Desloratadine: Package Insert / Prescribing Info
Package insert / product label
Dosage form: tablet
Drug class: Antihistamines
Medically reviewed by Drugs.com. Last updated on Jul 17, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
DESLORATADINE tablets, for oral use
Initial U.S. Approval: 2001
Indications and Usage for Desloratadine
Desloratadine Tablets are a histamine-1 (H1) receptor antagonist indicated for:
- •
- Seasonal Allergic Rhinitis: relief of nasal and non-nasal symptoms in patients 12 years of age and older. (1.1)
- •
- Perennial Allergic Rhinitis: relief of nasal and non-nasal symptoms in patients 12 years of age and older. (1.2)
- •
- Chronic Idiopathic Urticaria: symptomatic relief of pruritus, reduction in the number of hives, and size of hives in patients 12 years of age and older. (1.3)
Desloratadine Dosage and Administration
Dosage (by age):
Adults and Adolescents 12 Years of Age and Over:
- •
- Desloratadine Tablets - one 5 mg tablet once daily (2)
Dosage Forms and Strengths
- •
- Desloratadine Tablets - 5 mg (3)
Warnings and Precautions
- •
- Hypersensitivity reactions including rash, pruritus, urticaria, edema, dyspnea, and anaphylaxis have been reported. In such cases, stop Desloratadine Tablets at once and consider alternative treatments. (5.1)
Adverse Reactions/Side Effects
- •
- The most common adverse reactions (reported in ≥2% of adult and adolescent patients with allergic rhinitis and greater than placebo) were pharyngitis, dry mouth, myalgia, fatigue, somnolence, dysmenorrhea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Virtus Pharmaceuticals, LLC at 1-888-848-3593 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2020
Full Prescribing Information
1. Indications and Usage for Desloratadine
1.1 Seasonal Allergic Rhinitis
Desloratadine Tablets are indicated for the relief of the nasal and non-nasal symptoms of seasonal allergic rhinitis in patients 12 years of age and older.
2. Desloratadine Dosage and Administration
Desloratadine Tablets may be taken without regard to meals.
2.1 Adults and Adolescents 12 Years of Age and Over
The recommended dose of Desloratadine Tablets is one 5-mg tablet once daily.
2.5 Adults with Hepatic or Renal Impairment
In adult patients with liver or renal impairment, a starting dose of one 5 mg tablet every other day is recommended based on pharmacokinetic data. Dosing recommendation for children with liver or renal impairment cannot be made due to lack of data [see Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
Desloratadine Tablets are light blue round tablets debossed with "5" containing 5 mg desloratadine.
4. Contraindications
Desloratadine Tablets are contraindicated in patients who are hypersensitive to this medication or to any of its ingredients or to loratadine [see Warnings and Precautions (5.1) and Adverse Reactions (6.2)].
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Hypersensitivity reactions including rash, pruritus, urticaria, edema, dyspnea, and anaphylaxis have been reported after administration of desloratadine. If such a reaction occurs, therapy with Desloratadine Tablets should be stopped and alternative treatment should be considered. [See Adverse Reactions (6.2).]
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the label:
- •
- Hypersensitivity reactions. [See Warnings and Precautions (5.1).]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adults and Adolescents
Allergic Rhinitis: In multiple-dose placebo-controlled trials, 2834 patients ages 12 years or older received Desloratadine Tablets at doses of 2.5 mg to 20 mg daily, of whom 1655 patients received the recommended daily dose of 5 mg. In patients receiving 5 mg daily, the rate of adverse events was similar between Desloratadine and placebo-treated patients. The percent of patients who withdrew prematurely due to adverse events was 2.4% in the Desloratadine group and 2.6% in the placebo group. There were no serious adverse events in these trials in patients receiving desloratadine. All adverse events that were reported by greater than or equal to 2% of patients who received the recommended daily dose of Desloratadine Tablets (5 mg once daily), and that were more common with Desloratadine Tablets than placebo, are listed in Table 1.
| Adverse Event | Desloratadine Tablets 5 mg
(n=1655) | Placebo
(n=1652) |
|---|---|---|
|
Infections and Infestations
|
4.1% |
2.0% |
|
Nervous System Disorders
|
2.1% |
1.8% |
|
Gastrointestinal Disorders
|
3.0% |
1.9% |
|
Musculoskeletal and Connective Tissue Disorders
|
2.1% |
1.8% |
|
Reproductive System and Breast Disorders
|
2.1% |
1.6% |
|
General Disorders and Administration Site Conditions
|
2.1% |
1.2% |
The frequency and magnitude of laboratory and electrocardiographic abnormalities were similar in Desloratadine and placebo-treated patients. There were no differences in adverse events for subgroups of patients as defined by gender, age, or race.
Chronic Idiopathic Urticaria: In multiple-dose, placebo-controlled trials of chronic idiopathic urticaria, 211 patients ages 12 years or older received Desloratadine Tablets and 205 received placebo. Adverse events that were reported by greater than or equal to 2% of patients who received Desloratadine Tablets and that were more common with Desloratadine than placebo were (rates for Desloratadine and placebo, respectively): headache (14%, 13%), nausea (5%, 2%), fatigue (5%, 1%), dizziness (4%, 3%), pharyngitis (3%, 2%), dyspepsia (3%, 1%), and myalgia (3%, 1%).
Pediatrics
Two hundred and forty-six pediatric subjects 6 months to 11 years of age received Desloratadine Oral Solution for 15 days in three placebo-controlled clinical trials. Pediatric subjects aged 6 to 11 years received 2.5 mg once a day, subjects aged 1 to 5 years received 1.25 mg once a day, and subjects 6 to 11 months of age received 1.0 mg once a day.
In subjects 6 to 11 years of age, no individual adverse event was reported by 2 percent or more of the subjects.
In subjects 2 to 5 years of age, adverse events reported for Desloratadine and placebo in at least 2 percent of subjects receiving Desloratadine Oral Solution and at a frequency greater than placebo were fever (5.5%, 5.4%), urinary tract infection (3.6%, 0%) and varicella (3.6%, 0%).
In subjects 12 months to 23 months of age, adverse events reported for the Desloratadine product and placebo in at least 2 percent of subjects receiving Desloratadine Oral Solution and at a frequency greater than placebo were fever (16.9%, 12.9%), diarrhea (15.4%, 11.3%), upper respiratory tract infections (10.8%, 9.7%), coughing (10.8%, 6.5%), appetite increased (3.1%, 1.6%), emotional lability (3.1%, 0%), epistaxis (3.1%, 0%), parasitic infection (3.1%, 0%), pharyngitis (3.1%, 0%), rash maculopapular (3.1%, 0%).
In subjects 6 months to 11 months of age, adverse events reported for Desloratadine and placebo in at least 2 percent of subjects receiving Desloratadine Oral Solution and at a frequency greater than placebo were upper respiratory tract infections (21.2%, 12.9%), diarrhea (19.7%, 8.1%), fever (12.1%, 1.6%), irritability (12.1%, 11.3%), coughing (10.6%, 9.7%), somnolence (9.1%, 8.1%), bronchitis (6.1%, 0%), otitis media (6.1%, 1.6%), vomiting (6.1%, 3.2%), anorexia (4.5%, 1.6%), pharyngitis (4.5%, 1.6%), insomnia (4.5%, 0%), rhinorrhea (4.5%, 3.2%), erythema (3.0%, 1.6%), and nausea (3.0%, 0%).
There were no clinically meaningful changes in any electrocardiographic parameter, including the QTc interval. Only one of the 246 pediatric subjects receiving Desloratadine Oral Solution in the clinical trials discontinued treatment because of an adverse event.
6.2 Post-Marketing Experience
Because adverse events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. The following spontaneous adverse events have been reported during the marketing of desloratadine:
Cardiac disorders: tachycardia, palpitations
Respiratory, thoracic and mediastinal disorders: dyspnea
Skin and subcutaneous tissue disorders: rash, pruritus
Nervous system disorders: psychomotor hyperactivity, movement disorders (including dystonia, tics, and extrapyramidal symptoms), seizures (reported in patients with and without a known seizure disorder)
Immune system disorders: hypersensitivity reactions (such as urticaria, edema and anaphylaxis)
Investigations: elevated liver enzymes including bilirubin
Hepatobiliary disorders: hepatitis
Metabolism and nutrition disorders: increased appetite
Related/similar drugs
7. Drug Interactions
7.1 Inhibitors of Cytochrome P450 3A4
In controlled clinical studies co-administration of desloratadine with ketoconazole, erythromycin, or azithromycin resulted in increased plasma concentrations of desloratadine and 3 hydroxydesloratadine, but there were no clinically relevant changes in the safety profile of desloratadine. [See Clinical Pharmacology (12.3).]
7.2 Fluoxetine
In controlled clinical studies co-administration of desloratadine with fluoxetine, a selective serotonin reuptake inhibitor (SSRI), resulted in increased plasma concentrations of desloratadine and 3 hydroxydesloratadine, but there were no clinically relevant changes in the safety profile of desloratadine. [See Clinical Pharmacology (12.3).]
7.3 Cimetidine
In controlled clinical studies co-administration of desloratadine with cimetidine, a histamine H2-receptor antagonist, resulted in increased plasma concentrations of desloratadine and 3 hydroxydesloratadine, but there were no clinically relevant changes in the safety profile of desloratadine. [See Clinical Pharmacology (12.3).]
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
The limited available data with Desloratadine in pregnant women are not sufficient to inform a drug-associated risk for major birth defects and miscarriage. There are no adequate and well-controlled studies in pregnant women. Desloratadine given during organogenesis to pregnant rats was not teratogenic at the summed area under the concentration-time curve (AUC)-based exposures of desloratadine and its metabolite approximately 320 times that at the recommended human daily oral dose (RHD) of 5 mg/day. Desloratadine given during organogenesis to pregnant rabbits was not teratogenic at the AUC-based exposures of desloratadine approximately 230 times that at the RHD. Desloratadine given to pregnant rats during organogenesis through lactation resulted in reduced body weight and slow righting reflex of F1 pups at the summed AUC-based exposures of desloratadine and its metabolite approximately 70 times or greater than that at the RHD [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Desloratadine was given orally during organogenesis to pregnant rats at doses of 6, 24 and 48 mg/kg/day (approximately 50, 200 and 320 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD). No fetal malformations were present. Reduced fetal weights and skeletal variations noted at doses of 24 and 48 mg/kg/day were likely secondary to the maternal toxicities of reduced body weight gain and food consumption observed at the same doses. Desloratadine was also given orally during organogenesis to pregnant rabbits at doses of 15, 30 and 60 mg/kg/day (approximately 30, 70 and 230 times the AUC-based exposure of desloratadine at the RHD). No adverse effects to the fetus were noted. Reduced maternal body weight gain was noted in rabbits at 60 mg/kg/day. In a peri- and post-natal development study, desloratadine was given to rats orally during the perinatal (Gestation Day 6) through lactation periods (Postpartum Day 21) at doses of 3, 9 and 18 mg/kg/day. Reduced body weight and slow righting reflex were reported in F1 pups at doses of 9 mg/kg/day or greater (approximately 70 times or greater than the summed AUC-based exposure of desloratadine and its metabolite at the RHD). Desloratadine had no effect on F1 pup development at 3 mg/kg/day (approximately 10 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD). Maternal toxicities including reduced body weight gain and food consumption were noted at 18 mg/kg/day for F0 dams. F1 offspring were subsequently mated and there was no developmental toxicity for F2 pups observed.
8.2 Lactation
Risk Summary
Desloratadine passes into breast milk. There are not sufficient data on the effects of desloratadine on the breastfed infant or the effects of desloratadine on milk production. The decision should be made whether to discontinue nursing or to discontinue desloratadine, taking into account the developmental and health benefits of breastfeeding, the nursing mother's clinical need, and any potential adverse effects on the breastfed infant from desloratadine or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Infertility
There are no data available on human infertility associated with desloratadine.
There were no clinically relevant effects of desloratadine on female fertility in rats. A male specific decrease in fertility occurred at an oral desloratadine dose of 12 mg/kg or greater in rats (approximately 65 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD). Male fertility was unaffected at a desloratadine dose of 3 mg/kg (approximately 10 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD). [See Nonclinical Toxicology (13.1).]
8.4 Pediatric Use
The recommended dose of Desloratadine Oral Solution in the pediatric population is based on cross-study comparison of the plasma concentration of Desloratadine in adults and pediatric subjects. The safety of Desloratadine Oral Solution has been established in 246 pediatric subjects aged 6 months to 11 years in three placebo-controlled clinical studies. Since the course of seasonal and perennial allergic rhinitis and chronic idiopathic urticaria and the effects of Desloratadine are sufficiently similar in the pediatric and adult populations, it allows extrapolation from the adult efficacy data to pediatric patients. The effectiveness of Desloratadine Oral Solution in these age groups is supported by evidence from adequate and well-controlled studies of Desloratadine Tablets in adults. The safety and effectiveness of Desloratadine Tablets or Desloratadine Oral Solution have not been demonstrated in pediatric patients less than 6 months of age. [See Clinical Pharmacology (12.3).]
8.5 Geriatric Use
Clinical studies of desloratadine did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. [See Clinical Pharmacology (12.3).]
8.6 Renal Impairment
Dosage adjustment for patients with renal impairment is recommended [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Dosage adjustment for patients with hepatic impairment is recommended [see Dosage and Administration (2.5) and Clinical Pharmacology (12.3)].
9. Drug Abuse and Dependence
There is no information to indicate that abuse or dependency occurs with Desloratadine Tablets.
10. Overdosage
In the event of overdose, consider standard measures to remove any unabsorbed drug. Symptomatic and supportive treatment is recommended. Desloratadine and 3-hydroxydesloratadine are not eliminated by hemodialysis.
Information regarding acute overdosage is limited to experience from post-marketing adverse event reports and from clinical trials conducted during the development of the Desloratadine product. In a dose-ranging trial, at doses of 10 mg and 20 mg/day somnolence was reported.
In another study, no clinically relevant adverse events were reported in normal male and female volunteers who were given single daily doses of Desloratadine 45 mg for 10 days [see Clinical Pharmacology (12.2)].
11. Desloratadine Description
Desloratadine Tablets are light blue, round, tablets containing 5 mg desloratadine, an antihistamine, to be administered orally. Desloratadine Tablets also contain the following excipients: microcrystalline cellulose NF, pregelatinized starch NF, croscarmellose sodium NF, talc USP, zinc stearate, USP and FD&C Blue #2 HT 11-14%.
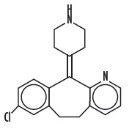
Desloratadine is a white to off-white powder that is slightly soluble in water, but very soluble in ethanol and propylene glycol. It has an empirical formula: C19H19ClN2 and a molecular weight of 310.8. The chemical name is 8-chloro-6,11-dihydro-11-(4-piperdinylidene)-5H-benzo[5,6]cyclohepta[1,2-b]pyridine and has the following structure:
12. Desloratadine - Clinical Pharmacology
12.1 Mechanism of Action
Desloratadine is a long-acting tricyclic histamine antagonist with selective H1-receptor histamine antagonist activity. Receptor binding data indicates that at a concentration of 2-3 ng/mL (7 nanomolar), desloratadine shows significant interaction with the human histamine H1-receptor. Desloratadine inhibited histamine release from human mast cells in vitro. Results of a radiolabeled tissue distribution study in rats and a radioligand H1-receptor binding study in guinea pigs showed that desloratadine did not readily cross the blood brain barrier. The clinical significance of this finding is unknown.
12.2 Pharmacodynamics
Wheal and Flare: Human histamine skin wheal studies following single and repeated 5-mg doses of desloratadine have shown that the drug exhibits an antihistaminic effect by 1 hour; this activity may persist for as long as 24 hours. There was no evidence of histamine-induced skin wheal tachyphylaxis within the desloratadine 5-mg group over the 28-day treatment period. The clinical relevance of histamine wheal skin testing is unknown.
Effects on QTc: Single daily doses of 45 mg were given to normal male and female volunteers for 10 days. All ECGs obtained in this study were manually read in a blinded fashion by a cardiologist. In Desloratadine-treated subjects, there was an increase in mean heart rate of 9.2 bpm relative to placebo. The QT interval was corrected for heart rate (QTc) by both the Bazett and Fridericia methods. Using the QTc (Bazett) there was a mean increase of 8.1 msec in Desloratadine-treated subjects relative to placebo. Using QTc (Fridericia) there was a mean increase of 0.4 msec in Desloratadine-treated subjects relative to placebo. No clinically relevant adverse events were reported.
12.3 Pharmacokinetics
Absorption
Following oral administration of a desloratadine 5 mg tablet once daily for 10 days to normal healthy volunteers, the mean time to maximum plasma concentrations (Tmax) occurred at approximately 3 hours post dose and mean steady state peak plasma concentrations (Cmax) and AUC of 4 ng/mL and 56.9 ng∙hr/mL were observed, respectively. Neither food nor grapefruit juice had an effect on the bioavailability (Cmax and AUC) of desloratadine.
The pharmacokinetic profile of Desloratadine Oral Solution was evaluated in a three-way crossover study in 30 adult volunteers. A single dose of 10 mL of Desloratadine Oral Solution containing 5 mg of desloratadine was bioequivalent to a single dose of 5 mg Desloratadine Tablet. Food had no effect on the bioavailability (AUC and Cmax) of Desloratadine Oral Solution.
Distribution
Desloratadine and 3-hydroxydesloratadine are approximately 82% to 87% and 85% to 89% bound to plasma proteins, respectively. Protein binding of desloratadine and 3-hydroxydesloratadine was unaltered in subjects with impaired renal function.
Metabolism
Desloratadine (a major metabolite of loratadine) is extensively metabolized to 3-hydroxydesloratadine, an active metabolite, which is subsequently glucuronidated. The enzyme(s) responsible for the formation of 3-hydroxydesloratadine have not been identified. Data from clinical trials indicate that a subset of the general population has a decreased ability to form 3-hydroxydesloratadine, and are poor metabolizers of desloratadine. In pharmacokinetic studies (n=3748), approximately 6% of subjects were poor metabolizers of desloratadine (defined as a subject with an AUC ratio of 3-hydroxydesloratadine to desloratadine less than 0.1, or a subject with a desloratadine half-life exceeding 50 hours). These pharmacokinetic studies included subjects between the ages of 2 and 70 years, including 977 subjects aged 2 to 5 years, 1575 subjects aged 6 to 11 years, and 1196 subjects aged 12 to 70 years. There was no difference in the prevalence of poor metabolizers across age groups. The frequency of poor metabolizers was higher in Blacks (17%, n=988) as compared to Caucasians (2%, n=1,462) and Hispanics (2%, n=1,063). The median exposure (AUC) to desloratadine in the poor metabolizers was approximately 6-fold greater than in the subjects who are not poor metabolizers. Subjects who are poor metabolizers of desloratadine cannot be prospectively identified and will be exposed to higher levels of desloratadine following dosing with the recommended dose of desloratadine. In multidose clinical safety studies, where metabolizer status was identified, a total of 94 poor metabolizers and 123 normal metabolizers were enrolled and treated with Desloratadine Oral Solution for 15-35 days. In these studies, no overall differences in safety were observed between poor metabolizers and normal metabolizers. Although not seen in these studies, an increased risk of exposure-related adverse events in patients who are poor metabolizers cannot be ruled out.
Elimination
The mean plasma elimination half-life of desloratadine was approximately 27 hours. Cmax and AUC values increased in a dose proportional manner following single oral doses between 5 and 20 mg. The degree of accumulation after 14 days of dosing was consistent with the half-life and dosing frequency. A human mass balance study documented a recovery of approximately 87% of the 14C-desloratadine dose, which was equally distributed in urine and feces as metabolic products. Analysis of plasma 3-hydroxydesloratadine showed similar Tmax and half-life values compared to desloratadine.
Special Populations
Geriatric Subjects: In older subjects (≥65 years old; n=17) following multiple-dose administration of Desloratadine Tablets, the mean Cmax and AUC values for desloratadine were 20% greater than in younger subjects (<65 years old). The oral total body clearance (CL/F) when normalized for body weight was similar between the two age groups. The mean plasma elimination half-life of desloratadine was 33.7 hr in subjects ≥65 years old. The pharmacokinetics for 3-hydroxydesloratadine appeared unchanged in older versus younger subjects. These age-related differences are unlikely to be clinically relevant and no dosage adjustment is recommended in elderly subjects.
Pediatric Subjects: In subjects 6 to 11 years old, a single dose of 5 mL of Desloratadine Oral solution containing 2.5 mg of desloratadine, resulted in desloratadine plasma concentrations similar to those achieved in adults administered a single 5-mg Desloratadine Tablet. In subjects 2 to 5 years old, a single dose of 2.5 mL of Desloratadine Oral solution containing 1.25 mg of desloratadine, resulted in desloratadine plasma concentrations similar to those achieved in adults administered a single 5-mg Desloratadine Tablet. However, the Cmax and AUC of the metabolite (3-hydroxydesloratadine) were 1.27 and 1.61 times higher for the 5-mg dose of Oral solution administered in adults compared to the Cmax and AUC obtained in children 2 to 11 years of age receiving 1.25-2.5 mg of Desloratadine Oral solution.
A single dose of either 2.5 mL or 1.25 mL of Desloratadine Oral solution containing 1.25 mg or 0.625 mg, respectively, of desloratadine was administered to subjects 6 to 11 months of age and 12 to 23 months of age. The results of a population pharmacokinetic analysis indicated that a dose of 1 mg for subjects aged 6 to 11 months and 1.25 mg for subjects 12 to 23 months of age is required to obtain desloratadine plasma concentrations similar to those achieved in adults administered a single 5-mg dose of Desloratadine Oral solution.
Renally Impaired: Desloratadine pharmacokinetics following a single dose of 7.5 mg were characterized in patients with mild (n=7; creatinine clearance 51-69 mL/min/1.73 m2), moderate (n=6; creatinine clearance 34-43 mL/min/1.73 m2), and severe (n=6; creatinine clearance 5-29 mL/min/1.73 m2) renal impairment or hemodialysis dependent (n=6) patients. In patients with mild and moderate renal impairment, median Cmax and AUC values increased by approximately 1.2- and 1.9-fold, respectively, relative to subjects with normal renal function. In patients with severe renal impairment or who were hemodialysis dependent, Cmax and AUC values increased by approximately 1.7- and 2.5-fold, respectively. Minimal changes in 3-hydroxydesloratadine concentrations were observed. Desloratadine and 3-hydroxydesloratadine were poorly removed by hemodialysis. Plasma protein binding of desloratadine and 3-hydroxydesloratadine was unaltered by renal impairment. Dosage adjustment for patients with renal impairment is recommended [see Dosage and Administration (2.5)].
Hepatically Impaired: Desloratadine pharmacokinetics were characterized following a single oral dose in patients with mild (n=4), moderate (n=4), and severe (n=4) hepatic impairment as defined by the Child-Pugh classification of hepatic function and 8 subjects with normal hepatic function. Patients with hepatic impairment, regardless of severity, had approximately a 2.4-fold increase in AUC as compared with normal subjects. The apparent oral clearance of desloratadine in patients with mild, moderate, and severe hepatic impairment was 37%, 36%, and 28% of that in normal subjects, respectively. An increase in the mean elimination half-life of desloratadine in patients with hepatic impairment was observed. For 3-hydroxydesloratadine, the mean Cmax and AUC values for patients with hepatic impairment were not statistically significantly different from subjects with normal hepatic function. Dosage adjustment for patients with hepatic impairment is recommended [see Dosage and Administration (2.5)].
Gender: Female subjects treated for 14 days with Desloratadine Tablets had 10% and 3% higher desloratadine Cmax and AUC values, respectively, compared with male subjects. The 3-hydroxydesloratadine Cmax and AUC values were also increased by 45% and 48%, respectively, in females compared with males. However, these apparent differences are not likely to be clinically relevant and therefore no dosage adjustment is recommended.
Race: Following 14 days of treatment with Desloratadine Tablets, the Cmax and AUC values for desloratadine were 18% and 32% higher, respectively, in Blacks compared with Caucasians. For 3-hydroxydesloratadine there was a corresponding 10% reduction in Cmax and AUC values in Blacks compared to Caucasians. These differences are not likely to be clinically relevant and therefore no dose adjustment is recommended.
Drug Interactions: In two controlled crossover clinical pharmacology studies in healthy male (n=12 in each study) and female (n=12 in each study) volunteers, desloratadine 7.5 mg (1.5 times the daily dose) once daily was coadministered with erythromycin 500 mg every 8 hours or ketoconazole 200 mg every 12 hours for 10 days. In three separate controlled, parallel group clinical pharmacology studies, desloratadine at the clinical dose of 5 mg has been coadministered with azithromycin 500 mg followed by 250 mg once daily for 4 days (n=18) or with fluoxetine 20 mg once daily for 7 days after a 23-day pretreatment period with fluoxetine (n=18) or with cimetidine 600 mg every 12 hours for 14 days (n=18) under steady-state conditions to normal healthy male and female volunteers. Although increased plasma concentrations (Cmax and AUC0-24 hrs) of desloratadine and 3-hydroxydesloratadine were observed (see Table 2), there were no clinically relevant changes in the safety profile of desloratadine, as assessed by electrocardiographic parameters (including the corrected QT interval), clinical laboratory tests, vital signs, and adverse events.
| Desloratadine | 3-Hydroxydesloratadine | |||
|---|---|---|---|---|
| Cmax | AUC0-24 hrs | Cmax | AUC0-24 hrs | |
|
Erythromycin |
+24% |
+14% |
+43% |
+40% |
|
Ketoconazole |
+45% |
+39% |
+43% |
+72% |
|
Azithromycin |
+15% |
+5% |
+15% |
+4% |
|
Fluoxetine |
+15% |
+0% |
+17% |
+13% |
|
Cimetidine |
+12% |
+19% |
-11% |
-3% |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity Studies
The carcinogenic potential of desloratadine was assessed using a loratadine study in rats and a desloratadine study in mice. In a 2-year study in rats, loratadine was administered in the diet at doses up to 25 mg/kg/day (approximately 45 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD). A significantly higher incidence of hepatocellular tumors (combined adenomas and carcinomas) was observed in males given 10 mg/kg/day of loratadine (approximately 10 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD) and in males and females given 25 mg/kg/day of loratadine. The clinical significance of these findings during long-term use of desloratadine is not known. In a 2-year dietary study in mice, males and females given up to 16 mg/kg/day and 32 mg/kg/day desloratadine, respectively (approximately 30 and 70 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD, respectively), did not show significant increases in the incidence of any tumors.
Genotoxicity Studies
In genotoxicity studies with desloratadine, there was no evidence of genotoxic potential in a reverse mutation assay (Salmonella/E. coli mammalian microsome bacterial mutagenicity assay) or in 2 assays for chromosomal aberrations (human peripheral blood lymphocyte clastogenicity assay and mouse bone marrow micronucleus assay).
Impairment of Fertility
In a female fertility study, desloratadine was given to female rats orally 14 days prior to and throughout mating until Gestation Day 7 at doses of 6, 12 and 24 mg/kg/day. An increase in preimplantation loss and a decrease in number of implantations and fetuses noted at 24 mg/kg (approximately 200 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD) was likely due to maternal toxicities including reduced body weight gain and food consumption. In a male fertility study in rats, desloratadine was given orally to male rats for 70 days prior to mating and throughout the mating period (total dosing period 106-108 days) at doses of 3, 12 and 40 mg/kg/day. Reduced body weight gain, food consumption, and absolute organ weights of testes, epididymis, and cauda epididymis were noted at 40 mg/kg/day. A male-specific decrease in fertility, demonstrated by reduced female conception rates, decreased sperm numbers and motility, and histopathologic changes in testes and epididymis, occurred at a dose of 12 mg/kg or greater (approximately 65 times or greater than the summed AUC-based exposure of desloratadine and its metabolite at the RHD). Desloratadine had no effect on male fertility in rats at 3 mg/kg/day (approximately 10 times the summed AUC-based exposure of desloratadine and its metabolite at the RHD).
14. Clinical Studies
14.1 Seasonal Allergic Rhinitis
The clinical efficacy and safety of Desloratadine Tablets were evaluated in over 2300 patients 12 to 75 years of age with seasonal allergic rhinitis. A total of 1838 patients received 2.5 to 20 mg/day of Desloratadine in 4 double-blind, randomized, placebo-controlled clinical trials of 2 to 4 weeks' duration conducted in the United States. The results of these studies demonstrated the efficacy and safety of Desloratadine 5 mg in the treatment of adult and adolescent patients with seasonal allergic rhinitis. In a dose-ranging trial, Desloratadine 2.5 to 20 mg/day was studied. Doses of 5, 7.5, 10, and 20 mg/day were superior to placebo; and no additional benefit was seen at doses above 5.0 mg. In the same study, an increase in the incidence of somnolence was observed at doses of 10 mg/day and 20 mg/day (5.2% and 7.6%, respectively), compared to placebo (2.3%).
In two 4-week studies of 924 patients (aged 15 to 75 years) with seasonal allergic rhinitis and concomitant asthma, Desloratadine Tablets 5 mg once daily improved rhinitis symptoms, with no decrease in pulmonary function. This supports the safety of administering Desloratadine Tablets to adult patients with seasonal allergic rhinitis with mild to moderate asthma.
Desloratadine Tablets 5 mg once daily significantly reduced the Total Symptom Score (the sum of individual scores of nasal and non-nasal symptoms) in patients with seasonal allergic rhinitis. See Table 3.
| Treatment Group
(n) | Mean Baseline*
(SEM) | Change from Baseline†
(SEM) | Placebo Comparison
(P-value) |
|---|---|---|---|
| SEM = Standard Error of the Mean | |||
|
|||
|
Desloratadine
|
14.2 (0.3) |
-4.3 (0.3) |
P<0.01 |
|
Placebo (173) |
13.7 (0.3) |
-2.5 (0.3) | |
There were no significant differences in the effectiveness of Desloratadine Tablets 5 mg across subgroups of patients defined by gender, age, or race.
14.2 Perennial Allergic Rhinitis
The clinical efficacy and safety of Desloratadine Tablets 5 mg were evaluated in over 1300 patients 12 to 80 years of age with perennial allergic rhinitis. A total of 685 patients received 5 mg/day of Desloratadine in two double-blind, randomized, placebo-controlled clinical trials of 4 weeks' duration conducted in the United States and internationally. In one of these studies Desloratadine Tablets 5 mg once daily was shown to significantly reduce the Total Symptom Score in patients with perennial allergic rhinitis (Table 4).
| Treatment Group
(n) | Mean Baseline*
(SEM) | Change from Baseline†
(SEM) | Placebo Comparison
(P-value) |
|---|---|---|---|
| SEM = Standard Error of the Mean | |||
|
|||
|
Desloratadine
|
12.37 (0.18) |
-4.06 (0.21) |
P=0.01 |
|
Placebo (337) |
12.30 (0.18) |
-3.27 (0.21) | |
14.3 Chronic Idiopathic Urticaria
The efficacy and safety of Desloratadine Tablets 5 mg once daily was studied in 416 chronic idiopathic urticaria patients 12 to 84 years of age, of whom 211 received Desloratadine. In two double-blind, placebo-controlled, randomized clinical trials of six weeks duration, at the pre-specified one-week primary time point evaluation, Desloratadine Tablets significantly reduced the severity of pruritus when compared to placebo (Table 5). Secondary endpoints were also evaluated, and during the first week of therapy Desloratadine Tablets 5 mg reduced the secondary endpoints, "Number of Hives" and the "Size of the Largest Hive," when compared to placebo.
| Treatment Group
(n) | Mean Baseline
(SEM) | Change from Baseline*
(SEM) | Placebo Comparison
(P-value) |
|---|---|---|---|
| Pruritus scored 0 to 3 where 0=no symptom to 3=maximal symptom | |||
| SEM = Standard Error of the Mean | |||
|
|||
|
Desloratadine
|
2.19 (0.04) |
-1.05 (0.07) |
P<0.01 |
|
Placebo (110) |
2.21 (0.04) |
-0.52 (0.07) | |
16. How is Desloratadine supplied
Desloratadine Tablets: Debossed "5", light blue, round tablets that are packaged in high-density polyethylene plastic bottles of 100 (NDC 69543-107-10) and 500 (NDC 69543-107-50).
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
17.1 Information for Patients
- •
- Patients should be instructed to use Desloratadine Tablets as directed.
- •
- As there are no food effects on bioavailability, patients can be instructed that Desloratadine Tablets may be taken without regard to meals.
- •
- Patients should be advised not to increase the dose or dosing frequency as studies have not demonstrated increased effectiveness at higher doses and somnolence may occur.
Manufactured by
Belcher Pharmaceuticals, LLC.
6911 Bryan Dairy Road, Suite 210
Largo, FL 33777
Manufactured for:
Virtus Pharmaceuticals, LLC
Langhorne, PA 19047
1-888-848-3593
MADE IN USA
Rev. 10/2020
L05I-VIR
PATIENT INFORMATION
Desloratadine Tablets
Read the Patient Information that comes with Desloratadine Tablets before you start taking it and each time you get a refill. There may be new information. This leaflet is a summary of the information for patients. Your doctor or pharmacist can give you additional information. This leaflet does not take the place of talking to your doctor about your medical condition or treatment.
What are Desloratadine Tablets?
Desloratadine Tablets are a prescription medicine that contains the medicine desloratadine (an antihistamine).
Desloratadine Tablets are used to help control the symptoms of:
- •
- seasonal allergic rhinitis (sneezing, stuffy nose, runny nose and itching of the nose) in people 12 years of age and older.
- •
- perennial allergic rhinitis (sneezing, stuffy nose, runny nose and itching of the nose) in people 12 years of age and older.
- •
- chronic idiopathic urticaria (long-term itching) and to reduce the number and size of hives in people 12 years of age and older.
Desloratadine Tablets, 5 mg is not for children younger than 12 years of age.
Who should not take Desloratadine Tablets?
Do not take Desloratadine Tablets if you:
- •
- are allergic to desloratadine or any of the ingredients in Desloratadine Tablets. See the end of this leaflet for a complete list of ingredients.
- •
- are allergic to loratadine (Alavert, Claritin).
Talk to your doctor before taking this medicine if you have any questions about whether or not to take this medicine.
What should I tell my doctor before taking Desloratadine Tablets?
Before you take Desloratadine Tablets, tell your doctor if you:
- •
- have liver or kidney problems.
- •
- have any other medical conditions.
- •
- are pregnant or plan to become pregnant. It is not known if Desloratadine will harm your unborn baby. Talk to your doctor if you are pregnant or plan to become pregnant.
- •
- are breast-feeding or plan to breast-feed. Desloratadine can pass into your breast milk. Talk to your doctor about the best way to feed your baby if you take Desloratadine.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. Desloratadine may affect the way other medicines work, and other medicines may affect how Desloratadine works. Especially tell your doctor if you take:
- •
- ketoconazole (Nizoral)
- •
- erythromycin (Ery-tab, Eryc, PCE)
- •
- azithromycin (Zithromax, Zmax)
- •
- antihistamines
- •
- fluoxetine (Prozac)
- •
- cimetidine (Tagamet)
Know the medicines you take. Keep a list of your medicines and show it to your doctor and pharmacist when you get a new medicine.
How should I take Desloratadine Tablets?
- •
- Take Desloratadine Tablets exactly as your doctor tells you to take it.
- •
- Do not change your dose of Desloratadine Tablets or take more often than prescribed.
- •
- Desloratadine Tablets can be taken with or without food.
- •
- If you take too many Desloratadine Tablets, call your doctor or get medical attention right away.
What are the possible side effects of Desloratadine Tablets?
Desloratadine Tablets may cause serious side effects, including:
- •
- Allergic reactions. Stop taking Desloratadine Tablets and call your doctor right away or get emergency help if you have any of these symptoms:
- o
- rash
- o
- itching
- o
- hives
- o
- swelling of your lips, tongue, face, and throat
- o
- shortness of breath or trouble breathing
The most common side effects of Desloratadine Tablets in adults and children 12 years of age and older with allergic rhinitis include:
- •
- sore throat
- •
- dry mouth
- •
- muscle pain
- •
- tiredness
- •
- sleepiness
- •
- menstrual pain
Increased sleepiness or tiredness can happen if you take more Desloratadine Tablets than your doctor prescribed to you.
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of Desloratadine Tablets. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to Virtus Pharmaceuticals, LLC at 1-888-848-3593 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
How should I store Desloratadine Tablets?
- •
- Store Desloratadine Tablets between 59°F to 86°F (15°C to 30°C).
- •
- Desloratadine Tablets are sensitive to heat. Do not store above 86°F (30°C).
- •
- Protect Desloratadine Tablets from moisture.
Keep Desloratadine Tablets, and all medicines out of the reach of children.
General information about Desloratadine Tablets
Medicines are sometimes prescribed for purposes other than those listed in a patient information leaflet. Do not use Desloratadine Tablets for a condition for which it was not prescribed. Do not give Desloratadine Tablets to other people, even if they have the same condition you have. It may harm them.
This Patient Information leaflet summarizes the most important information about Desloratadine Tablets. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about Desloratadine Tablets that is written for health professionals.
What are the ingredients in Desloratadine Tablets?
Active ingredient: Desloratadine
Inactive ingredients in Desloratadine Tablets:
microcrystalline cellulose NF, pregelatinized starch NF, croscarmellose sodium NF, talc USP, zinc stearate, USP and FD&C Blue #2 HT 11-14%.
Manufactured for:
Virtus Pharmaceuticals, LLC
Langhorne, PA 19047
1-888-848-3593
MADE IN USA
Rev. 10/2020
R-2010

| DESLORATADINE
desloratadine tablet |
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
|
|||||||||||||||||||||||||
| Labeler - Virtus Pharmaceuticals, LLC (079659493) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AJENAT PHARMACEUTICALS LLC | 119388729 | MANUFACTURE(69543-107) , ANALYSIS(69543-107) | |
Frequently asked questions
More about desloratadine
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (36)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: antihistamines
- Breastfeeding
- En español
