE(65847-205) , ANALYSIS(65847-205) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sandoz GmbH, A-6330 Schaftenau | 301698247 | MANUFACTURE(65847-205) | |
More about Natrecor (nesiritide)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: vasodilators
Patient resources
Related treatment guides
Copyright 2007 Scios LLC