Probuphine: Package Insert / Prescribing Info
Package insert / product label
Generic name: buprenorphine hydrochloride
Dosage form: implant
Medically reviewed by Drugs.com. Last updated on Mar 17, 2025.
The Probuphine brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
PROBUPHINE (buprenorphine) implant, for subdermal use, CIII
Initial U.S. Approval: 1981
WARNING: IMPLANT MIGRATION, PROTRUSION, EXPULSION, and NERVE DAMAGE ASSOCIATED WITH INSERTION and REMOVAL
See full prescribing information for complete boxed warning.
Recent Major Changes
Indications and Usage for Probuphine
PROBUPHINE contains buprenorphine, a partial opioid agonist. PROBUPHINE is indicated for the maintenance treatment of opioid dependence in patients who have achieved and sustained prolonged clinical stability on low-to-moderate doses of a transmucosal buprenorphine-containing product (i.e., doses of no more than 8 mg per day of Subutex or Suboxone sublingual tablet or generic equivalent). (1)
PROBUPHINE should be used as part of a complete treatment program to include counseling and psychosocial support. (1)
PROBUPHINE is not appropriate for new entrants to treatment and patients who have not achieved and sustained prolonged clinical stability, while being maintained on buprenorphine 8 mg per day or less of a Subutex or Suboxone sublingual tablet or generic equivalent. (1)
Probuphine Dosage and Administration
Prescription use of this product is limited under the Drug Addiction Treatment Act. (2.1)
Four PROBUPHINE implants are inserted subdermally in the upper arm for 6 months of treatment and are removed by the end of the sixth month. (2.2)
PROBUPHINE implants should not be used for additional treatment cycles after one insertion in each upper arm. (2.2)
Strongly consider prescribing naloxone at the time PROBUPHINE is initiated or renewed because patients being treated for opioid use disorder have the potential for relapse, putting them at risk for opioid overdose. (2.3)
PROBUPHINE implants must be inserted and removed by trained Healthcare Providers only. (2.4)
PROBUPHINE implants should be administered in patients who have achieved and sustained prolonged clinical stability on transmucosal buprenorphine. (2.5)
Examine the insertion site one week following insertion of PROBUPHINE implants for signs of infection or other problems. (2.6)
Dosage Forms and Strengths
Each PROBUPHINE implant is an ethylene vinyl acetate (EVA) implant, 26 mm in length and 2.5 mm in diameter, containing 74.2 mg of buprenorphine (equivalent to 80 mg of buprenorphine hydrochloride). (3)
Contraindications
Hypersensitivity to buprenorphine or any other ingredients in PROBUPHINE (e.g., EVA). (4)
Warnings and Precautions
- Serious Complications from Insertion and Removal: Rare but serious complications including nerve damage and migration resulting in embolism and death may result from improper insertion of drug implants inserted in the upper arm. Additional complications may include local migration, protrusion, and expulsion. Incomplete insertions or infections may lead to protrusion or expulsion. All Healthcare Providers must successfully complete a live training program on the insertion and removal procedures and become certified in the PROBUPHINE REMS program, prior to performing insertions or prescribing PROBUPHINE implants. (5.1, 5.2)
- Addiction, Abuse, and Misuse: Buprenorphine can be abused in a manner similar to other opioids. Monitor patients for conditions indicative of diversion or progression of opioid dependence and addictive behaviors. (5.3)
- Respiratory Depression: Life-threatening respiratory depression and death have occurred in association with buprenorphine use. Warn patients of the potential danger of self-administration of benzodiazepines or other CNS depressants while under treatment with PROBUPHINE. (5.4, 5.5)
- Neonatal Opioid Withdrawal Syndrome: Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy. (5.6)
- Adrenal Insufficiency: If diagnosed, treat with physiologic replacement of corticosteroids, and wean patient off of the opioid. (5.7)
- Unintentional Pediatric Exposure: In the event an implant protrudes or comes out, keep the implant away from children. Buprenorphine can cause severe, possibly fatal, respiratory depression in children. (5.8)
- Risk of Opioid Withdrawal with Abrupt Discontinuation: If treatment with PROBUPHINE is discontinued, monitor patients for withdrawal and treat appropriately. (5.9)
- Risk of Hepatitis, Hepatic Events: Monitor liver function tests prior to initiation and during treatment and evaluate suspected hepatic events. (5.10)
- Risk of Withdrawal in Patients Dependent on Full Agonist Opioids: Verify that patient is clinically stable on transmucosal buprenorphine and not dependent on full agonists before inserting PROBUPHINE. (5.12)
- Treatment of Emergent Acute Pain: Treat pain with a non-opioid analgesic whenever possible. If opioid therapy is required, monitor patients closely because higher doses may be required for analgesic effect. (5.13)
Adverse Reactions/Side Effects
Adverse events commonly associated with PROBUPHINE administration (>10% of subjects) were implant-site pain, pruritus, and erythema, as well as non-implant-site related events (≥5%) of headache, depression, constipation, nausea, vomiting, back pain, toothache, and oropharyngeal pain. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Titan at 1-844-859-6341 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
- Benzodiazepines: Use caution in prescribing PROBUPHINE for patients receiving benzodiazepines or other CNS depressants and warn patients against concomitant self-administration/misuse. (7)
- CYP3A4 Inhibitors and Inducers: Monitor patients starting or ending CYP3A4 inhibitors or inducers for potential over- or under-dosing. (7)
- Antiretrovirals: Patients who are on chronic buprenorphine treatment should have their dose monitored if NNRTIs are added to their treatment regimen. Monitor patients taking buprenorphine and atazanavir with and without ritonavir. Dose reduction of buprenorphine may be warranted. (7)
- Serotonergic Drugs: Concomitant use may result in serotonin syndrome. Discontinue PROBUPHINE if serotonin syndrome is suspected. (7)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2021
Full Prescribing Information
WARNING: IMPLANT MIGRATION, PROTRUSION, EXPULSION, and NERVE DAMAGE ASSOCIATED WITH INSERTION and REMOVAL
Risk Associated with Insertion and Removal
Insertion and removal of PROBUPHINE are associated with the risk of implant migration, protrusion, and expulsion resulting from the procedure. Rare but serious complications including nerve damage and migration resulting in embolism and death may result from improper insertion of drug implants inserted in the upper arm. Additional complications may include local migration, protrusion and expulsion. Incomplete insertions or infections may lead to protrusion or expulsion [see Warnings and Precautions (5.1)].
Because of the risks associated with insertion and removal, PROBUPHINE is available only through a restricted program called the PROBUPHINE REMS Program. All Healthcare Providers must successfully complete a live training program on the insertion and removal procedures and become certified, prior to performing insertions or prescribing PROBUPHINE implants. Patients must be monitored to ensure that PROBUPHINE is removed by a Healthcare Provider certified to perform insertions [see Warnings and Precautions (5.2)].
1. Indications and Usage for Probuphine
PROBUPHINE is indicated for the maintenance treatment of opioid dependence in patients who have achieved and sustained prolonged clinical stability on low-to-moderate doses of a transmucosal buprenorphine-containing product (i.e., doses of no more than 8 mg per day of Subutex or Suboxone sublingual tablet or generic equivalent).
PROBUPHINE should be used as part of a complete treatment program to include counseling and psychosocial support.
PROBUPHINE is not appropriate for new entrants to treatment and patients who have not achieved and sustained prolonged clinical stability, while being maintained on buprenorphine 8 mg per day or less of a Subutex or Suboxone sublingual tablet equivalent or generic equivalent.
2. Probuphine Dosage and Administration
2.1. Drug Addiction Treatment Act
Under the Drug Addiction Treatment Act (DATA) codified at 21 United States Code (U.S.C.) 823(g), use of this product in the treatment of opioid dependence is limited to Healthcare Providers who meet certain qualifying requirements, and who have notified the Secretary of Health and Human Services (HHS) of their intent to prescribe or dispense this product for the treatment of opioid dependence and have been assigned a unique identification number that must be included on every prescription.
2.2. Important Dosage and Administration Information
PROBUPHINE implants should be used only in patients who are opioid tolerant.
Each dose consists of four PROBUPHINE implants inserted subdermally in the inner side of the upper arm.
PROBUPHINE subdermal implants are intended to be in place for 6 months of treatment. Remove PROBUPHINE implants by the end of the sixth month.
New implants may be inserted subdermally in an area of the inner side of either upper arm that has not been previously used at the time of removal, if continued treatment is desired. If new implants are not inserted on the same day as the removal of implants, maintain patients on their previous dosage of transmucosal buprenorphine (i.e., the dose from which they were transferred to PROBUPHINE treatment) prior to additional PROBUPHINE treatment.
After one insertion in each arm, most patients should be transitioned back to a transmucosal buprenorphine-containing product for continued treatment. There is no experience with inserting additional implants into other sites in the arm to recommend an approach to a second insertion into a previously-used arm. Neither re-insertion into previously-used administration sites, nor into sites other than the upper arm, has been studied [see Dosage and Administration (2.4, 2.5, and 2.9), Warnings and Precautions (5.1)].
2.3. Patient Access to Naloxone for the Emergency Treatment of Opioid Overdose
Discuss the availability of naloxone for the emergency treatment of opioid overdose with the patient and caregiver. Because patients being treated for opioid use disorder have the potential for relapse, putting them at risk for opioid overdose, strongly consider prescribing naloxone for the emergency treatment of opioid overdose, both when initiating and renewing treatment with PROBUPHINE. Also consider prescribing naloxone if the patient has household members (including children) or other close contacts at risk for accidental ingestion or opioid overdose [see Warnings and Precautions (5.4)].
Advise patients and caregivers that naloxone may also be administered for a known or suspected overdose with buprenorphine, such as due to an accidental exposure of a household contact to a PROBUPHINE implant that has been expelled. Inform patients to call a Healthcare Provider immediately and follow protrusion or/and expulsion guidelines [see Patient Counseling Information (17)].
Higher than normal doses and repeated administration of naloxone may be necessary due to the long duration of action of PROBUPHINE and its affinity for the mu receptor.
Inform patients and caregivers of their options for obtaining naloxone as permitted by individual state naloxone dispensing and prescribing requirements or guidelines (e.g., by prescription, directly from a pharmacist, or as part of a community-based program) [see Patient Counseling Information (17)].
2.4. Healthcare Provider Training
All Healthcare Providers who intend to prescribe PROBUPHINE must successfully complete a live training program [see Warnings and Precautions (5.2)].
All Healthcare Providers performing insertions and/or removals of PROBUPHINE must successfully complete a live training program, and demonstrate procedural competency prior to inserting or removing the implants.
Information concerning the insertion and removal procedures can be obtained by calling 1-844-859-6341. The basis for successful use and subsequent removal of PROBUPHINE is a correct and carefully-performed subdermal insertion of the four implants in accordance with the instructions. As a prerequisite for participating in the live training program leading to certification, the Healthcare Provider must have performed at least one qualifying surgical procedure in the last 3 months. Qualifying procedures are those performed under local anesthesia using aseptic technique, and include, at a minimum, making skin incisions, or placing sutures [see Warnings and Precautions (5.2)].
2.5. Patient Selection
PROBUPHINE implants are only for use in patients who meet ALL of the following criteria:
- Achieved and sustained prolonged clinical stability on transmucosal buprenorphine
- Are currently on a maintenance dose of 8 mg per day or less of a Subutex or Suboxone sublingual tablet or its transmucosal buprenorphine product equivalent (the dose of transmucosal buprenorphine providing blood levels comparable or lower than the level provided by PROBUPHINE)
- Patients should not be tapered to a lower dose for the sole purpose of transitioning to PROBUPHINE.
- Stable transmucosal buprenorphine dose (of 8 mg per day or less of a sublingual Subutex tablet or Suboxone sublingual tablet or its transmucosal buprenorphine product equivalent) for three months or longer without any need for supplemental dosing or adjustments
Examples of acceptable doses of transmucosal buprenorphine include:
- Subutex (buprenorphine) sublingual tablet (generic equivalent) 8 mg or less
- Suboxone (buprenorphine and naloxone) sublingual tablet (generic equivalent) 8 mg/2 mg or less
- Bunavail (buprenorphine and naloxone) buccal film 4.2 mg/0.7 mg or less
- Zubsolv (buprenorphine and naloxone) sublingual tablets 5.7 mg/1.4 mg or less
Consider the following factors in determining clinical stability and suitability for PROBUPHINE treatment:
- period free from illicit opioid drug use
- stability of living environment
- participation in a structured activity/job
- consistency in participation in recommended behavioral therapy/peer support program
- consistency in compliance with clinic visit requirements
- minimal to no desire or need to use illicit opioids
- period without episodes of hospitalizations (addiction or mental health issues), emergency room visits, or crisis interventions
- social support system
2.6. Clinical Supervision
Examine the insertion site one week following insertion of PROBUPHINE for signs of infection or any problems with wound healing, including evidence of implant extrusion from the skin.
The recommended visit schedule for most patients is a frequency of no less than once-monthly for continued counseling and psychosocial support.
Although some patients may require occasional supplemental dosing with buprenorphine, patients should not be provided with prescriptions for transmucosal buprenorphine-containing products for as-needed use. Instead, patients who feel the need for supplemental dosing should be seen and evaluated promptly. Ongoing use of supplemental dosing with transmucosal buprenorphine indicates that the amount of buprenorphine delivered by PROBUPHINE is not adequate for stable maintenance. Consider use of alternate buprenorphine products for maintenance of treatment.
2.7. Insertion of PROBUPHINE
Preparation
Prior to inserting PROBUPHINE, carefully read the insertion instructions as well as the full prescribing information.
Before insertion of PROBUPHINE, confirm that:
- The patient does not have any contraindications for the use of PROBUPHINE [see Contraindications (4)].
- The patient has had a medical history and physical examination.
- The patient understands the benefits and risks of PROBUPHINE.
- The patient has received a copy of the Medication Guide included in the packaging.
- The patient does not have allergies to the antiseptic and anesthetic to be used during insertion.
Insert PROBUPHINE Under Aseptic Conditions.
The Following Equipment is Needed for Implant Insertion:
- An examination table for the patient to lie on
- Instrument stand, sterile tray
- Adequate lighting (e.g., headlamp)
- Sterile fenestrated drape
- Latex and talc-free sterile gloves
- EtOH prep
- Surgical marker
- Antiseptic solution (e.g., chlorhexidine)
- Local anesthetic (1% lidocaine with epinephrine 1:100,000)
- 5 mL syringe with 1.5 inch 25g needle
- Adson single tooth tissue forceps
- #15 blade scalpel
- ¼ inch thin adhesive strip (butterfly strip) (e.g., Steri-strip skin closures)
- 4×4 sterile gauze
- Adhesive bandages
- 3-inch pressure bandages
- Liquid adhesive (e.g., Mastisol)
- 4 PROBUPHINE implants
- 1 PROBUPHINE disposable applicator (Figure 1)
The applicator and its parts are shown in Figure 1.
Correctly performed subdermal insertion of the implants will facilitate their removal. Implants should be placed just under the skin to avoid the large blood vessels that lie in the subcutaneous deep tissue. If the implants are placed improperly, resulting in deep tissue placement, the implants will be more difficult to remove.
Figure 1
Insertion Procedure
Step 1. Have the patient lie on his/her back, with the intended arm flexed at the elbow and externally rotated, so that the hand is positioned next to the head (Figure 2).
Figure 2

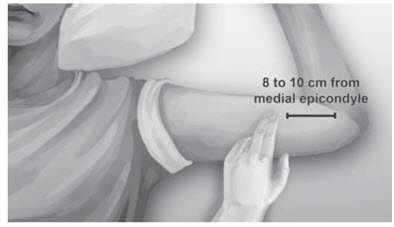
Step 2. Identify the insertion site, which is at the inner side of the upper arm about 8-10 cm (3-4 inches) above the medial epicondyle of the humerus in the sulcus between the biceps and triceps muscle. Having the patient flex the biceps muscle may facilitate identification of the site (Figure 3).
Figure 3
Step 3. Clean insertion site with alcohol prep pad prior to marking the skin.
Step 4. Mark the insertion site with the surgical marker. The implants will be inserted through a small 2.5 mm-3mm subdermal incision.
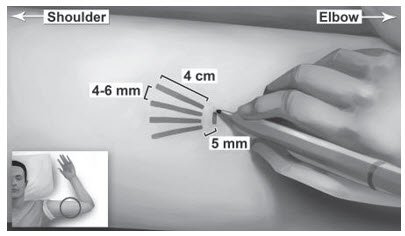
Step 5. Using the surgical marker, mark the channel tracks where each implant will be inserted by drawing 4 lines with each line 4 cm in length. The implants will be positioned in a close fan-shaped distribution 4-6 mm apart with the fan opening towards the shoulder (Figure 4). The closer the implants lie to each other at time of insertion, the more easily they can be removed. There should be at least 5 mm between the incision and the implant when the implant is properly positioned.
Figure 4
Step 6. Put on sterile gloves.
Step 7. Using aseptic technique, place the sterile equipment, PROBUPHINE implants and the applicator on the sterile field of the instrument stand. One applicator is used to insert all four implants.
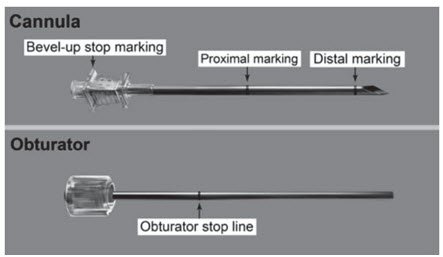
Step 8. Check applicator function by removing the obturator from the cannula and relocking it.
Step 9. Clean the insertion site with an antiseptic solution (e.g., chlorhexidine) using gentle repeated back-and-forth strokes for 30 seconds. When using triple swab stick applicators, use each swab stick sequentially within the 30 seconds. Allow the area to air dry for approximately 30 seconds and do not blot or wipe away.
Step 10. Apply the sterile drape to the arm of the patient.
Step 11. Anesthetize the insertion area at the incision site and just under the skin along the planned insertion channels using local anesthetic (for example, by injecting 5 mL lidocaine 1% with epinephrine 1:100,000).
Step 12. After determining that anesthesia is adequate and effective, make a shallow incision that is 2.5-3 mm in length.
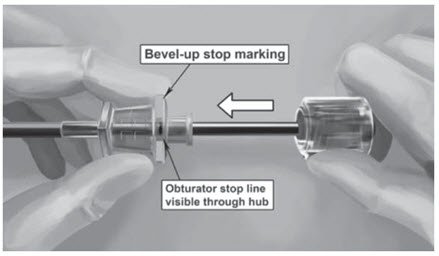
Step 13. Lift the edge of the incision opening with a toothed forceps. While applying counter-traction to the skin, insert only the tip of the applicator at a slight angle (no greater than 20 degrees), into the subdermal space (depth of 3-4 mm below the skin), with the bevel-up stop marking on the cannula facing upwards and visible with the obturator locked fully into the cannula (Figure 5).
Figure 5

Figure 6
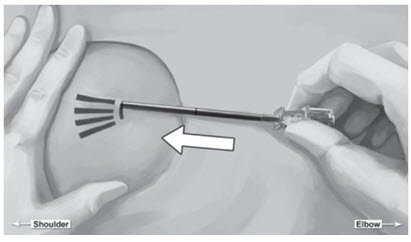
Figure 7
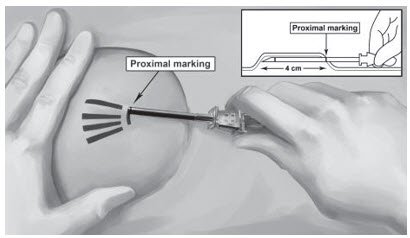
Step 14. Lower the applicator to a horizontal position, lift the skin up with the tip of the applicator but keep the cannula in the subdermal connective tissue (Figure 6). While tenting (lifting), gently advance the applicator subdermally along the channel marking on the skin until the proximal marking on the cannula just disappears into the incision (Figure 7).
Step 15. While holding the cannula in place, unlock the obturator and remove the obturator.
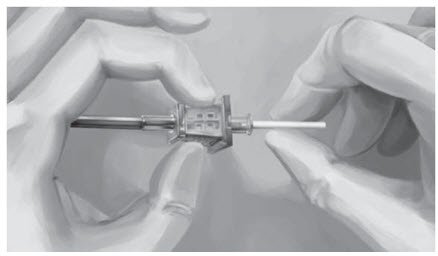
Step 16. Insert one implant into the cannula (Figure 8), re-insert the obturator, and gently push the obturator forward (mild resistance should be felt) until the obturator stop line is level with the bevel-up stop marking, which indicates the implant is positioned at the tip of the cannula (Figure 9). Do not force the implant beyond the end of the cannula with the obturator. There should be at least 5 mm between the incision and the implant when the implant is properly positioned.
| Figure 8 | Figure 9 |
|
|
|
Step 17. While holding the obturator fixed in place on the arm, retract the cannula along the obturator, leaving the implant in place (Figure 10). Note: do not push the obturator. By holding the obturator fixed in place on the arm and by retracting the cannula, the implant will be left in its correct subdermal position.
Figure 10
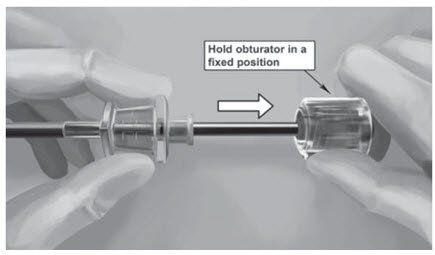
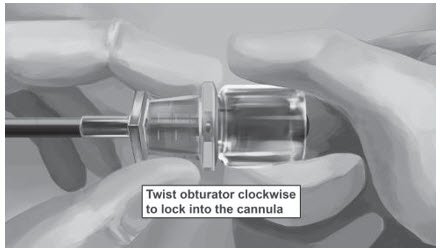
Step 18. Withdraw the cannula until the hub is flush with the obturator, and then twist the obturator clockwise to lock onto the cannula (Figure 11). Retract the applicator, bevel up, until the distal marking of the cannula is visualized at the incision opening (the sharp tip remaining in the subcutaneous space).
Figure 11
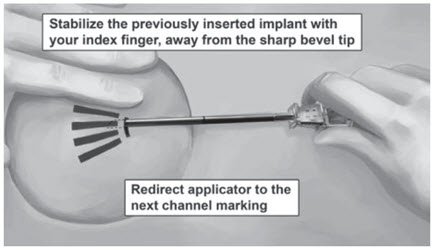
Step 19. Redirect the applicator to the next channel marking while stabilizing the previously inserted implant, with your index finger, away from the sharp tip (Figure 12). Follow steps 13 through 16 for the insertion of the three remaining implants through the same incision, placing implants in a close fan-shaped distribution 4-6 mm apart at the top of the implant. The applicator can now be removed.
Figure 12
Step 20. Always verify the presence of each implant by palpation of the patient's arm immediately after the insertion. By palpating both ends of the implant, you should be able to confirm the presence of the 26 mm implant (Figure 13). If you cannot feel each of the four implants, or are in doubt of their presence, use other methods to confirm the presence of the implant. Suitable methods to locate are: Ultrasound with a high frequency linear array transducer (10 MHz or greater) or Magnetic Resonance Imaging (MRI). Please note that the PROBUPHINE implants are not radiopaque and cannot be seen by X-ray or CT scan. If ultrasound and MRI fail, call 1-844-859-6341.
Figure 13

Step 21. Apply pressure to the incision site for approximately five minutes if necessary.
Step 22. Clean the incision site. Apply liquid adhesive to the skin margins and allow to dry before closing the incision with the ¼ inch thin adhesive strip (butterfly strip) (for example, Steri-strip skin closures).
Step 23. Place a small adhesive bandage over the insertion site.
Step 24. Apply a pressure bandage with sterile gauze to minimize bruising. The pressure bandage can be removed in 24 hours and the adhesive bandage can be removed in three to five days.
Step 25. Complete the PATIENT IDENTIFICATION CARD and give it to the patient to keep. Also, complete the PATIENT CHART STICKER and affix it to the patient medical record or scan or input into electronic medical record. Provide the patient with the Medication Guide and explain proper care of the insertion site.
Step 26. The applicator is for single use only. Dispose of the applicator in accordance with the Centers for Disease Control and Prevention guidelines for hazardous waste.
Step 27. Instruct the patient to apply an ice pack on his/her arm for 40 minutes every two hours for first 24 hours and as needed.
Step 28. Complete the PROBUPHINE REMS Insertion/Removal Log Form.
2.8. PROBUPHINE Removal Procedure
Before initiating the removal procedure, read the instructions for removal.
Identify the location of the implants by consulting the PATIENT IDENTIFICATION CARD and/or THE PATIENT CHART STICKER. The exact location of all implants in the arm (patients will have four implants) should be verified by palpation.
If all of the implants are not palpable, use other methods to confirm the presence of the implant(s). Non-palpable implants should always be located prior to attempted removal. Suitable methods to locate implants are: Ultrasound with a high frequency linear array transducer (10 MHz or greater) or Magnetic Resonance Imaging (MRI). Note that PROBUPHINE implants are not radiopaque and cannot be seen by X-ray or CT scan.
Report any event of failure to locate non-palpable implants using MRI or ultrasound, by calling 1-844-859-6341 for company surveillance purposes.
After localization of a non-palpable implant, removal should be performed under ultrasound guidance. Exploratory surgery without knowledge of the exact location of all implants is strongly discouraged.
There is a greater risk of injury to neural and vascular structures during removal of implants located deeper than the subdermal space. As the anatomical location of these structures must be taken into consideration during the removal of deeply inserted implants, the procedure should only be attempted by Healthcare Providers familiar with this anatomy. A surgical specialist consulted to assist with a difficult removal does not need to be certified in the REMS program.
Preparation
Before removal of PROBUPHINE, confirm that:
- The patient does not have allergies to the antiseptic or the anesthetic to be used. Implants should be removed under aseptic conditions.
The Following Equipment is Needed for Implant Removal:
- An examination table for the patient to lie on
- Instrument stand
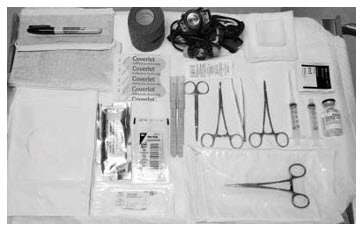
- Sterile tray
- Adequate lighting (e.g., headlamp)
- Sterile fenestrated drapes
- Latex and talc-free sterile gloves
- EtOH prep
- Antiseptic solution (e.g., chlorhexidine)
- Surgical marker
- Local anesthetic (e.g., 1% lidocaine with epinephrine 1:100,000)
- 5 mL syringe with 1.5 inch 25g needle
- Adson single tooth tissue forceps
- Mosquito forceps
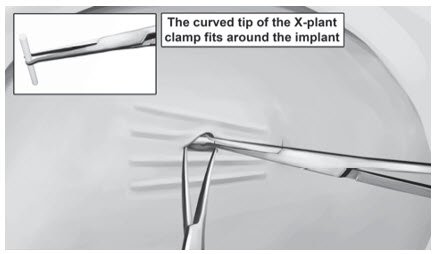
- Two X-plant clamps (vasectomy fixation clamps with 2.5 mm ring diameter)
- Iris scissors
- Needle driver
- #15 blade scalpel
- Sterile ruler
- 4×4 sterile gauze
- Adhesive bandage
- 3-inch pressure bandage
- Sutures (e.g., 4-0 Prolene™ with an FS-2 cutting needle) (may be absorbable)
Removal Procedure
Step 1. Have the patient lie on his/her back, with the implant arm flexed at the elbow and externally rotated, so that the hand is positioned next to the head.
Step 2. Reconfirm the location of the implants by palpation.
Step 3. Clean removal site with alcohol prep pad prior to marking the skin.
Step 4. Mark the location of the implants with a surgical marker. In addition, mark the location of the incision, parallel to the axis of the arm, between the second and third implants (Figure 14).
Figure 14
Step 5. Put on sterile gloves.
Step 6. Using aseptic technique, place the sterile equipment on the sterile field of the instrument stand.
Step 7. Clean the removal site with an antiseptic solution (e.g., chlorhexidine) using gentle repeated back and forth strokes for 30 seconds. When using triple swab stick applicators, use each swab stick sequentially within the 30 seconds. Allow the area to air dry for approximately 30 seconds and do not blot or wipe away.
Step 8. Apply the sterile drape to the arm of the patient.
Step 9. Anesthetize the incision site and the subcutaneous space containing the implants (for example, by injecting 5-7 mL lidocaine 1% with epinephrine 1:100,000). Separate needles may be used for the incision site and the subcutaneous injections. NOTE: Be sure to inject the local anesthetic just beneath the implants; this will effectively lift the implants toward the skin, facilitating removal of the implants.
Step 10. After determining that anesthesia is adequate and effective, make a 7-10 mm incision with a scalpel, parallel to the axis of the arm, between the second and third implants.
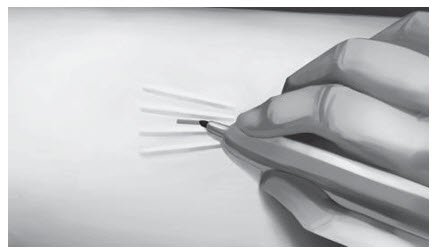
Step 11. Pick up the skin edge with Adson single-toothed tissue forceps and separate the tissues above and below the first visualized implant using an iris scissors or a curved mosquito forceps (Figure 15). Grasp the center of the implant with the X-plant clamp and apply gentle traction. Use the technique of spreading and closing with either the iris scissors or mosquito forceps to separate the fibrous tissue (Figure 16). If the implant is encapsulated use the scalpel to shave the tissue sheath and carefully dissect the tissue around the implant. The implant can then be removed.
| Figure 15 | Figure 16 |
 |  |
Step 12. Retract the next visible implant toward the incisional opening. You may see tenting of the skin at this point if the surrounding tissue is still adhering to the implant. Maintain gentle traction on the implant while you continue to dissect proximally and distally until the implant is free of all adhering tissue. At this point, you may require the use of your second X-plant clamp to remove the implant. If the implant is encapsulated use the scalpel to shave the tissue sheath and carefully dissect the tissue around the implant. The implant can then be removed.
Step 13. After removal of each implant confirm that the entire implant, which is 26 mm long, has been removed by measuring its length. If a partial implant (less than 26 mm) is removed, the remaining piece should be removed by following the same removal instructions. Follow steps 11 through 13 for the removal of the remaining implants through the same incision. Visual identification of whether an entire implant has been removed is unreliable. Therefore, it is important to measure the implant to ensure the entire implant has been removed.
Step 14. After removal of all four implants, clean the incision site.
Step 15. Close the incision with sutures.
Step 16. Place an adhesive bandage over the incision.
Step 17. Use the sterile gauze and apply gentle pressure for five minutes to the incision site to ensure hemostasis.
Step 18. Apply a pressure bandage with sterile gauze to minimize bruising. The pressure bandage can be removed in 24 hours and the adhesive bandage in three to five days.
Step 19. Counsel the patient on proper aseptic wound care. Instruct the patient to apply an ice pack to his/her arm for 40 minutes every two hours for first 24 hours and as needed.
Step 20. Schedule an appointment for the sutures to be removed.
Step 21. The removed implant contains a significant amount of residual buprenorphine. It must be handled with adequate security, accountability, and proper disposal, per facility procedure for a Schedule III drug product, and per applicable federal, state, and local regulations. Disposal of PROBUPHINE implants should also be in keeping with local, State and Federal regulations governing the disposal of pharmaceutical biohazardous waste.
Step 22. Complete the PROBUPHINE REMS Insertion/Removal Log Form.
If implant(s) or implant fragment(s) are not removed during a removal attempt, the patient should undergo imaging for localization as soon as is feasible. The subsequent removal attempt should be performed on the same day of localization. If localization and a second removal attempt are not performed on the same day as the initial removal attempt that necessitated imaging for localization, the wound should be closed with sutures in the interim.
2.9. Spontaneous Expulsion
If spontaneous expulsion of the implant occurs after insertion, the following steps should be taken:
- Schedule two appointments for the patient to return to the office of the inserting Healthcare Provider as soon as possible and to the office of the prescribing Healthcare Provider.
- Instruct the patient to place the implant in a plastic bag, store it safely out of reach of children, and to bring it to the Healthcare Provider office to determine whether the full implant has been expelled.
- If the patient returns the expelled implant, measure it to ensure that the entire implant was expelled (26 mm).
- Dispose of the removed implant in keeping with local, state, and federal regulations governing the disposal of pharmaceutical biohazardous waste, after measuring.
- Examine incision site for infection. If infected, treat appropriately and determine if remaining implants need to be removed.
- If the expelled implant is not intact, palpate the insertion location to identify the location of any remaining partial implant. Remove the remaining partial implant using the techniques described above.
- Call 1-844-859-6341 to obtain a new kit that will include four implants and return instructions for any unused implants.
- The prescribing Healthcare Provider must carefully monitor patient until the implant is replaced to evaluate for withdrawal or other clinical indicators that supplemental transmucosal buprenorphine may be needed.
- Schedule an appointment to insert replacement implant(s).
- Insert the replacement implant(s) in same arm either medially or laterally to in situ implants. Alternatively, replacement implant(s) may be inserted in the contralateral arm.
- Record the new serial number on the PROBUPHINE REMS Insertion/Removal Log Form.
2.10. Continuation of Therapy: Subsequent Insertion of PROBUPHINE in the Contralateral Arm
There is no clinical experience with insertion of PROBUPHINE beyond a single insertion in each arm. If continued treatment is desired at the end of the first six-month treatment cycle, PROBUPHINE implants may be replaced by new implants at the time of removal in the contralateral arm, following the insertion steps above to locate the appropriate insertion site.
If new implants are not inserted on the same day as the removal, patients should be maintained on their previous dose of transmucosal buprenorphine (i.e., the dose from which they were transferred to PROBUPHINE treatment) prior to additional PROBUPHINE treatment [see Dosage and Administration (2.6), Warnings and Precautions (5.1)].
There is no experience with inserting additional implants into other sites in the arm to recommend an approach to a second insertion into a previously-used arm. Neither re-insertion into previously used administration sites, nor into sites other than the upper arm, have been studied. It is important to avoid previously-implanted sites because the effect of scarring and fibrosis in previously-used insertion sites on either the effectiveness of PROBUPHINE or the safety of insertion have not been evaluated. After one insertion in each arm, additional cycles of treatment should only be considered if the potential benefits of continuing PROBUPHINE outweigh the potential risks of additional insertion and removal procedures, taking into account the experience of the Healthcare Provider with PROBUPHINE procedures and related procedures, and the clinical need of the patient for ongoing treatment with subdermal medication. In most cases, patients should be transitioned back to a transmucosal buprenorphine-containing product for continued treatment.
3. Dosage Forms and Strengths
Each PROBUPHINE implant is a sterile, single, off-white, soft, flexible, rod-shaped ethylene vinyl acetate (EVA) implant, 26 mm in length and 2.5 mm in diameter, containing 74.2 mg of buprenorphine (equivalent to 80 mg of buprenorphine hydrochloride).
4. Contraindications
PROBUPHINE is contraindicated in patients with a history of hypersensitivity to buprenorphine or any other ingredients in PROBUPHINE (e.g., EVA) [see Warnings and Precautions (5.11)].
5. Warnings and Precautions
5.1. Serious Complications From Insertion and Removal of PROBUPHINE
Rare but serious complications including nerve damage and migration resulting in embolism and death may result from improper insertion of drug implants inserted in the upper arm. Additional complications may include local migration, protrusion, and expulsion.
Insert PROBUPHINE in accordance with the instructions [see Indications and Usage (1), Dosage and Administration (2.2, 2.6)]. It is essential to insert PROBUPHINE subdermally so that each implant is palpable after insertion. It is also essential to confirm proper placement by palpation immediately after insertion. If PROBUPHINE is inserted too deeply (intramuscular or in the fascia), neural or vascular injury may occur.
Incomplete insertions or infections may lead to protrusion or expulsion [see Dosage and Administration (2.8)]. Accidental exposures to PROBUPHINE can result from protrusion or expulsion of the implants [see Warnings and Precautions (5.8)].
Improper insertion may lead to complicated removal if the implant is inserted too deeply, is not palpable, or has migrated. Deep insertions may lead to difficulty localizing the implant; additional surgical procedures may be required in order to remove the implant [see Dosage and Administration (2.6, 2.7)]. Injury to deeper neural or vascular structures in the arm may occur when removing deeply inserted implants.
All Healthcare Providers must successfully complete a live training program on the insertion and removal procedures and become certified in the PROBUPHINE REMS program, prior to performing insertions or prescribing PROBUPHINE implants. There are additional requirements and prerequisites that must be met to become certified to insert PROBUPHINE implants. Only Healthcare Providers who have performed a surgical procedure in the last 3 months and demonstrate competency in the PROBUPHINE procedures at the live training can become certified to perform insertions. Patients must be monitored to ensure that PROBUPHINE is removed by a Healthcare Provider certified to insert PROBUPHINE implants [see Warnings and Precautions (5.2)].
5.2. PROBUPHINE REMS Program
PROBUPHINE is available only through a restricted program under a REMS, called the PROBUPHINE REMS Program, because of the risk of complications of migration, protrusion and expulsion, and nerve damage associated with the insertion and removal of PROBUPHINE [see Warnings and Precautions (5.1)].
Notable requirements of the PROBUPHINE REMS Program include the following:
- Healthcare Providers who prescribe PROBUPHINE must be certified with the program by enrolling and completing live training
- Healthcare Providers who insert PROBUPHINE must
- meet the prerequisite requirements [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)]
- be certified with the program by enrolling and completing live training, including demonstrating competency in PROBUPHINE procedures
- Patients must be monitored to ensure that PROBUPHINE is removed by a Healthcare Provider certified to insert PROBUPHINE implants
- PROBUPHINE will only be distributed to certified prescribers through a restricted distribution program
Further information is available at www.PROBUPHINEREMS.com or 1-844-859-6341.
5.3. Addiction, Abuse, and Misuse
PROBUPHINE contains buprenorphine, a Schedule III controlled substance that can be abused in a manner similar to other opioids. Buprenorphine is sought by people with opioid use disorders and is subject to criminal diversion. Consider these risks and the patient's stability in treatment for opioid dependence when determining whether PROBUPHINE is appropriate for the patient. Monitor all patients receiving PROBUPHINE for conditions indicative of diversion or progression of opioid dependence and addictive behaviors.
5.4. Risk of Life-Threatening Respiratory and Central Nervous System (CNS) Depression
Buprenorphine, has been associated with life-threatening respiratory depression and death. Many, but not all, post-marketing reports regarding coma and death involved misuse by self-injection or were associated with the concomitant use of buprenorphine and benzodiazepines or other CNS depressants, including alcohol.
Warn patients of the potential danger of self-administration of benzodiazepines or other CNS depressants while under treatment with PROBUPHINE [see Warnings and Precautions (5.5), Drug Interactions (7), Patient Counseling Information (17)].
Use PROBUPHINE with caution in patients with compromised respiratory function (e.g., chronic obstructive pulmonary disease, cor pulmonale, decreased respiratory reserve, hypoxia, hypercapnia, or pre-existing respiratory depression).
Educate patients and caregivers on how to recognize respiratory depression and emphasize the importance of calling 911 or seeking emergency medical help right away in the event of a known or suspected overdose [see Patient Counseling Information (17)].
Opioids can cause sleep-related breathing disorders including central sleep apnea (CSA) and sleep-related hypoxemia. Opioid use increases the risk of CSA in a dose-dependent fashion. In patients who present with CSA, removal of PROBUPHINE may be required.
Patient Access to Naloxone for the Emergency Treatment of Opioid Overdose
Discuss the availability of naloxone for the emergency treatment of opioid overdose with the patient and caregiver.
Because patients being treated for opioid use disorder have the potential for relapse, putting them at risk for opioid overdose, strongly consider prescribing naloxone for the emergency treatment of opioid overdose, both when initiating and renewing treatment with PROBUPHINE. Also consider prescribing naloxone if the patient has household members (including children) or other close contacts at risk for accidental ingestion or opioid overdose [see Dosage and Administration (2.3)].
Advise patients and caregivers that naloxone may also be administered for a known or suspected overdose with buprenorphine, such as due to an accidental exposure of a household contact to a PROBUPHINE implant that is protruding or has been expelled. Inform patients to call a Healthcare Provider immediately and follow protrusion or/and expulsion guidelines. Higher than normal doses and repeated administration of naloxone may be necessary due to the long duration of action of PROBUPHINE and its affinity for the mu receptor [see Patient Counseling Information (17)].
Inform patients and caregivers of their options for obtaining naloxone as permitted by individual state naloxone dispensing and prescribing requirements or guidelines (e.g., by prescription, directly from a pharmacist, or as part of a community-based program).
Educate patients and caregivers on how to recognize respiratory depression and, if naloxone is prescribed, how to treat with naloxone. Emphasize the importance of calling 911 or seeking emergency medical help, even if naloxone is administered [see Patient Counseling Information (17)].
5.5. Managing Risks From Concomitant Use of Benzodiazepines or Other CNS Depressants With Buprenorphine
Concomitant use of buprenorphine and benzodiazepines or other CNS depressants increases the risk of adverse reactions including overdose and death. Medication-assisted treatment of opioid use disorder, however, should not be categorically denied to patients taking these drugs. Prohibiting or creating barriers to treatment can pose an even greater risk of morbidity and mortality due to the opioid use disorder alone.
As a routine part of orientation to buprenorphine treatment, educate patients about the risks of concomitant use of benzodiazepines, sedatives, opioid analgesics, and alcohol.
Develop strategies to manage use of prescribed or illicit benzodiazepines or other CNS depressants at initiation of buprenorphine treatment, or if it emerges as a concern during treatment. Adjustments to induction procedures and additional monitoring may be required. There is no evidence to support dose limitations or arbitrary caps of buprenorphine as a strategy to address benzodiazepines use in buprenorphine-treated patients. However, if a patient is sedated at the time of buprenorphine dosing, delay or omit the buprenorphine dose if appropriate.
Cessation of benzodiazepines or other CNS depressants is preferred in most cases of concomitant use. In some cases, monitoring in a higher level of care for taper may be appropriate. In others, gradually tapering a patient off of a prescribed benzodiazepine or other CNS depressant or decreasing to the lowest effective dose may be appropriate.
For patients in buprenorphine treatment, benzodiazepines are not the treatment of choice for anxiety or insomnia. Before co-prescribing benzodiazepines, ensure that patients are appropriately diagnosed and consider alternative medications and non-pharmacologic treatments to address anxiety or insomnia. Ensure that other Healthcare Providers prescribing benzodiazepines or other CNS depressants are aware of the patient's buprenorphine treatment and coordinate care to minimize the risks associated with concomitant use.
If concomitant use is warranted, strongly consider prescribing naloxone for the emergency treatment of opioid overdose, as is recommended for all patients in buprenorphine treatment for opioid use disorder [see Warnings and Precautions (5.4)].
In addition, take measures to confirm that patients are taking their medications as prescribed and are not diverting or supplementing with illicit drugs. Toxicology screening should test for prescribed and illicit benzodiazepines [see Drug Interactions (7)].
5.6. Neonatal Opioid Withdrawal Syndrome
Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy, whether that use is medically-authorized or illicit. Unlike opioid withdrawal syndrome in adults, NOWS may be life-threatening if not recognized and treated in the neonate. Healthcare professionals should observe newborns for signs of NOWS and manage accordingly [see Use in Specific Populations (8.1)].
Advise pregnant women receiving opioid addiction treatment with PROBUPHINE of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Use in Specific Populations 8.1]. This risk must be balanced against the risk of untreated opioid addiction, which often results in continued or relapsing illicit opioid use and is associated with poor pregnancy outcomes. Therefore, prescribers should discuss the importance and benefits of management of opioid addiction throughout pregnancy.
5.7. Adrenal Insufficiency
Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use. Presentation of adrenal insufficiency may include non-specific symptoms and signs including nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. If adrenal insufficiency is suspected, confirm the diagnosis with diagnostic testing as soon as possible. If adrenal insufficiency is diagnosed, treat with physiologic replacement doses of corticosteroids. Wean the patient off of the opioid to allow adrenal function to recover and continue corticosteroid treatment until adrenal function recovers. Other opioids may be tried as some cases reported use of a different opioid without recurrence of adrenal insufficiency. The information available does not identify any particular opioids as being more likely to be associated with adrenal insufficiency.
5.8. Unintentional Pediatric Exposure
Buprenorphine can cause severe, possibly fatal, respiratory depression in children who are accidentally exposed to it. Instruct patients to keep the expelled implant(s) away from others, especially children.
5.9. Risk of Opioid Withdrawal With Abrupt Discontinuation of PROBUPHINE Treatment
Buprenorphine is a partial agonist at the mu-opioid receptor and chronic administration produces physical dependence of the opioid type, characterized by withdrawal signs and symptoms upon abrupt discontinuation or rapid taper. The withdrawal syndrome is milder than that seen with full agonists, and may be delayed in onset [see Drug Abuse and Dependence (9.2, 9.3)]. If PROBUPHINE implants are not to be immediately replaced upon removal, maintain patients on their previous dosage of sublingual buprenorphine until PROBUPHINE treatment is resumed [see Dosage and Administration (2.2)]. Patients who elect to discontinue PROBUPHINE treatment should be monitored for withdrawal with consideration given to use of a tapering dose of transmucosal buprenorphine.
5.10. Risk of Hepatitis, Hepatic Events
Cases of cytolytic hepatitis and hepatitis with jaundice have been observed in individuals receiving sublingual buprenorphine for the treatment of opioid dependence, both in clinical trials and through post-marketing adverse event reports.
The spectrum of abnormalities ranges from transient asymptomatic elevations in hepatic transaminases to case reports of death, hepatic failure, hepatic necrosis, hepatorenal syndrome, and hepatic encephalopathy. In many cases, the presence of pre-existing liver enzyme abnormalities, infection with hepatitis B or hepatitis C virus, concomitant usage of other potentially hepatotoxic drugs, and ongoing injection drug abuse may have played a causative or contributory role. In other cases, insufficient data were available to determine the etiology of the abnormality. The possibility exists that buprenorphine had a causative or contributory role in the development of the hepatic abnormality in some cases. Liver function tests are recommended prior to initiation of treatment to establish a baseline. Periodic monitoring of liver function during treatment is also recommended. A biological and etiological evaluation is recommended when a hepatic event is suspected. Monitor patients with declining hepatic function for side effects resulting from increased exposure to buprenorphine. Patients may require removal of PROBUPHINE implants.
5.11. Hypersensitivity Reactions
Allergic reactions to buprenorphine and/or EVA are possible. Cases of hypersensitivity to sublingual buprenorphine have been reported both in clinical trials and in the post-marketing experience. Cases of bronchospasm, angioneurotic edema, and anaphylactic shock have been reported. The most common signs and symptoms include rashes, hives, and pruritus. A history of hypersensitivity to buprenorphine or EVA is a contraindication to PROBUPHINE use.
5.12. Precipitation of Opioid Withdrawal in Patients Dependent on Full Agonist Opioids
Because of the partial opioid agonist properties of buprenorphine, buprenorphine may precipitate opioid withdrawal signs and symptoms in persons who are currently physically dependent on full opioid agonists such as heroin, morphine, or methadone before the effects of the full opioid agonist have subsided. Verify that patients are clinically stable on transmucosal buprenorphine and not dependent on full agonists before inserting PROBUPHINE.
5.13. Risks Associated With Treatment of Emergent Acute Pain
While on PROBUPHINE, situations may arise where patients need acute pain management, or may require anesthesia. Treat patients receiving PROBUPHINE with a non-opioid analgesic whenever possible. Patients requiring opioid therapy for analgesia may be treated with a high-affinity full opioid analgesic under the supervision of a physician, with particular attention to respiratory function. Higher doses may be required for analgesic effect. Therefore, a higher potential for toxicity exists with opioid administration. If opioid therapy is required as part of anesthesia, patients should be continuously monitored in an anesthesia care setting by persons not involved in the conduct of the surgical or diagnostic procedure. The opioid therapy must be provided by individuals specifically trained in the use of anesthetic drugs and the management of the respiratory effects of potent opioids, specifically the establishment and maintenance of a patent airway and assisted ventilation.
5.14. Use in Patients With Impaired Hepatic Function
In a pharmacokinetic study with sublingual buprenorphine, buprenorphine plasma levels were found to be higher and the half-life was found to be longer in subjects with moderate and severe hepatic impairment, but not in subjects with mild hepatic impairment. The effect of hepatic impairment on the pharmacokinetics of implanted buprenorphine, such as PROBUPHINE, has not been studied.
Because PROBUPHINE cannot be titrated, patients with pre-existing moderate to severe hepatic impairment are not candidates for treatment with PROBUPHINE. Patients who develop moderate to severe hepatic impairment while being treated with PROBUPHINE should be monitored for signs and symptoms of toxicity or overdose caused by increased levels of buprenorphine, and patients may require removal of PROBUPHINE implants [see Use in Specific Populations (8.6), Clinical Pharmacology (12)].
5.15. Impairment of Ability to Drive and Operate Machinery
PROBUPHINE may impair the mental or physical abilities required for the performance of potentially dangerous tasks such as driving a car or operating machinery, especially for the first 24-48 hours following initial insertion. Caution patients about driving or operating hazardous machinery until they are reasonably certain that PROBUPHINE does not adversely affect their ability to engage in such activities.
5.16. Orthostatic Hypotension
PROBUPHINE may produce orthostatic hypotension in ambulatory patients.
5.17. Elevation of Cerebrospinal Fluid Pressure
Buprenorphine may elevate cerebrospinal fluid pressure and should be used with caution in patients with head injury, intracranial lesions, and other circumstances where cerebrospinal pressure may be increased. Buprenorphine can produce miosis and changes in the level of consciousness that may interfere with patient evaluation.
5.18. Elevation of Intracholedochal Pressure
Buprenorphine has been shown to increase intracholedochal pressure, as do other opioids, and thus should be administered with caution to patients with dysfunction of the biliary tract.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Serious Complications from Insertion and Removal of PROBUPHINE [see Warnings and Precautions (5.1)]
- Addiction, Abuse, and Misuse [see Warnings and Precautions (5.3)]
- Respiratory and CNS Depression [see Warnings and Precautions (5.4, 5.5)]
- Neonatal Opioid Withdrawal Syndrome [see Warnings and Precautions (5.6)]
- Adrenal Insufficiency [see Warnings and Precautions (5.7)]
- Opioid Withdrawal [see Warnings and Precautions (5.9, 5.12)]
- Hepatitis, Hepatic Events [see Warnings and Precautions (5.10)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.11)]
- Orthostatic Hypotension [see Warnings and Precautions (5.16)]
- Elevation of Cerebrospinal Fluid Pressure [see Warnings and Precautions (5.17)]
- Elevation of Intracholedochal Pressure [see Warnings and Precautions (5.18)]
- Infection [see Warnings and Precautions (5.20)]
6.1. Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of PROBUPHINE is supported by clinical trials using PROBUPHINE, and other trials using buprenorphine tablets and buprenorphine sublingual solutions. The safety of PROBUPHINE was evaluated in 349 opioid-dependent subjects across three double-blind trials (n=309) and two open-label extension studies (n=40). In these studies, there were a total of 258 subjects exposed to PROBUPHINE for at least 24 weeks and 82 subjects exposed for 48 weeks. The safety of the PROBUPHINE insertion and removal procedures has been evaluated in 568 unique subjects across the entire development program who received PROBUPHINE implants or placebo implants, with 507 subjects across the three double-blind trials, 40 subjects from two open-label extension trials, and 21 subjects from two phase 2 pharmacokinetic studies.
In total, safety data from clinical studies are available from over 3000 opioid-dependent subjects exposed to buprenorphine at doses in the range used in the treatment of opioid dependence.
Table 1 shows the non-implant-site related adverse events for PROBUPHINE and comparator groups in the three 6-month, double-blind, PROBUPHINE Phase 3 studies. Patients in the PROBUPHINE arm were treated with 4–5 implants and may have received supplemental sublingual buprenorphine. Patients in the Placebo/SL BPN comparator group had either regularly dosed or as-needed sublingual buprenorphine; some had placebo implants. Adverse events were categorized using the Medical Dictionary for Regulatory Activities (MedDRA, Version 17).
In Table 1, MedDRA High Level Group Terms (HLGT) reported in at least 5% of patients in the PROBUPHINE group and more commonly than in the comparator group, are listed at the Higher Level Group Term (HLGT) level along with subordinate Preferred Terms (PT) reported in ≥1% of PROBUPHINE patients (and at least 0.5% more frequent than comparator). Events involving the implant site, or insertion or removal procedures or complications are not included in the table below, but are shown in Table 2.
| System Organ Class | PROBUPHINE | Placebo / SL BPN * |
|---|---|---|
| High Level Group Term | (N=309) | (N=317) |
| MedDRA Preferred Term | n (%) † | n (%) † |
|
||
| GASTROINTESTINAL DISORDERS | ||
| GASTROINTESTINAL SIGNS AND SYMPTOMS | 42 (14) | 39 (12) |
| Nausea | 20 (6) | 15 (5) |
| Vomiting | 17 (6) | 11 (3) |
| Abdominal pain upper | 10 (3) | 7 (2) |
| Flatulence | 2 (1) | 1 (0.3) |
| GASTROINTESTINAL MOTILITY AND DEFAECATION CONDITIONS | 27 (9) | 23 (7) |
| Constipation | 20 (6) | 9 (3) |
| DENTAL AND GINGIVAL CONDITIONS | 16 (5) | 12 (4) |
| Toothache | 14 (5) | 10 (3) |
| GENERAL DISORDERS AND ADMINISTRATION SITE CONDITIONS | ||
| GENERAL SYSTEM DISORDERS NEC | 38 (12) | 26 (8) |
| Pain | 12 (4) | 9 (3) |
| Fatigue | 9 (3) | 4 (1) |
| Asthenia | 5 (2) | 1 (0.3) |
| Chest pain | 2 (1) | 0 |
| Local swelling | 2 (1) | 0 |
| BODY TEMPERATURE CONDITIONS | 14 (5) | 6 (2) |
| Pyrexia | 8 (3) | 4 (1) |
| Chills | 5 (2) | 2 (1) |
| Feeling cold | 2 (1) | 0 |
| INJURY, POISONING AND PROCEDURAL COMPLICATIONS | ||
| INJURIES NEC | 25 (8) | 23 (7) |
| Laceration | 8 (3) | 4 (1) |
| Excoriation | 6 (2) | 2 (1) |
| Scratch | 2 (1) | 0 |
| MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS | ||
| MUSCULOSKELETAL AND CONNECTIVE TISSUE DISORDERS NEC | 26 (8) | 23 (7) |
| Back pain | 18 (6) | 15 (5) |
| Pain in extremity | 8 (3) | 3 (1) |
| NERVOUS SYSTEM DISORDERS | ||
| HEADACHES | 42 (14) | 35 (11) |
| Headache | 39 (13) | 32 (10) |
| Migraine | 5 (2) | 3 (1) |
| NEUROLOGICAL DISORDERS NEC | 25 (8) | 16 (5) |
| Dizziness | 11 (4) | 7 (2) |
| Somnolence | 9 (3) | 1 (0.3) |
| Sedation | 3 (1) | 0 |
| Paresthesia | 2 (1) | 0 |
| PSYCHIATRIC DISORDERS | ||
| DEPRESSED MOOD DISORDERS AND DISTURBANCES | 20 (6) | 13 (4) |
| Depression | 20 (6) | 10 (3) |
| RESPIRATORY, THORACIC AND MEDIASTINAL DISORDERS | ||
| RESPIRATORY DISORDERS NEC | 31 (10) | 19 (6) |
| Oropharyngeal pain | 14 (5) | 10 (3) |
| Cough | 10 (3) | 4 (1) |
| Dyspnoea | 3 (1) | 1 (0.3) |
| SKIN AND SUBCUTANEOUS TISSUE DISORDERS | ||
| EPIDERMAL AND DERMAL CONDITIONS | 16 (5) | 6 (2) |
| Rash | 5 (2) | 2 (1) |
| Skin lesion | 2 (1) | 0 |
The following implant site-related adverse events were reported to occur by at least 2% of patients who received either PROBUPHINE or placebo implants in the pooled double-blind, PROBUPHINE Phase 3 studies:
| MedDRA Preferred Term | PROBUPHINE N=309 n (%) | Placebo implant N=198 n (%) | Total N=507 n (%) |
|---|---|---|---|
| Any Implant Site TEAE | 115 (37) | 54 (27) | 169 (33) |
| Individual Implant Site AE | |||
| Implant site pain | 39 (13) | 18 (9) | 57 (11) |
| Implant site pruritus | 38 (12) | 15 (8) | 53 (11) |
| Implant site erythema | 32 (10) | 13 (7) | 45 (9) |
| Implant site hematoma | 20 (7) | 15 (8) | 35 (7) |
| Implant site hemorrhage | 23 (7) | 10 (5) | 33 (7) |
| Implant site edema | 16 (5) | 5 (3) | 21 (4) |
The adverse event profile of buprenorphine in a transmucosal form (i.e., sublingual) was also characterized in the dose-controlled study of buprenorphine solution, over a range of doses in four months of treatment. The table below shows adverse events reported by at least 5% of subjects in any dose group in the dose-controlled study.
| Body System /Adverse Event (COSTART Terminology) | Buprenorphine Dose * | ||||
|---|---|---|---|---|---|
| Very Low *(N=184) | Low *
(N=180) | Moderate *
(N=186) | High *
(N=181) | Total *
(N=731) |
|
| N (%) | N (%) | N (%) | N (%) | N (%) | |
|
|||||
| Body as a Whole | |||||
| Abscess | 9 (5%) | 2 (1%) | 3 (2%) | 2 (1%) | 16 (2%) |
| Asthenia | 26 (14%) | 28 (16%) | 26 (14%) | 24 (13%) | 104 (14%) |
| Chills | 11 (6%) | 12 (7%) | 9 (5%) | 10 (6%) | 42 (6%) |
| Fever | 7 (4%) | 2 (1%) | 2 (1%) | 10 (6%) | 21 (3%) |
| Flu Syndrome | 4 (2%) | 13 (7%) | 19 (10%) | 8 (4%) | 44 (6%) |
| Headache | 51 (28%) | 62 (34%) | 54 (29%) | 53 (29%) | 220 (30%) |
| Infection | 32 (17%) | 39 (22%) | 38 (20%) | 40 (22%) | 149 (20%) |
| Injury Accidental | 5 (3%) | 10 (6%) | 5 (3%) | 5 (3%) | 25 (3%) |
| Pain | 47 (26%) | 37 (21%) | 49 (26%) | 44 (24%) | 177 (24%) |
| Pain Back | 18 (10%) | 29 (16%) | 28 (15%) | 27 (15%) | 102 (14%) |
| Withdrawal Syndrome | 45 (24%) | 40 (22%) | 41 (22%) | 36 (20%) | 162 (22%) |
| Digestive System | |||||
| Constipation | 10 (5%) | 23 (13%) | 23 (12%) | 26 (14%) | 82 (11%) |
| Diarrhea | 19 (10%) | 8 (4%) | 9 (5%) | 4 (2%) | 40 (5%) |
| Dyspepsia | 6 (3%) | 10 (6%) | 4 (2%) | 4 (2%) | 24 (3%) |
| Nausea | 12 (7%) | 22 (12%) | 23 (12%) | 18 (10%) | 75 (10%) |
| Vomiting | 8 (4%) | 6 (3%) | 10 (5%) | 14 (8%) | 38 (5%) |
| Nervous System | |||||
| Anxiety | 22 (12%) | 24 (13%) | 20 (11%) | 25 (14%) | 91 (12%) |
| Depression | 24 (13%) | 16 (9%) | 25 (13%) | 18 (10%) | 83 (11%) |
| Dizziness | 4 (2%) | 9 (5%) | 7 (4%) | 11 (6%) | 31 (4%) |
| Insomnia | 42 (23%) | 50 (28%) | 43 (23%) | 51 (28%) | 186 (25%) |
| Nervousness | 12 (7%) | 11 (6%) | 10 (5%) | 13 (7%) | 46 (6%) |
| Somnolence | 5 (3%) | 13 (7%) | 9 (5%) | 11 (6%) | 38 (5%) |
| Respiratory System | |||||
| Cough Increase | 5 (3%) | 11 (6%) | 6 (3%) | 4 (2%) | 26 (4%) |
| Pharyngitis | 6 (3%) | 7 (4%) | 6 (3%) | 9 (5%) | 28 (4%) |
| Rhinitis | 27 (15%) | 16 (9%) | 15 (8%) | 21 (12%) | 79 (11%) |
| Skin And Appendages | |||||
| Sweat | 23 (13%) | 21 (12%) | 20 (11%) | 23 (13%) | 87 (12%) |
| Special Senses | |||||
| Runny Eyes | 13 (7%) | 9 (5%) | 6 (3%) | 6 (3%) | 34 (5%) |
6.2. Postmarketing Experience
No post-marketing data exist at this time for PROBUPHINE. The most frequently reported post-marketing adverse event observed with sublingual buprenorphine was drug misuse or abuse. The most frequently reported post-marketing adverse event with buprenorphine/naloxone sublingual tablets was peripheral edema.
Serotonin syndrome: Cases of serotonin syndrome, a potentially life-threatening condition, have been reported during concomitant use of opioids with serotonergic drugs.
Adrenal insufficiency: Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use.
Anaphylaxis: Anaphylaxis has been reported with ingredients contained in PROBUPHINE.
Androgen deficiency: Cases of androgen deficiency have occurred with chronic use of opioids [see Clinical Pharmacology (12.2)].
Related/similar drugs
7. Drug Interactions
Table 4 includes clinically significant drug interactions with PROBUPHINE.
| Benzodiazepine and other Central Nervous System (CNS) Depressants | |
| Clinical Impact: | Due to additive pharmacologic effects, the concomitant use of benzodiazepines and other CNS depressants, including alcohol, increases the risk of respiratory depression, profound sedation, coma, and death. |
| Intervention: | Cessation of benzodiazepines or other CNS depressants is preferred in most cases of concomitant use. In some cases, monitoring in a higher level of care for taper may be appropriate. In others, gradually tapering a patient off of a prescribed benzodiazepine or other CNS depressant or decreasing to the lowest effective dose may be appropriate. Before co-prescribing benzodiazepines for anxiety or insomnia, ensure that patients are appropriately diagnosed and consider alternative medications and non-pharmacologic treatments [see Warnings and Precautions (5.4, 5.5)]. If concomitant use is warranted, strongly consider prescribing naloxone for the emergency treatment of opioid overdose, as is recommended for all patients in treatment for opioid use disorder [see Warnings and Precautions (5.4)]. |
| Examples: | Alcohol, benzodiazepines and other sedatives/hypnotics, anxiolytics, tranquilizers, muscle relaxants, general anesthetics, antipsychotics, and other opioids. |
| Inhibitors of CYP3A4 | |
| Clinical Impact: | The effects of coadministered CYP3A4 inhibitors on buprenorphine exposure in subjects treated with PROBUPHINE have not been studied and the effects may be dependent on the route of administration; however, such interactions have been established in studies using transmucosal buprenorphine. Buprenorphine is metabolized to norbuprenorphine primarily by CYP3A4; therefore, potential interactions may occur when PROBUPHINE is given concurrently with agents that affect CYP3A4 activity. The concomitant use of sublingual buprenorphine and CYP3A4 inhibitors can increase the plasma concentration of buprenorphine, resulting in increased or prolonged opioid effects. |
| Intervention: | Patients who transfer to PROBUPHINE treatment from a regimen of transmucosal buprenorphine used concomitantly with CYP3A4 inhibitors [e.g., azole antifungals such as ketoconazole, macrolide antibiotics such as erythromycin, and HIV protease inhibitors (e.g., ritonavir, indinavir, and saquinavir)] should be monitored to ensure that the plasma buprenorphine level provided by PROBUPHINE is adequate. If patients already on PROBUPHINE require newly-initiated treatment with CYP3A4 inhibitors, the patients should be monitored for signs and symptoms of over-medication. If the concomitant medication cannot be reduced or discontinued, it may be necessary to remove the PROBUPHINE implants and treat the patient with a formulation of buprenorphine that permits dose adjustments. Conversely, if a patient has been stabilized on PROBUPHINE in the setting of concomitant medication that is a CYP3A4 inhibitor, and the concomitant medication is discontinued, the patient should be monitored for withdrawal. If the dose of PROBUPHINE is not adequate in the absence of the concomitant medication, that patient should be transitioned back to a formulation of buprenorphine that permits dose adjustments [see Clinical Pharmacology (12.3)]. |
| Examples: | Macrolide antibiotics (e.g., erythromycin), azole-antifungal agents (e.g., ketoconazole), protease inhibitors (e.g., ritonavir) |
| CYP3A4 Inducers | |
| Clinical Impact: | The effects of coadministered CYP3A4 inducers on buprenorphine exposure in subjects treated with PROBUPHINE have not been studied and the effects may be dependent on the route of administration; however, such interactions have been established in studies using transmucosal buprenorphine. Buprenorphine is metabolized to norbuprenorphine primarily by CYP3A4; therefore, potential interactions may occur when PROBUPHINE is given concurrently with agents that affect CYP3A4 activity. CYP3A4 inducers may induce the metabolism of buprenorphine and, therefore, may cause increased clearance of the drug which could lead to a decrease in buprenorphine plasma concentrations, lack of efficacy or, possibly, development of an abstinence syndrome. |
| Intervention: | Patients who transfer to PROBUPHINE treatment from a regimen of transmucosal buprenorphine used concomitantly with CYP3A4 inducers should be monitored to ensure that the plasma buprenorphine level provided by PROBUPHINE is not excessive. If patients already on PROBUPHINE require newly-initiated treatment with CYP3A4 inducers, the patients should be monitored for withdrawal. If the dose of PROBUPHINE is not adequate in the absence of the concomitant medication, and the concomitant medication cannot be reduced or discontinued, that patient should be transitioned back to a formulation of buprenorphine that permits dose adjustments. Conversely, if a patient has been stabilized on PROBUPHINE in the setting of concomitant medication that is a CYP3A4 inducer, and the concomitant medication is discontinued, the patient should be monitored for signs and symptoms of over-medication. If the dose provided by PROBUPHINE is excessive in the absence of the concomitant inducer, it may be necessary to remove the PROBUPHINE implants and treat the patient with a formulation of buprenorphine that permits dose adjustments [see Clinical Pharmacology (12.3)]. |
| Examples: | Rifampin, carbamazepine, phenytoin, phenobarbital |
| Antiretrovirals: Non-nucleoside reverse transcriptase inhibitors (NNRTIs) | |
| Clinical Impact: | Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are metabolized principally by CYP3A4. Efavirenz, nevirapine, and etravirine are known CYP3A inducers, whereas delaviridine is a CYP3A inhibitor. Significant pharmacokinetic interactions between NNRTIs (e.g., efavirenz and delavirdine) and buprenorphine have been shown in clinical studies, but these pharmacokinetic interactions did not result in any significant pharmacodynamic effects. |
| Intervention: | Patients who are on PROBUPHINE treatment should have their dose monitored if NNRTIs are added to their treatment regimen. |
| Examples: | Efavirenz, nevirapine, etravirine, delavirdine |
| Antiretrovirals: Protease inhibitors (PIs) | |
| Clinical Impact: | Studies have shown some antiretroviral protease inhibitors (PIs) with CYP3A4 inhibitory activity (nelfinavir, lopinavir/ritonavir, ritonavir) have little effect on buprenorphine pharmacokinetic and no significant pharmacodynamic effects. Other PIs with CYP3A4 inhibitory activity (atazanavir and atazanavir/ritonavir) resulted in elevated levels of buprenorphine and norbuprenorphine, and patients in one study reported increased sedation. Symptoms of opioid excess have been found in post-marketing reports of patients receiving buprenorphine and atazanavir with and without ritonavir concomitantly. |
| Intervention: | If treatment with atazanavir with and without ritonavir must be initiated in a patient already treated with PROBUPHINE, the patient should be monitored for signs and symptoms of over-medication. It may be necessary to remove the PROBUPHINE implants and treat the patient with a formulation of buprenorphine that permits dose adjustments. |
| Examples: | Atazanavir, ritonavir |
| Antiretrovirals: Nucleoside reverse transcriptase inhibitors (NRTIs) | |
| Clinical Impact: | Nucleoside reverse transcriptase inhibitors (NRTIs) do not appear to induce or inhibit the P450 enzyme pathway, thus no interactions with buprenorphine are expected. |
| Intervention: | None |
| Serotonergic Drugs | |
| Clinical Impact: | The concomitant use of opioids with other drugs that affect the serotonergic neurotransmitter system has resulted in serotonin syndrome. |
| Intervention: | If concomitant use is warranted, carefully observe the patient, particularly during treatment initiation and dose adjustment. Discontinue PROBUPHINE if serotonin syndrome is suspected. |
| Examples: | Selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), triptans, 5-HT3 receptor antagonists, drugs that effect the serotonin neurotransmitter system (e.g., mirtazapine, trazodone, tramadol), certain muscle relaxants (i.e., cyclobenzaprine, metaxalone), monoamine oxidase (MAO) inhibitors (those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue). |
| Monoamine Oxidase Inhibitors (MAOIs) | |
| Clinical Impact: | MAOI interactions with opioids may manifest as serotonin syndrome or opioid toxicity (e.g., respiratory depression, coma). |
| Intervention: | The use of PROBUPHINE is not recommended for patients taking MAOIs or within 14 days of stopping such treatment. |
| Examples: | Phenelzine, tranylcypromine, linezolid |
| Muscle Relaxants | |
| Clinical Impact: | Buprenorphine may enhance the neuromuscular blocking action of skeletal muscle relaxants and produce an increased degree of respiratory depression. |
| Intervention: | Monitor patients receiving muscle relaxants and PROBUPHINE for signs of respiratory depression that may be greater than otherwise expected and decrease the dosage of PROBUPHINE and/or the muscle relaxant as necessary. Due to the risk of respiratory depression with concomitant use of skeletal muscle relaxants and opioids, strongly consider prescribing naloxone for the emergency treatment of opioid overdose [see Dosage and Administration (2.3), Warnings and Precautions (5.4, 5.5)]. |
| Diuretics | |
| Clinical Impact: | Opioids can reduce the efficacy of diuretics by inducing the release of antidiuretic hormone. |
| Intervention: | Monitor patients for signs of diminished diuresis and/or effects on blood pressure and increase the dosage of the diuretic as needed. |
| Anticholinergic Drugs | |
| Clinical Impact: | The concomitant use of anticholinergic drugs may increase the risk of urinary retention and/or severe constipation, which may lead to paralytic ileus. |
| Intervention: | Monitor patients for signs of urinary retention or reduced gastric motility when PROBUPHINE is used concomitantly with anticholinergic drugs. |
8. Use In Specific Populations
8.1. Pregnancy
Risk Summary
The data on use of buprenorphine, the active ingredient in PROBUPHINE implant, in pregnancy, are limited; however, these data do not indicate an increased risk of major malformations specifically due to buprenorphine exposure. There are limited data from randomized clinical trials in women maintained on buprenorphine that were not designed appropriately to assess the risk of major malformations [see Human Data].
Observational studies have reported on congenital malformations among buprenorphine-exposed pregnancies, but were also not designed appropriately to assess the risk of congenital malformations specifically due to buprenorphine exposure [see Human Data]. Adequate and well-controlled studies have not been conducted with PROBUPHINE or buprenorphine in pregnant women. Neonatal opioid withdrawal syndrome has been reported in the infants of women treated with buprenorphine sublingual tablets during pregnancy [see Clinical Considerations].
Reproductive and developmental studies in rats and rabbits identified adverse events at clinically relevant and higher doses. Embryo fetal death was observed in both rats and rabbits administered buprenorphine during the period of organogenesis at doses approximately 6 and 0.3-times, respectively, the human sublingual dose of 16 mg/day of buprenorphine. Pre- and post-natal development studies in rats demonstrated increased neonatal deaths at 0.3-times and above and dystocia at approximately 3-times the human sublingual dose of 16 mg/day of buprenorphine. No clear teratogenic effects were seen when buprenorphine was administered during organogenesis with a range of doses equivalent to or greater than the human sublingual dose of 16 mg/day of buprenorphine. However, increases in skeletal abnormalities were noted in rats administered buprenorphine daily during organogenesis at a dose approximately 0.6- and approximately equal to the human sublingual dose of 16 mg/day of buprenorphine, respectively. In a few studies, some events such as acephalous and omphalocele were also observed but these findings were not clearly treatment-related [see Animal Data]. Based on animal data, advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and embryo-fetal risk
Untreated opioid addiction in pregnancy is associated with adverse obstetrical outcomes such as low birth weight, preterm birth, and fetal death. In addition, untreated opioid addiction often results in continued or relapsing illicit opioid use.
Dose Adjustment during Pregnancy and the Postpartum Period
Dose adjustments of buprenorphine may be required during pregnancy, even if the patient was maintained on a stable dose prior to pregnancy. Withdrawal signs and symptoms should be monitored closely and the dose adjusted as necessary.
Fetal/neonatal adverse reactions
Neonatal opioid withdrawal syndrome may occur in newborn infants of mothers who are receiving treatment with PROBUPHINE implant.
Neonatal opioid withdrawal syndrome presents as irritability, hyperactivity and abnormal sleep pattern, high pitched cry, tremor, vomiting, diarrhea, and/or failure to gain weight. Signs of neonatal withdrawal usually occur in the first days after birth. The duration and severity of neonatal opioid withdrawal syndrome may vary. Observe newborns for signs of neonatal opioid withdrawal syndrome and manage accordingly [see Warnings and Precautions (5.6)].
Data
Human Data
Studies have been conducted to evaluate neonatal outcomes in women exposed to buprenorphine during pregnancy. Limited data from trials, observational studies, case series, and case reports on buprenorphine use in pregnancy do not indicate an increased risk of major malformations specifically due to buprenorphine. Several factors may complicate the interpretation of investigations of the children of women who take buprenorphine during pregnancy, including maternal use of illicit drugs, late presentation for prenatal care, infection, poor compliance, poor nutrition, and psychosocial circumstances. Interpretation of data is complicated further by the lack of information on untreated opioid-dependent pregnant women, who would be the most appropriate group for comparison. Rather, women on another form of opioid medication-assisted treatment, or women in the general population are generally used as the comparison group. However, women in these comparison groups may be different from women prescribed buprenorphine-containing products with respect to maternal factors that may lead to poor pregnancy outcomes.
In a multicenter, double-blind, randomized, controlled trial [Maternal Opioid Treatment: Human Experimental Research (MOTHER)] designed primarily to assess neonatal opioid withdrawal effects, opioid-dependent pregnant women were randomized to buprenorphine (n=86) or methadone (n=89) treatment, with enrollment at an average gestational age of 18.7 weeks in both groups. A total of 28 of the 86 women in the buprenorphine group (33%) and 16 of the 89 women in the methadone group (18%) discontinued treatment before the end of pregnancy.
Among women who remained in treatment until delivery, there was no difference between buprenorphine-treated and methadone-treated groups in the number of neonates requiring NOWS treatment or in the peak severity of NOWS. Buprenorphine-exposed neonates required less morphine (mean total dose, 1.1 mg vs 10.4 mg), had shorter hospital stays (10.0 days vs 17.5 days), and shorter duration of treatment for NOWS (4.1 days vs 9.9 days) compared to the methadone-exposed group. There were no differences between groups in other primary outcomes (neonatal head circumference), or secondary outcomes (weight and length at birth, preterm birth, gestational age at delivery, and 1-minute and 5-minute Apgar scores), or in the rates of maternal or neonatal adverse events. The outcomes among mothers who discontinued treatment before delivery and may have relapsed to illicit opioid use are not known. Because of the imbalance in discontinuation rates between the buprenorphine and methadone groups, the study findings are difficult to interpret.
Animal Data
The exposure margins listed below are based on body surface area comparisons (mg/m2) to the human sublingual dose of 16 mg buprenorphine via SUBOXONE sublingual tablet.
Effects on embryo-fetal development were studied in Sprague-Dawley rats and Russian white rabbits following oral (1:1) and intramuscular (IM; 3:2) administration of mixtures of buprenorphine and naloxone during the period of organogenesis. Following oral administration to rats, no teratogenic effects were observed at buprenorphine doses up to 250 mg/kg/day (estimated exposure approximately 150 times the human sublingual dose of 16 mg) in the presence of maternal toxicity (mortality). Following oral administration to rabbits, no teratogenic effects were observed at buprenorphine doses up to 40 mg/kg/day (estimated exposure approximately 50 times the human sublingual dose of 16 mg) in the absence of clear maternal toxicity. No definitive drug-related teratogenic effects were observed in rats and rabbits at IM doses up to 30 mg/kg/day (estimated exposure approximately 20 times and 35 times, respectively, the human sublingual does of 16 mg). Maternal toxicity resulting in mortality was noted in these studies in both rats and rabbits.
Acephalous was observed in one rabbit fetus from the low-dose group and omphalocele was observed in two rabbit fetuses from the same litter in the mid-dose group; no findings were observed in fetuses from the high-dose group. Maternal toxicity was seen in the high-dose group but not at the lower doses where the findings were observed. Following oral administration of buprenorphine to rats, dose-related post-implantation losses, evidenced by increases in the numbers of early resorptions with consequent reductions in the numbers of fetuses, were observed at doses of 10 mg/kg/day or greater (estimated exposure approximately 6 times the human sublingual dose of 16 mg). In the rabbit, increased post-implantation losses occurred at an oral dose of 40 mg/kg/day. Following IM administration in the rat and rabbit, post-implantation losses, as evidenced by decreases in live fetuses and increases in resorptions, occurred at 30 mg/kg/day.
Buprenorphine was not teratogenic in rats and rabbits after IM or subcutaneous (SC) doses of up to 5 mg/kg/day (estimated exposure was approximately 3 and 6 times, respectively, the human sublingual dose of 16 mg), after IV doses up to 0.8 mg/kg/day (estimated exposure was approximately 0.5 times and equal to, respectively, the human sublingual dose of 16 mg), or after oral doses up to 160 mg/kg/day in rats (estimated exposure was approximately 95 times the human daily sublingual dose of 16 mg) and 25 mg/kg/day in rabbits (estimated exposure was approximately 30 times the human daily sublingual dose of 16 mg). Significant increases in skeletal abnormalities (e.g., extra thoracic vertebra or thoracolumbar ribs) were noted in rats after SC administration of 1 mg/kg/day and up (estimated exposure was approximately 0.6 times the human sublingual does of 16 mg), but were not observed at oral doses up to 160 mg/kg/day. Increases in skeletal abnormalities in rabbits after IM administration of 5 mg/kg/day (estimated exposure was approximately 6 times the human daily sublingual dose of 16 mg) in the absence of maternal toxicity or oral administration of 1 mg/kg/day or greater (estimated exposure was approximately equal to the human sublingual dose of 16 mg) were not statistically significant.
In rabbits, buprenorphine produced statistically significant pre-implantation losses at oral doses of 1 mg/kg/day or greater and post-implantation losses that were statistically significant at IV doses of 0.2 mg/kg/day or greater (estimated exposure approximately 0.3 times the human daily sublingual dose of 16 mg. No maternal toxicity was noted at doses causing post-implantation loss in this study.
Dystocia was noted in pregnant rats treated intramuscularly with buprenorphine from Gestation Day 14 through Lactation Day 21 at 5 mg/kg/day (approximately 3 times the human sublingual dose of 16 mg). Fertility, pre-, and post-natal development studies with buprenorphine in rats indicated increases in neonatal mortality after oral doses of 0.8 mg/kg/day and up (approximately 0.5 times the human daily sublingual dose of 16 mg), after IM doses of 0.5 mg/kg/day and up (approximately 0.3 times the human sublingual dose of 16 mg), and after SC doses of 0.1 mg/kg/day and up (approximately 0.06 times the human sublingual dose of 16 mg). An apparent lack of milk production during these studies likely contributed to the decreased pup viability and lactation indices. Delays in the occurrence of righting reflex and startle response were noted in rat pups at an oral dose of 80 mg/kg/day (approximately 50 times the human sublingual dose of 16 mg).
8.2. Lactation
Risk Summary
Based on two studies in 13 lactating women maintained on sublingual buprenorphine treatment, buprenorphine and its metabolite norbuprenorphine were present in low levels in human milk and available data have not shown adverse reactions in breastfed infants. The development and health benefits of breastfeeding should be considered along with the mother's clinical need for buprenorphine treatment and any potential adverse effects on the breastfed child from the drug or from the underlying maternal condition.
Clinical Considerations
Advise breastfeeding women taking buprenorphine products to monitor the infant for increased drowsiness and breathing difficulties.
Data
Data were consistent from two studies (N=13) of breastfeeding infants whose mothers were maintained on sublingual doses of buprenorphine ranging from 2.4 to 24 mg/day, showing that the infants were exposed to less than 1% of the maternal daily dose.
In a study of six lactating women who were taking a median sublingual buprenorphine dose of 0.29 mg/kg/day 5 to 8 days after delivery, breast milk provided a median infant dose of 0.42 mcg/kg/day of buprenorphine and 0.33 mcg/kg/day of norbuprenorphine, equal to 0.2% and 0.12% respectively, of the maternal weight-adjusted dose (relative dose/kg (%) of norbuprenorphine was calculated from the assumption that buprenorphine and norbuprenorphine are equipotent).
Data from a study of seven lactating women who were taking a median sublingual buprenorphine dose of 7 mg/day an average of 1.12 months after delivery indicated that the mean milk concentrations (Cavg) of buprenorphine and norbuprenorphine were 3.65 mcg/L and 1.94 mcg/L respectively. Based on the study data, and assuming milk consumption of 150 mL/kg/day, an exclusively breastfed infant would receive an estimated mean absolute infant dose (AID) of 0.55 mcg/kg/day of buprenorphine and 0.29 mcg/kg/day of norbuprenorphine, or a mean relative infant dose (RID) of 0.38% and 0.18%, respectively, of the maternal weight-adjusted dose.
8.3. Females and Males of Reproductive Potential
Infertility
Dietary administration of buprenorphine in the rat at dose levels of 500 ppm or greater (equivalent to approximately 47 mg/kg/day or greater; estimated exposure approximately 22 times the highest daily exposure from PROBUPHINE on an AUC basis) produced a reduction in fertility demonstrated by reduced female conception rates [see Nonclinical Pharmacology (13.1)].
Chronic use of opioids may cause reduced fertility in females and males of reproductive potential. It is not known whether these effects on fertility are reversible [see Adverse Reactions (6.2), Clinical Pharmacology (12.2)].
8.4. Pediatric Use
The safety and effectiveness of PROBUPHINE have not been established in children or adolescents younger than 16 years of age.
8.5. Geriatric Use
Clinical studies of PROBUPHINE did not include subjects over the age of 65. Other reported clinical experience with buprenorphine has not identified differences in responses between the geriatric and younger patients. Due to possible decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy in geriatric patients, the decision to prescribe PROBUPHINE should be made cautiously in individuals 65 years of age or older and these patients should be monitored for signs and symptoms of toxicity or overdose.
8.6. Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of sublingual buprenorphine has been evaluated in a pharmacokinetic study. While no clinically significant changes have been observed in subjects with mild hepatic impairment, the plasma levels have been shown to be higher and half-life values have been shown to be longer for buprenorphine in subjects with moderate and severe hepatic impairment.
The effect of hepatic impairment on the pharmacokinetics of implanted buprenorphine, such as PROBUPHINE, has not been studied. Since the drug is extensively metabolized, the plasma levels can be expected to be higher in patients with moderate and severe hepatic impairment. Because PROBUPHINE cannot be titrated, patients with pre-existing moderate to severe hepatic impairment are not candidates for treatment with PROBUPHINE. Monitor patients who develop moderate or severe hepatic impairment while being treated with PROBUPHINE for signs and symptoms of toxicity or overdose caused by increased levels of buprenorphine. If signs and symptoms of toxicity or overdose are observed, removal of PROBUPHINE implants may be required [see Dosage and Administration (2.5), Clinical Pharmacology (12.3)].
9. Drug Abuse and Dependence
9.1. Controlled Substance
PROBUPHINE contains buprenorphine, a Schedule III controlled substance under the Controlled Substances Act.
Under the Drug Addiction Treatment Act (DATA) codified at 21 United States Code (U.S.C.) 823(g), use of this product in the treatment of opioid dependence is limited to Healthcare Providers who meet certain qualifying requirements, and who have notified the Secretary of Health and Human Services (HHS) of their intent to prescribe or dispense this product for the treatment of opioid dependence and have been assigned a unique identification number that must be included on every prescription.
9.2. Abuse
Buprenorphine, like morphine and other opioids, has the potential for being abused and is subject to criminal diversion. Each PROBUPHINE implant contains 74.2 mg of buprenorphine and can come out or protrude, resulting in the potential for accidental exposure or intentional misuse, abuse, and diversion. Healthcare Providers should contact their state professional licensing board or state controlled substances authority for information on how to prevent and detect misuse, abuse, and diversion of buprenorphine.
Abuse of buprenorphine poses a risk of overdose and death. This risk is increased with the concomitant abuse of buprenorphine and alcohol and other substances, especially benzodiazepines.
Proper assessment of the patient, periodic re-evaluation of therapy, and proper handling and storage of PROBUPHINE are appropriate measures that help to limit misuse, abuse, and diversion of opioid drugs.
Monitor all patients receiving PROBUPHINE and provide or refer patients who have conditions indicative of diversion or progression of opioid dependence and addictive behaviors to more intensive and structured treatment for substance use.
9.3. Dependence
Buprenorphine is a partial agonist at the mu-opioid receptor and chronic administration produces physical dependence of the opioid type, characterized by moderate withdrawal signs and symptoms upon abrupt discontinuation or rapid taper. The withdrawal syndrome is typically milder than seen with full agonists and may be delayed in onset.
Patients treated with PROBUPHINE who experience a delay between removal of implants and insertion of new implants should be maintained on their previous dose of sublingual buprenorphine. Patients who elect to discontinue PROBUPHINE treatment without continuing on other buprenorphine treatment should be monitored for withdrawal. Some or all of the following can characterize this syndrome: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, myalgia, and mydriasis. Other signs and symptoms also may develop, including: irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, or increased blood pressure, respiratory rate, or heart rate [see Warnings and Precautions (5.9)].
Neonatal opioid withdrawal syndrome (NOWS) is an expected and treatable outcome of prolonged use of opioids during pregnancy [see Warnings and Precautions (5.6)].
10. Overdosage
Clinical Presentation
The manifestations of acute buprenorphine overdose include pinpoint pupils, sedation, hypotension, respiratory depression, and death.
Treatment of Overdose
In case of overdose, priorities are the re-establishment of a patent and protected airway and institution of assisted ventilation, if needed. Employ other supportive measures (including oxygen, vasopressors) in the management of circulatory shock and pulmonary edema as indicated. Cardiac arrest or arrhythmias will require advanced life support techniques.
The opioid antagonist naloxone is a specific antidote to respiratory depression resulting from opioid overdose. Naloxone may be of value for the management of buprenorphine overdose. Higher than normal doses and repeated administration may be necessary.
Clinicians should consider the potential role and contribution of buprenorphine, other CNS depressant drugs, and other opioids in a patient's clinical presentation, in determining whether the implants should be removed. In an emergency situation, the removal procedure can be performed by a surgeon who is not certified in the REMS.
11. Probuphine Description
PROBUPHINE (buprenorphine) implant is a sterile, single, off-white, soft, flexible rod-shaped drug product. It is 26 mm in length and 2.5 mm in diameter. Each implant contains 74.2 mg buprenorphine (equivalent to 80 mg buprenorphine hydrochloride) and ethylene vinyl acetate (EVA). PROBUPHINE is designed to be implanted subdermally by a trained medical professional and to provide sustained delivery of buprenorphine for up to six months.
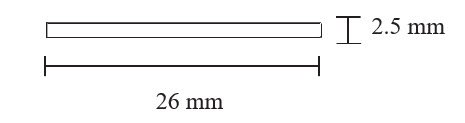
PROBUPHINE implant (not to scale)
The drug substance used is buprenorphine hydrochloride (HCl), an opioid partial agonist.
Chemically, buprenorphine HCl is 6,14-Ethenomorphinan-7-methanol, 17-(cyclopropylmethyl)-a- (1,1-dimethylethyl)-4,5-epoxy-18,19-dihydro-3-hydroxy-6-methoxy-a-methyl, hydrochloride,[5a,7a(S)]. It has the following chemical structure:
Buprenorphine HCl has the molecular formula C29H41NO4 HCl and the molecular weight is 504.10.
12. Probuphine - Clinical Pharmacology
12.1. Mechanism of Action
PROBUPHINE implants contain buprenorphine HCl. Buprenorphine is a partial agonist at the mu-opioid receptor and an antagonist at the kappa-opioid receptor.
12.2. Pharmacodynamics
Four PROBUPHINE implants deliver circulating drug blood levels comparable to the average plasma concentrations observed following daily doses of: 8 mg Subutex or Suboxone tablet equivalent.
Subjective Effects
Comparisons of buprenorphine to full opioid agonists such as methadone and hydromorphone suggest that sublingual buprenorphine produces typical opioid agonist effects that are limited by a ceiling effect.
In opioid-experienced subjects who were not physically dependent, acute sublingual doses of buprenorphine/naloxone tablets produced opioid agonist effects that reached a maximum between doses of 8/2 mg and 16/4 mg buprenorphine/naloxone. Opioid agonist ceiling effects were also observed in a double-blind, parallel-group, dose-ranging comparison of single doses of buprenorphine sublingual solution (1, 2, 4, 8, 16, or 32 mg), placebo, and a full agonist control at various doses. The treatments were given in ascending dose order at intervals of at least one week to 16 opioid-experienced subjects who were not physically dependent. Both active drugs produced typical opioid agonist effects. For all measures for which the drugs produced an effect, buprenorphine produced a dose-related response. However, in each case, there was a dose that produced no further effect. In contrast, the highest dose of the full agonist control always produced the greatest effects. Agonist objective rating scores remained elevated for the higher doses of buprenorphine (8-32 mg) longer than for the lower doses and did not return to baseline until 48 hours after drug administration. The onset of effects appeared more rapidly with buprenorphine than with the full agonist control, with most doses nearing peak effect after 100 minutes for buprenorphine, compared with 150 minutes for the full agonist control.
Physiological Effects
Buprenorphine in IV (2, 4, 8, 12, and 16 mg) and sublingual (12 mg) doses has been administered to opioid-experienced subjects who were not physically dependent, to examine cardiovascular, respiratory, and subjective effects at doses comparable to those used for treatment of opioid dependence. Compared with placebo, there were no statistically significant differences among any of the treatment conditions for blood pressure, heart rate, respiratory rate, O2 saturation, or skin temperature across time. Systolic BP was higher in the 8 mg group than placebo (3-hour AUC values). Minimum and maximum effects were similar across all treatments. Subjects remained responsive to low voice and responded to computer prompts. Some subjects showed irritability, but no other changes were observed. The respiratory effects of sublingual buprenorphine were compared with the effects of methadone in a double-blind, parallel-group, dose-ranging comparison of single doses of buprenorphine sublingual solution (1, 2, 4, 8, 16, or 32 mg) and oral methadone (15, 30, 45, or 60 mg) in non-dependent, opioid-experienced volunteers. In this study, hypoventilation not requiring medical intervention was reported more frequently after buprenorphine doses of 4 mg and higher than after methadone. Both drugs decreased O2 saturation to the same degree.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticotropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans [see Adverse Reactions (6.2)]. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon.
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date [see Adverse Reactions (6.2)].
12.3. Pharmacokinetics
Absorption
After PROBUPHINE insertion, an initial buprenorphine peak was observed and the median Tmax occurred at 12 hours after insertion. After the initial buprenorphine peak, the plasma buprenorphine concentrations decreased slowly and steady-state plasma buprenorphine concentrations were reached by approximately Week 4. Mean steady-state plasma buprenorphine concentrations were approximately 0.5 to 1 ng/mL and were maintained for approximately 20 weeks (Week 4 through Week 24) in a 24-week treatment period. At steady state, the buprenorphine concentrations were stable and comparable to the trough buprenorphine concentration of 8 mg per day sublingual buprenorphine at steady state.
In one pharmacokinetics study (Figure 17), subjects received 16 mg per day sublingual buprenorphine for a minimum of 5 consecutive days, followed by 4 implants of PROBUPHINE (totally 320 mg buprenorphine hydrochloride). Overall peak plasma buprenorphine concentrations were markedly lower after PROBUPHINE insertion than after dosing with 16 mg per day sublingual buprenorphine. The steady-state buprenorphine AUC0-24 value after 4 implants of PROBUPHINE on Day 28 was 19.6 ± 33.7 ng*hr/mL, 31% of the steady-state buprenorphine AUC0-24 value of 16 mg per day sublingual administration (62.7 ± 36.4 ng*hr/mL). The average steady-state buprenorphine concentration of PROBUPHINE on Day 28 was approximately 0.82 ng/mL, 8% of the peak concentration (10.4 ± 13.4 ng/mL), and 52% of the trough concentration (1.58 ± 0.60 ng/mL) of 16 mg per day sublingual buprenorphine at steady state.
The figure below shows the steady state buprenorphine concentration of 16 mg per day sublingual buprenorphine on Day -1, the initial buprenorphine concentration after PROBUPHINE insertion on Day 1, and steady state buprenorphine after PROBUPHINE insertion on Day 28.
Figure 17: Buprenorphine concentration versus time profiles after daily administration of 16 mg sublingual buprenorphine for 5 days (Day -5 to Day -1), followed by 4 implants of PROBUPHINE (totally 320 mg buprenorphine hydrochloride) on Day 1.
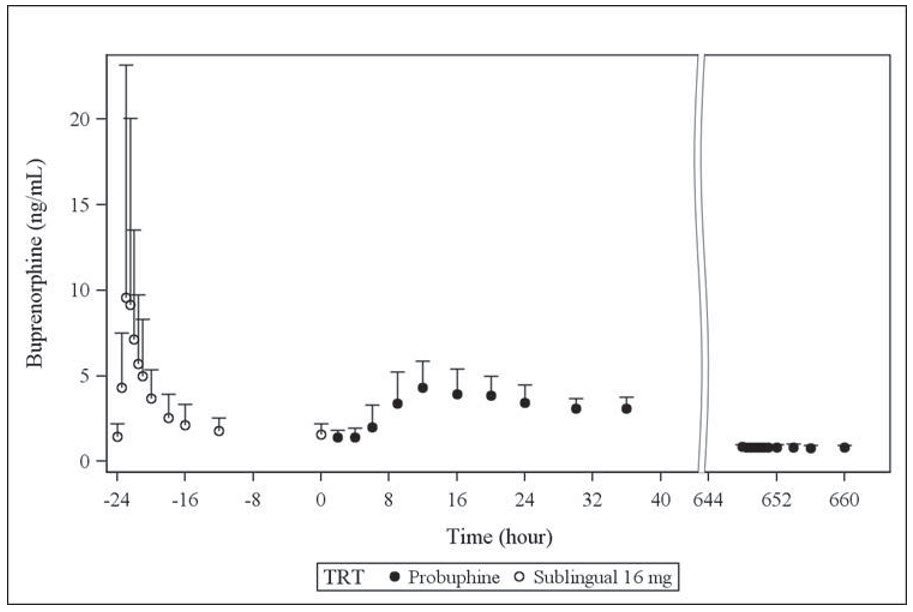
Distribution
Buprenorphine is approximately 96% protein bound, primarily to alpha and beta globulin.
Elimination
Metabolism
Buprenorphine undergoes both N-dealkylation to norbuprenorphine and glucuronidation. The N- dealkylation pathway is mediated primarily by the CYP3A4. Norbuprenorphine, the major metabolite, can further undergo glucuronidation. Norbuprenorphine has been found to bind opioid receptors in vitro; however, it has not been studied clinically for opioid-like activity.
Excretion
A mass balance study of buprenorphine showed complete recovery of radiolabel in urine (30%) and feces (69%) collected up to 11 days after dosing. Almost all of the dose was accounted for in terms of buprenorphine, norbuprenorphine, and two unidentified buprenorphine metabolites. In urine, most of the buprenorphine and norbuprenorphine was conjugated (buprenorphine, 1% free and 9.4% conjugated; norbuprenorphine, 2.7% free and 11% conjugated). In feces, almost all of the buprenorphine and norbuprenorphine were free (buprenorphine, 33% free and 5% conjugated; norbuprenorphine, 21% free and 2% conjugated). Based on all studies performed with buprenorphine/naloxone, buprenorphine has a mean elimination half-life from plasma ranging from 24 to 48 hours.
Specific Populations
Hepatic Impairment
The effect of hepatic impairment on the pharmacokinetics of implanted buprenorphine product, such as PROBUPHINE, has not been studied.
The disposition of buprenorphine was determined in a pharmacokinetics study after administering a 2.0/0.5 mg buprenorphine/naloxone sublingual tablet in subjects with varied degrees of hepatic impairment as indicated by Child-Pugh criteria. The disposition of buprenorphine in patients with hepatic impairment was compared to disposition in subjects with normal hepatic function. In subjects with mild hepatic impairment, the changes in mean Cmax, AUC0-last, and half-life values of buprenorphine were not clinically significant. For subjects with moderate and severe hepatic impairment, mean Cmax, AUC0-last, and half-life values of buprenorphine were increased.
Drug Interaction Studies
CYP3A4 Inhibitors and Inducers
Buprenorphine is metabolized to norbuprenorphine primarily by cytochrome CYP3A4; therefore, potential interactions may occur when PROBUPHINE is given concurrently with agents that affect CYP3A4 activity. The effects of co-administered CYP3A4 inducers or inhibitors have been established in studies using transmucosal buprenorphine; the effects on buprenorphine exposure in patients treated with PROBUPHINE have not been studied, and the effects may be dependent on the route of administration.
Buprenorphine has been found to be a CYP2D6 and CYP3A4 inhibitor and its major metabolite, norbuprenorphine, has been found to be a moderate CYP2D6 inhibitor in in vitro studies employing human liver microsomes [see Drug Interactions (7)].
13. Nonclinical Toxicology
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Carcinogenicity studies testing PROBUPHINE have not been completed.
Carcinogenicity studies of buprenorphine were conducted in Sprague-Dawley rats and CD-1 mice. Buprenorphine was administered in the diet for 27 months to rats at equivalent doses of 0.6, 5.5, and 56 mg/kg body weight/day (approximately 2, 13, and 99 times the steady state exposure from PROBUPHINE on an AUC basis). A statistically significant dose-related increase in Leydig cell tumors occurred. In an 86-week study in CD-1 mice, buprenorphine was not carcinogenic when administered in the diet at equivalent doses up to 100 mg/kg body weight/day (approximately 53 times the steady state exposure from PROBUPHINE on an AUC basis).
Mutagenicity
Buprenorphine was studied in a series of tests utilizing gene, chromosome, and DNA interactions in both prokaryotic and eukaryotic systems. Results were negative in yeast (Saccharomyces cerevisiae) for recombinant, gene convertant, or forward mutations; negative in Bacillus subtilis "rec" assay; negative for clastogenicity in Chinese hamster ovary, bone marrow, and spermatogonia cells; and negative in the mouse lymphoma L5178Y assay.
Results were equivocal in the Ames test: negative in studies in two laboratories, but positive for frame shift mutation at a high dose (5 mg/plate) in a third study. Results were positive in the Green-Tweats (E. coli) survival test, positive in a DNA synthesis inhibition (DSI) test with testicular tissue from mice, for both in vivo and in vitro incorporation of [3H]thymidine, and positive in unscheduled DNA synthesis test using testicular cells from mice.
Impairment of Fertility
Dietary administration of buprenorphine in the rat at dose levels of 500 ppm or greater (equivalent to approximately 47 mg/kg/day or greater; estimated exposure approximately 22 times the highest human daily dose from PROBUPHINE on an AUC basis) produced a reduction in fertility demonstrated by reduced female conception rates. A dietary dose of 100 ppm (equivalent to approximately 10 mg/kg/day; estimated exposure approximately 18 times the recommended human daily dose from PROBUPHINE on an AUC basis) had no adverse effect on fertility.
Reproduction studies of buprenorphine in rats demonstrated no evidence of impaired fertility at daily oral doses up to 80 mg/kg/day (estimated exposure approximately 100 times the human daily SL dose of 8 mg on a mg/m2 basis) or up to 5 mg/kg/day IM or SC (estimated exposure was approximately 12 times the human daily SL dose of 8 mg on a mg/m2 basis for IM dosing and 18 times the steady state exposure from PROBUPHINE on an AUC basis for SC dosing).
14. Clinical Studies
The efficacy of PROBUPHINE was demonstrated in one randomized double-blind, double-dummy study in adults who met DSM-IV-TR criteria for opioid dependence as their primary diagnosis, and were considered clinically stable, on a sublingual buprenorphine dose of no more than 8 mg per day, by their treating Healthcare Provider. Healthcare Providers attested to their patient's clinical stability and endorsed criteria forming the basis for that determination on a clinical stability checklist that included the following factors:
- –
- no reports of any illicit opioid use
- –
- no reports of significant withdrawal symptoms
- –
- reports of low to no desire/need to use illicit opioids
- –
- no episodes of hospitalizations (addiction or mental health issues), emergency room visits, or crisis interventions in the past 90 days
- –
- stable living environment, participation in a structured activity/job that contributes to the community, consistent participation in recommended cognitive behavioral therapy/peer support program
- –
- consistent compliance with clinic visit requirements
In addition to the treating Healthcare Provider's determination of clinical stability, patients were also on a sublingual buprenorphine dose of no more than 8 mg per day as a Suboxone tablet or equivalent and had no positive urine toxicology results for illicit opioids for the last 90 days, and were intended to have been on sublingual buprenorphine treatment for at least the last 6 months prior to randomization. Most of the study subjects endorsed prescription opioid pain relievers as their primary opioid of abuse.
In this study, clinically-stable subjects on maintenance treatment with no more than 8 mg per day sublingual buprenorphine were randomized 1:1 to either PROBUPHINE (4 implants) or treatment as usual with their pre-randomization dose of sublingual buprenorphine. A total of 87 were treated with PROBUPHINE and received placebo sublingual tablets; 89 were treated with sublingual buprenorphine/naloxone tablets and received placebo implants. Patients were seen monthly for six months and were also required to provide four randomly-scheduled urine samples for toxicology. Efficacy was evaluated through urine toxicology screening and patient self-report to detect opioid use, over the 6-month treatment period. Supplemental dosing with open-label sublingual buprenorphine/naloxone tablets was permitted as clinically indicated.
The table below illustrates the proportion of patients who successfully maintained clinical stability on their assigned treatment. Missing samples were considered to be evidence of opioid use, and only patients with no evidence of opioid use were adjudicated as maintaining stability. Although the protocol permitted supplemental buprenorphine use for patients in both arms, the use of supplemental dosing in patients assigned to the sublingual treatment arm is consistent with treatment as usual, which includes dose adjustments as needed. The use of supplemental buprenorphine in patients on PROBUPHINE, which cannot be titrated, may be interpreted to indicate that the dose of buprenorphine provided by PROBUPHINE was inadequate for that patient (to maintain stability) and so patients who required supplemental dosing were not included in the table as successfully maintained, even if they did not have evidence of opioid use.
| PROBUPHINE Only (no supplemental dosing) (N=87) | Treatment as Usual (sublingual buprenorphine) (N=89) | Treatment Difference (95% CI) |
|---|---|---|
| 55 (63%) | 57 (64%) | -1% (-15%, 13%) |
Additionally, there were 11 patients in the PROBUPHINE arm who required supplemental sublingual buprenorphine, but had no evidence of opioid use. Of these, one required supplemental dosing only at the end of the implantation period, potentially indicating the need for early replacement of implants.
Two additional studies in patients who were new entrants to buprenorphine treatment suggested that PROBUPHINE should not be used for patients who are new entrants to buprenorphine treatment or who have not achieved and sustained prolonged clinical stability on low to moderate doses of a transmucosal buprenorphine-containing product, i.e., doses of no more than 8 mg per day of a Subutex or Suboxone sublingual tablet or generic equivalent, because the dose appears to be too low to be effective in these populations.
16. How is Probuphine supplied
One PROBUPHINE implant kit consists of four individually packaged sterile implants and one individually packaged sterile disposable applicator. Each implant is 26 mm in length and 2.5 mm in diameter and contains 74.2 mg of buprenorphine (equivalent to 80 mg of buprenorphine hydrochloride).
One applicator kit consists of one sterile, single patient use, disposable PROBUPHINE applicator.
Store PROBUPHINE at 20 to 25°C (68 to 77°F); excursions permitted at 15 to 30°C (59-86°F) [see USP Controlled Room Temperature].
Store PROBUPHINE in accordance with Federal and State controlled substance laws and regulations. Contact state controlled substances authority for information on how to store and prevent diversion of this product.
The PROBUPHINE implant is a Schedule III drug product. Handle with adequate security and accountability. Expired implants should be properly disposed, per facility procedure for a Schedule III drug product, and per applicable federal, state, and local regulations.
NDC code for set of four implants is 52440-0100-14.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Instruct patients to read the Medication Guide each time PROBUPHINE is implanted because new information may be available.
Risks Relating to the Insertion and Removal Procedure [see Warnings and Precautions (5.1)], Accidental Overdose, Misuse and Abuse if an Implant Comes Out or Protrudes from the Skin [see Warnings and Precautions (5.3)].
- Inform patients that there are risks associated with the insertion and removal of PROBUPHINE, including:
- –
- Migration with potential for embolism or nerve damage
- –
- Expulsion or protrusion
- –
- Injury to nerves or blood vessels
- –
- Infection at the insertion or removal site
- –
- Removal complications
- Implants may be difficult to locate if they were inserted too deeply, or if patients manipulate them, or have gained significant weight since insertion.
- Special procedures, tests, or a referral to a specialist may be needed to remove the implants if they are difficult to locate.
- Inform patients that there is a risk to others of accidental overdose, misuse, and abuse if the implant comes out of the arm.
- Inform patients that appropriate care of their incision is important to reduce the risk of complications associated with the insertion of PROBUPHINE.
- Inform patients to call a Healthcare Provider immediately if they experience any of the following:
- –
- The implant protrudes or is expelled.
- –
- Bleeding or symptoms of infection, such as excessive or worsening itching, pain, irritation or redness, or swelling at the insertion site.
- –
- Symptoms suggesting the implant has migrated, such as numbness or weakness, or shortness of breath.
- –
- Symptoms of numbness or weakness in the arm after insertion or removal procedure.
- Inform patients that if the PROBUPHINE implants protrude or are expelled, they should:
- –
- Wash their hands if they have touched the PROBUPHINE implants.
- –
- Cover the area where the implants were inserted with a clean bandage.
- –
- Not allow others to touch or use the PROBUPHINE implants, since this could be very dangerous.
- –
- Put the implants in a plastic bag and take the implants to a Healthcare Provider right away.
- –
- Keep the implants away from others, especially children.
- –
- Protect the implants from theft until they can take them to their doctor.
- Inform patients that improper removal by a non-Healthcare Provider carries the risk of implant-site infection and premature removal may induce opioid withdrawal symptoms.
- Inform patients that there are risks associated with minor surgical procedures, such as:
- –
- Itching, pain, irritation or redness, swelling, bleeding, or bruising at the insertion or removal site.
- –
- Scarring around the incision site.
Life-Threatening Respiratory Depression
Educate patients and caregivers on how to recognize respiratory depression and emphasize the importance of calling 911 or seeking emergency medical help right away in the event of a known or suspected overdose [see Warnings and Precautions (5.4)].
Patient Access to Naloxone for the Emergency Treatment of Opioid Overdose
Because patients being treated for opioid use disorder are at risk for relapse, discuss the importance of having access to naloxone with the patient and caregiver. Also discuss the importance of having access to naloxone if there are household members (including children) or other close contacts at risk for accidental ingestion or opioid overdose.
Inform patients and caregivers of the options for obtaining naloxone as permitted by individual state naloxone dispensing and prescribing requirements or guidelines (e.g., by prescription, directly from a pharmacist, or as part of a community-based program).
Educate patients and caregivers on how to recognize the signs and symptoms of an opioid overdose.
Explain to patients and caregivers that naloxone's effects are temporary, and that they must call 911 or seek emergency medical help right away in all cases of known or suspected opioid overdose, even if naloxone is administered. Repeat administration may be necessary, particularly for overdose involving buprenorphine, because naloxone is often not effective at the doses available for patient access [see Dosage and Administration (2.3), Warnings and Precautions (5.4), Overdosage (10)].
If naloxone is prescribed, also advise patients and caregivers:
- How to treat with naloxone in the event of an opioid overdose
- To tell family and friends about their naloxone and to keep it in a place where family and friends can easily access it in an emergency
- To read the Patient Information (or other educational material) that will come with their naloxone. Emphasize the importance of doing this before an opioid emergency happens, so the patient and caregiver will know what to do.
Interaction with Benzodiazepines and other CNS Depressants
Inform patients and caregivers that potentially fatal additive effects may occur if PROBUPHINE is used with benzodiazepines or other CNS depressants, including alcohol, and not to use these concomitantly unless supervised by a health care provider [see Warnings and Precautions (5.5), Drug Interactions (7)].
Serotonin Syndrome
Inform patients that PROBUPHINE could cause a rare but potentially life-threatening condition resulting from concomitant administration of serotonergic drugs. Warn patients of the symptoms of serotonin syndrome and to seek medical attention right away if symptoms develop. Instruct patients to inform their physicians if they are taking, or plan to take serotonergic medications [see Drug Interactions 7].
Adrenal Insufficiency
Inform patients that PROBUPHINE could cause adrenal insufficiency, a potentially life-threatening condition. Adrenal insufficiency may present with non-specific symptoms and signs such as nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. Advise patients to seek medical attention if they experience a constellation of these symptoms [see Warnings and Precautions (5.7)].
Anaphylaxis
Inform patients that anaphylaxis has been reported with ingredients contained in PROBUPHINE. Advise patients how to recognize such a reaction and when to seek medical attention [see Warnings and Precautions (5.11)].
Driving or Operating Heavy Machinery
Caution patients that PROBUPHINE may impair the mental or physical abilities required for the performance of potentially dangerous tasks such as driving or operating hazardous machinery.
Instruct patients not to drive or operate hazardous machinery until they are reasonably certain that PROBUPHINE therapy does not adversely affect their ability to engage in such activities [see Warnings and Precautions (5.15)].
Dependence and Withdrawal
Inform patients that PROBUPHINE can cause drug dependence and that withdrawal signs and symptoms may occur when the medication is discontinued [see Warnings and Precautions (5.9)].
Orthostatic Hypotension
Inform patients that, like other opioids, PROBUPHINE may produce orthostatic hypotension in ambulatory individuals [see Warnings and Precautions (5.16)].
Drug Interactions
Instruct patients to inform their Healthcare Providers of any other prescription medications, over-the-counter medications, or herbal preparations that are prescribed or currently being used [see Drug Interactions (7)].
Pregnancy
Neonatal Opioid Withdrawal Syndrome
Advise women that if they are pregnant while being treated with PROBUPHINE, the baby may have signs of withdrawal at birth and that withdrawal is treatable [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)].
Embryo Fetal Toxicity
Advise women of childbearing potential who become pregnant or are planning to become pregnant to consult their Healthcare Provider regarding the possible effects of using PROBUPHINE during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Warn patients that buprenorphine passes into breast milk. Advise the nursing mother taking buprenorphine to monitor the infant for increased drowsiness and breathing difficulties. [see Use in Specific Populations (8.2)].
Infertility
Inform patients that chronic use of opioids may cause reduced fertility. It is not known whether these effects on fertility are reversible [see Adverse Reactions (6.2)].
Emergency Analgesia
Advise patients to instruct their family members to, in the event of emergency, inform the treating physician or emergency room staff that the patient is physically dependent on an opioid and that the patient is being treated with PROBUPHINE [see Warnings and Precautions (5.13)].
PROBUPHINE REMS Program
PROBUPHINE is available only through a restricted program called the PROBUPHINE REMS Program [see Warnings and Precautions (5.2)]. Inform the patient of the following notable requirements:
- PROBUPHINE is not available in retail pharmacies.
- PROBUPHINE must be inserted or removed only in the facility of a certified prescriber.
Distributed by:
Titan Pharmaceuticals, Inc.
South San Francisco, CA 94080
PROAW00009-R2 03/21
US Patent No: 7736665
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 03/2021 | |||
| Medication Guide
PROBUPHINE® (pro-bú-feen) (buprenorphine) implant (CIII) |
||||
| Read this Medication Guide before starting PROBUPHINE and each time PROBUPHINE is inserted. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider. Talk to your healthcare provider if you have questions about PROBUPHINE. Share the important information in this Medication Guide with members of your household. Carry your Patient Identification Card with you at all times. |
||||
What is the most important information I should know about PROBUPHINE?
|
||||
|
|
|||
Call your healthcare provider right away if:
|
||||
|
||||
|
|
|||
| These can be signs of an overdose or other serious problems. | ||||
|
||||
| What is PROBUPHINE?
PROBUPHINE is an implant that contains the medicine buprenorphine. PROBUPHINE is used to treat certain adults who are addicted to (dependent on) opioid drugs (either prescription or illegal). PROBUPHINE is part of a complete treatment program that also includes counseling and behavioral therapy.
|
||||
| PROBUPHINE is a controlled substance (CIII) because it contains buprenorphine that can be a target for people who abuse prescription medicines or street drugs. If it comes out of your arm, keep the implant in a safe and secure place away from others, especially children. Tell your healthcare provider if you are living in a household where there are small children. If a child is accidentally exposed to PROBUPHINE, get emergency help or call 911 right away. Protect the implants from theft until you can return them to your healthcare provider. Never give your PROBUPHINE to anyone else, because it may cause death or harm them. Selling or giving away PROBUPHINE is against the law. | ||||
| Who should not use PROBUPHINE?
Do not use PROBUPHINE if you are allergic to buprenorphine or any ingredients in PROBUPHINE. See the end of this Medication Guide for a list of ingredients in PROBUPHINE. |
||||
PROBUPHINE may not be right for you. Before starting PROBUPHINE, tell your healthcare provider about all of your medical conditions, including:
|
||||
How are the PROBUPHINE implants inserted and removed?
|
||||
| What should I do if PROBUPHINE implant sticks out or comes out?
If a PROBUPHINE implant sticks out or comes out of your skin:
|
||||
What should I avoid while being treated with PROBUPHINE?
|
||||
| What are the possible side effects of PROBUPHINE? PROBUPHINE can cause serious side effects, including:
|
||||
|
|
|||
Common risks with minor surgical procedure include:
Call your doctor for medical advice about side effects. You may also report side effects to the FDA at 1-800-FDA-1088. |
||||
| General information about PROBUPHINE
This Medication Guide summarizes important information about PROBUPHINE. If you would like more information, talk to your healthcare provider. You can ask your healthcare provider for information that is written for healthcare professionals. |
||||
| What are the ingredients in PROBUPHINE?
Active ingredient: buprenorphine Inactive ingredient: ethylene vinyl acetate (EVA). Distributed by Titan Pharmaceuticals, Inc., South San Francisco, CA 94080, USA. PROAW00008-R2 03/21 For more information, go to www.PROBUPHINE.com or call 1-844-859-6341. |
||||
PRINCIPAL DISPLAY PANEL - 74.2 mg Pouch Carton
NDC 52440-0100-14
Probuphine®
(buprenorphine) implant
74.2 mg per implant
CIII
Implant Kit
Contents:
- Four (4) PROBUPHINE Implants,
74.2 mg (buprenorphine) per implant
(equivalent to 80 mg buprenorphine HCl) - One (1) Applicator
- Prescribing Information
- Instructions for Use
- Medication Guide
- Patient ID Card
- Patient Chart Sticker
For Subdermal Administration Only
For questions, call 1-844-859-6341
Dispense the accompanying Medication Guide to each patient.
Dispose of PROBUPHINE implants in keeping with local, state, and federal
regulations governing the disposal of pharmaceutical biohazardous waste.
Rx only
Distributed by: Titan Pharmaceuticals, Inc., South San Francisco, CA 94080
PROAW00001-R0 07/18
122371
| PROBUPHINE
buprenorphine hydrochloride implant |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Titan Pharmaceuticals, Inc. (807182720) |
| Registrant - Titan Pharmaceuticals, Inc. (807182720) |
Frequently asked questions
- What are the different brands of buprenorphine?
- Is buprenorphine an opiate / narcotic?
- Is Probuphine better than Sublocade?
- When was the Probuphine implant discontinued?
- Is Probuphine covered by insurance?
- What drugs cause pinpoint pupils?
- How long does buprenorphine stay in your system?
- How long does opioid withdrawal last?
More about Probuphine (buprenorphine)
- Check interactions
- Compare alternatives
- Reviews (3)
- Latest FDA alerts (5)
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Breastfeeding
Patient resources
Professional resources
Other brands
Belbuca, Butrans, Sublocade, Brixadi, Buprenex