Retavase: Package Insert / Prescribing Info
Package insert / product label
Generic name: reteplase, recombinant
Dosage form: injection
Drug class: Thrombolytics
Medically reviewed by Drugs.com. Last updated on Sep 26, 2024.
On This Page
Highlights of Prescribing Information
RETAVASE (reteplase) for injection, for intravenous use
Initial U.S. Approval: 1996
Indications and Usage for Retavase
RETAVASE is a tissue plasminogen activator (tPA) indicated for treatment of acute ST-elevation myocardial infarction (STEMI) to reduce the risk of death and heart failure. (1)
Limitation of Use: The risk of stroke may outweigh the benefit produced by thrombolytic therapy in patients whose STEMI puts them at low risk for death or heart failure. (1)
Retavase Dosage and Administration
Dosage Forms and Strengths
For Injection: 10 units as a lyophilized powder in single-use vials for reconstitution co-packaged with Sterile Water for Injection, USP in 10 mL prefilled syringe. (3)
Contraindications
Warnings and Precautions
Adverse Reactions/Side Effects
The most common adverse reaction (>5%) is bleeding. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Chiesi USA, Inc. at 1-888-661-9260 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pediatric Use: Safety and effectiveness have not been established. (8.4)
Revised: 9/2024
Full Prescribing Information
1. Indications and Usage for Retavase
RETAVASE is indicated for use in acute ST-elevation myocardial infarction (STEMI) to reduce the risk of death and heart failure.
Limitation of Use: The risk of stroke may outweigh the benefit produced by thrombolytic therapy in patients whose STEMI puts them at low risk for death or heart failure.
2. Retavase Dosage and Administration
2.1 Dosing Information and Administration
As soon as possible after the onset of STEMI, administer 10 units intravenously over 2 minutes. Administer a second dose of 10 units 30 minutes after the first dose.
2.2 Reconstitution
Reconstitute RETAVASE immediately before administration.
Reconstitute RETAVASE only with the supplied Sterile Water for Injection. Slight foaming upon reconstitution may occur; if necessary allow the vial to stand undisturbed for several minutes to allow dissipation of any large bubbles. Prior to administration, inspect the product for particulate matter and discoloration.
Use aseptic technique throughout.
Step 1: Open the package containing the reconstitution spike. Remove the protective cap from the luer lock port of the reconstitution spike and remove the protective cap on the end of the sterile 10 mL pre-filled syringe. Remove the protective flip-cap from one vial of RETAVASE.
Step 2: Clean the rubber closure with an alcohol wipe (not contained within kit).
Step 3: Connect the sterile pre-filled syringe to the reconstitution spike.
Step 4: Remove the protective cap from the spike end of the reconstitution spike and firmly insert the spike into the vial of RETAVASE.
Step 5: Connect the syringe plunger to the sterile 10 mL pre-filled syringe by screwing the plunger into the rubber stopper.
Step 6: Transfer the 10 mL of Sterile Water for Injection through the reconstitution spike into the vial of RETAVASE.
Step 7: With the reconstitution spike and empty pre-filled syringe still attached to the vial, swirl the vial gently until the contents are fully dissolved, may take up to 2 minutes. DO NOT SHAKE. Upon reconstitution, the solution should be clear and colorless. Discard if particulate matter or discoloration is observed. The resulting solution concentration is 1 unit/mL and delivers 10 mL (10 units reteplase).
Step 8: Disconnect the empty pre-filled syringe from the reconstitution spike and connect the plastic, graduated syringe to the reconstitution spike that is still attached to the vial.
Step 9: Withdraw 10 mL of RETAVASE reconstituted solution into the graduated syringe. A small amount of solution will remain in the vial due to overfill. Detach the graduated syringe from the reconstitution spike.
3. Dosage Forms and Strengths
For Injection: 10 units as a lyophilized powder in single-use vials for reconstitution co-packaged with Sterile Water for Injection, USP in 10 mL prefilled syringe.
4. Contraindications
Because thrombolytic therapy increases the risk of bleeding, RETAVASE is contraindicated in the following situations:
- Active internal bleeding
- Recent stroke
- Intracranial or intraspinal surgery or serious head trauma within 3 months
- Intracranial conditions that increase the risk of bleeding (e.g. neoplasms, arteriovenous malformation, or aneurysms)
- Bleeding diathesis
- Current severe uncontrolled hypertension
5. Warnings and Precautions
5.1 Bleeding
RETAVASE can cause significant and sometimes fatal bleeding. Avoid intramuscular injections and other trauma to a patient administered RETAVASE. Minimize venipunctures. Avoid puncturing noncompressible veins, such as the internal jugular and subclavian. If an arterial puncture is necessary, use an upper extremity vessel that is accessible to manual compression, apply pressure for at least 30 minutes, and monitor the puncture site closely. As fibrin is lysed during RETAVASE therapy, bleeding from recent puncture sites or other recent trauma may occur.
Should serious bleeding (not controllable by local pressure) occur, terminate concomitant anticoagulant therapy. Withhold the second RETAVASE dose if serious bleeding occurs before it is administered.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions have been reported with RETAVASE administration. Signs and symptoms observed included rash, pruritis, erythema, glossal (tongue) edema, hypotension and respiratory distress. If an anaphylactoid reaction occurs, withhold the second dose of RETAVASE and initiate appropriate therapy.
5.3 Cholesterol Embolization
Cholesterol embolism has been reported in patients treated with thrombolytic agents. Cholesterol embolism may present with livedo reticularis, “purple toe” syndrome, acute renal failure, gangrenous digits, hypertension, pancreatitis, myocardial infarction, cerebral infarction, spinal cord infarction, retinal artery occlusion, bowel infarction, and rhabdomyolysis and can be fatal. It is also associated with invasive vascular procedures (e.g., cardiac catheterization, angiography, vascular surgery) and/or anticoagulant therapy.
5.4 Drug/Laboratory Test Interactions
Coagulation tests and measures of fibrinolytic activity are unreliable during RETAVASE therapy unless specific precautions are taken to prevent in vitro artifacts. When present in blood at pharmacologic concentrations, RETAVASE remains active under in vitro conditions, which can result in degradation of fibrinogen in blood samples removed for analysis.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in the other sections of the label:
- Bleeding [see Contraindications (4) and Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Cholesterol Embolization [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Bleeding
The most frequent adverse reaction associated with RETAVASE is bleeding.
- Intracranial hemorrhage [see Clinical Studies (14)]
In the INJECT clinical trial, the overall rate of in-hospital, intracranial hemorrhage was 0.8%. The risk for intracranial hemorrhage is increased in patients with advanced age (2.2% among patients >70 years old) or with elevated blood pressure (2.4% among patients with systolic blood pressure >160 mmHg).
- Other types of hemorrhage
The incidence of other types of bleeding events in clinical studies of RETAVASE varied depending upon the use of arterial catheterization or other invasive procedures and whether the study was performed in Europe or the USA. The overall incidence of any bleeding event in patients treated with RETAVASE in clinical studies (n = 3,805) was 21.1%. The rates for bleeding events, regardless of severity, for the 10 + 10 unit RETAVASE regimen from controlled clinical studies are summarized in Table 1.
Table 1: RETAVASE Hemorrhage Rates
| Bleeding Site | INJECT | RAPID 1 and RAPID 2 | |
|---|---|---|---|
| Europe N = 2,965 | USA N = 210 | Europe N =113 | |
|
|||
|
Injection Site* |
4.6% |
48.6% |
19.5% |
|
Gastrointestinal |
2.5% |
9.0% |
1.8% |
|
Genitourinary |
1.6% |
9.5% |
0.9% |
|
Anemia, site unknown |
2.6% |
1.4% |
0.9% |
In these studies the severity and sites of bleeding events were similar for RETAVASE and the comparison thrombolytic agents.
Allergic Reactions
Among the 2,965 patients receiving RETAVASE in the INJECT trial, serious allergic reactions were noted in 3 patients, with one patient experiencing dyspnea and hypotension.
Among the 9,938 patients that received RETAVASE in a postmarketing clinical study, 8 patients experienced allergic and/or anaphylactoid reactions. Signs and symptoms observed included rash, pruritis, erythema, glossal (tongue) edema, hypotension, and respiratory distress.
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Limited published data with RETAVASE use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. In animal reproduction studies, reteplase administered to pregnant rabbits resulted in hemorrhaging in the genital tract, leading to abortions in mid-gestation in doses 3 times the human dose; however, there was no evidence of fetal anomalies in rats at doses up to 15 times the human dose.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Maternal Adverse Reactions
The most common complication of thrombolytic therapy is bleeding and pregnancy may increase this risk.
Data
Animal Data
Reteplase was administered to pregnant rabbits in doses 3 times the human dose (0.86 units/kg) resulting in hemorrhaging in the genital tract, leading to abortions in mid-gestation. In animal developmental studies in rats at Reteplase doses up to 15 times the human dose (4.31 units/kg), there was no evidence of fetal anomalies.
11. Retavase Description
Reteplase is a non-glycosylated deletion mutein of tissue plasminogen activator (tPA), containing the kringle 2 and the protease domains of human tPA. Reteplase contains 355 of the 527 amino acids of native tPA (amino acids 1-3 and 176-527). Reteplase is produced by recombinant DNA technology in E. coli. The protein is isolated as inactive inclusion bodies from E. coli, converted into its active form by an in vitro folding process and purified by chromatographic separation. The molecular weight of Reteplase is 39,571 daltons.
Potency is expressed in units (U) using a reference standard which is specific for RETAVASE and is not comparable with units used for other thrombolytic agents.
RETAVASE (reteplase) for Injection is a sterile, white, lyophilized powder for intravenous injection after reconstitution with Sterile Water for Injection, USP (without preservatives). Following reconstitution with 10 mL of Sterile Water for Injection, the resulting concentration is 1 unit/mL to allow for delivery of 10 mL (10 units reteplase). The pH is 6.0 ± 0.3. RETAVASE is supplied with overfill to ensure sufficient drug for administration of each 10 unit injection.
Each single-use vial delivers:
|
Reteplase |
10 units |
|
Dipotassium Hydrogen Phosphate |
131 mg |
|
Phosphoric Acid |
49.3 mg |
|
Polysorbate 80 |
5 mg |
|
Sucrose |
350 mg |
|
Tranexamic Acid |
8 mg |
12. Retavase - Clinical Pharmacology
12.1 Mechanism of Action
RETAVASE is a recombinant plasminogen activator which catalyzes the cleavage of endogenous plasminogen to generate plasmin. Plasmin in turn degrades the fibrin matrix of the thrombus, thereby exerting its thrombolytic action.
12.2 Pharmacodynamics
In a controlled trial, 36 of 56 patients treated for myocardial infarction had a decrease in fibrinogen levels to below 100 mg/dL by 2 hours following the administration of RETAVASE as two intravenous injections (10 + 10 unit) in which 10 units was followed 30 minutes later by a second dose of 10 units. The mean fibrinogen level returned to the baseline value by 48 hours.
14. Clinical Studies
RETAVASE was evaluated in three controlled clinical studies comparing RETAVASE to other thrombolytic agents. In all three studies, patients were treated with aspirin (initial doses of 160 mg to 350 mg and subsequent doses of 75 mg to 350 mg) and heparin (a 5,000 unit IV bolus prior to the administration of RETAVASE or control, followed by a 1000 unit/hour continuous IV infusion for at least 24 hours).
The INJECT study compared RETAVASE to streptokinase on mortality rates at 35 days following acute ST-elevation myocardial infarction (STEMI). INJECT was a double-blind study in which 6,010 patients with no more than 12 hours of chest pain consistent with coronary ischemia and either ST segment elevation or bundle branch block on ECG were randomized 1:1 to RETAVASE (10 + 10 unit) or streptokinase (1.5 million units over 60 minutes). Patients with cerebrovascular or other bleeding risks or with systolic blood pressure >200 mm Hg or diastolic blood pressure >100 mm Hg were excluded from enrollment. The study was designed to determine whether the effect of RETAVASE on survival was noninferior to that of streptokinase by ruling out with 95% confidence that 35-day mortality among RETAVASE patients was no more than 1% higher than among streptokinase patients. The results of the primary endpoint (mortality at 35 days), 6-month mortality, and selected other in-hospital endpoints are shown in Table 2.
Table 2: INJECT Study: Selected Results
| Endpoint | RETAVASE N=2,965 | Streptokinase N = 2,971 | RETAVASE-Streptokinase ∆(95% CI) |
|---|---|---|---|
|
|||
|
35-day mortality* |
8.9% |
9.4% |
-0.5 (-2.0, 0.9) |
|
6-month mortality |
11.0% |
12.1% |
-1.1 (-2.7, 0.6) |
|
Cardiogenic shock |
4.6% |
5.8% |
-1.2 (-2.4, -0.1) |
|
Heart failure in-hospital |
24.8% |
28.1% |
-3.3 (-5.6, -1.1) |
|
Any stroke in-hospital |
1.4% |
1.1% |
0.3 (-0.3, 0.8) |
|
Intracranial hemorrhage in-hospital |
0.8% |
0.4% |
0.4 (0.0, 0.8) |
More patients treated with RETAVASE experienced hemorrhagic strokes than did patients treated with streptokinase. An exploratory analysis indicated that the incidence of intracranial hemorrhage was higher among older patients or those with elevated blood pressure.
The other two studies (RAPID 1 and RAPID 2) compared coronary artery patency of RETAVASE to two regimens of alteplase in patients with STEMI. In RAPID 1 patients within 6 hours of the onset of symptoms were randomized to open-label administration of one of three regimens of RETAVASE (doses of 10 + 10 unit, 15 unit, or 10 + 5 unit) or to alteplase (100 mg over 3 hours). In RAPID 2 patients within 12 hours of the onset of symptoms were randomized to open-label administration of either RETAVASE (10 + 10 unit) or alteplase (100 mg over 1.5 hours). The primary endpoint for both studies was patency of the infarct-related artery 90 minutes after initiation of therapy. Interpretation of coronary angiograms was blinded.
A higher percentage of subjects administered RETEVASE had complete flow (TIMI grade 3) and partial or complete flow (TIMI grades 2 or 3) compared to both regimens of alteplase. The relationship between coronary artery patency and clinical efficacy has not been established.
In both clinical trials the re-occlusion rates were similar for RETAVASE and alteplase.
Table 3: RAPID 1 and RAPID 2 Studies: Angiographic Results
| 90 minute patency rates | RAPID 2 | RAPID 1* | ||||
|---|---|---|---|---|---|---|
| RETAVASE(10 +10 unit)N = 157 | Alteplase(100 mg over 1.5 hours)N = 146 | p- value | RETAVASE(10 +10 unit)N = 142 | Alteplase(100 mg over 3 hours)N = 145 | p- value | |
|
||||||
|
TIMI 2 or 3 |
83% |
73% |
0.03 |
85% |
77% |
0.08 |
|
TIMI 3 |
60% |
45% |
0.01 |
63% |
49% |
0.02 |
16. How is Retavase supplied
RETAVASE (reteplase) for Injection is supplied as a sterile, preservative-free, lyophilized powder in 10 unit vials without a vacuum, in the following packaging configurations:
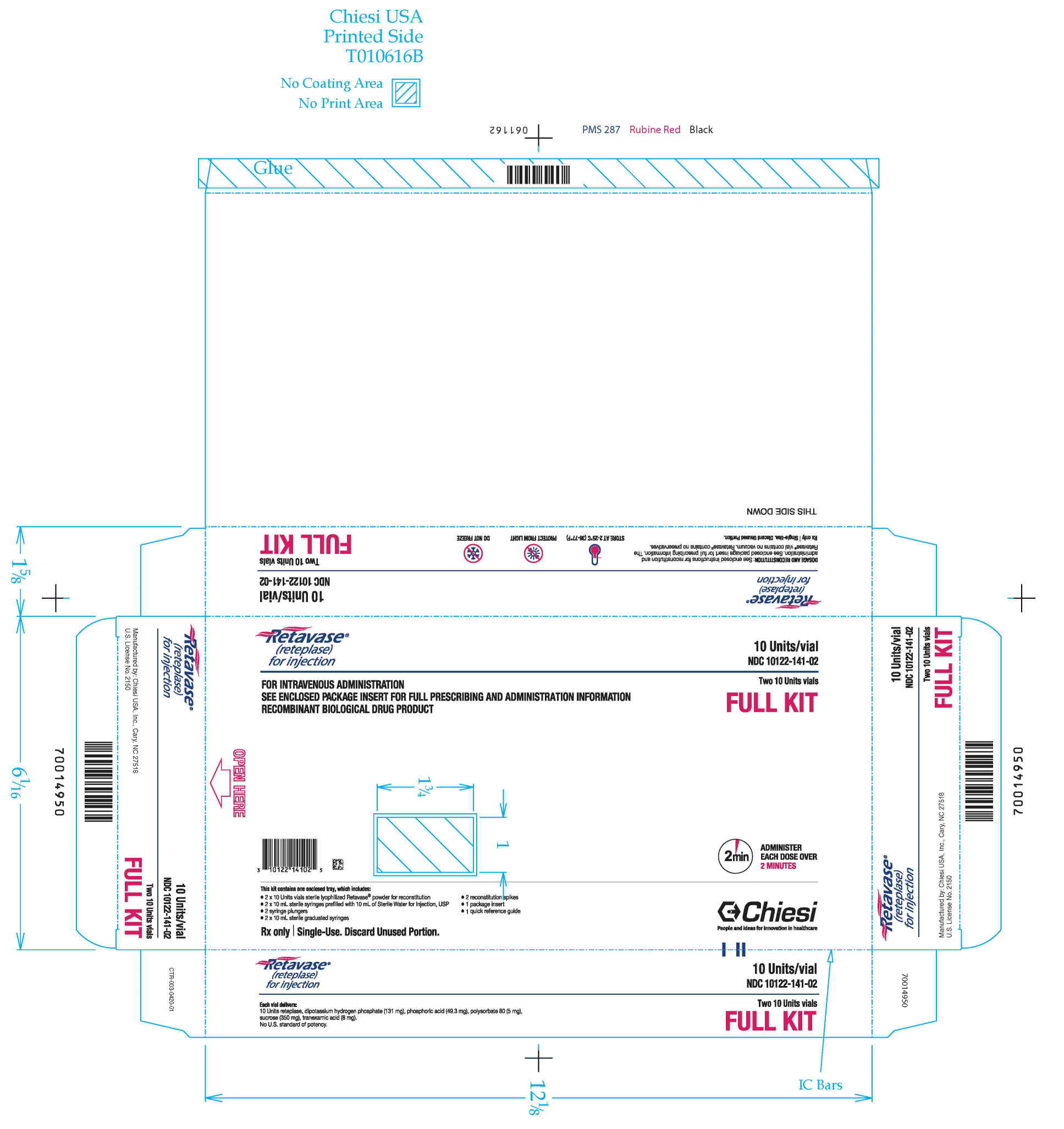
RETAVASE Kit (NDC 10122-141-02): 2 single-use RETAVASE vials 10 units, 2 single-use prefilled syringes for reconstitution (10 mL Sterile Water for Injection, USP), 2 syringe plungers, 2 sterile 10 mL graduated syringes, 2 sterile reconstitution spikes, 1 quick reference guide and 1 package insert.
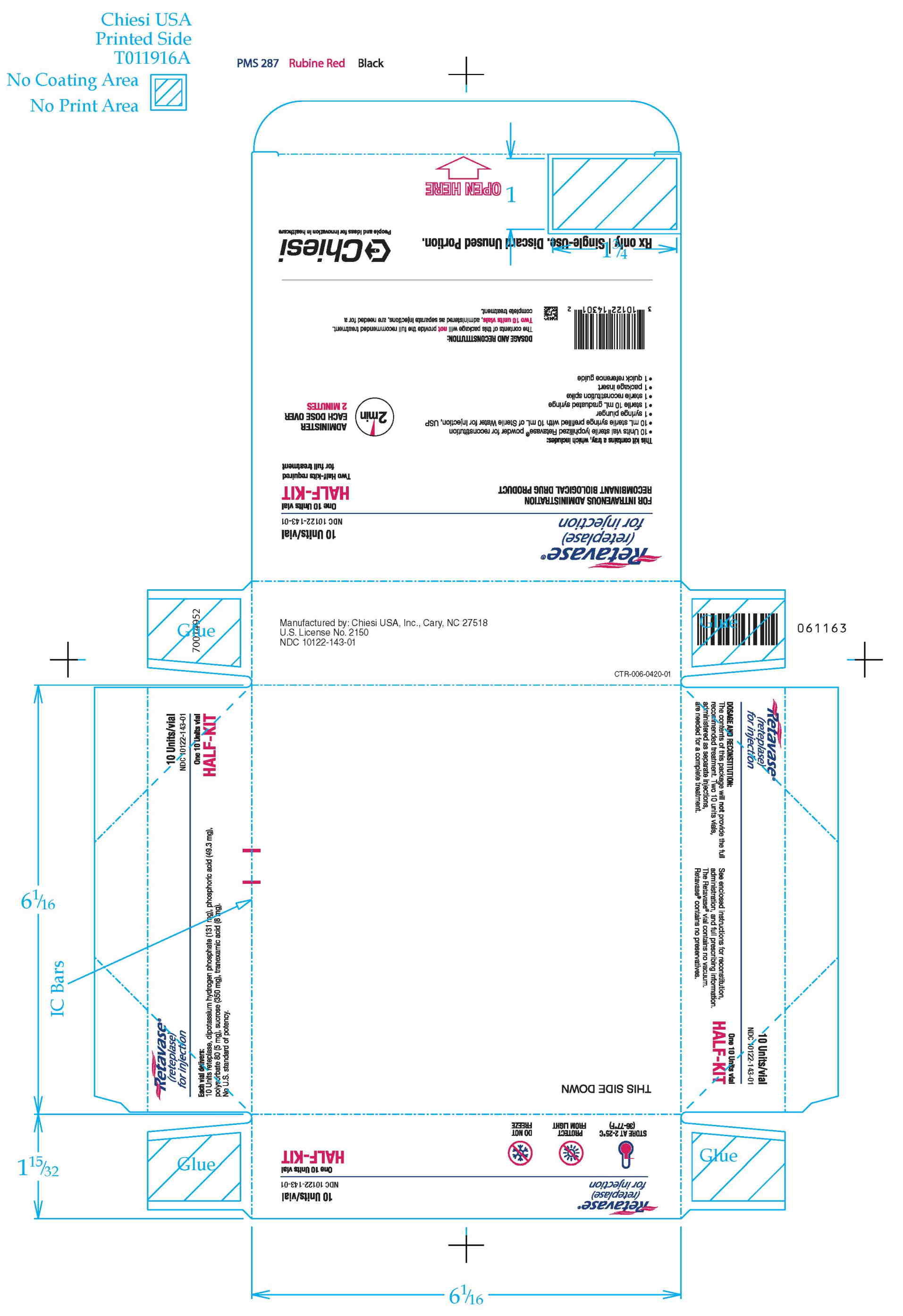
RETAVASE Half-Kit (NDC 10122-143-01): 1 single-use RETAVASE vial 10 units, 1 single-use prefilled syringe for reconstitution (10 mL Sterile Water for Injection, USP), 1 syringe plunger, 1 sterile 10 mL graduated syringe, 1 sterile reconstitution spike, 1 quick reference guide and 1 package insert.
Storage: Store RETAVASE at 2°C to 25°C (36°F to 77°F). The box should remain sealed until use to protect the lyophilisate from exposure to light.
Manufactured by:
Chiesi USA, Inc.
Cary, NC 27518
U.S. License No. 2150
Retavase® manufactured at Patheon Italia, S.p.A., Monza, Italy 20900.
To report an adverse event, record the lot number and call Medical Information at 1-888-661-9260.
RETAVASE® is a registered trademark of Chiesi USA, Inc.
The trademarks Streptase®, Activase®, and Actilyse® referenced herein are the property of their respective owners and are not affiliated with, connected to, or sponsored by Chiesi USA, Inc.
CTR-001-0422-03-SPL-1
| RETAVASE
reteplase kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| RETAVASE
reteplase kit |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Chiesi USA, Inc. (088084228) |
| Registrant - Chiesi USA, Inc. (088084228) |
Biological Products Related to Retavase
Find detailed information on biosimilars for this medication.
Frequently asked questions
More about Retavase (reteplase)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: thrombolytics
- En español