Ibrance Tablets: Package Insert / Prescribing Info
Package insert / product label
Generic name: palbociclib
Dosage form: tablet, film coated
Drug class: CDK 4/6 inhibitors
Medically reviewed by Drugs.com. Last updated on May 9, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
IBRANCE® (palbociclib) tablets, for oral use
Initial U.S. Approval: 2015
Indications and Usage for Ibrance Tablets
IBRANCE is a kinase inhibitor indicated:
- •
- for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer in combination with:
- •
- in combination with inavolisib and fulvestrant for the treatment of adult patients with endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer, as detected by an FDA-approved test, following recurrence on or after completing adjuvant endocrine therapy. (1)
Ibrance Tablets Dosage and Administration
IBRANCE tablets are taken orally with or without food in combination with an aromatase inhibitor, fulvestrant, or inavolisib and fulvestrant. (2)
Dosage Forms and Strengths
Tablets: 125 mg, 100 mg, and 75 mg. (3)
Contraindications
None. (4)
Warnings and Precautions
- •
- Neutropenia: Monitor complete blood count prior to start of IBRANCE therapy and at the beginning of each cycle, as well as on Day 15 of the first 2 cycles, and as clinically indicated. (2.2, 5.1)
- •
- Interstitial Lung Disease (ILD)/Pneumonitis: Severe and fatal cases of ILD/pneumonitis have been reported. Monitor for pulmonary symptoms of ILD/pneumonitis. Interrupt IBRANCE immediately in patients with suspected ILD/pneumonitis. Permanently discontinue IBRANCE if severe ILD/pneumonitis occurs. (5.2)
- •
- Embryo-Fetal Toxicity: IBRANCE can cause fetal harm. Advise patients of potential risk to a fetus and to use effective contraception. (5.3, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥20%) in combination with either letrozole or fulvestrant, including laboratory abnormalities, were white blood cell count decreased, neutrophils decreased, blood creatinine increased, hemoglobin decreased, platelets decreased, infections, aspartate aminotransferase increased, alanine aminotransferase increased, fatigue, nausea, stomatitis, diarrhea, and alopecia. (6.1)
The most common adverse reactions (incidence ≥20%) in combination with inavolisib and fulvestrant, including laboratory abnormalities, were neutrophils decreased, hemoglobin decreased, fasting glucose increased, platelets decreased, lymphocytes decreased, stomatitis, diarrhea, calcium decreased, fatigue, potassium decreased, creatinine increased, alanine aminotransferase (ALT) increased, nausea, sodium decreased, magnesium decreased, rash, decreased appetite, COVID-19 infection, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- •
- CYP3A Inhibitors: Avoid concurrent use of IBRANCE with strong CYP3A inhibitors. If the strong inhibitor cannot be avoided, reduce the IBRANCE dose. (2.2, 7.1)
- •
- CYP3A Inducers: Avoid concurrent use of IBRANCE with strong CYP3A inducers. (7.2)
- •
- CYP3A Substrates: The dose of sensitive CYP3A4 substrates with narrow therapeutic indices may need to be reduced when given concurrently with IBRANCE. (7.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
Full Prescribing Information
1. Indications and Usage for Ibrance Tablets
IBRANCE is indicated for the treatment of adult patients with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative advanced or metastatic breast cancer in combination with:
- •
- an aromatase inhibitor as initial endocrine-based therapy; or
- •
- fulvestrant in patients with disease progression following endocrine therapy.
IBRANCE is indicated in combination with inavolisib and fulvestrant for the treatment of adult patients with endocrine-resistant, PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer, as detected by an FDA-approved test, following recurrence on or after completing adjuvant endocrine therapy.
2. Ibrance Tablets Dosage and Administration
2.1 Recommended Dose and Schedule
The recommended dose of IBRANCE is a 125 mg tablet taken orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a complete cycle of 28 days. IBRANCE tablet may be taken with or without food [see Clinical Pharmacology (12.3)].
Administer the recommended dose of an aromatase inhibitor when given with IBRANCE. Please refer to the Full Prescribing Information for the aromatase inhibitor being used.
When given with IBRANCE, the recommended dose of fulvestrant is 500 mg administered on Days 1, 15, 29, and once monthly thereafter. Please refer to the Full Prescribing Information of fulvestrant.
Refer to the Full Prescribing Information for inavolisib and fulvestrant for dosing information.
Advise patients to take their dose of IBRANCE at approximately the same time each day.
If the patient vomits or misses a dose, an additional dose should not be taken. The next prescribed dose should be taken at the usual time. IBRANCE tablets should be swallowed whole (do not chew, crush, or split them prior to swallowing). Tablets should not be ingested if they are broken, cracked, or otherwise not intact.
Pre/perimenopausal women treated with the combination IBRANCE plus an aromatase inhibitor or fulvestrant therapy or IBRANCE plus inavolisib and fulvestrant should also be treated with luteinizing hormone-releasing hormone (LHRH) agonists according to current clinical practice standards.
For men treated with combination IBRANCE plus aromatase inhibitor or IBRANCE plus inavolisib and fulvestrant therapy, consider treatment with an LHRH agonist according to current clinical practice standards.
2.2 Dose Modification
The recommended dose modifications for adverse reactions are listed in Tables 1, 2, and 3.
| Dose Level | Dose |
|---|---|
|
|
|
Recommended starting dose |
125 mg/day |
|
First dose reduction |
100 mg/day |
|
Second dose reduction |
75 mg/day* |
| Grading according to CTCAE 4.0. CTCAE=Common Terminology Criteria for Adverse Events; LLN=lower limit of normal. |
|
|
Monitor complete blood counts prior to the start of IBRANCE therapy and at the beginning of each cycle, as well as on Day 15 of the first 2 cycles, and as clinically indicated. |
|
|
For patients who experience a maximum of Grade 1 or 2 neutropenia in the first 6 cycles, monitor complete blood counts for subsequent cycles every 3 months, prior to the beginning of a cycle and as clinically indicated. |
|
|
CTCAE Grade |
Dose Modifications |
|
Grade 1 or 2 |
No dose adjustment is required. |
|
Grade 3 |
Day 1 of cycle: |
|
Grade 3 neutropenia† with fever ≥38.5 °C and/or infection |
At any time: |
|
At any time: |
|
| CTCAE Grade | Dose Modifications |
|---|---|
| Grading according to CTCAE 4.0. CTCAE=Common Terminology Criteria for Adverse Events. |
|
|
Grade 1 or 2 |
No dose adjustment is required. |
|
Grade ≥3 non-hematologic toxicity (if persisting despite optimal medical treatment) |
Withhold until symptoms resolve to:
Resume at the next lower dose. |
Permanently discontinue IBRANCE in patients with severe interstitial lung disease (ILD)/pneumonitis.
Refer to the Full Prescribing Information for coadministered endocrine therapy and/or inavolisib dose adjustment guidelines in the event of toxicity and other relevant safety information or contraindications.
Dose Modifications for Use With Strong CYP3A Inhibitors
Avoid concomitant use of strong CYP3A inhibitors and consider an alternative concomitant medication with no or minimal CYP3A inhibition. If patients must be coadministered a strong CYP3A inhibitor, reduce the IBRANCE dose to 75 mg once daily. If the strong inhibitor is discontinued, increase the IBRANCE dose (after 3 to 5 half-lives of the inhibitor) to the dose used prior to the initiation of the strong CYP3A inhibitor [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
Dose Modifications for Hepatic Impairment
No dose adjustment is required for patients with mild or moderate hepatic impairment (Child-Pugh classes A and B). For patients with severe hepatic impairment (Child-Pugh class C), the recommended dose of IBRANCE is 75 mg once daily for 21 consecutive days followed by 7 days off treatment to comprise a complete cycle of 28 days [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
125 mg tablets: Oval, light purple, film-coated tablets debossed with "Pfizer" on one side and "PBC 125" on the other side.
100 mg tablets: Oval, green, film-coated tablets debossed with "Pfizer" on one side and "PBC 100" on the other side.
75 mg tablets: Round, light purple, film-coated tablets debossed with "Pfizer" on one side and "PBC 75" on the other side.
5. Warnings and Precautions
5.1 Neutropenia
Neutropenia was the most frequently reported adverse reaction in PALOMA-2 with an incidence of 80% and PALOMA-3 with an incidence of 83%. A Grade ≥3 decrease in neutrophil counts was reported in 66% of patients receiving IBRANCE plus letrozole in PALOMA-2 and 66% of patients receiving IBRANCE plus fulvestrant in PALOMA-3. In PALOMA-2 and PALOMA-3, the median time to first episode of any grade neutropenia was 15 days and the median duration of Grade ≥3 neutropenia was 7 days [see Adverse Reactions (6.1)].
Monitor complete blood counts prior to starting IBRANCE therapy and at the beginning of each cycle, as well as on Day 15 of the first 2 cycles, and as clinically indicated. Dose interruption, dose reduction, or delay in starting treatment cycles is recommended for patients who develop Grade 3 or 4 neutropenia [see Dosage and Administration (2.2)].
Febrile neutropenia has been reported in 1.8% of patients exposed to IBRANCE across PALOMA-2 and PALOMA-3. One death due to neutropenic sepsis was observed in PALOMA-3. Physicians should inform patients to promptly report any episodes of fever [see Patient Counseling Information (17)].
5.2 Interstitial Lung Disease (ILD)/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD) and/or pneumonitis can occur in patients treated with cyclin-dependent kinase 4/6 (CDK4/6) inhibitors, including IBRANCE when taken in combination with endocrine therapy.
Across clinical trials (PALOMA-1, PALOMA-2, PALOMA-3), 1% of IBRANCE-treated patients had ILD/pneumonitis of any grade, 0.1% had Grade 3 or 4 and no fatal cases were reported. Additional cases of ILD/pneumonitis have been observed in the postmarketing setting, with fatalities reported [see Adverse Reactions (6.2)].
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis (e.g. hypoxia, cough, dyspnea). In patients who have new or worsening respiratory symptoms and are suspected to have developed pneumonitis, interrupt IBRANCE immediately and evaluate the patient. Permanently discontinue IBRANCE in patients with severe ILD or pneumonitis [see Dosage and Administration (2.2)].
5.3 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, IBRANCE can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of palbociclib to pregnant rats and rabbits during organogenesis resulted in embryo-fetal toxicity at maternal exposures that were ≥4 times the human clinical exposure based on area under the curve (AUC). Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with IBRANCE and for at least 3 weeks after the last dose [see Use in Specific Populations (8.1 and 8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Neutropenia [see Warnings and Precautions (5.1)]
- •
- ILD/Pneumonitis [see Warnings and Precautions (5.2)]
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
PALOMA-2: IBRANCE plus Letrozole
Patients with estrogen receptor (ER)-positive, HER2-negative advanced or metastatic breast cancer for initial endocrine-based therapy
The safety of IBRANCE (125 mg/day) plus letrozole (2.5 mg/day) versus placebo plus letrozole was evaluated in PALOMA-2. The data described below reflect exposure to IBRANCE in 444 out of 666 patients with ER-positive, HER2-negative advanced breast cancer who received at least 1 dose of IBRANCE plus letrozole in PALOMA-2. The median duration of treatment for IBRANCE plus letrozole was 19.8 months while the median duration of treatment for placebo plus letrozole arm was 13.8 months.
Dose reductions due to an adverse reaction of any grade occurred in 36% of patients receiving IBRANCE plus letrozole. No dose reduction was allowed for letrozole in PALOMA-2.
Permanent discontinuation associated with an adverse reaction occurred in 43 of 444 (10%) patients receiving IBRANCE plus letrozole and in 13 of 222 (6%) patients receiving placebo plus letrozole. Adverse reactions leading to permanent discontinuation for patients receiving IBRANCE plus letrozole included neutropenia (1.1%) and alanine aminotransferase increase (0.7%).
The most common adverse reactions (≥10%) of any grade reported in patients in the IBRANCE plus letrozole arm by descending frequency were neutropenia, infections, leukopenia, fatigue, nausea, alopecia, stomatitis, diarrhea, anemia, rash, asthenia, thrombocytopenia, vomiting, decreased appetite, dry skin, pyrexia, and dysgeusia.
The most frequently reported Grade ≥3 adverse reactions (≥5%) in patients receiving IBRANCE plus letrozole by descending frequency were neutropenia, leukopenia, infections, and anemia.
Adverse reactions (≥10%) reported in patients who received IBRANCE plus letrozole or placebo plus letrozole in PALOMA-2 are listed in Table 4.
| IBRANCE plus Letrozole
(N=444) | Placebo plus Letrozole
(N=222) |
|||||
|---|---|---|---|---|---|---|
| Adverse Reaction | All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
| Grading according to CTCAE 4.0. CTCAE=Common Terminology Criteria for Adverse Events; N=number of patients; N/A=not applicable; |
||||||
|
||||||
|
Infections and infestations | ||||||
|
Infections* |
60† |
6 |
1 |
42 |
3 |
0 |
|
Blood and lymphatic system disorders | ||||||
|
Neutropenia |
80 |
56 |
10 |
6 |
1 |
1 |
|
Leukopenia |
39 |
24 |
1 |
2 |
0 |
0 |
|
Anemia |
24 |
5 |
<1 |
9 |
2 |
0 |
|
Thrombocytopenia |
16 |
1 |
<1 |
1 |
0 |
0 |
|
Metabolism and nutrition disorders | ||||||
|
Decreased appetite |
15 |
1 |
0 |
9 |
0 |
0 |
|
Nervous system disorders | ||||||
|
Dysgeusia |
10 |
0 |
0 |
5 |
0 |
0 |
|
Gastrointestinal disorders | ||||||
|
Stomatitis‡ |
30 |
1 |
0 |
14 |
0 |
0 |
|
Nausea |
35 |
<1 |
0 |
26 |
2 |
0 |
|
Diarrhea |
26 |
1 |
0 |
19 |
1 |
0 |
|
Vomiting |
16 |
1 |
0 |
17 |
1 |
0 |
|
Skin and subcutaneous tissue disorders | ||||||
|
Alopecia |
33§ |
N/A |
N/A |
16¶ |
N/A |
N/A |
|
Rash# |
18 |
1 |
0 |
12 |
1 |
0 |
|
Dry skin |
12 |
0 |
0 |
6 |
0 |
0 |
|
General disorders and administration site conditions | ||||||
|
Fatigue |
37 |
2 |
0 |
28 |
1 |
0 |
|
Asthenia |
17 |
2 |
0 |
12 |
0 |
0 |
|
Pyrexia |
12 |
0 |
0 |
9 |
0 |
0 |
Clinically relevant adverse reactions in <10% of patients who received IBRANCE plus letrozole in PALOMA-2 included epistaxis (9%), lacrimation increased (6%), dry eye (4.1%), vision blurred (3.6%), and febrile neutropenia (2.5%).
| IBRANCE plus Letrozole
(N=444) | Placebo plus Letrozole
(N=222) |
|||||
|---|---|---|---|---|---|---|
| Laboratory Abnormality | All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
| N=number of patients; WBC=white blood cells. | ||||||
|
WBC decreased |
97 |
35 |
1 |
25 |
1 |
0 |
|
Blood creatinine increased |
96 |
2 |
<1 |
91 |
0 |
0 |
|
Neutrophils decreased |
95 |
56 |
12 |
20 |
1 |
1 |
|
Hemoglobin decreased |
78 |
6 |
0 |
42 |
2 |
0 |
|
Platelets decreased |
63 |
1 |
1 |
14 |
0 |
0 |
|
Aspartate aminotransferase increased |
52 |
3 |
0 |
34 |
1 |
0 |
|
Alanine aminotransferase increased |
43 |
2 |
<1 |
30 |
0 |
0 |
PALOMA-3: IBRANCE plus Fulvestrant
Patients with HR-positive, HER2-negative advanced or metastatic breast cancer who have had disease progression on or after prior adjuvant or metastatic endocrine therapy
The safety of IBRANCE (125 mg/day) plus fulvestrant (500 mg) versus placebo plus fulvestrant was evaluated in PALOMA-3. The data described below reflect exposure to IBRANCE in 345 out of 517 patients with HR-positive, HER2-negative advanced or metastatic breast cancer who received at least 1 dose of IBRANCE plus fulvestrant in PALOMA-3. The median duration of treatment for IBRANCE plus fulvestrant was 10.8 months while the median duration of treatment for placebo plus fulvestrant arm was 4.8 months.
Dose reductions due to an adverse reaction of any grade occurred in 36% of patients receiving IBRANCE plus fulvestrant. No dose reduction was allowed for fulvestrant in PALOMA-3.
Permanent discontinuation associated with an adverse reaction occurred in 19 of 345 (6%) patients receiving IBRANCE plus fulvestrant, and in 6 of 172 (3%) patients receiving placebo plus fulvestrant. Adverse reactions leading to discontinuation for those patients receiving IBRANCE plus fulvestrant included fatigue (0.6%), infections (0.6%), and thrombocytopenia (0.6%).
The most common adverse reactions (≥10%) of any grade reported in patients in the IBRANCE plus fulvestrant arm by descending frequency were neutropenia, leukopenia, infections, fatigue, nausea, anemia, stomatitis, diarrhea, thrombocytopenia, vomiting, alopecia, rash, decreased appetite, and pyrexia.
The most frequently reported Grade ≥3 adverse reactions (≥5%) in patients receiving IBRANCE plus fulvestrant in descending frequency were neutropenia and leukopenia.
Adverse reactions (≥10%) reported in patients who received IBRANCE plus fulvestrant or placebo plus fulvestrant in PALOMA-3 are listed in Table 6.
| Adverse Reaction | IBRANCE plus Fulvestrant
(N=345) | Placebo plus Fulvestrant
(N=172) |
||||
|---|---|---|---|---|---|---|
| All Grades | Grade 3 | Grade 4 | All Grades | Grade 3 | Grade 4 | |
| % | % | % | % | % | % | |
| Grading according to CTCAE 4.0. CTCAE=Common Terminology Criteria for Adverse Events; N=number of patients; N/A=not applicable. |
||||||
|
||||||
|
Infections and infestations |
||||||
|
Infections* |
47† |
3 |
1 |
31 |
3 |
0 |
|
Blood and lymphatic system disorders |
||||||
|
Neutropenia |
83 |
55 |
11 |
4 |
1 |
0 |
|
Leukopenia |
53 |
30 |
1 |
5 |
1 |
1 |
|
Anemia |
30 |
4 |
0 |
13 |
2 |
0 |
|
Thrombocytopenia |
23 |
2 |
1 |
0 |
0 |
0 |
|
Metabolism and nutrition disorders |
||||||
|
Decreased appetite |
16 |
1 |
0 |
8 |
1 |
0 |
|
Gastrointestinal disorders |
||||||
|
Nausea |
34 |
0 |
0 |
28 |
1 |
0 |
|
Stomatitis‡ |
28 |
1 |
0 |
13 |
0 |
0 |
|
Diarrhea |
24 |
0 |
0 |
19 |
1 |
0 |
|
Vomiting |
19 |
1 |
0 |
15 |
1 |
0 |
|
Skin and subcutaneous tissue disorders |
||||||
|
Alopecia |
18§ |
N/A |
N/A |
6¶ |
N/A |
N/A |
|
Rash# |
17 |
1 |
0 |
6 |
0 |
0 |
|
General disorders and administration site conditions |
||||||
|
Fatigue |
41 |
2 |
0 |
29 |
1 |
0 |
|
Pyrexia |
13 |
<1 |
0 |
5 |
0 |
0 |
Clinically relevant adverse reactions in <10% of patients who received IBRANCE plus fulvestrant in PALOMA-3 included asthenia (8%), dysgeusia (7%), epistaxis (7%), lacrimation increased (6%), dry skin (6%), vision blurred (6%), dry eye (3.8%), and febrile neutropenia (0.9%).
| Laboratory Abnormality | IBRANCE plus Fulvestrant
(N=345) | Placebo plus Fulvestrant
(N=172) |
||||
|---|---|---|---|---|---|---|
| All Grades
% | Grade 3
% | Grade 4
% | All Grades
% | Grade 3
% | Grade 4
% |
|
| N=number of patients; WBC=white blood cells. | ||||||
|
WBC decreased |
99 |
45 |
1 |
26 |
0 |
1 |
|
Neutrophils decreased |
96 |
56 |
11 |
14 |
0 |
1 |
|
Blood creatinine increased |
95 |
1 |
0 |
82 |
0 |
0 |
|
Hemoglobin decreased |
78 |
3 |
0 |
40 |
2 |
0 |
|
Platelets decreased |
62 |
2 |
1 |
10 |
0 |
0 |
|
Aspartate aminotransferase increased |
43 |
4 |
0 |
48 |
4 |
0 |
|
Alanine aminotransferase increased |
36 |
2 |
0 |
34 |
0 |
0 |
INAVO120: IBRANCE plus Inavolisib and Fulvestrant
Adults with PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease
The safety of the combination of IBRANCE plus inavolisib and fulvestrant was evaluated in a randomized, double-blind, placebo-controlled study (INAVO120) in 324 patients with PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer [see Clinical Studies (14)].
Patients received either IBRANCE 125 mg orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a cycle of 28 days plus fulvestrant in combination with inavolisib (n=162) or placebo (n=162). The median duration of treatment for IBRANCE plus inavolisib and fulvestrant was 9 months (range: 0 to 39 months).
Serious adverse reactions occurred in 24% of patients who received IBRANCE plus inavolisib and fulvestrant. Serious adverse reactions occurring in ≥1% of patients receiving IBRANCE plus inavolisib and fulvestrant included anemia (1.9%), diarrhea (1.2%), and urinary tract infection (1.2%).
Fatal adverse reactions occurred in 3.7% of patients who received IBRANCE plus inavolisib and fulvestrant, including (0.6% each) acute coronary syndrome, cerebral hemorrhage, cerebrovascular accident, COVID-19 infection, and gastrointestinal hemorrhage.
Permanent discontinuation of IBRANCE associated with an adverse reaction occurred in 8 of 162 (4.9%) patients receiving IBRANCE plus inavolisib and fulvestrant and in 0 of 162 patients receiving IBRANCE plus placebo and fulvestrant. Adverse reactions leading to discontinuation of IBRANCE in patients receiving IBRANCE plus inavolisib and fulvestrant were neutropenia, infections, alanine aminotransferase increased, gastric ulcer, intestinal perforation, hyperglycemia, type 2 diabetes mellitus, bone pain, musculoskeletal pain, transitional cell carcinoma, and acute kidney injury (0.6% each).
Dose reduction of IBRANCE due to an adverse reaction occurred in 38% of patients receiving IBRANCE plus inavolisib and fulvestrant and in 30% of patients receiving IBRANCE plus placebo and fulvestrant. Adverse reactions leading to dose reductions of IBRANCE in ≥2% patients receiving IBRANCE plus inavolisib and fulvestrant were neutropenia (30%), leukopenia (6%), and thrombocytopenia (3.7%).
Dose interruption of IBRANCE due to an adverse reaction occurred in 71% of patients receiving IBRANCE plus inavolisib and fulvestrant and in 61% of patients receiving IBRANCE plus placebo and fulvestrant. Adverse reactions leading to dose interruption of IBRANCE in ≥2% patients receiving IBRANCE plus inavolisib and fulvestrant were neutropenia (56%), infections (29%), leukopenia (12%), stomatitis (4.9%), anemia (6%), thrombocytopenia (4.3%), diarrhea (3.7%), pyrexia (3.1%), alanine aminotransferase increased (2.5%), hyperglycemia (2.5%), and nausea (2.5%).
The most common (≥20%) adverse reactions, including laboratory abnormalities, were decreased neutrophils, decreased hemoglobin, increased fasting glucose, decreased platelets, decreased lymphocytes, stomatitis, diarrhea, decreased calcium, fatigue, decreased potassium, increased creatinine, increased alanine aminotransferase (ALT), nausea, decreased sodium, decreased magnesium, rash, decreased appetite, COVID-19 infection, and headache.
Adverse reactions and laboratory abnormalities in INAVO120 are summarized in Table 8 and Table 9, respectively.
Table 8. Adverse Reactions (≥10% with ≥5% [All Grades] or ≥2% [Grade 3-4] Higher Incidence in the IBRANCE plus inavolisib and fulvestrant Arm) in INAVO120
| Adverse Reaction | IBRANCE plus Inavolisib and Fulvestrant
(N=162) | IBRANCE plus Placebo and Fulvestrant
(N=162) |
||
|---|---|---|---|---|
| All Grades
(%) | Grade 3-4
(%) | All Grades
(%) | Grade 3-4
(%) |
|
|
Gastrointestinal Disorders |
||||
|
Stomatitis* |
51 |
6† |
27 |
0 |
|
Diarrhea |
48 |
3.7† |
16 |
0 |
|
Nausea |
28 |
0.6† |
17 |
0 |
|
Vomiting |
15 |
0.6† |
4.9 |
1.2† |
|
General Disorders and Administration Site Conditions |
||||
|
Fatigue |
38 |
1.9† |
25 |
1.2† |
|
Skin and Subcutaneous Tissue Disorders |
||||
|
Rash‡ |
26 |
0 |
19 |
0 |
|
Alopecia |
19 |
0 |
6 |
0 |
|
Dry skin§ |
13 |
0 |
4.3 |
0 |
|
Metabolism and Nutrition Disorders |
||||
|
Decreased appetite |
24 |
0 |
9 |
0 |
|
Infections and Infestations |
||||
|
COVID-19 infection |
23 |
1.9 |
10 |
0.6 |
|
Urinary tract infection‡ |
15 |
1.2† |
9 |
0 |
|
Nervous System Disorders |
||||
|
Headache‡ |
22 |
0 |
14 |
0 |
|
Investigations |
||||
|
Decreased weight |
17 |
3.7† |
0.6 |
0 |
Clinically relevant adverse reactions occurring in <10% of patients who received the triplet combination of IBRANCE plus inavolisib and fulvestrant included abdominal pain, dry eye, dysgeusia, and dyspepsia.
Table 9. Select Laboratory Abnormalities (≥10% with a ≥2% [All Grades or Grade 3-4] Higher Incidence in the IBRANCE plus Inavolisib and Fulvestrant Arm) in INAVO120
| ALT=alanine aminotransferase. | ||||
|
||||
|
Laboratory Abnormality |
IBRANCE plus Inavolisib and Fulvestrant* |
IBRANCE plus Placebo and Fulvestrant† |
||
|
All Grades (%) |
Grade 3-4 (%) |
All Grades (%) |
Grade 3-4 (%) |
|
|
Hematology |
||||
|
Neutrophils (total, absolute) decreased |
95 |
82 |
97 |
79 |
|
Hemoglobin decreased |
88 |
8‡ |
85 |
2.5‡ |
|
Platelets decreased |
8 |
16 |
71 |
3.7 |
|
Lymphocytes (absolute) decreased |
72 |
9 |
68 |
14 |
|
Chemistry |
||||
|
Glucose (fasting) increased§ |
85 |
12 |
43 |
0 |
|
Calcium decreased |
42 |
3.1 |
32 |
3.7 |
|
Potassium decreased |
38 |
6 |
21 |
0.6‡ |
|
Creatinine increased |
38 |
1.9‡ |
30 |
1.2‡ |
|
ALT increased |
34 |
3.1‡ |
29 |
1.2‡ |
|
Sodium decreased |
28 |
2.5‡ |
19 |
2.5 |
|
Magnesium decreased |
27 |
0.6 |
21 |
0 |
|
Lipase (fasting) increased |
16 |
1.4‡ |
7 |
0 |
Other Clinical Trials Experience
The following adverse reaction has been reported following administration of IBRANCE: venous thromboembolism.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of IBRANCE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory Disorders: Interstitial lung disease (ILD)/non-infectious pneumonitis
Skin and Subcutaneous Tissue Disorders: Palmar-plantar erythrodysesthesia syndrome (PPES)
Related/similar drugs
7. Drug Interactions
Palbociclib is primarily metabolized by CYP3A and sulfotransferase (SULT) enzyme SULT2A1. In vivo, palbociclib is a time-dependent inhibitor of CYP3A.
7.1 Agents That May Increase Palbociclib Plasma Concentrations
Effect of CYP3A Inhibitors
Coadministration of a strong CYP3A inhibitor (itraconazole) increased the plasma exposure of palbociclib in healthy subjects by 87%. Avoid concomitant use of strong CYP3A inhibitors (e.g., clarithromycin, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, and voriconazole). Avoid grapefruit or grapefruit juice during IBRANCE treatment. If coadministration of IBRANCE with a strong CYP3A inhibitor cannot be avoided, reduce the dose of IBRANCE [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
7.2 Agents That May Decrease Palbociclib Plasma Concentrations
Effect of CYP3A Inducers
Coadministration of a strong CYP3A inducer (rifampin) decreased the plasma exposure of palbociclib in healthy subjects by 85%. Avoid concomitant use of strong CYP3A inducers (e.g., phenytoin, rifampin, carbamazepine, enzalutamide, and St John's Wort) [see Clinical Pharmacology (12.3)].
7.3 Drugs That May Have Their Plasma Concentrations Altered by Palbociclib
Coadministration of midazolam with multiple doses of IBRANCE increased the midazolam plasma exposure by 61%, in healthy subjects, compared to administration of midazolam alone. The dose of the sensitive CYP3A substrate with a narrow therapeutic index (e.g., alfentanil, cyclosporine, dihydroergotamine, ergotamine, everolimus, fentanyl, pimozide, quinidine, sirolimus, and tacrolimus) may need to be reduced, as IBRANCE may increase its exposure [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, IBRANCE can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data in pregnant women to inform the drug-associated risk. In animal reproduction studies, administration of palbociclib to pregnant rats and rabbits during organogenesis resulted in embryo-fetal toxicity at maternal exposures that were ≥4 times the human clinical exposure based on AUC (see Data). Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Data
Animal Data
In a fertility and early embryonic development study in female rats, palbociclib was administered orally for 15 days before mating through to Day 7 of pregnancy, which did not cause embryo toxicity at doses up to 300 mg/kg/day with maternal systemic exposures approximately 4 times the human exposure (AUC) at the recommended dose.
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of palbociclib up to 300 mg/kg/day and 20 mg/kg/day, respectively, during the period of organogenesis. The maternally toxic dose of 300 mg/kg/day was fetotoxic in rats, resulting in reduced fetal body weights. At doses ≥100 mg/kg/day in rats, there was an increased incidence of a skeletal variation (increased incidence of a rib present at the seventh cervical vertebra). At the maternally toxic dose of 20 mg/kg/day in rabbits, there was an increased incidence of skeletal variations, including small phalanges in the forelimb. At 300 mg/kg/day in rats and 20 mg/kg/day in rabbits, the maternal systemic exposures were approximately 4 and 9 times the human exposure (AUC) at the recommended dose, respectively.
CDK4/6 double knockout mice have been reported to die in late stages of fetal development (gestation Day 14.5 until birth) due to severe anemia. However, knockout mouse data may not be predictive of effects in humans due to differences in degree of target inhibition.
8.2 Lactation
Risk Summary
There is no information regarding the presence of palbociclib in human milk, its effects on milk production, or the breastfed infant. Because of the potential for serious adverse reactions in breastfed infants from IBRANCE, advise a lactating woman not to breastfeed during treatment with IBRANCE and for 3 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Based on animal studies, IBRANCE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Females of reproductive potential should have a pregnancy test prior to starting treatment with IBRANCE.
Contraception
Females
IBRANCE can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with IBRANCE and for at least 3 weeks after the last dose.
Males
Because of the potential for genotoxicity, advise male patients with female partners of reproductive potential to use effective contraception during treatment with IBRANCE and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on animal studies, IBRANCE may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of IBRANCE in pediatric patients have not been studied.
Altered glucose metabolism (glycosuria, hyperglycemia, decreased insulin) associated with changes in the pancreas (islet cell vacuolation), eye (cataracts, lens degeneration), kidney (tubule vacuolation, chronic progressive nephropathy) and adipose tissue (atrophy) were identified in a 27 week repeat-dose toxicology study in rats that were immature at the beginning of the studies and were most prevalent in males at oral palbociclib doses ≥30 mg/kg/day (approximately 11 times the adult human exposure [AUC] at the recommended dose). Some of these findings (glycosuria/hyperglycemia, pancreatic islet cell vacuolation, and kidney tubule vacuolation) were present with lower incidence and severity in a 15 week repeat-dose toxicology study in immature rats. Altered glucose metabolism or associated changes in the pancreas, eye, kidney and adipose tissue were not identified in a 27-week repeat-dose toxicology study in rats that were mature at the beginning of the study and in dogs in repeat-dose toxicology studies up to 39 weeks duration.
Toxicities in teeth independent of altered glucose metabolism were observed in rats. Administration of 100 mg/kg palbociclib for 27 weeks (approximately 15 times the adult human exposure [AUC] at the recommended dose) resulted in abnormalities in growing incisor teeth (discolored, ameloblast degeneration/necrosis, mononuclear cell infiltrate). Other toxicities of potential concern to pediatric patients have not been evaluated in juvenile animals.
8.5 Geriatric Use
Of 444 patients who received IBRANCE in PALOMA-2, 181 patients (41%) were ≥65 years of age and 48 patients (11%) were ≥75 years of age. Of 347 patients who received IBRANCE in PALOMA-3, 86 patients (25%) were ≥65 years of age and 27 patients (8%) were ≥75 years of age. No overall differences in safety or effectiveness of IBRANCE were observed between these patients and younger patients.
8.6 Hepatic Impairment
No dose adjustment is required in patients with mild or moderate hepatic impairment (Child-Pugh classes A and B). For patients with severe hepatic impairment (Child-Pugh class C), the recommended dose of IBRANCE is 75 mg once daily for 21 consecutive days followed by 7 days off treatment to comprise a complete cycle of 28 days [see Dosage and Administration (2.2)]. Based on a pharmacokinetic trial in subjects with varying degrees of hepatic function, the palbociclib unbound exposure (unbound AUCINF) decreased by 17% in subjects with mild hepatic impairment (Child-Pugh class A), and increased by 34% and 77% in subjects with moderate (Child-Pugh class B) and severe (Child-Pugh class C) hepatic impairment, respectively, relative to subjects with normal hepatic function. Peak palbociclib unbound exposure (unbound Cmax) increased by 7%, 38% and 72% for mild, moderate and severe hepatic impairment, respectively, relative to subjects with normal hepatic function [see Clinical Pharmacology (12.3)].
Review the Full Prescribing Information for the aromatase inhibitor or fulvestrant for dose modifications related to hepatic impairment.
8.7 Renal Impairment
No dose adjustment is required in patients with mild, moderate, or severe renal impairment (CrCl >15 mL/min).
Based on a pharmacokinetic trial in subjects with varying degrees of renal function, the total palbociclib exposure (AUCINF) increased by 39%, 42%, and 31% with mild (60 mL/min ≤ CrCl <90 mL/min), moderate (30 mL/min ≤ CrCl <60 mL/min), and severe (CrCl <30 mL/min) renal impairment, respectively, relative to subjects with normal renal function. Peak palbociclib exposure (Cmax) increased by 17%, 12%, and 15% for mild, moderate, and severe renal impairment, respectively, relative to subjects with normal renal function.
The pharmacokinetics of palbociclib have not been studied in patients requiring hemodialysis [see Clinical Pharmacology (12.3)].
11. Ibrance Tablets Description
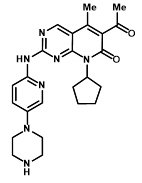
IBRANCE tablets for oral administration contain 125 mg, 100 mg, or 75 mg of palbociclib, a kinase inhibitor. The molecular formula for palbociclib is C24H29N7O2. The molecular weight is 447.54 daltons. The chemical name is 6-acetyl-8-cyclopentyl-5-methyl-2-{[5-(piperazin-1-yl)pyridin-2-yl]amino}pyrido[2,3-d]pyrimidin-7(8H)-one, and its structural formula is:
Palbociclib is a yellow to orange powder. At or below pH 4, palbociclib behaves as a high-solubility compound. Above pH 4, the solubility of the drug substance reduces significantly.
Inactive Ingredients: Microcrystalline cellulose, colloidal silicon dioxide, crospovidone, magnesium stearate, succinic acid, HPMC 2910/hypromellose, titanium dioxide, triacetin, and FD&C Blue #2/Indigo Carmine Aluminum Lake. In addition, the 75 mg and 125 mg tablets contain red iron oxide and the 100 mg tablets contain yellow iron oxide.
12. Ibrance Tablets - Clinical Pharmacology
12.1 Mechanism of Action
Palbociclib is an inhibitor of cyclin-dependent kinases (CDK) 4 and 6. Cyclin D1 and CDK4/6 are downstream of signaling pathways which lead to cellular proliferation. In vitro, palbociclib reduced cellular proliferation of estrogen receptor (ER)-positive breast cancer cell lines by blocking progression of the cell from G1 into S phase of the cell cycle. Treatment of breast cancer cell lines with the combination of palbociclib and antiestrogens leads to decreased retinoblastoma (Rb) protein phosphorylation resulting in reduced E2F expression and signaling, and increased growth arrest compared to treatment with each drug alone. In vitro treatment of ER-positive breast cancer cell lines with the combination of palbociclib and antiestrogens led to increased cell senescence compared to each drug alone, which was sustained for up to 6 days following palbociclib removal and was greater if antiestrogen treatment was continued. In vivo studies using a patient-derived ER-positive breast cancer xenograft model demonstrated that the combination of palbociclib and letrozole increased the inhibition of Rb phosphorylation, downstream signaling, and tumor growth compared to each drug alone.
Human bone marrow mononuclear cells treated with palbociclib in the presence or absence of an anti-estrogen in vitro did not become senescent and resumed proliferation following palbociclib withdrawal.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of palbociclib on the QT interval corrected for heart rate (QTc) was evaluated using time-matched electrocardiograms (ECGs) evaluating the change from baseline and corresponding pharmacokinetic data in 77 patients with breast cancer. Palbociclib had no large effect on QTc (i.e., >20 ms) at 125 mg once daily for 21 consecutive days followed by 7 days off treatment to comprise a complete cycle of 28 days.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of palbociclib were characterized in patients with solid tumors including advanced breast cancer and in healthy subjects.
Absorption
The maximum observed concentration (Cmax) of palbociclib is generally observed between 4 to 12 hours (time to reach maximum concentration, Tmax) following oral administration of IBRANCE tablets. The mean absolute bioavailability of IBRANCE after an oral 125 mg dose is 46%. In the dosing range of 25 mg to 225 mg, the AUC and Cmax increased proportionally with dose in general. Steady state was achieved within 8 days following repeated once daily dosing. With repeated once daily administration, palbociclib accumulated with a median accumulation ratio of 2.4 (range 1.5 to 4.2).
Food Effect: The area under the concentration-time curve from zero to infinity (AUCINF) and Cmax of palbociclib increased by 22% and 26%, respectively, when IBRANCE tablets were given with a high-fat, high-calorie meal (approximately 800 to 1000 calories with 150, 250, and 500 to 600 calories from protein, carbohydrate, and fat, respectively), and by 9% and 10%, respectively, when IBRANCE tablets were given with a moderate-fat, standard-calorie meal (approximately 500 to 700 calories with 75 to 105, 250 to 350 and 175 to 245 calories from protein, carbohydrate, and fat, respectively), compared to IBRANCE tablets given under overnight fasted conditions.
Distribution
Binding of palbociclib to human plasma proteins in vitro was approximately 85%, with no concentration dependence over the concentration range of 500 ng/mL to 5000 ng/mL. The mean fraction unbound (fu) of palbociclib in human plasma in vivo increased incrementally with worsening hepatic function. There was no obvious trend in the mean palbociclib fu in human plasma in vivo with worsening renal function. The geometric mean apparent volume of distribution (Vz/F) was 2583 L with a coefficient of variation (CV) of 26%.
Metabolism
In vitro and in vivo studies indicated that palbociclib undergoes hepatic metabolism in humans. Following oral administration of a single 125 mg dose of [14C]palbociclib to humans, the primary metabolic pathways for palbociclib involved oxidation and sulfonation, with acylation and glucuronidation contributing as minor pathways. Palbociclib was the major circulating drug-derived entity in plasma (23%). The major circulating metabolite was a glucuronide conjugate of palbociclib, although it only represented 1.5% of the administered dose in the excreta. Palbociclib was extensively metabolized with unchanged drug accounting for 2.3% and 6.9% of radioactivity in feces and urine, respectively. In feces, the sulfamic acid conjugate of palbociclib was the major drug-related component, accounting for 26% of the administered dose. In vitro studies with human hepatocytes, liver cytosolic and S9 fractions, and recombinant SULT enzymes indicated that CYP3A and SULT2A1 are mainly involved in the metabolism of palbociclib.
Elimination
The geometric mean apparent oral clearance (CL/F) of palbociclib was 63.1 L/hr (29% CV), and the mean (± standard deviation) plasma elimination half-life was 29 (±5) hours in patients with advanced breast cancer. In 6 healthy male subjects given a single oral dose of [14C] palbociclib, a median of 91.6% of the total administered radioactive dose was recovered in 15 days; feces (74.1% of dose) was the major route of excretion, with 17.5% of the dose recovered in urine. The majority of the material was excreted as metabolites.
Specific Populations
Age, Gender, and Body Weight
Based on a population pharmacokinetic analysis in 183 patients with cancer (50 male and 133 female patients, age range from 22 to 89 years, and body weight range from 37.9 to 123 kg), gender had no effect on the exposure of palbociclib, and age and body weight had no clinically important effect on the exposure of palbociclib.
Pediatric Population
Pharmacokinetics of IBRANCE have not been evaluated in patients <18 years of age.
Hepatic Impairment
Data from a pharmacokinetic trial in subjects with varying degrees of hepatic impairment indicate that palbociclib unbound AUCINF decreased 17% in subjects with mild hepatic impairment (Child-Pugh class A), and increased by 34% and 77% in subjects with moderate (Child-Pugh class B) and severe (Child-Pugh class C) hepatic impairment, respectively, relative to subjects with normal hepatic function. Palbociclib unbound Cmax increased by 7%, 38% and 72% for mild, moderate and severe hepatic impairment, respectively, relative to subjects with normal hepatic function. In addition, based on a population pharmacokinetic analysis that included 183 patients, where 40 patients had mild hepatic impairment based on National Cancer Institute (NCI) classification (total bilirubin ≤ ULN and AST > ULN, or total bilirubin >1.0 to 1.5 × ULN and any AST), mild hepatic impairment had no effect on the exposure of palbociclib, further supporting the findings from the dedicated hepatic impairment study.
Renal Impairment
Data from a pharmacokinetic trial in subjects with varying degrees of renal impairment indicate that palbociclib AUCINF increased by 39%, 42%, and 31% with mild (60 mL/min ≤ CrCl < 90 mL/min), moderate (30 mL/min ≤ CrCl <60 mL/min), and severe (CrCl <30 mL/min) renal impairment, respectively, relative to subjects with normal renal function. Peak palbociclib exposure (Cmax) increased by 17%, 12%, and 15% for mild, moderate, and severe renal impairment, respectively, relative to subjects with normal renal function. In addition, based on a population pharmacokinetic analysis that included 183 patients where 73 patients had mild renal impairment and 29 patients had moderate renal impairment, mild and moderate renal impairment had no effect on the exposure of palbociclib. The pharmacokinetics of palbociclib have not been studied in patients requiring hemodialysis.
Drug Interactions
In vitro data indicate that CYP3A and SULT enzyme SULT2A1 are mainly involved in the metabolism of palbociclib. Palbociclib is a weak time-dependent inhibitor of CYP3A following daily 125 mg dosing to steady state in humans. In vitro, palbociclib is not an inhibitor of CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, and 2D6, and is not an inducer of CYP1A2, 2B6, 2C8, and 3A4 at clinically relevant concentrations.
CYP3A Inhibitors: Data from a drug interaction trial in healthy subjects (N=12) indicate that coadministration of multiple 200 mg daily doses of itraconazole with a single 125 mg IBRANCE dose increased palbociclib AUCINF and the Cmax by approximately 87% and 34%, respectively, relative to a single 125 mg IBRANCE dose given alone [see Drug Interactions (7.1)].
CYP3A Inducers: Data from a drug interaction trial in healthy subjects (N=15) indicate that coadministration of multiple 600 mg daily doses of rifampin, a strong CYP3A inducer, with a single 125 mg IBRANCE dose decreased palbociclib AUCINF and Cmax by 85% and 70%, respectively, relative to a single 125 mg IBRANCE dose given alone. Data from a drug interaction trial in healthy subjects (N=14) indicate that coadministration of multiple 400 mg daily doses of modafinil, a moderate CYP3A inducer, with a single 125 mg IBRANCE dose decreased palbociclib AUCINF and Cmax by 32% and 11%, respectively, relative to a single 125 mg IBRANCE dose given alone [see Drug Interactions (7.2)].
CYP3A Substrates: Palbociclib is a weak time-dependent inhibitor of CYP3A following daily 125 mg dosing to steady state in humans. In a drug interaction trial in healthy subjects (N=26), coadministration of midazolam with multiple doses of IBRANCE increased the midazolam AUCINF and the Cmax values by 61% and 37%, respectively, as compared to administration of midazolam alone [see Drug Interactions (7.3)].
Gastric pH Elevating Medications: In a drug interaction trial in healthy subjects, coadministration of a single 125 mg IBRANCE tablet with multiple doses of the proton pump inhibitor (PPI) rabeprazole under overnight fasted conditions had no effect on the rate and extent of absorption of palbociclib when compared to a single 125 mg IBRANCE tablet administered alone. Given the reduced effect on gastric pH of H2-receptor antagonists and local antacids compared to PPIs, an effect of these classes of acid reducing agents on palbociclib exposure is not expected.
Effect of Palbociclib on Transporters: In vitro evaluations indicated that palbociclib has a low potential to inhibit the activities of drug transporters organic anion transporter (OAT)1, OAT3, organic cation transporter (OCT)2, and organic anion transporting polypeptide (OATP)1B1, OATP1B3 at clinically relevant concentrations. In vitro, palbociclib has the potential to inhibit OCT1 at clinically relevant concentrations, as well as the potential to inhibit P-glycoprotein (P-gp) or breast cancer resistance protein (BCRP) in the gastrointestinal tract at the proposed dose.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Palbociclib was assessed for carcinogenicity in a 6-month transgenic mouse study and in a 2-year rat study. Oral administration of palbociclib for 2 years resulted in an increased incidence of microglial cell tumors in the central nervous system of male rats at a dose of 30 mg/kg/day (approximately 8 times the human clinical exposure based on AUC). There were no neoplastic findings in female rats at doses up to 200 mg/kg/day (approximately 5 times the human clinical exposure based on AUC). Oral administration of palbociclib to male and female rasH2 transgenic mice for 6 months did not result in increased incidence of neoplasms at doses up to 60 mg/kg/day.
Palbociclib was aneugenic in Chinese Hamster Ovary cells in vitro and in the bone marrow of male rats at doses ≥100 mg/kg/day for 3 weeks. Palbociclib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay and was not clastogenic in the in vitro human lymphocyte chromosome aberration assay.
In a fertility study in female rats, palbociclib did not affect mating or fertility at any dose up to 300 mg/kg/day (approximately 4 times human clinical exposure based on AUC) and no adverse effects were observed in the female reproductive tissues in repeat-dose toxicity studies up to 300 mg/kg/day in the rat and 3 mg/kg/day in the dog (approximately 6 times and similar to human exposure [AUC], at the recommended dose, respectively).
The adverse effects of palbociclib on male reproductive function and fertility were observed in the repeat-dose toxicology studies in rats and dogs and a male fertility study in rats. In repeat-dose toxicology studies, palbociclib-related findings in the testis, epididymis, prostate, and seminal vesicle at ≥30 mg/kg/day in rats and ≥0.2 mg/kg/day in dogs included decreased organ weight, atrophy or degeneration, hypospermia, intratubular cellular debris, and decreased secretion. Partial reversibility of male reproductive organ effects was observed in the rat and dog following a 4- and 12-week non-dosing period, respectively. These doses in rats and dogs resulted in approximately ≥10 and 0.1 times, respectively, the exposure [AUC] in humans at the recommended dose. In the fertility and early embryonic development study in male rats, palbociclib caused no effects on mating but resulted in a slight decrease in fertility in association with lower sperm motility and density at 100 mg/kg/day with projected exposure levels [AUC] of 20 times the exposure in humans at the recommended dose.
14. Clinical Studies
PALOMA-2: IBRANCE plus Letrozole
Patients with ER-positive, HER2-negative advanced or metastatic breast cancer for initial endocrine-based therapy
PALOMA-2 was an international, randomized, double-blind, parallel-group, multicenter study of IBRANCE plus letrozole versus placebo plus letrozole conducted in postmenopausal women with ER-positive, HER2-negative advanced breast cancer who had not received previous systemic treatment for their advanced disease. A total of 666 patients were randomized 2:1 to IBRANCE plus letrozole or placebo plus letrozole. Randomization was stratified by disease site (visceral versus non-visceral), disease-free interval (de novo metastatic versus ≤12 months from the end of adjuvant treatment to disease recurrence versus >12 months from the end of adjuvant treatment to disease recurrence), and nature of prior (neo)adjuvant anticancer therapies (prior hormonal therapies versus no prior hormonal therapy). IBRANCE was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment. Patients received study treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. The major efficacy outcome of the study was investigator-assessed progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST). Additional efficacy outcome measures were confirmed overall response rate (ORR) as assessed by the investigator according to RECIST Version 1.1 and overall survival (OS).
Patients enrolled in this study had a median age of 62 years (range 28 to 89). The majority of patients were White (78%), and most patients had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1 (98%). Forty-eight percent of patients had received chemotherapy and 56% had received antihormonal therapy in the neoadjuvant or adjuvant setting prior to their diagnosis of advanced breast cancer. Thirty-seven percent of patients had no prior systemic therapy in the neoadjuvant or adjuvant setting. The majority of patients (97%) had metastatic disease. Twenty-three percent of patients had bone only disease, and 49% of patients had visceral disease.
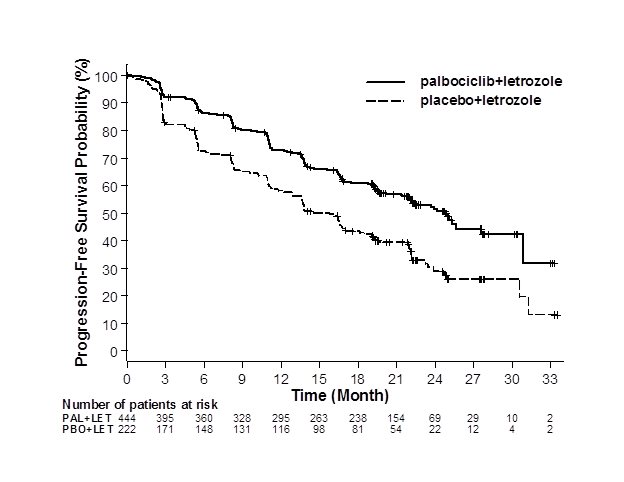
Major efficacy results from PALOMA-2 are summarized in Table 10 and Figure 1. Consistent results were observed across patient subgroups of disease-free interval (DFI), disease site, and prior therapy. The treatment effect of the combination on PFS was also supported by an independent review of radiographs. Based on the prespecified final OS analysis conducted after 435 events, OS was not statistically significant.
| IBRANCE plus Letrozole | Placebo plus Letrozole | |
|---|---|---|
| CI=confidence interval; ITT=Intent-to-Treat; N=number of patients; NE=not estimable; OS=Overall survival; PFS=Progression‑free survival. | ||
|
Progression-free survival for ITT (investigator assessment) |
N=444 |
N=222 |
|
Number of PFS events (%) |
194 (43.7) |
137 (61.7) |
|
Median progression-free survival |
24.8 (22.1, NE) |
14.5 (12.9, 17.1) |
|
Hazard ratio (95% CI) and p-value | ||
|
Objective Response for patients with measurable disease (investigator assessment) |
N=338 |
N=171 |
|
Objective response rate‡ (%, 95% CI) |
55.3 (49.9, 60.7) |
44.4 (36.9, 52.2) |
|
Overall survival for ITT |
N=444 |
N=222 |
|
Number of OS events (%) |
287 (64.6) |
148 (66.7) |
|
Median OS (months, 95% CI) |
53.8 (49.8, 59.2) |
49.8 (42.3, 56.4) |
|
Hazard ratio (95% CI) and p‑value | ||
PALOMA-3: IBRANCE plus Fulvestrant
Patients with HR-positive, HER2-negative advanced or metastatic breast cancer who have had disease progression on or after prior adjuvant or metastatic endocrine-therapy
PALOMA-3 was an international, randomized, double-blind, parallel-group, multicenter study of IBRANCE plus fulvestrant versus placebo plus fulvestrant conducted in women with HR-positive, HER2-negative advanced breast cancer, regardless of their menopausal status, whose disease progressed on or after prior endocrine therapy. A total of 521 pre/postmenopausal women were randomized 2:1 to IBRANCE plus fulvestrant or placebo plus fulvestrant and stratified by documented sensitivity to prior hormonal therapy, menopausal status at study entry (pre/peri versus postmenopausal), and presence of visceral metastases. IBRANCE was given orally at a dose of 125 mg daily for 21 consecutive days followed by 7 days off treatment. Pre/perimenopausal women were enrolled in the study and received the LHRH agonist goserelin for at least 4 weeks prior to and for the duration of PALOMA-3. Patients continued to receive assigned treatment until objective disease progression, symptomatic deterioration, unacceptable toxicity, death, or withdrawal of consent, whichever occurred first. The major efficacy outcome of the study was investigator-assessed PFS evaluated according to RECIST 1.1.
Patients enrolled in this study had a median age of 57 years (range 29 to 88). The majority of patients on study were White (74%), all patients had an ECOG PS of 0 or 1, and 80% were postmenopausal. All patients had received prior systemic therapy, and 75% of patients had received a previous chemotherapy regimen. Twenty-five percent of patients had received no prior therapy in the metastatic disease setting, 60% had visceral metastases, and 23% had bone only disease.
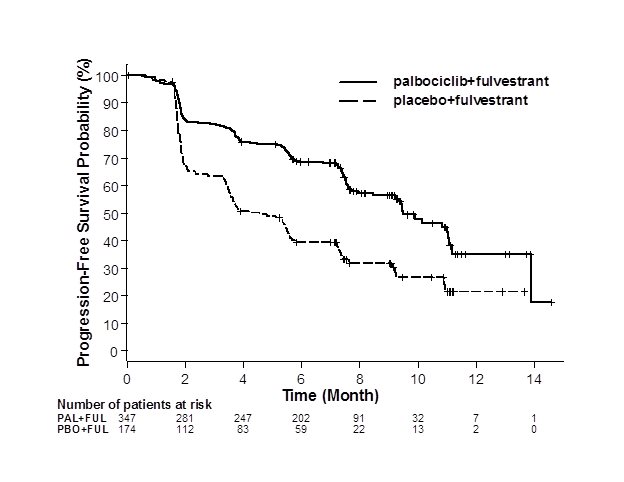
The results from the investigator-assessed PFS and final OS from PALOMA-3 are summarized in Table 11. The relevant Kaplan-Meier plots are shown in Figures 2 and 3, respectively. Consistent PFS results were observed across patient subgroups of disease site, sensitivity to prior hormonal therapy, and menopausal status. After a median follow-up time of 45 months, the final OS results were not statistically significant.
| IBRANCE plus Fulvestrant | Placebo plus Fulvestrant | |
|---|---|---|
| CI=confidence interval; ITT=Intent-to-Treat; N=number of patients; OS=overall survival; PFS=progression-free survival. | ||
|
Progression-free survival for ITT
|
N=347 |
N=174 |
|
Number of PFS events (%) |
145 (41.8) |
114 (65.5) |
|
Median PFS (months, 95% CI) |
9.5 (9.2, 11.0) |
4.6 (3.5, 5.6) |
|
Hazard ratio (95% CI) and p-value |
0.461 (0.360, 0.591), p<0.0001 |
|
|
Objective Response for patients with measurable disease
|
N=267 |
N=138 |
|
Objective response rate* (%, 95% CI) |
24.6 (19.6, 30.2) |
10.9 (6.2, 17.3) |
|
Overall survival for ITT |
N=347 |
N=174 |
|
Number of OS events (%) |
201 (57.9) |
109 (62.6) |
|
Median OS (months, 95% CI) |
34.9 (28.8, 40.0) |
28.0 (23.6, 34.6) |
|
Hazard ratio (95% CI) and p-value | ||
|
Figure 2. Kaplan-Meier Plot of Progression-Free Survival – PALOMA-3 (Investigator Assessment, Intent-to-Treat Population) |
||
 | ||
|
FUL=fulvestrant; PAL=palbociclib; PBO=placebo. | ||
|
Figure 3. Kaplan-Meier Plot of Overall Survival (Intent-to-Treat Population) – PALOMA-3 |
||
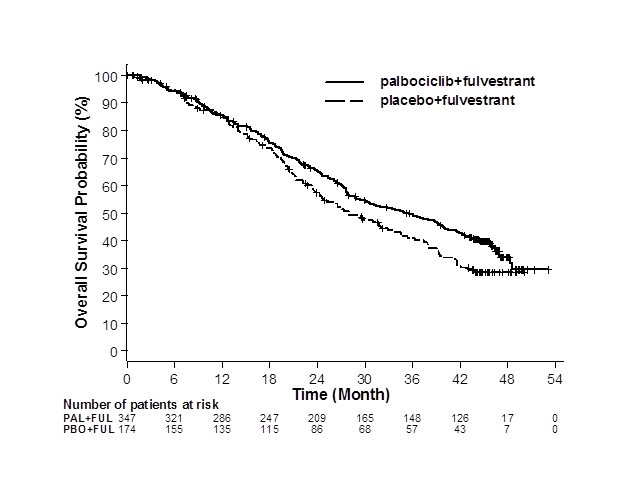 | ||
|
FUL=fulvestrant; PAL=palbociclib; PBO=placebo. | ||
INAVO120: IBRANCE plus Inavolisib and Fulvestrant
Adults with PIK3CA-mutated, HR-positive, HER2-negative, locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease
INAVO120 (NCT04191499) was a randomized (1:1), double-blind, placebo-controlled trial evaluating the efficacy of inavolisib in combination with IBRANCE and fulvestrant in adult patients with endocrine-resistant PIK3CA-mutated, HR-positive, HER2-negative (defined as IHC 0 or 1+, or IHC 2+/ISH-), locally advanced or metastatic breast cancer whose disease progressed during or within 12 months of completing adjuvant endocrine therapy and who have not received prior systemic therapy for locally advanced or metastatic disease. Randomization was stratified by presence of visceral disease (yes or no), endocrine resistance (primary or secondary), and geographic region (North America/Western Europe, Asia, other).
Primary endocrine resistance was defined as relapse while on the first 2 years of adjuvant endocrine therapy (ET) and secondary endocrine resistance was defined as relapse while on adjuvant ET after at least 2 years or relapse within 12 months of completing adjuvant ET.
Patients were required to have a HbA1C <6% and fasting blood glucose <126 mg/dL. The study excluded patients with Type 1 diabetes mellitus or Type 2 diabetes mellitus requiring ongoing anti-hyperglycemic treatment at the start of study treatment.
PIK3CA mutation status was prospectively determined in a central laboratory using the FoundationOne® Liquid CDx assay on plasma-derived circulating tumor DNA (ctDNA) or in local laboratories using various validated polymerase chain reaction (PCR) or next-generation sequencing (NGS) assays on tumor tissue or plasma. All patients were required to provide both a freshly collected pre-treatment blood sample and a tumor tissue sample for central evaluation and determination of PIK3CA mutation(s) status.
Patients received either inavolisib 9 mg (n=161) or placebo (n=164) orally once daily, in combination with IBRANCE 125 mg orally once daily for 21 consecutive days followed by 7 days off treatment to comprise a cycle of 28 days, and fulvestrant 500 mg administered intramuscularly on Cycle 1, Days 1 and 15, and then on Day 1 of every 28-day cycle. Patients received treatment until disease progression or unacceptable toxicity. In addition, all pre/perimenopausal women and men received an LHRH agonist throughout therapy.
The baseline demographic and disease characteristics were: median age 54 years (range: 27 to 79 years); 98% female, of which 39% were pre/perimenopausal; 59% White, 38% Asian, 2.5% Unknown, 0.6% Black or African American; 6% Hispanic or Latino; and ECOG PS of 0 (63%) or 1 (36%). Tamoxifen (57%) and aromatase inhibitors (50%) were the most commonly used adjuvant endocrine therapies. Sixty-four percent of patients were considered to have secondary endocrine resistance. Eighty-three percent of patients had received prior chemotherapy (in the neo/adjuvant setting) and 1.2% of patients had been treated with a CDK4/6 inhibitor.
The major efficacy outcome measure was investigator (INV)-assessed PFS per RECIST version 1.1. Additional efficacy outcome measures included OS, INV-assessed ORR, and INV-assessed duration of response (DOR).
Efficacy results are summarized in Table 12 and Figure 4. INV-assessed PFS results were supported by consistent results from a blinded independent central review (BICR) assessment. At the time of the PFS analysis, OS data were not mature with 30% deaths in the overall population.
Table 12. Efficacy Results in Patients with Locally Advanced or Metastatic Breast Cancer in INAVO120
| CI=confidence interval; CR=complete response; DOR=duration of response; N=number of patients; PR=partial response. | ||
|
Efficacy Endpoint |
IBRANCE plus Inavolisib and Fulvestrant N=161 |
IBRANCE plus Placebo and Fulvestrant N=164 |
|
Patients with event (%) |
82 (51) |
113 (69) |
|
Median, months (95% CI) |
15.0 (11.3, 20.5) |
7.3 (5.6, 9.3) |
|
Hazard ratio (95% CI) |
0.43 (0.32, 0.59) |
|
|
p-value |
<0.0001 |
|
|
Patients with CR or PR (%) |
94 (58) |
41 (25) |
|
95% CI |
(50, 66) |
(19, 32) |
|
Duration of Response† |
||
|
Median DOR, months (95% CI) |
18.4 (10.4, 22.2) |
9.6 (7.4, 16.6) |
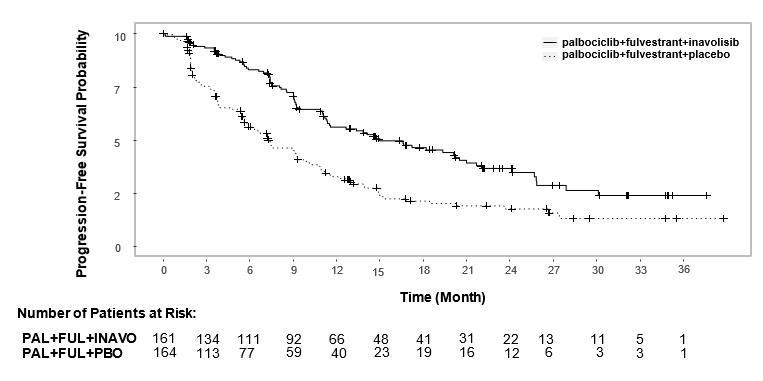
16. How is Ibrance Tablets supplied
IBRANCE is supplied in the following strengths and package configurations:
| Package Configuration | Tablet Strength (mg) | NDC | Tablet Description |
|---|---|---|---|
|
Monthly box containing 3 weekly blister packs of 7 tablets each (21 tablets total) |
125 |
NDC 0069-0688-03 |
Oval, light purple, film-coated tablets debossed with "Pfizer" on one side and "PBC 125" on the other side. |
|
Monthly box containing 3 weekly blister packs of 7 tablets each (21 tablets total) |
100 |
NDC 0069-0486-03 |
Oval, green, film-coated tablets debossed with "Pfizer" on one side and "PBC 100" on the other side. |
|
Monthly box containing 3 weekly blister packs of 7 tablets each (21 tablets total) |
75 |
NDC 0069-0284-03 |
Round, light purple, film-coated tablets debossed with "Pfizer" on one side and "PBC 75" on the other side. |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Myelosuppression/Infection
- •
- Advise patients to immediately report any signs or symptoms of myelosuppression or infection, such as fever, chills, dizziness, shortness of breath, weakness, or any increased tendency to bleed and/or to bruise [see Warnings and Precautions (5.1)].
Interstitial Lung Disease/Pneumonitis
- •
- Advise patients to immediately report new or worsening respiratory symptoms [see Warnings and Precautions (5.2)].
Drug Interactions
- •
- Grapefruit may interact with IBRANCE. Patients should not consume grapefruit products while on treatment with IBRANCE.
- •
- Inform patients to avoid strong CYP3A inhibitors and strong CYP3A inducers.
- •
- Advise patients to inform their healthcare providers of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products [see Drug Interactions (7)].
Dosing and Administration
- •
- Inform patients that IBRANCE tablets may be taken with or without food.
- •
- If the patient vomits or misses a dose, an additional dose should not be taken. The next prescribed dose should be taken at the usual time. IBRANCE tablets should be swallowed whole (do not chew, crush, or split them prior to swallowing). No tablet should be ingested if it is broken, cracked, or otherwise not intact.
- •
- Pre/perimenopausal women treated with IBRANCE should also be treated with LHRH agonists [see Dosage and Administration (2.1)].
- •
- When IBRANCE is used in combination, refer to the other products’ Full Prescribing Information for dosing and administration information [see Dosage and Administration (2.1, 2.2)].
Pregnancy, Lactation, and Infertility
- •
- Embryo-Fetal Toxicity
- o
- Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with IBRANCE therapy and for at least 3 weeks after the last dose. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.3) and Use in Specific Populations (8.1 and 8.3)].
- o
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment with IBRANCE and for at least 3 months after the last dose [see Use in Specific Populations (8.3)].
- •
- Lactation: Advise women not to breastfeed during treatment with IBRANCE and for 3 weeks after the last dose [see Use in Specific Populations (8.2)].
- •
- Infertility: Inform males of reproductive potential that IBRANCE may cause infertility and to consider sperm preservation before taking IBRANCE [see Use in Specific Populations (8.3)].
This product's labeling may have been updated. For full prescribing information, please visit www.pfizer.com. For medical information about IBRANCE, please visit www.pfizermedinfo.com or call 1-800-438-1985.

LAB-1371-6.0
| This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: April 2025 | ||
|
PATIENT INFORMATION
|
||
|
What is the most important information I should know about IBRANCE? |
||
|
IBRANCE may cause serious side effects, including: |
||
|
Low white blood cell counts (neutropenia). Low white blood cell counts are very common when taking IBRANCE and may cause serious infections that can lead to death. Your healthcare provider should check your white blood cell counts before and during treatment. |
||
|
If you develop low white blood cell counts during treatment with IBRANCE, your healthcare provider may stop your treatment, decrease your dose, or may tell you to wait to begin your treatment cycle. Tell your healthcare provider right away if you have signs and symptoms of low white blood cell counts or infections such as fever and chills. |
||
|
Lung problems (pneumonitis). IBRANCE may cause severe or life-threatening inflammation of the lungs during treatment that can lead to death. Tell your healthcare provider right away if you have any new or worsening symptoms, including:
|
||
|
Your healthcare provider may interrupt or stop treatment with IBRANCE completely if your symptoms are severe. |
||
|
See "What are the possible side effects of IBRANCE?" for more information about side effects. |
||
|
What is IBRANCE? |
||
|
IBRANCE is a prescription medicine used: In adults to treat hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer that has spread to other parts of the body (metastatic) in combination with:
In adults with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative and with an abnormal phosphatidylinositol-3-kinase catalytic subunit alpha (PIK3CA) gene breast cancer that has spread to nearby tissue or lymph nodes (locally advanced), or to other parts of the body (metastatic) in combination with:
|
||
|
It is not known if IBRANCE is safe and effective in children. |
||
|
What should I tell my healthcare provider before taking IBRANCE? Before taking IBRANCE, tell your healthcare provider about all of your medical conditions, including if you:
|
||
|
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. IBRANCE and other medicines may affect each other causing side effects. Know the medicines you take. Keep a list of them to show your healthcare provider or pharmacist when you get a new medicine. |
||
|
How should I take IBRANCE?
|
||
|
IBRANCE may cause serious side effects. See "What is the most important information I should know about IBRANCE?" |
||
|
The most common side effects of IBRANCE when used with either letrozole or fulvestrant include:
|
||
|
|
|
|
|
|
|
The most common side effects when inavolisib is added to IBRANCE plus fulvestrant include:
IBRANCE may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider about family planning options before starting IBRANCE if this is a concern for you. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. |
||
|
These are not all of the possible side effects of IBRANCE. |
||
|
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
|
How should I store IBRANCE?
|
||
|
Keep IBRANCE and all medicines out of the reach of children. |
||
|
General information about the safe and effective use of IBRANCE |
||
|
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use IBRANCE for a condition for which it was not prescribed. Do not give IBRANCE to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for more information about IBRANCE that is written for health professionals. |
||
|
What are the ingredients in IBRANCE? |
||
|
Active ingredient: palbociclib |
||
|
Inactive ingredients: microcrystalline cellulose, colloidal silicon dioxide, crospovidone, magnesium stearate, succinic acid, HPMC 2910/hypromellose, titanium dioxide, triacetin, and FD&C Blue #2/Indigo Carmine Aluminum Lake. In addition, the 75 mg and 125 mg tablets contain red iron oxide and the 100 mg tablets contain yellow iron oxide.
|
||
 |
||
|
LAB-1372-6.0 For more information, go to www.pfizer.com or call 1-800-438-1985. |
||
PRINCIPAL DISPLAY PANEL - Topper Card
Your Monthly Box
Includes 3 weekly packs of
IBRANCE® (palbociclib) medication.
Fill in the dates below to help track your treatment cycle.
Treatment Cycle
Week 1 Week 2 Week 3
IBRANCE
Week 4
No IBRANCE
My Treatment Cycle Tracker
I start my Week 1
treatment cycle on:
________ / ______
MONTH DAY
After Week 4, I will start my
next treatment cycle on:
________ / ______
MONTH DAY
PAA119239
PRINCIPAL DISPLAY PANEL - 75 mg Tablet Dose Pack
Pfizer
NDC 0069-0284-07
IBRANCE®
(palbociclib)
tablets
75 mg per tablet
This weekly pack contains:
7 tablets
Rx only

PRINCIPAL DISPLAY PANEL - 75 mg Tablet Dose Pack Carton
Pfizer
NDC 0069-0284-03
IBRANCE®
(palbociclib)
tablets
75 mg
21 tablets
Monthly Box Contains: 3 individual weekly
packs. Each pack contains 7 IBRANCE tablets
(75 mg per tablet).
Rx only
Safety
Label
PRINCIPAL DISPLAY PANEL - 100 mg Tablet Dose Pack
Pfizer
NDC 0069-0486-07
IBRANCE®
(palbociclib)
tablets
100 mg per tablet
This weekly pack contains:
7 tablets
Rx only

PRINCIPAL DISPLAY PANEL - 100 mg Tablet Dose Pack Carton
Pfizer
NDC 0069-0486-03
IBRANCE®
(palbociclib)
tablets
100 mg
21 tablets
Monthly Box Contains: 3 individual weekly
packs. Each pack contains 7 IBRANCE tablets
(100 mg per tablet).
Rx only
Safety
Label
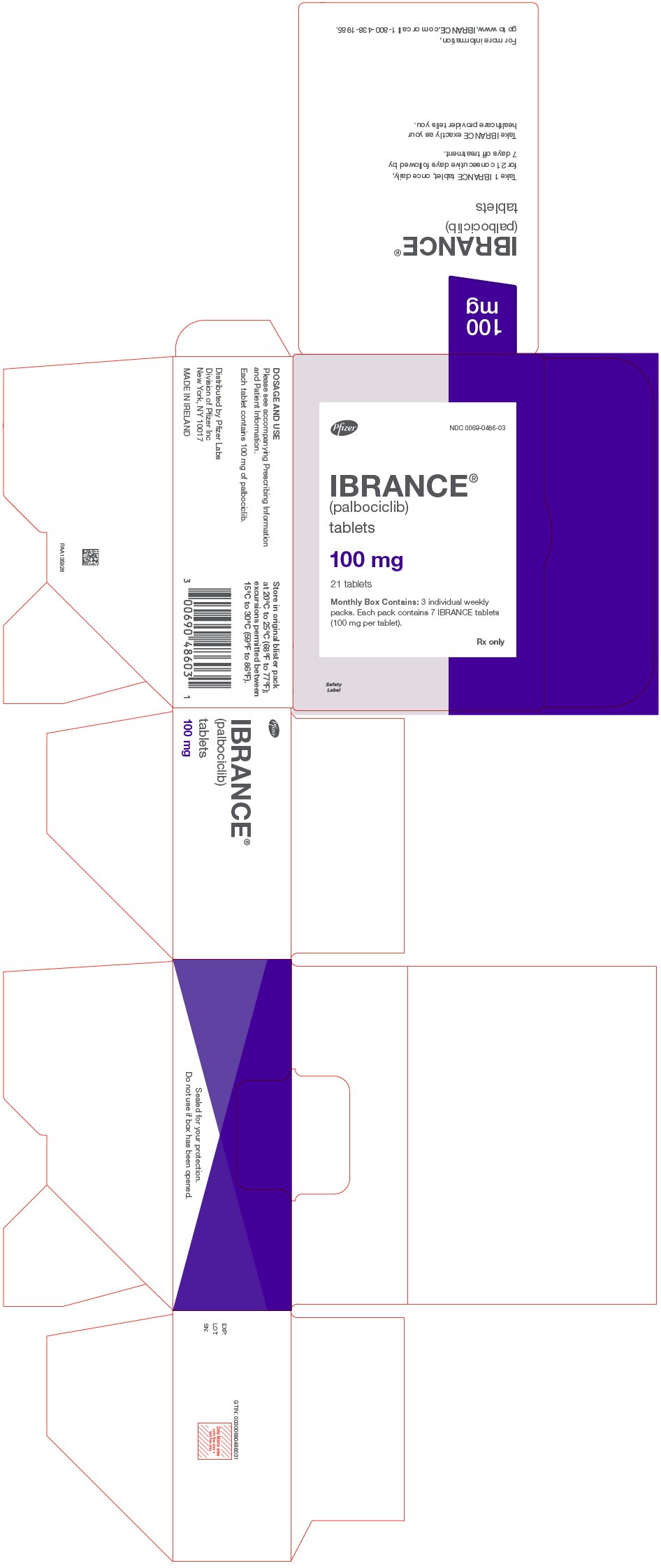

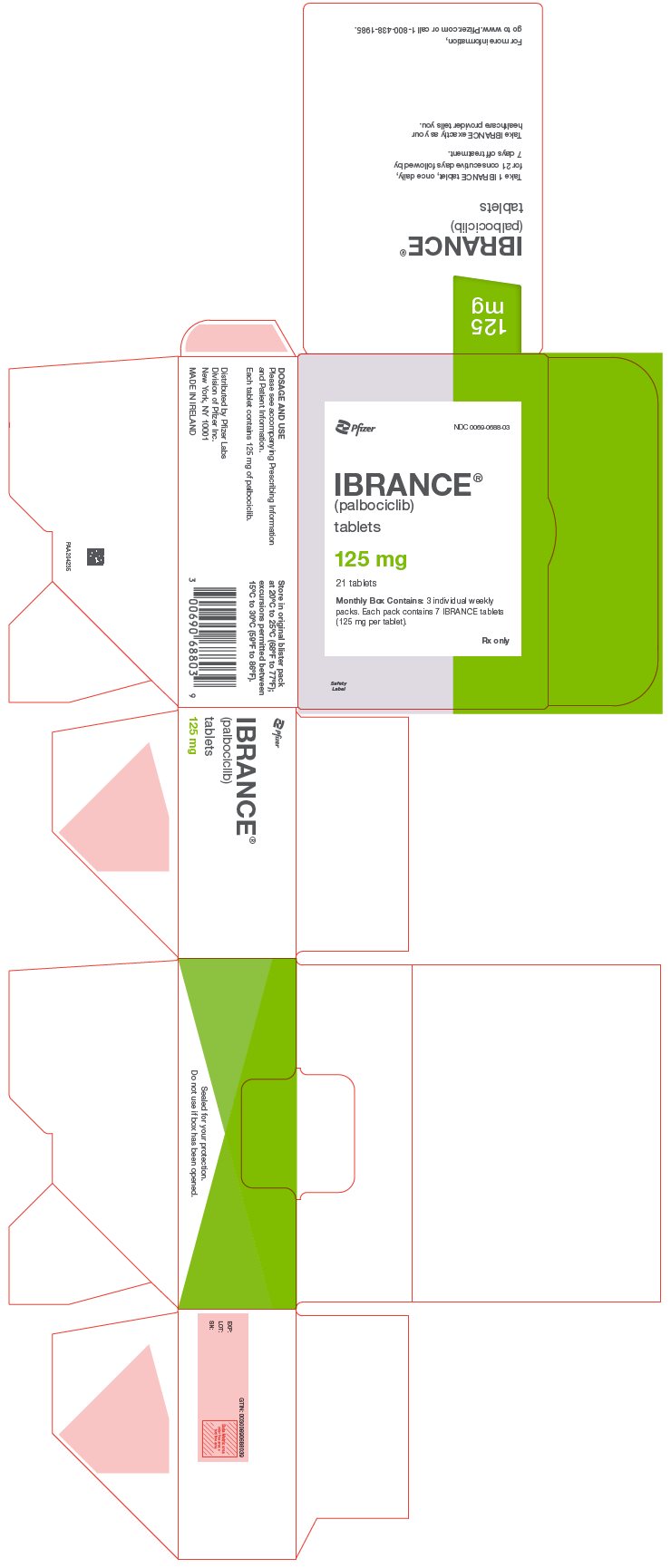
| IBRANCE
palbociclib tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| IBRANCE
palbociclib tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| IBRANCE
palbociclib tablet, film coated |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Ireland Pharmaceuticals Unlimited Company | 985052076 | ANALYSIS(0069-0284, 0069-0486, 0069-0688) , API MANUFACTURE(0069-0284, 0069-0486, 0069-0688) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Manufacturing Deutschland GmbH | 341970073 | ANALYSIS(0069-0284, 0069-0486, 0069-0688) , MANUFACTURE(0069-0284, 0069-0486, 0069-0688) , PACK(0069-0284, 0069-0486, 0069-0688) , LABEL(0069-0284, 0069-0486, 0069-0688) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Asia Manufacturing Pte Ltd | 936889401 | API MANUFACTURE(0069-0284, 0069-0486, 0069-0688) , ANALYSIS(0069-0284, 0069-0486, 0069-0688) | |
Frequently asked questions
- How long does Ibrance extend life?
- Is Verzenio better than Ibrance?
- What are 10 key Ibrance side effects to watch out for?
- Will insurance pay for the cost of Ibrance?
- Can you take Verzenio after Ibrance fails?
- How long can you take Ibrance?
- Is Ibrance a form of chemo?
- How common is hair loss with Ibrance?
- How effective is Ibrance?
More about Ibrance (palbociclib)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (85)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- FDA approval history
- Drug class: CDK 4/6 inhibitors
- Breastfeeding
- En español