Iclusig: Package Insert / Prescribing Info
Package insert / product label
Generic name: ponatinib hydrochloride
Dosage form: tablet, film coated
Drug classes: BCR-ABL tyrosine kinase inhibitors, Multikinase inhibitors, VEGF/VEGFR inhibitors
Medically reviewed by Drugs.com. Last updated on Jan 8, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
ICLUSIG® (ponatinib) tablets, for oral use
Initial U.S. Approval: 2012
WARNING: ARTERIAL OCCLUSIVE EVENTS, VENOUS THROMBOEMBOLIC EVENTS, HEART FAILURE, and HEPATOTOXICITY
See full prescribing information for complete boxed warning.
- Arterial occlusive events (AOEs), including fatalities, have occurred in ICLUSIG-treated patients. AOEs included fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced these events. Monitor for evidence of AOEs. Interrupt or discontinue ICLUSIG based on severity. Consider benefit-risk to guide a decision to restart ICLUSIG. (2.2, 5.1)
- Venous thromboembolic events (VTEs) have occurred in ICLUSIG-treated patients. Monitor for evidence of VTEs. Interrupt or discontinue ICLUSIG based on severity (2.2, 5.2).
- Heart failure, including fatalities, occurred in ICLUSIG-treated patients. Monitor for heart failure and manage patients as clinically indicated. Interrupt or discontinue ICLUSIG for new or worsening heart failure (2.2, 5.3).
- Hepatotoxicity, liver failure and death have occurred in ICLUSIG-treated patients. Monitor liver function tests. Interrupt or discontinue ICLUSIG based on severity (2.2, 5.4).
Recent Major Changes
Indications and Usage for Iclusig
ICLUSIG is a kinase inhibitor indicated for the treatment of adult patients with:
Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia (Ph+ ALL)
- Newly diagnosed Ph+ ALL, in combination with chemotherapy. (1)
This indication is approved under accelerated approval based on minimal residual disease (MRD)-negative complete remission (CR) at the end of induction. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). - As monotherapy in Ph+ ALL for whom no other kinase inhibitors are indicated or T315I-positive Ph+ ALL. (1)
Chronic Myeloid Leukemia (CML)
- Chronic phase (CP) CML with resistance or intolerance to at least two prior kinase inhibitors. (1)
- Accelerated phase (AP) or blast phase (BP) CML for whom no other kinase inhibitors are indicated. (1)
- T315I-positive CML (chronic phase, accelerated phase, or blast phase). (1)
Limitations of Use: ICLUSIG is not indicated and is not recommended for the treatment of patients with newly diagnosed CP-CML. (5.7)
Iclusig Dosage and Administration
- Recommended Dosage in Newly Diagnosed Ph+ ALL: Starting dose is 30 mg orally once daily in combination with chemotherapy, with a reduction to 15 mg once daily upon achievement of MRD-negative (≤0.01% BCR::ABL1/ABL1) CR at the end of induction. (2.1)
- Recommended Dosage in Monotherapy for Ph+ ALL for Whom No Other Kinase Inhibitors are Indicated or T315I-positive Ph+ ALL: Starting dose is 45 mg orally once daily. (2.1)
- Recommended Dosage in CP-CML: Starting dose is 45 mg orally once daily with a reduction to 15 mg once daily upon achievement of ≤1% BCR::ABL1IS. (2.1)
- Recommended Dosage in AP-CML and BP-CML: Starting dose is 45 mg orally once daily. (2.1)
- Hepatic Impairment: See the Full Prescribing Information for dosage modifications for hepatic impairment. (2.4)
- ICLUSIG may be taken with or without food. (2.1)
Dosage Forms and Strengths
Tablets: 10 mg, 15 mg, 30 mg, and 45 mg. (3)
Contraindications
None. (4)
Warnings and Precautions
- Hypertension: Monitor blood pressure and manage hypertension as clinically indicated. Interrupt, dose reduce or stop ICLUSIG if hypertension is not medically controlled. (2.2, 5.5)
- Pancreatitis: Monitor serum lipase. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on severity. Evaluate for pancreatitis when lipase elevation is accompanied by abdominal symptoms. (2.2, 5.6)
- Neuropathy: Monitor for symptoms of peripheral and cranial neuropathy. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity. (2.2, 5.8)
- Ocular Toxicity: Conduct comprehensive eye exams at baseline and periodically during treatment. (5.9)
- Hemorrhage: Monitor for hemorrhage and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity. (2.2, 5.10)
- Fluid Retention: Monitor for fluid retention and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity. (2.2, 5.11)
- Cardiac Arrhythmias: Monitor for signs or symptoms of arrhythmias and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity. (5.12)
- Myelosuppression: Obtain complete blood counts every 2 weeks for the first 3 months and then monthly or as clinically indicated. If ANC less than 1 × 109/L or platelets less than 50 × 109/L, interrupt ICLUSIG until ANC at least 1.5 × 109/L and platelets at least 75 × 109/L, then resume at same or reduced dose. (2.2, 5.13)
- Tumor Lysis Syndrome: Ensure adequate hydration and correct elevated uric acid levels prior to initiating ICLUSIG. (5.14)
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Interrupt ICLUSIG until resolution. The safety of resumption of ICLUSIG in patients upon resolution of RPLS is unknown. (5.15)
- Impaired Wound Healing and Gastrointestinal Perforation: Withhold ICLUSIG for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of ICLUSIG after resolution of wound healing complications has not been established. (5.16)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.17, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (occurring in >20% of patients) are:
- ICLUSIG as a single agent: rash and related conditions, arthralgia, abdominal pain, headache, constipation, dry skin, hypertension, fatigue, fluid retention and edema, pyrexia, nausea, pancreatitis/lipase elevation, hemorrhage, anemia, hepatic dysfunction and AOEs. The most common Grade 3 or 4 laboratory abnormalities (>20%) are platelet count decreased, neutrophil cell count decreased, and white blood cell decreased. (6.1)
- ICLUSIG in combination with chemotherapy: hepatic dysfunction, arthralgia, rash and related conditions, headache, pyrexia, abdominal pain, constipation, fatigue, nausea, oral mucositis, hypertension, pancreatitis/lipase elevation, neuropathy peripheral, hemorrhage, febrile neutropenia, fluid retention and edema, vomiting, paresthesia, and cardiac arrhythmias. The most common Grade 3 or 4 laboratory abnormalities (>20%) are decreased white blood cell count, decreased neutrophil cell count, decreased platelet count, decreased lymphocyte cell count, decreased hemoglobin, increased lipase, and increased alanine aminotransferase. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-844-817-6468 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 9/2024
Full Prescribing Information
WARNING: ARTERIAL OCCLUSIVE EVENTS, VENOUS THROMBOEMBOLIC EVENTS, HEART FAILURE, and HEPATOTOXICITY
Arterial Occlusive Events:
- Arterial occlusive events (AOEs), including fatalities, have occurred in ICLUSIG-treated patients. AOEs included fatal myocardial infarction, stroke, stenosis of large arterial vessels of the brain, severe peripheral vascular disease, and the need for urgent revascularization procedures. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced these events. Monitor for evidence of AOEs. Interrupt or discontinue ICLUSIG based on severity. Consider benefit-risk to guide a decision to restart ICLUSIG [see Dosage and Administration (2.2), Warnings and Precautions (5.1)].
Venous Thromboembolic Events:
- Venous thromboembolic events (VTEs) have occurred in ICLUSIG-treated patients. Monitor for evidence of VTEs. Interrupt or discontinue ICLUSIG based on severity [see Dosage and Administration (2.2), Warnings and Precautions (5.2)].
Heart Failure:
- Heart failure, including fatalities, occurred in ICLUSIG-treated patients. Monitor for heart failure and manage patients as clinically indicated. Interrupt or discontinue ICLUSIG for new or worsening heart failure [see Dosage and Administration (2.2), Warnings and Precautions (5.3)].
Hepatotoxicity:
- Hepatotoxicity, liver failure and death have occurred in ICLUSIG-treated patients. Monitor liver function tests. Interrupt or discontinue ICLUSIG based on severity [see Dosage and Administration (2.2), Warnings and Precautions (5.4)].
1. Indications and Usage for Iclusig
ICLUSIG is indicated for the treatment of adult patients with:
Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia (Ph+ ALL)
- Newly diagnosed Ph+ ALL in combination with chemotherapy.
This indication is approved under accelerated approval based on minimal residual disease (MRD)-negative complete remission (CR) at the end of induction [see Clinical Studies (14)]. Continued approval for this indication may be contingent upon verification of clinical benefit in a confirmatory trial(s). - As monotherapy in Ph+ ALL for whom no other kinase inhibitors are indicated or T315I-positive Ph+ ALL.
Chronic Myeloid Leukemia (CML)
- Chronic phase (CP) CML with resistance or intolerance to at least two prior kinase inhibitors.
- Accelerated phase (AP) or blast phase (BP) CML for whom no other kinase inhibitors are indicated.
- T315I-positive CML (chronic phase, accelerated phase, or blast phase).
Limitations of Use: ICLUSIG is not indicated and is not recommended for the treatment of patients with newly diagnosed CP-CML [see Warnings and Precautions (5.7)].
2. Iclusig Dosage and Administration
2.1 Recommended Dosage
Newly Diagnosed Ph+ ALL
The recommended starting dosage of ICLUSIG in combination with chemotherapy is 30 mg orally once daily with a reduction to 15 mg orally once daily upon achievement of MRD-negative (≤0.01% BCR::ABL1/ABL1) CR at the end of induction. Continue ICLUSIG in combination with chemotherapy for up to 20 cycles until loss of response or unacceptable toxicity [see Clinical Studies (14)].
For a description of dosing of agents administered in combination with ICLUSIG, [see Clinical Studies (14)].
Monotherapy for Ph+ ALL for Whom No Other Kinase Inhibitors Are Indicated or T315I-positive Ph+ ALL
The optimal dose of ICLUSIG has not been identified.
The recommended starting dosage of ICLUSIG is 45 mg orally once daily. Continue ICLUSIG until loss of response or unacceptable toxicity.
Consider discontinuing ICLUSIG if response has not occurred by 3 months.
CP-CML
The recommended starting dosage of ICLUSIG is 45 mg orally once daily with a reduction to 15 mg orally once daily upon achievement of ≤1% BCR::ABL1IS. Patients with loss of response can re-escalate the dose of ICLUSIG to a previously tolerated dosage of 30 mg or 45 mg orally once daily. Continue ICLUSIG until loss of response at the re-escalated dose or unacceptable toxicity.
Consider discontinuing ICLUSIG if hematologic response has not occurred by 3 months.
AP-CML and BP-CML
The optimal dose of ICLUSIG has not been identified.
The recommended starting dosage of ICLUSIG is 45 mg orally once daily. Consider reducing the dose of ICLUSIG for patients with accelerated phase (AP) CML who have achieved a major cytogenetic response. Continue ICLUSIG until loss of response or unacceptable toxicity.
Consider discontinuing ICLUSIG if response has not occurred by 3 months.
2.2 Dosage Modifications for Adverse Reactions
Recommended dosage modifications of ICLUSIG for adverse reactions are provided in Table 1 and recommended dose reductions of ICLUSIG for adverse reactions are presented in Table 2.
| Adverse Reaction | Severity | ICLUSIG Dosage Modifications |
|---|---|---|
| Based on CTCAE v5.0: Grade 1 mild, Grade 2 moderate, Grade 3 severe, Grade 4 life-threatening ULN = Upper Limit of Normal for the lab; AOE = Arterial Occlusive Event; VTE = Venous Thromboembolic Event; ANC = absolute neutrophil count |
||
| AOE: cardiovascular or cerebrovascular [see Warnings and Precautions (5.1)] | Grade 1 | Interrupt ICLUSIG until resolved, then resume at same dose. |
| Grade 2 | Interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. Discontinue ICLUSIG if recurrence. |
|
| Grade 3 or 4 | Discontinue ICLUSIG. | |
| AOE: peripheral vascular and other or VTE [see Warnings and Precautions (5.1, 5.2)] | Grade 1 | Interrupt ICLUSIG until resolved, then resume at same dose. |
| Grade 2 | Interrupt ICLUSIG until Grade 0 or 1, then resume at same dose. If recurrence, interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. |
|
| Grade 3 | Interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. Discontinue ICLUSIG if recurrence. |
|
| Grade 4 | Discontinue ICLUSIG. | |
| Heart Failure [see Warnings and Precautions (5.3)] | Grade 2 or 3 | Interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. Discontinue ICLUSIG if recurrence. |
| Grade 4 | Discontinue ICLUSIG. | |
| Hepatotoxicity [see Warnings and Precautions (5.4)] | AST or ALT greater than 3 times ULN | Interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. |
| AST or ALT at least 3 times ULN concurrent with bilirubin greater than 2 times ULN and alkaline phosphatase less than 2 times ULN | Discontinue ICLUSIG. | |
| Pancreatitis and Elevated Lipase [see Warnings and Precautions (5.6)] | Serum lipase greater than 1 to 1.5 times ULN | Consider interrupting ICLUSIG until resolution, then resume at same dose. |
| Serum lipase greater than 1.5 to 2 times ULN, 2 to 5 times ULN and asymptomatic, or asymptomatic radiologic pancreatitis | Interrupt ICLUSIG until Grade 0 or 1 (less than 1.5 times ULN), then resume at next lower dose. | |
| Serum lipase greater than 2 to 5 times ULN and symptomatic, symptomatic Grade 3 pancreatitis, or serum lipase greater than 5 times ULN and asymptomatic | Interrupt ICLUSIG until complete resolution of symptoms and after recovery of lipase elevation Grade 0 or 1, then resume at next lower dose. | |
| Symptomatic pancreatitis and serum lipase greater than 5 times ULN | Discontinue ICLUSIG. | |
| Myelosuppression [see Warnings and Precautions (5.13)] | ANC less than 1 × 109/L or Platelets less than 50 × 109/L | Interrupt ICLUSIG until ANC at least 1.5 × 109/L and platelet at least 75 × 109/L, then resume at same dose. If recurrence, interrupt ICLUSIG until resolution, then resume at next lower dose. |
| Other Non-hematologic Adverse Reactions [see Warnings and Precautions (5.5, 5.8, 5.10, 5.11, 5.12)] | Grade 1 | Interrupt ICLUSIG until resolved, then resume at same dose. |
| Grade 2 | Interrupt ICLUSIG until Grade 0 or 1, then resume at same dose. If recurrence, interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. |
|
| Grade 3 or 4 | Interrupt ICLUSIG until Grade 0 or 1, then resume at next lower dose. Discontinue ICLUSIG if recurrence. |
|
| Dose Reduction | Dosage for Patients with CP-CML | Dosage for Patients with AP-CML, BP-CML, and Ph+ ALL Monotherapy | Dosage for Patients with Newly Diagnosed Ph+ ALL |
|---|---|---|---|
| First | 30 mg orally once daily | 30 mg orally once daily | 15 mg orally once daily |
| Second | 15 mg orally once daily | 15 mg orally once daily | 10 mg orally once daily |
| Third | 10 mg orally once daily | Permanently discontinue ICLUSIG in patients unable to tolerate 15 mg orally once daily. | Permanently discontinue ICLUSIG in patients unable to tolerate 10 mg orally once daily. |
| Subsequent Reduction | Permanently discontinue ICLUSIG in patients unable to tolerate 10 mg orally once daily. |
2.3 Dosage Modification for Coadministration of Strong CYP3A Inhibitors
Avoid coadministration of ICLUSIG with strong CYP3A inhibitors. If coadministration of a strong CYP3A inhibitor cannot be avoided, reduce the dosage of ICLUSIG as recommended in Table 3.
After the strong CYP3A inhibitor has been discontinued for 3 to 5 elimination half-lives, resume the ICLUSIG dosage that was tolerated prior to initiating the strong CYP3A inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
| Current ICLUSIG Dosage | Recommended ICLUSIG Dosage with a Strong CYP3A Inhibitor |
|---|---|
| 45 mg orally once daily | 30 mg orally once daily |
| 30 mg orally once daily | 15 mg orally once daily |
| 15 mg orally once daily | 10 mg orally once daily |
| 10 mg orally once daily | Avoid coadministration of ICLUSIG with a strong CYP3A inhibitor |
2.4 Dosage for Patients with Hepatic Impairment
For patients with CP-CML, AP-CML, BP-CML, and Ph+ ALL receiving monotherapy, reduce the starting dose of ICLUSIG from 45 mg orally once daily to 30 mg orally once daily in patients with pre-existing hepatic impairment (Child-Pugh A, B, or C).
For patients with newly diagnosed Ph+ ALL, no dosage adjustment is recommended when administering ICLUSIG to patients with mild hepatic impairment (Child-Pugh A). Closely monitor patients with moderate or severe hepatic impairment (Child-Pugh B or C) and modify the ICLUSIG dosage in the event of adverse reactions [see Dosage and Administration (2.2), Use in Specific Populations (8.6)].
3. Dosage Forms and Strengths
Tablets, film-coated:
- 10 mg of ponatinib: Oval, white to off-white, biconvex, debossed "NZ" on one side and plain on the other side
- 15 mg of ponatinib: Round, white, biconvex, debossed "A5" on one side and plain on the other side
- 30 mg of ponatinib: Round, white, biconvex, debossed "C7" on one side and plain on the other side
- 45 mg of ponatinib: Round, white, biconvex, debossed "AP4" on one side and plain on the other side
5. Warnings and Precautions
5.1 Arterial Occlusive Events
Arterial occlusive events (AOEs), including fatalities, occurred in patients who received ICLUSIG [see Adverse Reactions (6.1)].
In PhALLCON, 6% of 163 patients experienced AOEs, of which 3.1%, 1.8%, and 1.2% experienced cardiovascular, cerebrovascular, and peripheral vascular AOEs, respectively. The median time to onset of the first AOE was 11.3 months (range: 8 days to 2.8 years). Grade 3 or 4 AOEs occurred in 3.7% of patients; the most frequent Grade 3 or 4 AOEs were myocardial infarction (1.2%), peripheral arterial occlusive disease (1.2%), angina pectoris and cerebrovascular accident (0.6% each). Fatal AOE of sudden death occurred in 1 patient (0.6%). AOEs were more frequent with increasing age [see Use in Specific Populations (8.5)].
In PhALLCON, patients with uncontrolled hypertension, hypertriglyceridemia, or diabetes were excluded. Patients with clinically significant, uncontrolled, or active cardiovascular disease, including any history of myocardial infarction, peripheral vascular infarction, revascularization procedure, venous thromboembolism, clinically significant atrial/ventricular tachyarrhythmias, unstable angina, or congestive heart failure within the 6 months prior to the first dose of ICLUSIG, were also excluded.
In OPTIC, 94 patients who received a starting dose of 45 mg (45 mg → 15 mg), 14% experienced AOEs, of which 7%, 4.3%, and 2.1% experienced cardiovascular, cerebrovascular or peripheral vascular AOEs, respectively. The median time to onset of the first cardiovascular, cerebrovascular, or peripheral vascular event was 4.7 months (range: 12 days to 2.1 years), 11.7 months (range: 15 days to 1.6 years), and 3.6 months (range: 23 days to 6.3 months), respectively. Grade 3 or 4 AOEs occurred in 6% of patients; the most frequent Grade 3 or 4 AOEs were myocardial infarction, acute coronary syndrome, arterial thrombosis, ischemic stroke, ischemic cerebral infarction, and unstable angina (1.1% each). Fatal AOEs occurred in 2 patients (2.1%); both of which were sudden death. AOEs were more frequent with increasing age [see Use in Specific Populations (8.5)].
In OPTIC, patients with uncontrolled hypertension or diabetes and patients with clinically significant, uncontrolled, or active cardiovascular disease, including any history of myocardial infarction, peripheral vascular infarction, revascularization procedure, congestive heart failure, venous thromboembolism, or clinically significant atrial/ventricular arrhythmias, were excluded. In PACE, patients with uncontrolled hypertriglyceridemia and patients with clinically significant or active cardiovascular disease, including any history of clinically significant atrial/ventricular arrhythmias or history of myocardial infarction, unstable angina, or congestive heart failure within the 3 months prior to the first dose of ICLUSIG, were excluded [see Adverse Reactions (6.1)]. Consider whether the benefits of ICLUSIG are expected to exceed the risks.
In PACE, 26% of 449 patients experienced AOEs, of which 15%, 7%, and 11% experienced cardiovascular, cerebrovascular, and peripheral vascular AOEs, respectively. Some patients experienced recurrent or multisite vascular occlusion. The median time to onset of the first cardiovascular, cerebrovascular, and peripheral vascular AOEs was 1 year (range: 1 day to 4.1 years), 1.4 years (range: 2 days to 4.5 years), and 2 years (range: 10 days to 4.9 years), respectively. Grade 3 or 4 AOEs occurred in 14% of patients; the most frequent Grade 3 or 4 AOEs were peripheral arterial occlusive disease (3.1%), myocardial infarction (2%), coronary artery disease (1.6%), and cerebral infarction (1.6%). Fatal AOEs occurred in 9 patients (2%); the most frequent fatal AOE was cardiac arrest (0.9%).
In PACE, fatal and life-threatening AOEs occurred within 2 weeks of starting treatment at 45 mg, and at dose levels as low as 15 mg per day. Patients with and without cardiovascular risk factors, including patients age 50 years or younger, experienced AOEs. AOEs were more frequent with increasing age [see Use in Specific Populations (8.5)] and in patients with history of ischemia, hypertension, diabetes, or hypercholesterolemia. The most common risk factors in patients with AOEs were history of hypertension (67%; 77/115), hypercholesterolemia (59%; 68/115), and non-ischemic cardiac disease (43%; 49/115).
In PACE, patients developed heart failure concurrent or subsequent to a myocardial ischemic event [see Warnings and Precautions (5.3)]. Patients required revascularization procedures (coronary, cerebrovascular, and peripheral arterial). ICLUSIG caused stenosis over multiple segments in major arterial vessels that supply the brain (e.g., carotid, vertebral, middle cerebral artery). Patients developed digital or distal extremity necrosis and required amputations. Renal artery stenosis associated with worsening, labile or treatment-resistant hypertension occurred in some ICLUSIG-treated patients [see Warnings and Precautions (5.5)].
Monitor for evidence of AOEs. Interrupt, then resume at the same or decreased dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)]. Consider benefit-risk to guide a decision to restart ICLUSIG.
5.2 Venous Thromboembolic Events
Serious or severe VTEs have occurred in patients who received ICLUSIG.
In PhALLCON, VTEs occurred in 12% of 163 patients, including serious or severe (Grade 3 or 4) in 3.1%. VTEs included deep vein thrombosis (6%), superficial vein thrombosis (2.5%), embolism (1.8%), pulmonary embolism and thrombosis (1.2% each), and jugular vein thrombosis and retinal vein occlusion (0.6% each). The median time to onset of the first VTE event was 2.5 months (range: 6 days to 1.8 years).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, 1 patient experienced a VTE (Grade 1 retinal vein occlusion).
In PACE, VTEs occurred in 6% of 449 patients, including serious or severe (Grade 3 or 4) in 5.8%. VTEs included deep venous thrombosis (2.2%), pulmonary embolism (1.8%), superficial thrombophlebitis (0.7%), retinal vein occlusion (0.7%), and retinal vein thrombosis (0.4%) with vision loss. VTEs occurred in 10% of the 62 patients with BP-CML, 9% of the 32 patients with Ph+ ALL, 6% of the 270 patients with CP-CML, and 3.5% of the 85 patients with AP-CML.
Monitor for evidence of VTEs. Interrupt, then resume at the same or decreased dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)].
5.3 Heart Failure
Fatal, serious or severe heart failure events have occurred in patients who received ICLUSIG.
In PhALLCON, heart failure occurred in 6% of 163 patients; 1.2% experienced serious or severe (Grade 3 or 4) heart failure. The most frequently reported heart failure event (>1 patient) was increased brain natriuretic peptide (BNP) (2.5%).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, heart failure occurred in 13% of patients; 1.1% experienced serious or severe (Grade 3 or 4) heart failure. The most frequently reported heart failure events (>1 patient each) were left ventricular hypertrophy (3.2%) and BNP increased (3.2%).
Fatal or serious heart failure occurred in PACE. Heart failure occurred in 9% of 449 patients; 7% experienced serious or severe (Grade 3 or higher) heart failure. The most frequently reported heart failure events (≥2%) were congestive cardiac failure (3.1%), decreased ejection fraction (2.9%), and cardiac failure (2%).
Monitor patients for signs or symptoms consistent with heart failure and manage heart failure as clinically indicated. Interrupt, then resume at reduced dose or discontinue ICLUSIG for new or worsening heart failure [see Dosage and Administration (2.2)].
5.4 Hepatotoxicity
ICLUSIG can cause hepatotoxicity, including liver failure and death. Fulminant hepatic failure leading to death occurred in 3 patients, with hepatic failure occurring within 1 week of starting ICLUSIG in one of these patients. These fatal cases occurred in patients with BP-CML or Ph+ ALL treated with monotherapy.
In PhALLCON, hepatotoxicity occurred in 66% of 163 patients; 30% experienced Grade 3 or 4 hepatotoxicity. The median time to onset of hepatotoxicity was 15 days (range: 1 day to 10 months). The most frequent hepatotoxic events were elevated alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyl transferase (GGT), bilirubin and alkaline phosphatase, decreased albumin and decreased blood fibrinogen. In 6% of the 73 patients who reported ALT or AST elevation, the elevations were not resolved by the date of the last follow-up.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, hepatotoxicity occurred in 28% of patients; 6% experienced Grade 3 or 4 hepatotoxicity. The median time to onset of hepatotoxicity was 1.9 months, with a range of 3 days to 4.1 years. The most frequent hepatotoxic events were elevations of ALT, AST, alkaline phosphatase, and GGT. In 29% of the 21 patients who reported ALT or AST elevation, the event was not resolved by the date of last follow-up.
In PACE, hepatotoxicity occurred in 32% of 449 patients; 13% experienced Grade 3 or 4 hepatotoxicity. The median time to onset of hepatotoxicity was 3.1 months, with a range of 1 day to 4.9 years. The most frequent hepatotoxic events were elevations of ALT, AST, GGT, bilirubin, and alkaline phosphatase. In 9% of the 88 patients who reported ALT or AST elevation, the event was not resolved by the date of last follow-up.
Monitor liver function tests at baseline, then at least monthly or as clinically indicated. Interrupt, then resume at reduced dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)].
5.5 Hypertension
Serious or severe hypertension, including hypertensive crisis, has occurred in patients who received ICLUSIG.
In PhALLCON, hypertension occurred in 34% of 163 patients; 14% experienced serious or severe hypertension. Based on vital signs data, Grade 1 blood pressure elevation occurred in 15 out of 60 (25%) patients with normal initial blood pressure, Grade 2 occurred in 67 out of 134 (50%) patients with initial blood pressure of less than Grade 2, and Grade 3 occurred in 63 out of 160 (39%) patients with an initial blood pressure of less than Grade 3.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, hypertension events were reported in 32% of patients; 12% experienced serious or severe hypertension. Based on vital signs data, Grade 1 blood pressure elevation occurred in 8 out of 18 (44%) patients with normal initial blood pressure, Grade 2 occurred in 28 out of 81 (35%) patients with initial blood pressure of less than Grade 2, and Grade 3 occurred in 18 out of 92 (20%) patients with initial blood pressure of less than Grade 3. Three patients (3.2%) experienced hypertensive crisis.
In PACE, hypertension events were reported in 32% of 449 patients; 13% experienced serious or severe hypertension. Any post-baseline elevation of systolic or diastolic BP of Grade 2 or higher in patients with normal baseline blood pressure occurred in 44% of 449 patients. Grade 1 BP elevation occurred in 26%, Grade 2 in 45%, and Grade 3 in 26%. Two patients (<1%) experienced Grade 4 hypertension (hypertensive crisis).
Patients may require urgent clinical intervention for hypertension associated with confusion, headache, chest pain, or shortness of breath [see Adverse Reactions (6.1)]. Monitor blood pressure at baseline and as clinically indicated and manage hypertension as clinically indicated. Interrupt, dose reduce, or stop ICLUSIG if hypertension is not medically controlled [see Dosage and Administration (2.2)]. For significant worsening, labile or treatment-resistant hypertension, interrupt ICLUSIG and consider evaluating for renal artery stenosis.
5.6 Pancreatitis
Serious or severe pancreatitis has occurred in patients who received ICLUSIG.
In PhALLCON, pancreatitis occurred in 34% of 163 patients; 15% experienced serious or severe (Grade 3 or 4) pancreatitis. The median time to onset of pancreatitis was 8 days (range: 1 day to 2 years). In 7 patients with clinical pancreatitis that led to dose modification, pancreatitis resolved within 3 weeks. Laboratory abnormalities of amylase elevations occurred in 25% of patients, while lipase elevations occurred in 60% of patients.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, pancreatitis occurred in 23% of patients; 15% experienced serious or severe (Grade 3 or 4) pancreatitis. Pancreatitis resulted in discontinuation in 1.1% of patients and interruption and/or dose reduction in 20% of patients. The median time to onset of pancreatitis was 23 days (range: 3 days to 5.6 months). In two patients with clinical pancreatitis that led to dose modification or treatment discontinuation, pancreatitis resolved within 2 weeks. Laboratory abnormalities of amylase elevation occurred in 11% of patients, while lipase elevation occurred in 34% of patients.
In PACE, pancreatitis occurred in 26% of 449 patients; 17% experienced serious or severe (Grade 3 or 4) pancreatitis. Pancreatitis resulted in discontinuation in 0.4% of patients and interruption and/or dose reduction in 17% of patients. The median time to onset of pancreatitis was 29 days (range: 1 day to 4 years). Nineteen of the 28 cases of clinical pancreatitis that led to dose modification or treatment discontinuation resolved within 2 weeks. Laboratory abnormalities of amylase elevations occurred in 18% of patients, while lipase elevations occurred in 39% of patients.
Monitor serum lipase every 2 weeks for the first 2 months and then monthly thereafter or as clinically indicated. Consider additional serum lipase monitoring in patients with a history of pancreatitis or alcohol abuse. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on severity [see Dosage and Administration (2.2)]. Evaluate for pancreatitis when lipase elevation is accompanied by abdominal symptoms.
5.7 Increased Toxicity in Newly Diagnosed Chronic Phase CML
In a prospective randomized clinical trial in the first line treatment of newly diagnosed patients with CP-CML, single agent ICLUSIG 45 mg once daily increased the risk of serious adverse reactions 2-fold compared to single agent imatinib 400 mg once daily. The median exposure to treatment was less than 6 months. The trial was halted for safety.
Arterial and venous thrombosis and occlusions occurred at least twice as frequently in the ICLUSIG arm compared to the imatinib arm. Compared to imatinib-treated patients, ICLUSIG-treated patients exhibited a greater incidence of myelosuppression, pancreatitis, hepatotoxicity, cardiac failure, hypertension, and skin and subcutaneous tissue disorders. ICLUSIG is not indicated and is not recommended for the treatment of patients with newly diagnosed CP-CML.
5.8 Neuropathy
In PhALLCON, peripheral neuropathy occurred in 68% of 163 patients; 3.1% experienced Grade 3 or 4 peripheral neuropathy. The most frequent peripheral neuropathies were neuropathy peripheral (33%), paresthesia (22%), and peripheral sensory neuropathy (12%). The median time to onset of peripheral neuropathy was 1.1 month (range: 1 day to 17.2 months). Cranial neuropathy was reported in 0.6% of 163 patients.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, neuropathy occurred in 9% of patients. Peripheral neuropathy occurred in 6% of patients. The most frequently reported peripheral neuropathies were hypoesthesia (2.1%), muscular weakness (2.1%), and paresthesia (2.1%). Cranial neuropathy developed in 2 patients. The median time to onset of peripheral neuropathy and cranial neuropathy was 7.7 months (range: 1.5 months to 1.4 years) and 2.1 years (range: Day 1 to 4.2 years), respectively.
In PACE, neuropathy occurred in 22% of patients; 2.4% experienced Grade 3 or 4 neuropathy. Peripheral neuropathy occurred in 20% of 449 patients; 1.8% experienced Grade 3 or 4 peripheral neuropathy. The most frequent peripheral neuropathies were paresthesia (5%), neuropathy peripheral (4.5%), and hypoesthesia (3.6%). Cranial neuropathy developed in 3% of patients; 0.7% were Grade 3 or 4. The median time to onset of peripheral neuropathy and cranial neuropathy was 5.3 months (range: 1 day to 4.6 years) and 1.2 years (range: 18 days to 4 years), respectively.
Monitor patients for symptoms of neuropathy, such as hypoesthesia, hyperesthesia, paresthesia, discomfort, a burning sensation, neuropathic pain or weakness. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)].
5.9 Ocular Toxicity
Serious ocular toxicities leading to blindness or blurred vision have occurred in ICLUSIG-treated patients.
In PhALLCON, ocular toxicities occurred in 33% of 163 patients; 1.8% experienced a serious or severe ocular toxicity. The most frequent ocular toxicities were blurred vision and dry eye. Retinal toxicities occurred in 4.3% of patients; 0.6% experienced a Grade 3 retinal vein occlusion. The most frequent retinal toxicity event (>1 patient) was retinal hemorrhage (1.8%).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, ocular toxicities occurred in 11% of patients; 1.1% experienced a serious or severe ocular toxicity. The most frequent ocular toxicities were blurred vision and eye pain. Retinal toxicities, including age-related macular degeneration and retinal vein occlusion, occurred in 2.1% of patients.
In PACE, ocular toxicities occurred in 30% of 449 patients; 3.6% experienced a serious or severe ocular toxicity. The most frequent ocular toxicities were dry eye, blurred vision, and eye pain. Retinal toxicities occurred in 3.6% of patients. The most frequent retinal toxicities were macular edema, retinal vein occlusion, retinal hemorrhage, and vitreous floaters (0.7% each).
Conduct comprehensive eye exams at baseline and periodically during treatment.
5.10 Hemorrhage
Fatal and serious hemorrhage events have occurred in patients who received ICLUSIG.
In PhALLCON, hemorrhage occurred in 31% of 163 patients; 2.5% experienced a serious hemorrhage. Intracranial hemorrhage was the most frequently reported serious hemorrhage, occurring in 1.2% of patients.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, hemorrhage occurred in 12% of patients; 1 patient experienced a serious subdural hematoma.
In PACE, hemorrhage occurred in 28% of 449 patients; 6% experienced a serious hemorrhage and 1.3% experienced a fatal hemorrhage. The incidence of serious bleeding events was higher in patients with AP-CML, BP-CML, and Ph+ ALL. Gastrointestinal hemorrhage and subdural hematoma were the most frequently reported serious hemorrhages, each occurring in 0.9% of patients. Most hemorrhages occurred in patients with Grade 4 thrombocytopenia [see Warnings and Precautions (5.13)].
Monitor for hemorrhage and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)].
5.11 Fluid Retention
Fatal and serious fluid retention events have occurred in patients who received ICLUSIG.
In PhALLCON, fluid retention occurred in 24% of 163 patients; 1.2% experienced serious fluid retention, including pericardial effusion (1.2%). The most frequent occurrences of fluid retention were peripheral edema (11%) and pleural effusion (6%).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, fluid retention occurred in 5% of patients. The most frequent fluid retention events were peripheral edema (2.1%) and pleural effusion (2.1%).
In PACE, fluid retention events occurred in 33% of 449 patients; 4.5% experienced serious fluid retention. One instance of brain edema was fatal. Serious fluid retention included pleural effusion (1.6%), pericardial effusion (1.6%), and angioedema (0.4%). The most frequent fluid retention events were peripheral edema (17%), pleural effusion (9%), pericardial effusion (4.2%) and peripheral swelling (3.8%).
Monitor for fluid retention and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity [see Dosage and Administration (2.2)].
5.12 Cardiac Arrhythmias
In PhALLCON, cardiac arrhythmia events occurred in 22% of 163 patients; 2.5% experienced Grade 3 or 4 cardiac arrhythmias, including tachycardia, syncope, atrial fibrillation and supraventricular tachycardia (0.6%, each).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, cardiac arrhythmias occurred in 16% of patients; 4.3% experienced Grade 3 or 4 cardiac arrhythmias. Grade 3 or 4 cardiac arrhythmias included atrial fibrillation, cardio-respiratory arrest, supraventricular extrasystoles, and syncope.
In PACE, cardiac arrhythmias occurred in 20% of 449 patients; 7% experienced Grade 3 or 4 cardiac arrhythmias. Ventricular arrhythmias occurred in 3.4% of the 89 patients who reported an arrhythmia, with one event being Grade 3 or 4. Symptomatic bradyarrhythmias that led to pacemaker implantation occurred in 1% of patients. Atrial fibrillation was the most frequent cardiac arrhythmia (8%), with 3.3% being Grade 3 or 4. Other Grade 3 or 4 arrhythmia events included syncope (2%), tachycardia and bradycardia (0.4% each), and QT interval prolongation, atrial flutter, sinus bradycardia, supraventricular tachycardia, ventricular tachycardia, atrial tachycardia, atrioventricular block complete, cardio-respiratory arrest, loss of consciousness, and sinus node dysfunction (0.2% each). For 31 patients, the arrythmia led to hospitalization.
Monitor for signs and symptoms suggestive of slow heart rate (fainting, dizziness) or rapid heart rate (chest pain, palpitations or dizziness) and manage patients as clinically indicated. Interrupt, then resume at the same or reduced dose or discontinue ICLUSIG based on recurrence/severity.
5.13 Myelosuppression
In PhALLCON, neutropenia occurred in 66% (Grade 3 or 4 occurred in 63%), thrombocytopenia occurred in 65% (Grade 3 or 4 occurred in 62%) and anemia occurred in 53% (Grade 3 or 4 occurred in 38%) of 163 patients. The median time to onset of Grade 3 or 4 myelosuppression was 27 days (range: 1 day to 9.2 months).
In OPTIC, of the 94 patients who received a starting dose of 45 mg, neutropenia occurred in 55% (Grade 3 or 4 occurred in 22%), thrombocytopenia occurred in 65% (Grade 3 or 4 occurred in 31%), and anemia occurred in 35% of patients (Grade 3 or 4 occurred in 14%). The median time to onset of Grade 3 or 4 myelosuppression was 1.4 months (range: 1 day to 1.2 years).
In PACE, neutropenia occurred in 56% (Grade 3 or 4 occurred in 34%), thrombocytopenia occurred in 63% (Grade 3 or 4 occurred in 40%), and anemia occurred in 52% of patients (Grade 3 or 4 occurred in 20%). The incidence of myelosuppression was greater in patients with AP-CML, BP-CML, and Ph+ ALL than in patients with CP-CML. Severe myelosuppression (Grade 3 or 4) was observed early in treatment, with a median onset time of 29 days (range: 1 day to 4.1 years).
Obtain complete blood counts every 2 weeks for the first 3 months and then monthly or as clinically indicated. If ANC less than 1 × 109/L or platelets less than 50 × 109/L, interrupt ICLUSIG until ANC at least 1.5 × 109/L and platelets at least 75 × 109/L, then resume at same or reduced dose [see Dosage and Administration (2.2)].
5.14 Tumor Lysis Syndrome
In PhALLCON, serious tumor lysis syndrome (TLS) developed in 0.6% of 163 patients. Hyperuricemia occurred in 10% of patients.
In OPTIC, of the 94 patients who received a starting dose of 45 mg, serious TLS developed in 1.1% of patients. Hyperuricemia occurred in 2.1% of patients.
In PACE, serious TLS developed in 0.4% of 449 patients. One case occurred in a patient with advanced AP-CML and 1 case occurred in a patient with BP-CML. Hyperuricemia occurred in 7% of patients.
Ensure adequate hydration and treat high uric acid levels prior to initiating ICLUSIG.
5.15 Reversible Posterior Leukoencephalopathy Syndrome
Reversible posterior leukoencephalopathy syndrome (RPLS; also known as Posterior Reversible Encephalopathy Syndrome) has been reported in patients who received ICLUSIG. Patients can present with hypertension, seizure, headache, decreased alertness, altered mental functioning, vision loss, and other visual and neurological disturbances. Magnetic resonance imaging (MRI) is necessary to confirm the diagnosis. Interrupt ICLUSIG until resolution. The safety of resumption of ICLUSIG in patients upon resolution of RPLS is unknown.
5.16 Impaired Wound Healing and Gastrointestinal Perforation
Impaired wound healing occurred in patients receiving ICLUSIG [see Adverse Reactions (6.2)]. Withhold ICLUSIG for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of ICLUSIG after resolution of wound healing complications has not been established.
Gastrointestinal perforation or fistula occurred in patients receiving ICLUSIG [see Adverse Reactions (6.2)]. Permanently discontinue in patients with gastrointestinal perforation.
5.17 Embryo-Fetal Toxicity
Based on its mechanism of action and findings from animal studies, ICLUSIG can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of ponatinib to pregnant rats during organogenesis caused adverse developmental effects at exposures lower than human exposures at the maximum recommended human dose of 45 mg/day. Advise pregnant women of the potential risk to the fetus. Advise females of reproductive potential to use effective contraception during treatment with ICLUSIG and for 3 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Arterial Occlusive Events [see Warnings and Precautions (5.1)]
- Venous Thromboembolic Events [see Warnings and Precautions (5.2)]
- Heart Failure [see Warnings and Precautions (5.3)]
- Hepatotoxicity [see Warnings and Precautions (5.4)]
- Hypertension [see Warnings and Precautions (5.5)]
- Pancreatitis [see Warnings and Precautions (5.6)]
- Neuropathy [see Warnings and Precautions (5.8)]
- Ocular Toxicity [see Warnings and Precautions (5.9)]
- Hemorrhage [see Warnings and Precautions (5.10)]
- Fluid Retention [see Warnings and Precautions (5.11)]
- Cardiac Arrhythmias [see Warnings and Precautions (5.12)]
- Myelosuppression [see Warnings and Precautions (5.13)]
- Tumor Lysis Syndrome [see Warnings and Precautions (5.14)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.15)]
- Impaired Wound Healing and Gastrointestinal Perforation [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The most common adverse reactions identified in the Highlights of the Prescribing Information are based on two safety populations. The first is from a pooled safety population of 543 patients with CML or resistant or intolerant Ph+ ALL (OPTIC and PACE studies) who received ICLUSIG as a single agent at a starting dose of 45 mg orally once daily. In this pooled safety population, the most common (>20%) adverse reactions were rash and related conditions, arthralgia, abdominal pain, headache, constipation, dry skin, hypertension, fatigue, fluid retention and edema, pyrexia, nausea, pancreatitis/lipase elevation, hemorrhage, anemia, hepatic dysfunction, and AOEs. The most common Grade 3 or 4 laboratory abnormalities (>20%) were platelet count decreased, neutrophil cell count decreased, and white blood cell decreased.
The second safety population is from163 patients with newly diagnosed Ph+ ALL (PhALLCON study) who received ICLUSIG in combination with chemotherapy at a starting dose of 30 mg orally once daily. The most common adverse reactions (>20%) included hepatic dysfunction, arthralgia, rash and related conditions, headache, pyrexia, abdominal pain, constipation, fatigue, nausea, oral mucositis, hypertension, pancreatitis/lipase elevation, neuropathy peripheral, hemorrhage, febrile neutropenia, fluid retention and edema, vomiting, paresthesia, and cardiac arrhythmias. The most common Grade 3 or 4 laboratory abnormalities (>20%) included decreased white blood cell count, decreased neutrophil cell count, decreased platelet count, decreased lymphocyte cell count, decreased hemoglobin, increased lipase, and increased ALT.
Newly Diagnosed Ph+ ALL
The safety of ICLUSIG was evaluated in PhALLCON, a randomized, active-controlled, multicenter trial conducted in patients with newly diagnosed Ph+ ALL [see Clinical Studies (14)]. Patients received ICLUSIG (n=163) or imatinib 600 mg (n=81) in combination with reduced-intensity chemotherapy followed by continued treatment with ICLUSIG or imatinib as a single agent (imatinib in combination with chemotherapy is not an approved regimen in adult patients). In the ICLUSIG arm, patients received a starting dosage of ICLUSIG 30 mg orally once daily in combination with chemotherapy, with a reduction to 15 mg orally once daily upon achievement of MRD-negative CR at the end of induction. The median duration of exposure was 9.0 months (range: <1 month to 4.2 years) in the ICLUSIG arm and 5.2 months (range: <1 month to 4.4 years) in the imatinib arm.
Patients with uncontrolled hypertension, hypertriglyceridemia, or diabetes and patients with clinically significant, uncontrolled, or active cardiovascular disease, including any history of myocardial infarction, peripheral vascular infarction, revascularization procedure, venous thromboembolism, clinically significant atrial/ventricular tachyarrhythmias, history of myocardial infarction, unstable angina, or congestive heart failure within the 6 months prior to the first dose of ICLUSIG, were excluded.
Serious adverse reactions occurred in 63% of patients receiving ICLUSIG in combination with chemotherapy. Serious adverse reactions in >2% of patients included febrile neutropenia (18%), pyrexia (6%), thrombocytopenia (4.3%), sepsis (3.7%), septic shock (3.7%), anemia (2.5%), hemorrhage (2.5%), neutropenia (2.5%), pancreatitis (2.5%), peripheral neuropathy (2.5%), pneumonia (2.5%) and acute kidney injury (2.5%). Fatal adverse reactions occurred in 6% of patients who received ICLUSIG in combination with chemotherapy, including sepsis (3.7%), sudden death, pneumonitis and respiratory failure (0.6%, each).
Permanent discontinuation of ICLUSIG due to adverse reactions occurred in 13% of patients. Adverse reactions resulting in permanent discontinuation of ICLUSIG in >2% of patients included arterial occlusive events and sepsis.
Dosage modifications (dose interruption or reduction) of ICLUSIG due to adverse reactions occurred in 71% of patients. Adverse reactions leading to dose interruption or reduction of ICLUSIG in >5% of patients included increased ALT, neutropenia, increased lipase, thrombocytopenia, increased AST, febrile neutropenia, and abdominal pain.
Table 4 summarizes the adverse reactions in patients receiving ICLUSIG or imatinib in combination with chemotherapy in PhALLCON.
| Adverse Reaction | ICLUSIG 30 mg → 15 mg with Chemotherapy (n = 163) | Imatinib 600 mg with Chemotherapy (n = 81) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| Graded using CTCAE v5.0 | ||||
|
||||
| Hepatobiliary Disorders | ||||
| Hepatotoxicity | 66 | 30 | 57 | 14 |
| Musculoskeletal and Connective Tissue Disorders | ||||
| Arthralgia* | 47 | 4.3 | 35 | 1.2 |
| Myalgia | 13 | 1.2 | 10 | 1.2 |
| Nervous System Disorders | ||||
| Headache | 45 | 1.8 | 43 | 1.2 |
| Neuropathy peripheral | 33 | 1.2 | 24 | 1.2 |
| Paresthesia | 22 | 0 | 10 | 0 |
| Peripheral sensory neuropathy | 12 | 0 | 12 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rash and related conditions | 47 | 1.2 | 33 | 1.2 |
| Gastrointestinal Disorders | ||||
| Abdominal pain† | 43 | 4.9 | 28 | 0 |
| Constipation | 41 | 0.6 | 21 | 1.2 |
| Nausea | 37 | 3.1 | 52 | 7 |
| Oral mucositis | 35 | 4.9 | 30 | 10 |
| Pancreatitis/lipase elevation | 34 | 15 | 37 | 20 |
| Vomiting | 24 | 1.2 | 40 | 2.5 |
| Diarrhea | 20 | 0 | 35 | 2.5 |
| General Disorders | ||||
| Pyrexia | 44 | 4.3 | 26 | 2.5 |
| Fatigue or asthenia | 40 | 2.5 | 38 | 3.7 |
| Fluid retention and edema | 24 | 0.6 | 48 | 3.7 |
| Vascular Disorders | ||||
| Hypertension | 34 | 14 | 15 | 7 |
| Hemorrhage | 31 | 1.8 | 30 | 7 |
| Venous thromboembolic events | 12 | 3.1 | 10 | 2.5 |
| Blood and Lymphatic System Disorders | ||||
| Febrile neutropenia | 28 | 25 | 22 | 20 |
| Metabolism and Nutrition Disorders | ||||
| Impaired glucose tolerance | 20 | 4.9 | 20 | 9 |
| Hyperlipidemia | 16 | 1.2 | 15 | 1.2 |
| Decreased appetite | 10 | 0 | 19 | 3.7 |
| Cardiac Disorders | ||||
| Cardiac arrhythmias | 22 | 2.5 | 17 | 6 |
| Infections | ||||
| Sepsis‡ | 17 | 12 | 15 | 11 |
| Pneumonia | 11 | 7 | 11 | 6 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||
| Cough | 17 | 0 | 6 | 0 |
| Dyspnea | 13 | 1.2 | 4.9 | 2.5 |
Clinically relevant adverse reactions in ≤10% of patients receiving ICLUSIG with chemotherapy: urinary tract infection (10%), arterial occlusive events (6%), cardiac failure (6%), and acute kidney injury (4.3%).
Table 5 summarizes the laboratory abnormalities in PhALLCON for patients who received ICLUSIG or imatinib in combination with chemotherapy.
| Laboratory Abnormality | ICLUSIG 30 mg → 15 mg with Chemotherapy (n = 163) | Imatinib 600 mg with Chemotherapy (n = 81) |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| ALT = alanine aminotransferase, AST = aspartate aminotransferase Graded using CTCAE v5.0 |
||||
| Hematologic Laboratory Tests | ||||
| White blood cell decreased | 79 | 71 | 78 | 70 |
| Lymphocyte cell count decreased | 77 | 61 | 94 | 89 |
| Neutrophil cell count decreased | 66 | 63 | 57 | 53 |
| Platelet count decreased | 65 | 62 | 64 | 53 |
| Hemoglobin decreased | 53 | 38 | 59 | 49 |
| Liver Function Tests | ||||
| ALT increased | 69 | 21 | 62 | 7 |
| AST increased | 53 | 7 | 48 | 6 |
| Alkaline phosphatase increased | 44 | 1.2 | 24 | 0 |
| Total bilirubin increased | 25 | 0.6 | 24 | 0 |
| Direct bilirubin increased | 24 | 4.3 | 24 | 1.2 |
| Pancreatic Enzymes | ||||
| Lipase increased | 60 | 24 | 78 | 38 |
| Amylase increased | 25 | 6 | 35 | 7 |
| Chemistry | ||||
| Calcium decreased | 67 | 3.1 | 69 | 4.9 |
| Phosphate decreased | 58 | 16 | 85 | 36 |
| Potassium decreased | 44 | 10 | 74 | 25 |
| Albumin decreased | 42 | 1.8 | 56 | 0 |
| Glucose increased | 34 | 2.5 | 38 | 2.5 |
| Creatinine increased | 34 | 3.7 | 48 | 4.9 |
| Sodium decreased | 32 | 3.1 | 35 | 3.7 |
| Potassium increased | 31 | 3.7 | 12 | 0 |
| Magnesium decreased | 15 | 0.6 | 31 | 2.5 |
Previously Treated CP-CML
The safety of ICLUSIG was evaluated in OPTIC [see Clinical Studies (14)]. Patients received one of three starting doses of ICLUSIG: 45 mg orally once daily (n=94), 30 mg orally once daily (n=94), or 15 mg orally once daily (n=94). Patients with uncontrolled hypertension or diabetes and patients with clinically significant, uncontrolled, or active cardiovascular disease, including any history of myocardial infarction, peripheral vascular infarction, revascularization procedure, congestive heart failure, venous thromboembolism, or clinically significant atrial/ventricular arrhythmias, were excluded. Only the safety information for the recommended starting dosage (45 mg) is described below. Patients who received a starting dose of ICLUSIG 45 mg orally once daily had a mandatory dose reduction to 15 mg once daily upon achievement of ≤1% BCR::ABLIS. Of these patients, 76% were exposed for 1 year or longer and 38% were exposed for greater than two years. The median time to the response-based dose reduction to 15 mg was 6.4 months (range: 3.1 months to 1.8 years).
Serious adverse reactions occurred in 34% of patients who received ICLUSIG at a starting dose of 45 mg. Serious adverse reactions in >2% of patients included AOEs (9%; of which 2.1% were sudden death), cardiac arrhythmias (6%), thrombocytopenia (5%), pyrexia (4.3%), anemia (3.2%), abdominal pain (3.2%), atrial fibrillation (2.1%), pancreatitis/lipase elevation (2.1%), neutropenia (2.1%), and hypertension (2.1%). Fatal adverse reactions occurred in 2 patients (2.1%), both of which were sudden death.
Permanent discontinuation of ICLUSIG due to an adverse reaction occurred in 19% of patients who received ICLUSIG at a starting dose of 45 mg. Adverse reactions which resulted in permanent discontinuation in >2% of patients included AOEs, thrombocytopenia, hypertension, and sudden death.
Dose modifications (dose interruption or reductions) of ICLUSIG due to an adverse reaction occurred in 71% of patients who received ICLUSIG at a starting dose of 45 mg. Adverse reactions which required dose interruptions or reductions in >5% of patients included thrombocytopenia, pancreatitis/lipase elevation, neutropenia, hepatic dysfunction, rash and related conditions, and anemia.
The most common (>20%) adverse reactions were rash and related conditions, hypertension, arthralgia, hyperlipidemia, hepatic dysfunction, pancreatitis/lipase elevation, and abdominal pain. The most common (>20%) Grade 3 or 4 laboratory abnormalities were platelet count decreased and neutrophil cell count decreased.
Table 6 summarizes the adverse reactions in OPTIC for patients who received ICLUSIG at a starting dose of 45 mg.
| Adverse Reaction | ICLUSIG 45 mg → 15 mg (N = 94) |
|
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) |
|
| Graded using CTCAE v5.0 | ||
|
||
| Skin and Subcutaneous Tissue Disorders | ||
| Rash and related conditions | 51 | 3.2 |
| Dry skin | 12 | 0 |
| Vascular Disorders | ||
| Hypertension | 32 | 12 |
| Arterial occlusive events | 14 | 6 |
| Hemorrhage | 12 | 2.1 |
| Musculoskeletal and Connective Tissue Disorders | ||
| Arthralgia* | 30 | 0 |
| Metabolism and Nutrition Disorders | ||
| Hyperlipidemia† | 28 | 2.1 |
| Gastrointestinal Disorders | ||
| Abdominal pain‡ | 25 | 3.2 |
| Pancreatitis/lipase elevation | 23 | 15 |
| Constipation | 11 | 0 |
| Hepatobiliary Disorders | ||
| Hepatotoxicity | 28 | 6 |
| Nervous System Disorders | ||
| Headache | 17 | 0 |
| General Disorders and Administration Site Conditions | ||
| Pyrexia | 16 | 1.1 |
| Fatigue or asthenia | 10 | 1.1 |
| Cardiac Disorders | ||
| Cardiac arrhythmias | 16 | 4.3 |
| Cardiac failure | 13 | 1.1 |
Clinically relevant adverse reactions in <10% of patients who received ICLUSIG at a starting dose of 45 mg: neuropathy (9%), fluid retention and edema (5%), and hypothyroidism (3.2%).
Table 7 summarizes the laboratory abnormalities in OPTIC for patients who received ICLUSIG at a starting dose of 45 mg.
| Laboratory Abnormality | ICLUSIG 45 mg → 15 mg (N = 94) |
|
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) |
|
| ALT = alanine aminotransferase, AST = aspartate aminotransferase Graded using CTCAE v5.0 (except glucose increased which is graded using CTCAE v4.03) |
||
| Hematologic Laboratory Tests | ||
| Platelet count decreased | 65 | 31 |
| White blood cell decreased | 56 | 13 |
| Neutrophil cell count decreased | 55 | 22 |
| Lymphocyte decreased | 42 | 7 |
| Hemoglobin decreased | 35 | 14 |
| Liver Function Tests | ||
| ALT increased | 49 | 1.1 |
| AST increased | 40 | 0 |
| Alkaline phosphatase increased | 23 | 1.1 |
| Chemistry | ||
| Glucose increased | 48 | 1.1 |
| Triglycerides increased | 44 | 3.2 |
| Phosphate decreased | 27 | 3.2 |
| Bicarbonate decreased | 27 | 0 |
| Pancreatic Enzymes | ||
| Lipase increased | 34 | 12 |
Previously Treated CML or Ph+ ALL
The safety of ICLUSIG was evaluated in PACE [see Clinical Studies (14)]. Eligible patients had CML or Ph+ ALL whose disease was considered to be resistant or intolerant to prior kinase inhibitor, including those with the BCR::ABL T315I mutation. Patients with uncontrolled hypertriglyceridemia and patients with clinically significant or active cardiovascular disease, including any history of clinically significant atrial/ventricular arrhythmias or history of myocardial infarction, unstable angina, or congestive heart failure within the 3 months prior to the first dose of ICLUSIG, were excluded. Patients received a starting dose of ICLUSIG 45 mg orally once daily (N=449). Dose reductions to 30 mg orally once daily or 15 mg orally once daily were allowed for the management of adverse reactions. After approximately 2 years of follow-up, patients who were still taking a 45 mg orally once daily dose were recommended to undergo a dose reduction in response to the continued occurrence of AOEs and VTEs in the clinical trial [see Warnings and Precautions (5.1)]. At study completion (60 months of follow-up), the median duration of treatment with ICLUSIG was 32 months in patients with CP-CML, 19 months in patients with AP-CML, 2.9 months in patients with BP-CML, and 2.7 months in patients with Ph+ ALL.
Serious adverse reactions occurred in 69% of patients who received ICLUSIG. Serious adverse reactions in >2% of patients included AOEs (20%), pneumonia (10%), cardiac arrhythmias (8%), pancreatitis/lipase elevation (7%), abdominal pain (6%), cardiac failure (6%), hemorrhage (6%), sepsis (5%), VTEs (5%), fluid retention and edema (4.5%), pyrexia (4.5%), secondary malignancies (5%), anemia (3.3%), hypertension (3.1%), thrombocytopenia (3.1%), febrile neutropenia (2.9%), cellulitis (2.7%), and arthralgia (2.2%). Fatal adverse reactions occurred in 9% of patients who received ICLUSIG; the most frequent fatal adverse reactions were AOEs (2%), sepsis (1.6%), and hemorrhage (1.3%).
Permanent discontinuation of ICLUSIG due to an adverse reaction occurred in 21% of CP-CML, 12% of AP-CML, 15% of BP-CML, and 9% of Ph+ ALL patients. The most frequent adverse reactions that led to treatment discontinuation were thrombocytopenia (4.5%) and AOEs (4%).
Dose interruption of ICLUSIG for more than 3 days due to an adverse reaction occurred in 71% of patients and dose reduction of ICLUSIG due to an adverse reaction occurred in 68% of patients. Adverse reactions which required a dosage interruption or dose reduction in >5% of patients included thrombocytopenia (31%), pancreatitis/lipase elevation (17%), abdominal pain (14%), rash and related conditions (14%), neutropenia (14%), hepatic dysfunction (12%), AOEs (10%), arthralgia (8%), anemia (7%), ALT increased (6%), and AST increased (5%).
The most common (>20%) non-hematologic adverse reactions were rash and related conditions, arthralgia, abdominal pain, fatigue, constipation, headache, dry skin, fluid retention and edema, hepatic dysfunction, hypertension, pyrexia, nausea, hemorrhage, pancreatitis/lipase elevation, AOEs, diarrhea, vomiting, and myalgia.
Table 8 summarizes the adverse reactions in PACE.
| Adverse Reaction | CP-CML (N = 270) | AP-CML (N = 85) | BP-CML (N = 62) | Ph+ ALL (N = 32) |
||||
|---|---|---|---|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
| Graded using CTCAE v4.03. | ||||||||
|
||||||||
| Skin and Subcutaneous Tissue Disorders | ||||||||
| Rash and related conditions | 75 | 9 | 68 | 12 | 55 | 7 | 50 | 3.1 |
| Dry skin | 42 | 3.3 | 32 | 1.2 | 26 | 1.6 | 25 | 0 |
| Alopecia | 8 | 0 | 11 | 0 | 8 | 0 | 6 | 0 |
| Musculoskeletal and Connective Tissue Disorders | ||||||||
| Arthralgia | 61 | 9 | 58 | 6 | 52 | 4.8 | 41 | 0 |
| Myalgia | 24 | 1.1 | 21 | 0 | 18 | 0 | 6 | 0 |
| Muscle spasms | 14 | 0 | 7 | 0 | 4.8 | 0 | 13 | 0 |
| Bone pain | 14 | 0.4 | 13 | 1.2 | 11 | 3 | 9 | 3 |
| Musculoskeletal pain | 11 | 1.5 | 7 | 0 | 8.1 | 0 | 6 | 3 |
| Gastrointestinal Disorders | ||||||||
| Abdominal pain | 54 | 11 | 49 | 9 | 45 | 13 | 34 | 6 |
| Constipation | 42 | 2.6 | 29 | 2.4 | 27 | 0 | 53 | 3.1 |
| Pancreatitis/lipase elevation | 32 | 19 | 21 | 15 | 19 | 16 | 9 | 6 |
| Nausea | 29 | 0.7 | 32 | 0 | 34 | 1.6 | 22 | 0 |
| Diarrhea | 20 | 0.7 | 29 | 2.4 | 24 | 3.2 | 13 | 3.1 |
| Vomiting | 19 | 1.5 | 27 | 0 | 27 | 1.6 | 25 | 0 |
| Oral mucositis* | 16 | 1.1 | 20 | 1.2 | 24 | 0 | 9 | 3.1 |
| General Disorders | ||||||||
| Fatigue or asthenia | 44 | 3.7 | 47 | 8 | 36 | 4.8 | 34 | 3.1 |
| Fluid retention and edema | 31 | 3.7 | 37 | 3.5 | 32 | 4.8 | 41 | 6 |
| Pyrexia | 26 | 1.1 | 40 | 7 | 37 | 3.2 | 25 | 0 |
| Chills | 8 | 0 | 12 | 0 | 13 | 1.6 | 9 | 0 |
| Nervous System Disorders | ||||||||
| Headache | 43 | 3.3 | 31 | 1.2 | 31 | 3.2 | 25 | 0 |
| Neuropathy | 26 | 3.3 | 18 | 2.4 | 13 | 0 | 13 | 0 |
| Dizziness | 17 | 0.4 | 11 | 0 | 4.8 | 0 | 3.1 | 0 |
| Vascular Disorders | ||||||||
| Hypertension† | 42 | 30 | 53 | 28 | 48 | 6 | 31 | 25 |
| Arterial occlusive events | 31 | 17 | 22 | 12 | 13 | 10 | 13 | 6 |
| Hemorrhage | 23 | 3 | 38 | 12 | 37 | 8 | 31 | 13 |
| Hepatobiliary Disorders | ||||||||
| Hepatotoxicity | 32 | 10 | 39 | 14 | 34 | 19 | 16 | 13 |
| Cardiac Disorders | ||||||||
| Cardiac arrhythmias | 19 | 7 | 17 | 4.7 | 24 | 8 | 25 | 6 |
| Cardiac failure | 9 | 5 | 8 | 4.7 | 16 | 10 | 6 | 3.1 |
| Respiratory, Thoracic, and Mediastinal Disorders | ||||||||
| Cough‡ | 19 | 0 | 24 | 0 | 21 | 0 | 6 | 0 |
| Dyspnea§ | 19 | 3 | 20 | 3.5 | 23 | 6 | 16 | 0 |
| Infections | ||||||||
| Upper respiratory tract infection¶ | 14 | 1.1 | 13 | 0 | 13 | 1.6 | 3.1 | 0 |
| Urinary tract infection# | 12 | 2.2 | 14 | 3.5 | 1.6 | 1.6 | 9 | 0 |
| Nasopharyngitis | 12 | 0 | 18 | 0 | 3.2 | 0 | 3.1 | 0 |
| Pneumonia | 8 | 4.8 | 18 | 11 | 18 | 13 | 22 | 16 |
| Cellulitis | 4.4 | 1.9 | 8 | 3.5 | 13 | 4.8 | 0 | 0 |
| SepsisÞ | 2.6 | 1.9 | 11 | 6 | 18 | 6 | 28 | 25 |
| Metabolism and Nutrition Disorders | ||||||||
| Decreased appetite | 13 | 0.4 | 14 | 1.2 | 8 | 0 | 31 | 0 |
| Hyperlipidemia | 13 | 0.7 | 7 | 0 | 3.2 | 0 | 3.1 | 0 |
| Investigations | ||||||||
| Weight decreased | 10 | 0.4 | 9 | 0 | 4.8 | 0 | 13 | 0 |
| Psychiatric Disorders | ||||||||
| Insomnia | 11 | 0 | 13 | 0 | 11 | 0 | 13 | 0 |
| Anxiety | 4.8 | 0 | 18 | 0 | 8 | 0 | 6 | 0 |
| Blood and Lymphatic System Disorders | ||||||||
| Febrile neutropenia | 1.1 | 1.1 | 4.7 | 4.7 | 13 | 13 | 25 | 25 |
Clinically relevant adverse reactions occurring in ≤10% of patients: impaired glucose tolerance (9%)1, venous thromboembolic events (6%)1, secondary malignancies (6%)1, and hypothyroidism (3%).
- 1
- Grouped terms: secondary malignancies includes basal cell carcinoma, squamous cell carcinoma of the skin, melanoma, chronic myelomonocytic leukemia, colon cancer, epithelioid mesothelioma, large cell lung cancer recurrent, lung neoplasm, malignant ascites, myelodysplastic syndrome, neuroendocrine carcinoma metastatic, non-Hodgkin lymphoma, pancreatic cancer, thyroid neoplasm, vulval cancer; venous thromboembolic events includes deep vein thrombosis, pulmonary embolism, retinal vein occlusion, retinal vein thrombosis, superficial thrombophlebitis, venous embolism, veno-occlusive liver disease, portal vein thrombosis; impaired glucose tolerance includes blood glucose increased, diabetes mellitus, glucose tolerance impaired, glycosylated hemoglobin increased, hyperglycemia, insulin resistance, and type 2 diabetes mellitus
Tables 9 and 10 summarize the Grade 3 or 4 hematologic laboratory abnormalities or all grades non-hematologic abnormalities in PACE.
| Laboratory Abnormality | CP-CML (N = 270) (%) | AP-CML (N = 85) (%) | BP-CML (N = 62) (%) | Ph+ ALL (N = 32) (%) |
|---|---|---|---|---|
|
||||
| Hematology | ||||
| Platelet count decreased | 35 | 49 | 45 | 47 |
| Neutrophil cell count decreased | 23 | 52 | 48 | 59 |
| White blood cell decreased | 12 | 37 | 48 | 63 |
| Lymphocyte decreased | 10 | 25 | 32 | 19 |
| Hemoglobin decreased | 8 | 31 | 52 | 34 |
| Laboratory Abnormality | Pooled Safety Population (N = 449) |
|
|---|---|---|
| All Grades*
(%) | Grade 3 or 4 (%) |
|
| ALT = alanine aminotransferase, AST = aspartate aminotransferase | ||
|
||
| Chemistry | ||
| Glucose increased | 54 | 7 |
| Phosphate decreased | 34 | 10 |
| Calcium decreased | 30 | 0.9 |
| Sodium decreased | 27 | 4.9 |
| Creatinine increased | 21 | 0.2 |
| Potassium increased | 20 | 2.2 |
| Bicarbonate decreased | 20 | 0.2 |
| Liver Function Tests | ||
| ALT increased | 41 | 6 |
| Alkaline phosphatase increased | 40 | 2 |
| AST increased | 35 | 3.6 |
| Albumin decreased | 28 | 0.2 |
| Bilirubin increased | 13 | 0.9 |
| Pancreatic Enzymes | ||
| Lipase increased | 40 | 14 |
| Amylase increased | 18 | 3.6 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of ICLUSIG. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
Blood and Lymphatic System Disorders: Thrombotic microangiopathy
Endocrine Disorders: Hyperthyroidism
Gastrointestinal Disorders: Gastrointestinal perforation, fistula
Metabolism and Nutrition Disorders: Dehydration
Nervous System Disorders: Reversible posterior leukoencephalopathy syndrome (RPLS)
Skin and Subcutaneous Tissue Disorders: Severe cutaneous reaction (e.g., Erythema multiforme, Stevens-Johnson syndrome), impaired wound healing, panniculitis (including erythema nodosum)
Vascular Disorders: Arterial (including aortic) aneurysms, dissections, and rupture
Related/similar drugs
7. Drug Interactions
7.1 Effects of Other Drugs on ICLUSIG
Strong CYP3A Inhibitors
Coadministration of ICLUSIG with a strong CYP3A inhibitor increases ponatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the risk of ICLUSIG adverse reactions. Avoid coadministration of ICLUSIG with strong CYP3A inhibitors. If coadministration of ICLUSIG with strong CYP3A inhibitors cannot be avoided, reduce the ICLUSIG dosage [see Dosage and Administration (2.3)].
Strong CYP3A Inducers
Coadministration of ICLUSIG with a strong CYP3A inducer decreases ponatinib plasma concentrations [see Clinical Pharmacology (12.3)]. Avoid coadministration of ICLUSIG with strong CYP3A inducers unless the benefit outweighs the risk of decreased ponatinib exposure. Monitor patients for reduced efficacy. Selection of concomitant medication with no or minimal CYP3A induction potential is recommended.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action [see Clinical Pharmacology (12.1)], ICLUSIG can cause fetal harm when administered to a pregnant woman. There are no available data on ICLUSIG use in pregnant women. In animal reproduction studies, oral administration of ponatinib to pregnant rats during organogenesis caused adverse developmental effects at doses lower than human exposures at the maximum recommended human dose of 45 mg/day (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
Ponatinib was studied for effects on embryo-fetal development in pregnant rats given oral doses of 0.3 mg/kg/day, 1 mg/kg/day, and 3 mg/kg/day during organogenesis (25 rats per group). At the maternally toxic dose of 3 mg/kg/day (equivalent to the AUC in patients receiving the maximum recommended dose of 45 mg/day), ponatinib caused embryo-fetal toxicity as shown by increased resorptions, reduced body weight, external alterations, multiple soft tissue and skeletal alterations, and reduced ossification. Embryo-fetal toxicities also were observed at 1 mg/kg/day (approximately 24% the AUC in patients receiving the maximum recommended dose of 45 mg/day) and involved multiple fetal soft tissue and skeletal alterations, including reduced ossification.
8.2 Lactation
Risk Summary
There are no data on the presence of ponatinib in human milk, the effects on the breastfed child, or on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ICLUSIG and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
ICLUSIG can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating ICLUSIG.
Infertility
Females
Based on animal data, ponatinib may impair fertility in females of reproductive potential [see Nonclinical Toxicology (13.1)]. It is not known whether these effects on fertility are reversible.
8.4 Pediatric Use
Safety and effectiveness of ICLUSIG have not been established in pediatric patients.
Juvenile Animal Toxicity Data
A juvenile toxicity study in 15 day old rats was conducted with daily oral gavage administration of ponatinib at 0.75 mg/kg/day, 1.5 mg/kg/day, or 3 mg/kg/day for 21 days. There were no adverse effects of ponatinib on juvenile rat developmental parameters (vaginal opening, preputial separation or bone measurements) observed in this study. Once daily oral administration of 3 mg/kg/day ponatinib to juvenile rats beginning on Day 15 postpartum (pp) resulted in mortality related to inflammatory effects after 6 to 7 days following initiation of treatment. The dose of 3 mg/kg/day is approximately 0.32 times the maximum recommended human dose of 45 mg/day on a mg/m2 basis for a child.
8.5 Geriatric Use
Of the 163 patients with Ph+ALL who received ICLUSIG in PhALLCON, 21% were 65 years and older and 7% were 75 years and older. Overall, no differences in efficacy of ICLUSIG were observed between patients 65 years of age or older compared to younger patients. AOEs occurred in 21% (7/34) of patients 65 years and older and 2.3% (3/129) of patients less than 65 years of age.
Of the 94 patients with CP-CML who received ICLUSIG at a starting dose of 45 mg in OPTIC, 17% were 65 years and older and 2.1% were 75 years and older. Patients aged 65 years and older had a lower ≤1% BCR::ABL1IS rate at 12 months (27%) as compared with patients less than 65 years of age (47%). AOEs occurred in 38% (6/16) of patients 65 years and older and 9% (7/78) of patients less than 65 years of age [see Warnings and Precautions (5.1)].
Of the 449 patients who received ICLUSIG in PACE, 35% were 65 years and older and 8% were 75 years and older. In patients with CP-CML, patients aged 65 years and older had a lower major cytogenetic response rate (40%) as compared with patients less than 65 years of age (65%). In patients with AP-CML, BP-CML, and Ph+ ALL, patients aged 65 years and older had a similar hematologic response rate (45%) as compared with patients less than 65 years of age (44%). AOEs occurred in 35% (54/155) of patients 65 years and older and in 21% (61/294) of patients less than 65 years of age [see Warnings and Precautions (5.1)].
Patients aged 65 years or older are more likely to experience adverse reactions including vascular occlusion, decreased platelet count, peripheral edema, increased lipase, dyspnea, asthenia, muscle spasms, and decreased appetite. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
Patients with hepatic impairment are more likely to experience adverse reactions compared to patients with normal hepatic function. For patients with CP-CML, AP-CML, BP-CML, and Ph+ ALL receiving monotherapy, reduce the starting dose of ICLUSIG for patients with pre-existing hepatic impairment (Child-Pugh A, B, or C). For patients with newly diagnosed Ph+ ALL, dosage adjustment is not recommended when administering ICLUSIG to patients with mild hepatic impairment (Child-Pugh A). Clinical data in patients with newly diagnosed Ph+ ALL with pre-existing moderate or severe hepatic impairment (Child-Pugh B or C) is not available and patients should be closely monitored for potential increased incidence of adverse reactions. Modify the ICLUSIG dosage in the event of adverse reactions [see Dosage and Administration (2.2, 2.4), Clinical Pharmacology (12.3)]. The safety of multiple doses, or doses higher than 30 mg, has not been studied in patients with hepatic impairment.
10. Overdosage
Overdoses with ICLUSIG were reported in clinical trials. One patient was estimated to have been administered 540 mg via nasogastric tube. Two hours after the overdosage, the patient had an uncorrected QT interval of 520 ms. Subsequent ECGs showed normal sinus rhythm with uncorrected QT intervals of 480 ms and 400 ms. The patient died 9 days after the overdosage from pneumonia and sepsis. Another patient self-administered 165 mg on Cycle 1 Day 2. The patient experienced fatigue and non-cardiac chest pain on Day 3. Multiple doses of 90 mg per day for 12 days in a patient resulted in pneumonia, systemic inflammatory response, atrial fibrillation, and a moderate pericardial effusion.
In the event of an overdosage, stop ICLUSIG, observe the patient and provide supportive treatment as appropriate.
11. Iclusig Description
Ponatinib is a kinase inhibitor. The chemical name for ponatinib hydrochloride is 3-(imidazo[1,2-b]pyridazin-3-ylethynyl)-4-methyl-N-{4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl}benzamide hydrochloride. The molecular formula is C29H28ClF3N6O which corresponds to a formula weight of 569.02 g/mol. Its structure is shown below:
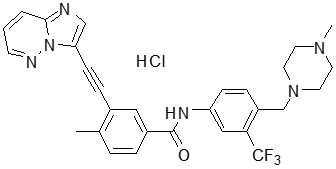
Ponatinib HCl is an off-white to yellow powder with pKa of 2.77 and 7.8. The solubility of ponatinib in pH 1.7, 2.7, and 7.5 buffers is 7790 mcg/mL, 3.44 mcg/mL, and 0.16 mcg/mL, respectively, indicating a decrease in solubility with increasing pH. Each tablet for oral administration contains 10 mg, 15 mg, 30 mg or 45 mg of ponatinib equivalent to 10.68 mg, 16.03 mg, 32.05 mg, and 48.08 mg of ponatinib hydrochloride with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, sodium starch glycolate (type B), colloidal silicon dioxide, magnesium stearate and a tablet coating. The tablet coating consists of talc, polyethylene glycol, polyvinyl alcohol, and titanium dioxide.
12. Iclusig - Clinical Pharmacology
12.1 Mechanism of Action
Ponatinib is a kinase inhibitor. Ponatinib inhibited the in vitro tyrosine kinase activity of ABL and T315I mutant ABL with IC50 concentrations of 0.4 nM and 2.0 nM, respectively. Ponatinib inhibited the in vitro activity of additional kinases with IC50 concentrations between 0.1 nM and 20 nM, including members of the VEGFR, PDGFR, FGFR, EPH receptors and SRC families of kinases, and KIT, RET, TIE2, and FLT3. Ponatinib inhibited the in vitro viability of cells expressing native or mutant BCR::ABL, including T315I. In mice, treatment with ponatinib reduced the size of tumors expressing native or T315I mutant BCR::ABL when compared to controls.
12.2 Pharmacodynamics
In PACE, the dose intensity-safety relationship indicated that there are significant increases in Grade ≥3 adverse reactions (hypertension, thrombocytopenia, pancreatitis, neutropenia, rash, ALT increase, AST increase, lipase increase, myelosuppression) over the dose range of 15 mg to 45 mg. In addition to dose, increased age and history of ischemia, hypertension, diabetes, or hypercholesterolemia were also contributory factors to a higher incidence of AOEs.
In OPTIC, an exposure-response relationship between ponatinib exposure and molecular response rate at 12 months was observed. A relationship between higher ponatinib exposures and higher incidence of adverse reactions, including thrombocytopenia (Grade ≥3) and AOEs, was observed.
In vitro, there was no significant inhibition of platelet aggregation with ponatinib at concentrations seen clinically and up to 0.7 mcg/mL (1.23 μM).
Cardiac Electrophysiology
The QT interval prolongation potential of ICLUSIG was assessed in 39 patients with cancer who received ICLUSIG 30 mg, 45 mg, or 60 mg (0.67 to 1.33 times the approved maximum recommended starting dose) orally once daily. No large mean increase (i.e., >20 msec) in QTc interval was detected.
12.3 Pharmacokinetics
Ponatinib administered to patients with cancer exhibited approximately dose proportional increases in both steady-state Cmax and AUC over the dose range of 2 mg to 60 mg (0.04 to 1.33 times the approved maximum recommended starting dose). The mean (CV%) Cmax and AUC(0-24) of ICLUSIG 45 mg orally once daily at presumed steady-state in patients with advanced hematologic malignancies were 73 ng/mL (74%) and 1253 ng∙hr/mL (73%), respectively. The mean (CV%) Cmax and AUC(0-24) of ICLUSIG 30 mg orally once daily at presumed steady-state in patients with advanced hematologic malignancies were 65 ng/mL (28%) and 1080 ng∙hr/mL (29%), respectively. Exposure increased by approximately 90% (median) [range: 20% to 440%] between the first dose and presumed steady-state.
Absorption
The absolute bioavailability of ponatinib is unknown. Peak concentrations of ponatinib are observed within 6 hours after ICLUSIG oral administration.
Effect of Food
Following ingestion of either a high-fat (approximately 900 to 1000 calories with approximately 150, 250, and 500 to 600 calories derived from protein, carbohydrate, and fat, respectively) or low-fat meal (approximately 547 calories with approximately 56, 428 and 63 calories derived from protein, carbohydrate, and fat, respectively) by 22 healthy volunteers, plasma ponatinib exposures (AUC and Cmax) were not different when compared to fasting conditions.
Distribution
Ponatinib is greater than 99% bound to plasma proteins in vitro. There was no plasma protein binding displacement of ponatinib (145 nM) in vitro by other highly protein bound medications (ibuprofen, nifedipine, propranolol, salicylic acid, and warfarin).
The mean (CV%) apparent steady-state volume of distribution is 1,223 liters (102%) following oral administration of ICLUSIG 45 mg orally once daily for 28 days in patients with cancer.
Elimination
The mean (range) terminal elimination half-life of ponatinib was approximately 24 (12 to 66) hours following ICLUSIG 45 mg orally once daily for 28 days in patients with cancer.
Specific Populations
No clinically significant differences in the pharmacokinetics of ponatinib were observed based on age (19 to 85 years), body weight (41 to 152 kg), and mild to moderate renal impairment (creatinine clearance 30 to 89 mL/min, estimated by the Cockcroft-Gault equation).
Patients with Renal Impairment
ICLUSIG has not been studied in patients with severe renal impairment. Although renal excretion is not a major route of ponatinib elimination, the potential for severe renal impairment to affect hepatic elimination has not been determined.
Patients with Hepatic Impairment
A single 30 mg oral dose of ICLUSIG was administered to subjects with normal hepatic function and to subjects with mild (Child-Pugh A), moderate (Child-Pugh B), and severe (Child-Pugh C) hepatic impairment. Compared to subjects with normal hepatic function, there was no trend of increased ponatinib exposure in subjects with hepatic impairment. There was an increased incidence of adverse reactions (e.g., gastrointestinal disorders, including a case of severe pancreatitis) in subjects with hepatic impairment compared to subjects with normal hepatic function.
Drug Interaction Studies
Clinical Studies
Strong CYP3A Inhibitors: Coadministration of ponatinib with multiple doses of ketoconazole (strong CYP3A inhibitor) increased the ponatinib AUC0-INF by 78% and Cmax by 47%.
In Vitro Studies
CYP Enzymes: Ponatinib does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A, or CYP2D6 and does not induce CYP1A2, CYP2B6, or CYP3A.
Transporter Systems: Ponatinib is a weak substrate for both P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Ponatinib is not a substrate for organic anion transporting polypeptides (OATP1B1, OATP1B3) and organic cation transporter 1 (OCT1).
Ponatinib inhibits P-gp, BCRP, and bile salt export pump (BSEP). Ponatinib does not inhibit OATP1B1, OATP1B3, OCT1, OCT2, or the organic anion transporters OAT1 and OAT3.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2 year carcinogenicity study, male and female rats were administered daily oral doses of ponatinib of 0.05 mg/kg/day, 0.1 mg/kg/day, 0.2 mg/kg/day and 0.2 mg/kg/day, 0.4 mg/kg/day, and 0.8 mg/kg/day, respectively. Exposures in animals at the highest dose tested were 0.3- to 0.8-fold the human exposure (based on AUC) at doses of 15 mg and 45 mg daily. Ponatinib induced a statistically significant increase in malignant squamous neoplasms of the clitoral gland in females at 0.8 mg/kg/day.
Ponatinib was not mutagenic in a bacterial mutagenesis (Ames) assay, was not clastogenic in a chromosome aberration assay in human lymphocytes, nor was it clastogenic in an in vivo mouse micronucleus assay at oral doses up to 2000 mg/kg.
Ponatinib may impair female fertility. In a fertility study in male and female rats, female fertility parameters were reduced at 1.5 mg/kg/day with exposure equivalent to 0.43 times and 1.23 times of human daily steady-state AUC at the maximum recommended human dose of 45 mg/day (AUC = 1296 h∙ng/mL) and 15 mg/day (451.8 h∙ng/mL), respectively. Evidence of pre- and post-implantation loss of embryos was observed in female rats. Although there were no effects on male fertility parameters in the rat fertility study, repeat dose toxicology studies in monkeys showed degeneration of epithelium of the testes in monkeys at exposures approximately 3.3 times the plasma drug exposure (AUC) in patients receiving the maximum recommended human dose of 45 mg/day.
14. Clinical Studies
Newly Diagnosed Ph+ ALL
The efficacy of ICLUSIG in combination with chemotherapy was evaluated in PhALLCON (NCT03589326), a randomized, active-controlled, multicenter, open-label trial of 245 patients with newly diagnosed Ph+ ALL. Randomization was stratified by age at the time of induction therapy (18 to <45 years; ≥45 to <60 years; and ≥60 years). Patients were randomized (2:1) to receive either ICLUSIG 30 mg orally once daily (n=164) or imatinib 600 mg orally once daily (n=81) in combination with chemotherapy (imatinib in combination with chemotherapy is an unapproved regimen in adult patients). The ICLUSIG dose was reduced to 15 mg once daily after completion of the induction phase and achievement of MRD-negative complete remission (CR). If a patient lost MRD negativity at any time after dose reduction to 15 mg, re-escalation to 30 mg once daily was allowed. Only patients who achieved CR or CR with incomplete hematologic recovery (CRi) with MRD-negativity at the end of induction could continue study treatment at the investigator’s discretion.
Per protocol, patients were allowed to receive one cycle of optional prephase therapy excluding TKI prior to randomization to manage the acute disease during the screening period.
Patients were randomized to receive either ICLUSIG or imatinib in combination with 20 cycles of chemotherapy, followed by ICLUSIG or imatinib as single-agent therapy (ICLUSIG or imatinib as single-agent after chemotherapy for newly diagnosed Ph+ ALL is not an approved regimen). Each cycle lasted 28 days.
-
Induction (Cycles 1 to 3): ICLUSIG 30 mg or imatinib 600 mg once daily in combination with:
- Vincristine: 1.4 mg/m2 IV on Days 1 and 14; capped at 2 mg and
- Dexamethasone: <60 years old – 40 mg orally on Days 1 to 4 and Days 11 to 14; ≥60 years old: 20 mg orally on Days 1 to 4 and Days 11 to 14.
-
Consolidation (Cycles 4 to 9, alternating methotrexate and cytarabine): ICLUSIG 30 mg (or decreased to 15 mg if in MRD-negative CR) or imatinib 600 mg once daily in combination with:
- Methotrexate (Cycles 4, 6, and 8): <60 years old – 1000 mg/m2 IV on Day 1; ≥60 years old – 250 mg/m2 IV, Day 1 or
- Cytarabine (Cycles 5, 7, and 9): <60 years old – 1000 mg/m2 IV every 12 hours on Days 1, 3, and 5; ≥60 years old – 250 mg/m2 IV every 12 hours on Days 1, 3, and 5
-
Maintenance (Cycles 10 to 20): ICLUSIG 30 mg (or decreased to 15 mg if in MRD-negative CR) or imatinib 600 mg once daily in combination with:
- Vincristine: 1.4 mg/m2 IV on Day 1 of each cycle; capped at 2 mg and
- Prednisone: <60 years old – 200 mg orally on Days 1 to 5; ≥60 to 69 years old – 100 mg orally on Days 1 to 5; ≥70 years old – 50 mg orally on Days 1 to 5
Following combination therapy, patients continued to receive ICLUSIG or imatinib as single-agent therapy until relapse from CR, progressive disease (PD), hematopoietic stem cell transplantation (HSCT), start of alternative therapy, or unacceptable toxicity.
The demographics and baseline disease characteristics of the randomized population are described in Table 11.
| Patient Characteristics at Entry | ICLUSIG 30 mg → 15 mg with Chemotherapy (N = 164) | Imatinib 600 mg with Chemotherapy (N = 81) |
|---|---|---|
|
||
| Age (years) | ||
| Median, years (range) | 54 (19 to 82) | 52 (19 to 75) |
| Age Category, n (%) | ||
| 18 to <45 years | 58 (35%) | 29 (36%) |
| 45 to <60 years | 45 (27%) | 22 (27%) |
| ≥60 years | 61 (37%) | 30 (37%) |
| Sex, n (%) | ||
| Female | 90 (55%) | 43 (53%) |
| Race, n (%) | ||
| White | 104 (63%) | 62 (77%) |
| Not reported | 28 (17%) | 2 (3%) |
| Asian | 20 (12%) | 11 (14%) |
| Black or African American | 9 (5%) | 4 (5%) |
| ECOG Performance Status, n (%) | ||
| 0 | 72 (44%) | 33 (41%) |
| 1 | 85 (52%) | 43 (53%) |
| 2 | 7 (4%) | 5 (6%) |
| Baseline BCR::ABL1 Dominant Variant | ||
| p190 | 114 (70%) | 53 (65%) |
| p210 | 40 (24%) | 25 (31%) |
| Undetermined/not tested | 10 (6%) | 3 (4%) |
| Prephase Therapy* | 74 (45%) | 41 (51%) |
| Comorbidities, n (%) | ||
| Hypertension | 58 (35%) | 30 (37%) |
| Diabetes | 39 (24%) | 24 (30%) |
| Dyslipidemia | 29 (18%) | 23 (28%) |
Among 244 treated patients, 96% completed induction (96% ICLUSIG, 95% imatinib), 84% received at least one cycle of consolidation (89% ICLUSIG, 75% imatinib), and 31% initiated maintenance (36% ICLUSIG, 21% imatinib). After completing combination therapy, 21% of patients received ICLUSIG and 9% received imatinib as single-agent therapy. The overall rate of HSCT was 34% (56/164) in the ICLUSIG arm versus 48% (39/81) in the imatinib arm.
Efficacy was based on the MRD-negative CR rate at the end of induction. The analysis population for MRD-negative CR included 232 randomized patients who had a baseline BCR::ABL1 dominant variant of p190 or p210 as determined by central laboratory tests (154 patients in the ICLUSIG arm and 78 in the imatinib arm). Efficacy results are summarized in Table 12.
| ICLUSIG 30 mg → 15 mg with Chemotherapy (N = 154) | Imatinib 600 mg with Chemotherapy (N = 78) |
|
|---|---|---|
| MRD: minimal residual disease; CR: complete remission (complete response); BCR::ABL1: breakpoint cluster region-Abelson. | ||
|
||
| MRD-negative CR* at End of Induction | ||
| Achieved at the end of induction % (n/N) | 30% (46/154) | 12% (9/78) |
| Risk difference (95% CI)† | 0.18 (0.08, 0.28) | |
| p-value† | 0.0004 | |
| CR‡ at End of Induction % (n/N) | 79% (122/154) | 63% (49/78) |
The median duration of follow-up for overall survival was 20.4 months (95% CI: 18.4, 23.9) in the ICLUSIG arm and 18.1 months (95% CI: 13.9, 24.3) in the imatinib arm.
In the subset of patients who did not receive prephase therapy, MRD-negative CR at the end of induction was achieved by 31% of patients in the ICLUSIG arm compared to 16% of patients in the imatinib arm and CR at the end of induction was achieved by 84% and 61%, respectively.
Chronic Phase (CP) CML
The efficacy of ICLUSIG was evaluated in OPTIC (NCT02467270), a dose-optimization trial. Eligible patients had CP-CML whose disease was considered to be resistant or resistant/intolerant to at least 2 prior kinase inhibitors or who have the T315I mutation. T315I mutation testing was performed on peripheral blood by Sanger Sequencing of the p190 or p210 BCR::ABL region. Resistance in CP-CML while on a prior kinase inhibitor was defined as failure to achieve either a complete hematologic response (by 3 months), a minor cytogenetic response (by 6 months), or a major cytogenetic response (by 12 months), or development of a new BCR::ABL1 kinase domain mutation or new clonal evolution. Patients were required to have >1% BCR::ABL1IS (by real-time polymerase chain reaction) at trial entry. Patients received one of three starting dosages: 45 mg orally once daily, 30 mg orally once daily, or 15 mg orally once daily. Patients who received a starting dose of 45 mg or 30 mg had a dose reduction to 15 mg once daily upon achieving ≤1% BCR::ABL1IS. The major efficacy outcome measure was ≤1% BCR::ABL1IS at 12 months. The median duration of follow-up for the 45 mg cohort (N=94) was 27.0 months. Only the efficacy results for the recommended starting dose of 45 mg are described below.
A total of 282 patients received ICLUSIG: 94 received a starting dose of 45 mg, 94 received a starting dose of 30 mg, and 94 received a starting dose of 15 mg. Baseline demographic characteristics are described in Table 13 for patients who received a starting dose of 45 mg.
| Patient Characteristics at Entry | ICLUSIG 45 mg → 15 mg (N = 94) |
|---|---|
| Age | |
| Median years (range) | 46 (19 to 81) |
| Sex, n (%) | |
| Male | 50 (53%) |
| Race, n (%) | |
| White | 73 (78%) |
| Asian | 16 (17%) |
| Other/Unknown | 4 (4%) |
| Black or African American | 1 (1%) |
| ECOG Performance Status, n (%) | |
| ECOG 0 or 1 | 93 (99%) |
| Disease History | |
| Median time from diagnosis to first dose, years (range) | 5.5 (1 to 21) |
| Resistant to Prior Kinase Inhibitor, n (%) | 92 (98%) |
| Presence of one or more BCR::ABL kinase domain mutations, n (%) | 41 (44%) |
| Number of Prior Kinase Inhibitors, n (%) | |
| 1 | 1 (1%) |
| 2 | 43 (46%) |
| ≥3 | 50 (53%) |
| T315I mutation at baseline | 25 (27%) |
| Comorbidities | |
| Hypertension | 29 (31%) |
| Diabetes | 5 (5%) |
| Hypercholesterolemia | 3 (3%) |
| History of ischemic heart disease | 3 (3%) |
Efficacy results are summarized in Table 14.
| ICLUSIG 45 mg → 15 mg (N = 93)* |
|
|---|---|
|
|
| Molecular Response at 12 months† | |
| Overall ≤1% BCR::ABL1IS Rate | |
| % (n/N) | 44% (41/93) |
| (95% CI)‡ | (34%, 55%) |
| Patients with T315I mutation | |
| % (n/N) | 44% (11/25) |
| (95% CI) | (24%, 65%) |
| Patients without T315I mutation | |
| % (n/N) | 44% (29/66)§ |
| (95% CI) | (32%, 57%) |
| Cytogenetic Response by 12 months | |
| Major (MCyR)¶ | |
| % (n/N) | 48% (44/91)# |
| (95% CI) | (38%, 59%) |
| Patients with T315I mutation | |
| % (n/N) | 52% (13/25) |
| (95% CI) | (31%, 72%) |
| Patients without T315I mutation | |
| % (n/N) | 46% (30/65)Þ |
| (95% CI) | (34%, 59%) |
Of the 45 patients who had a dose reduction after achieving ≤1% BCR::ABL1IS, 28 patients (62%) maintained their response at the reduced dose for at least 90 days. Of these 28 patients, 18 patients (64%) maintained the response for at least one year. Median duration of response (MR2) was not reached.
Chronic Phase (CP), Accelerated Phase (AP), Blast Phase (BP) CML and Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia (Ph+ ALL)
The efficacy of ICLUSIG was evaluated in PACE (NCT01207440), a single-arm, open-label, international, multicenter trial. Eligible patients had CML and Ph+ ALL whose disease was considered to be resistant or intolerant to a prior kinase inhibitor. Patients were assigned to one of six cohorts based on disease phase (CP-CML, AP-CML, or BP-CML/Ph+ ALL), resistance or intolerance (R/I) to prior kinase inhibitors, and the presence of the T315I mutation. T315I mutation testing was performed on peripheral blood by Sanger Sequencing of the p190 or p210 BCR::ABL region.
Resistance in CP-CML while on a prior kinase inhibitor was defined as failure to achieve either a complete hematologic response (by 3 months), a minor cytogenetic response (by 6 months), or a major cytogenetic response (by 12 months). Patients with CP-CML who experienced a loss of response or development of a kinase domain mutation in the absence of a complete cytogenetic response or progression to AP-CML or BP-CML at any time on a prior kinase inhibitor were also considered resistant.
Resistance in AP-CML, BP-CML, and Ph+ ALL was defined as failure to achieve either a major hematologic response (by 3 months in AP-CML, and by 1 month in BP-CML and Ph+ ALL), loss of major hematologic response (at any time), or development of a kinase domain mutation in the absence of a complete major hematologic response while on a prior kinase inhibitor. Intolerance was defined as the discontinuation of a prior kinase inhibitor due to toxicities despite optimal management in the absence of a complete cytogenetic response in patients with CP-CML or major hematologic response for patients with AP-CML, BP-CML, or Ph+ ALL.
Patients were administered a starting dose of ICLUSIG 45 mg orally once daily.
The major efficacy outcome measure for patients with CP-CML was major cytogenetic response (MCyR), which included complete and partial cytogenetic responses (CCyR and PCyR). The major efficacy outcome measure for patients with AP-CML, BP-CML, and Ph+ ALL was major hematologic response (MaHR), defined as either a complete hematologic response (CHR) or no evidence of leukemia (NEL).
The trial enrolled 449 patients, of which 444 were eligible for efficacy analysis: 267 patients with CP-CML (R/I Cohort: N=203, T315I: N=64), 83 patients with AP-CML, 62 patients with BP-CML, and 32 patients with Ph+ ALL. Five patients were not eligible for efficacy analysis due to lack of confirmation of T315I mutation status, and these patients had not received prior dasatinib or nilotinib.
At study completion, the median duration of follow-up for the trial (all cohorts) was 40.5 months (range: 0.1 months to 79.5 months). The median duration of treatment was 35 months for patients with CP-CML, 21.1 months for patients with AP-CML, 3.2 months for patients with BP-CML and 2.9 months for patients with Ph+ ALL. Baseline demographic characteristics are described in Table 15.
| Patient Characteristics at Entry | Efficacy Population (N = 444) |
|---|---|
|
|
| Age | |
| Median, years (range) | 59 (18 to 94) |
| Sex, n (%) | |
| Male | 236 (53%) |
| Race, n (%) | |
| White | 349 (79%) |
| Asian | 57 (13%) |
| Black or African American | 25 (6%) |
| Other/Unknown | 13 (3%) |
| ECOG Performance Status, n (%) | |
| ECOG = 0 or 1 | 409 (92%) |
| Disease History | |
| Median time from diagnosis to first dose, years (range) | 6.1 (0.3 to 29) |
| Resistant to Prior Kinase Inhibitor, n (%) | 374 (88%) |
| Presence of one or more BCR::ABL kinase domain mutations*, n (%) | 244 (55%) |
| Number of Prior Kinase Inhibitor, n (%) | |
| 1 | 29 (7%) |
| 2 | 166 (37%) |
| ≥3 | 249 (56%) |
| T315I mutation at baseline | 128 (29%) |
| Comorbidities | |
| Hypertension | 159 (35%) |
| Diabetes | 57 (13%) |
| Hypercholesterolemia | 100 (22%) |
| History of ischemic disease | 67 (15%) |
Efficacy results are summarized in Table 16 and Table 17.
| Overall (N = 267) | Cohort | ||
|---|---|---|---|
| R/I Cohort (N = 203) | T315I Cohort (N = 64) |
||
|
|||
| Cytogenetic Response | |||
| Major* (MCyR) (95% CI) | 55% (49%, 62%) | 51% (44%, 58%) | 70% (58%, 81%) |
| Complete (CCyR) (95% CI) | 46% (40%, 52%) | 40% (33%, 47%) | 66% (53%, 77%) |
| Major Molecular Response†
(95% CI) | 40% (35%, 47%) | 35% (28%, 42%) | 58% (45%, 70%) |
In patients with CP-CML who achieved MCyR or MMR, the median time to response was 3 months (range: 1.8 to 12.3 months) and 6 months (range: 2 to 60.2 months), respectively. With a minimum follow-up of 60 months, the median durations of MCyR (range: 1 day to 70.1 months) and MMR (range: 1 day to 67.8 months) had not yet been reached.
| AP-CML Overall (N = 83) | BP-CML Overall (N = 62) | Ph+ ALL Overall (N = 32) |
|
|---|---|---|---|
|
|||
| Hematologic Response | |||
| Major* (MaHR) | 57% | 31% | 41% |
| (95% CI) | (45%, 68%) | (20%, 44%) | (24%, 59%) |
| Complete† (CHR) | 51% | 21% | 34% |
| (95% CI) | (39%, 62%) | (12%, 33%) | (19%, 53%) |
The median time to MaHR in patients with AP-CML, BP-CML, and Ph+ ALL was 0.8 months (range: 0.4 to 6.3 months), 1.0 month (range: 0.4 to 4 months), and 0.7 months (range: 0.4 to 6 months), respectively. The median duration of MaHR for patients with AP-CML, BP-CML, and Ph+ ALL was 14 months (range: 1.3 to 74.3 months), 6.5 months (range: 1.9 to 64.7 months), and 3.5 months (range: 1.9 to 13.7 months), respectively.
16. How is Iclusig supplied
ICLUSIG tablets are available in the following configurations.
| Strength | NDC Number | Description | Presentation |
|---|---|---|---|
| 10 mg | 63020-536-30 | oval, white to off-white, biconvex film-coated tablets with debossed "NZ" on one side and plain on the other side | 30 tablets in a wide-mouth white high density polyethylene (HDPE) bottle with a desiccant canister and induction sealed child resistant closure. |
| 15 mg | 63020-535-30 | round, white, biconvex film-coated tablets with debossed "A5" on one side and plain on the other side | 30 tablets in a wide-mouth white high density polyethylene (HDPE) bottle with a desiccant canister and induction sealed child resistant closure. |
| 63020-535-60 | 60 tablets in a wide-mouth white high density polyethylene (HDPE) bottle with a desiccant canister and induction sealed child resistant closure. | ||
| 30 mg | 63020-533-30 | round, white, biconvex film-coated tablets with debossed "C7" on one side and plain on the other side | 30 tablets in a wide-mouth white high density polyethylene (HDPE) bottle with a desiccant canister and induction sealed child resistant closure. |
| 45 mg | 63020-534-30 | round, white, biconvex film-coated tablets with debossed "AP4" on one side and plain on the other side | 30 tablets in a wide-mouth white high density polyethylene (HDPE) bottle with a desiccant canister and induction sealed child resistant closure. |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Arterial Occlusive Events and Venous Thromboembolic Events
Inform patients that serious arterial thromboses (including arterial stenosis sometimes requiring revascularization) and VTEs have occurred. Advise patients to immediately contact their healthcare provider with any symptoms suggestive of a blood clot such as chest pain, shortness of breath, weakness on one side of the body, speech problems, leg pain, or leg swelling [see Warnings and Precautions (5.1, 5.2)].
Heart Failure and Cardiac Arrhythmias
Inform patients of the possibility of heart failure, and abnormally slow or fast heart rates. Advise patients to contact their healthcare provider if they experience symptoms such as shortness of breath, chest pain, palpitations, dizziness, or fainting [see Warnings and Precautions (5.3, 5.12)].
Hepatotoxicity
Inform patients of the possibility of developing liver function abnormalities and serious hepatic toxicity. Advise patients to immediately contact their healthcare provider if signs of liver failure occur, including jaundice, anorexia, bleeding or bruising [see Warnings and Precautions (5.4)].
Hypertension
Inform patients of the possibility of new or worsening of existing hypertension. Advise patients to contact their healthcare provider for elevated blood pressure or if symptoms of hypertension occur including confusion, headache, dizziness, chest pain, or shortness of breath [see Warnings and Precautions (5.5)].
Pancreatitis
Inform patients of the possibility of developing pancreatitis that may be accompanied by nausea, vomiting, abdominal pain, or abdominal discomfort, and to promptly report these symptoms [see Warnings and Precautions (5.6)].
Neuropathy
Inform patients of the possibility of developing peripheral or cranial neuropathy while being treated with ICLUSIG. Advise patients to report symptoms of neuropathy, such as hypoesthesia, hyperesthesia, paresthesia, discomfort, a burning sensation, neuropathic pain, or weakness [see Warnings and Precautions (5.8)].
Ocular Toxicity
Inform patients of the possibility of ocular toxicity while being treated with ICLUSIG. Advise patients to report symptoms of ocular toxicity, such as blurred vision, dry eye, or eye pain [see Warnings and Precautions (5.9)].
Hemorrhage
Inform patients of the possibility of serious bleeding and to immediately contact their healthcare provider with any signs or symptoms suggestive of hemorrhage such as unusual bleeding or easy bruising [see Warnings and Precautions (5.10)].
Fluid Retention
Inform patients of the possibility of developing fluid retention and to contact their healthcare provider for symptoms such as leg swelling, abdominal swelling, weight gain, or shortness of breath [see Warnings and Precautions (5.11)].
Myelosuppression
Inform patients of the possibility of developing low blood cell counts; inform patients to report immediately should fever develop, particularly in association with any suggestion of infection [see Warnings and Precautions (5.13)].
Tumor Lysis Syndrome
Inform patients of the possibility of developing TLS and to immediately contact their healthcare provider for any signs or symptoms associated with TLS [see Warnings and Precautions (5.14)]. Advise patients to be adequately hydrated when taking ICLUSIG to reduce the risk of TLS.
Reversible Posterior Leukoencephalopathy Syndrome (RPLS – also known as Posterior Reversible Encephalopathy Syndrome)
Inform patients of the possibility of developing Reversible Posterior Leukoencephalopathy Syndrome while being treated with ICLUSIG. Advise patients to report symptoms such as seizure, headache, decreased alertness, altered mental functioning, vision loss, and other visual and neurological disturbances [see Warnings and Precautions (5.15)].
Impaired Wound Healing and Gastrointestinal Perforation
Inform patients that impaired wound healing and gastrointestinal fistula or perforation have been reported. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.16)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy. Advise females of reproductive potential to use effective contraception during treatment with ICLUSIG and for 3 weeks after the last dose [see Warnings and Precautions (5.17), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with ICLUSIG and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise females of reproductive potential of the potential for reduced fertility from ICLUSIG [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Instructions for Taking ICLUSIG
Advise patients to take ICLUSIG exactly as prescribed and not to change their dose or to stop taking ICLUSIG unless they are told to do so by their healthcare provider. ICLUSIG may be taken with or without food. ICLUSIG tablets should be swallowed whole. Patients should not cut, crush or dissolve the tablets.
Patients should not take two doses at the same time to make up for a missed dose.
Advise patients not to drink grapefruit juice or eat grapefruit as it may increase the amount of ICLUSIG in their blood and therefore increase their risk of adverse reactions.
Distributed by:
Takeda Pharmaceuticals America, Inc.
Cambridge, MA 02142
ICLUSIG and  are registered trademarks of ARIAD Pharmaceuticals, Inc.
are registered trademarks of ARIAD Pharmaceuticals, Inc.
TAKEDA and  are registered trademarks of Takeda Pharmaceutical Company Limited.
are registered trademarks of Takeda Pharmaceutical Company Limited.
©2024 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
ICL348 R11
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: September 2024 | ||||
| MEDICATION GUIDE ICLUSIG® (eye-CLUE-sig) (ponatinib) tablets |
|||||
| What is the most important information I should know about ICLUSIG? ICLUSIG can cause serious side effects, including: Blood clots or blockage in your blood vessels (arteries and veins). Blood clots or blockage in your blood vessels may lead to heart attack, stroke, or death. A blood clot or blockage in your blood vessels can prevent proper blood flow to your heart, brain, bowels (intestines), legs, eyes, and other parts of your body. You may need emergency surgery or treatment in a hospital. Get medical help right away if you get any of the following symptoms: |
|||||
|
|
||||
| Blood clots or blockage in your blood vessels can happen in people with or without risk factors for heart and blood vessel disease, including people 50 years of age or younger. The most common risk factors for these problems are a history of high blood pressure (hypertension), high cholesterol, and heart disease. Blood clots or blockages in your blood vessels happen more often in people as they get older, and in people with a history of decreased blood flow, high blood pressure, diabetes, or high cholesterol. Heart problems. ICLUSIG can cause heart problems, including heart failure which can be serious and may lead to death. Heart failure means your heart does not pump blood well enough. ICLUSIG can also cause irregular, slow, or fast heartbeats and heart attack. Your healthcare provider will check you for heart problems during your treatment with ICLUSIG. Get medical help right away if you get any of the following symptoms: shortness of breath, chest pain, fast or irregular heartbeats, dizziness, or feel faint. Liver problems. ICLUSIG can cause liver problems, including liver failure, which can be severe and may lead to death. Your healthcare provider will do blood tests before and during your treatment with ICLUSIG to check for liver problems. Get medical help right away if you get any of these symptoms of liver problems during treatment:
|
|||||
| What is ICLUSIG?
ICLUSIG is a prescription medicine used to treat adults who have:
It is not known if ICLUSIG is safe and effective in children. |
|||||
Before you take ICLUSIG, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||
|
|
||||
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||||
How should I take ICLUSIG?
|
|||||
| What are the possible side effects of ICLUSIG? ICLUSIG may cause serious side effects, including:
|
|||||
|
|
||||
|
|||||
|
|
||||
|
|||||
|
|
|
|||
| The most common side effects of ICLUSIG when given with chemotherapy include: | |||||
|
|
|
|||
| Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with ICLUSIG if you have certain side effects. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of ICLUSIG. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||||
| How should I store ICLUSIG?
Store ICLUSIG at room temperature between 68°F to 77°F (20°C to 25°C). Keep ICLUSIG and all medicines out of the reach of children. |
|||||
| General information about the safe and effective use of ICLUSIG
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use ICLUSIG for a condition for which it was not prescribed. Do not give ICLUSIG to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ICLUSIG that is written for health professionals. |
|||||
|
What are the ingredients in ICLUSIG? |
|||||
PRINCIPAL DISPLAY PANEL - 15 mg Tablet Bottle Label
NDC 63020-535-30
ICLUSIG®
(ponatinib) tablets
15 mg
Each tablet contains 15 mg ponatinib
equivalent to 16.03 mg ponatinib HCl
Dispense Attached Medication Guide
30 tablets
Rx only
Takeda

PRINCIPAL DISPLAY PANEL - 45 mg Tablet Bottle Label
NDC 63020-534-30
ICLUSIG®
(ponatinib) tablets
45 mg
Each tablet contains 45 mg ponatinib
equivalent to 48.08 mg ponatinib HCl
Dispense Attached Medication Guide
Takeda



| ICLUSIG
ponatinib hydrochloride tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| ICLUSIG
ponatinib hydrochloride tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| ICLUSIG
ponatinib hydrochloride tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| ICLUSIG
ponatinib hydrochloride tablet, film coated |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Takeda Pharmaceuticals America, Inc. (039997266) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ash Stevens LLC | 049265333 | ANALYSIS(63020-535, 63020-534, 63020-533, 63020-536) , API MANUFACTURE(63020-535, 63020-534, 63020-533, 63020-536) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Micron Technologies, Inc. | 015966157 | ANALYSIS(63020-535, 63020-534, 63020-533, 63020-536) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Catalent Pharma Solutions, LLC | 014167995 | ANALYSIS(63020-535, 63020-534, 63020-533, 63020-536) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sharp Packaging Services, LLC | 143696495 | PACK(63020-535, 63020-534) , LABEL(63020-535, 63020-534) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Inc. | 240769596 | ANALYSIS(63020-533, 63020-534, 63020-535, 63020-536) , MANUFACTURE(63020-533, 63020-534, 63020-535, 63020-536) , PACK(63020-533, 63020-534, 63020-535, 63020-536) , LABEL(63020-533, 63020-534, 63020-535, 63020-536) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Takeda Ireland Limited | 988980314 | ANALYSIS(63020-535, 63020-534, 63020-533) , MANUFACTURE(63020-535, 63020-534, 63020-533) , PACK(63020-535, 63020-534, 63020-533) , LABEL(63020-533, 63020-534, 63020-535) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Almac Sciences (Ireland) Limited | 985822621 | ANALYSIS(63020-535, 63020-534, 63020-533) | |
More about Iclusig (ponatinib)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: BCR-ABL tyrosine kinase inhibitors
- Breastfeeding
- En español
