Nucynta Solution: Package Insert / Prescribing Info
Package insert / product label
Generic name: tapentadol hydrochloride
Dosage form: oral solution
Drug class: Opioids (narcotic analgesics)
Medically reviewed by Drugs.com. Last updated on Mar 25, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
NUCYNTA® (tapentadol) oral solution C-II
Initial U.S. Approval: 2008
WARNING: RISK OF MEDICATION ERRORS, ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; NEONATAL OPIOID WITHDRAWAL SYNDROME; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
See full prescribing information for complete boxed warning.
- Ensure accuracy when prescribing, dispensing, and administering NUCYNTA oral solution. Dosing errors due to confusion between mg and mL, and other tapentadol oral solution of different concentrations can result in accidental overdose and death. (2.1, 5.1)
- NUCYNTA oral solution exposes users to risks of addiction, abuse, and misuse, which can lead to overdose and death. Assess patient’s risk before prescribing and monitor regularly for these behaviors and conditions. (5.2)
- Serious, life-threatening, or fatal respiratory depression may occur. Monitor closely, especially upon initiation or following a dose increase. (5.3)
- Accidental ingestion of NUCYNTA oral solution, especially by children, can result in a fatal overdose of tapentadol. (5.3)
- Prolonged use of NUCYNTA oral solution during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated. If prolonged opioid use is required in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available. (5.4)
- Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death. Reserve concomitant prescribing for use in patients for whom alternative treatment options are inadequate; limit dosages and durations to the minimum required; and follow patients for signs and symptoms of respiratory depression and sedation (5.5, 7).
Recent Major Changes
Indications and Usage for Nucynta Solution
NUCYNTA oral solution is an opioid analgesic indicated for the management of acute pain severe enough to require an opioid analgesic and for which alternative treatments are inadequate. (1)
Limitations of Use (1)
Because of the risks of addiction, abuse, and misuse with opioids, even at recommended doses, reserve NUCYNTA oral solution for use in patients for whom alternative treatment options [e.g., non-opioid analgesics or opioid combination products]:
- Have not been tolerated, or are not expected to be tolerated,
- Have not provided adequate analgesia, or are not expected to provide adequate analgesia
Nucynta Solution Dosage and Administration
- Use the lowest effective dosage for the shortest duration consistent with individual patient treatment goals. (2.1)
- Individualize dosing according to the severity of pain, patient response, prior analgesic experience, and risk factors for addiction, abuse, and misuse. (2.1)
- Initiate treatment with NUCYNTA oral solution with or without food at a dose of 2.5 mL (50 mg), 3.75 mL (75 mg), or 5 mL (100 mg) every 4 to 6 hours depending upon pain intensity. On the first day of dosing, the second dose may be administered as soon as one hour after the first dose, if adequate pain relief is not attained with the first dose. Subsequent dosing is 2.5 mL (50 mg), 3.75 mL (75 mg), or 5 mL (100 mg) every 4 to 6 hours and should be adjusted to maintain adequate analgesia with acceptable tolerability. Daily doses greater than 700 mg on the first day of therapy and 600 mg on subsequent days have not been studied and are, therefore, not recommended. (2.2)
- Moderate Hepatic Impairment: Initiate treatment with 50 mg no more than once every 8 hours (maximum of three doses in 24 hours). Monitor closely for respiratory and central nervous system depression. (2.3)
- Do not stop NUCYNTA oral solution abruptly in a physically dependent patient. (2.5)
Dosage Forms and Strengths
Oral Solution: 20 mg/mL (3)
Contraindications
- Significant respiratory depression (4)
- Acute or severe bronchial asthma in an unmonitored setting or in absence of resuscitative equipment. (4)
- Known or gastrointestinal obstruction, including suspected paralytic ileus. (4)
- Hypersensitivity to tapentadol. (4)
- Concurrent use of monoamine oxidase inhibitors (MAOIs) or use of MAOIs within the last 14 days. (4)
Warnings and Precautions
- Life-Threatening Respiratory Depression in Patients with Chronic Pulmonary Disease or in Elderly, Cachectic, or Debilitated Patients: Monitor closely, particularly during initiation and titration. (5.6)
- Serotonin Syndrome: Potentially life-threatening condition could result from concomitant serotonergic drug administration. Discontinue NUCYNTA oral solution if serotonin syndrome is suspected. (5.7)
- Adrenal Insufficiency: If diagnosed, treat with physiologic replacement of corticosteroids, and wean patient off of the opioid. (5.8)
- Severe Hypotension: Monitor during dosage initiation and titration. Avoid use of NUCYNTA oral solution in patients with circulatory shock. (5.9)
- Risks of Use in Patients with Increased Intracranial Pressure, Brain Tumors, Head Injury, or Impaired Consciousness: Monitor for sedation and respiratory depression. Avoid use of NUCYNTA oral solution in patients with impaired consciousness or coma. (5.10)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence ≥10%) were nausea, dizziness, vomiting and somnolence. (6.1)
To report SUSPECTED ADVERSE REACTIONS, Depomed, Inc. at 1-866-458-6389 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Drug Interactions
- Mixed Agonist/Antagonist and Partial Agonist Opioids Analgesics: Avoid use with NUCYNTA oral solution because they reduce analgesic effect of NUCYNTA oral solution or precipitate withdrawal symptoms. (7)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2016
Full Prescribing Information
WARNING: RISK OF MEDICATION ERRORS, ADDICTION, ABUSE, AND MISUSE; LIFE-THREATENING RESPIRATORY DEPRESSION; ACCIDENTAL INGESTION; NEONATAL OPIOID WITHDRAWAL SYNDROME; and RISKS FROM CONCOMITANT USE WITH BENZODIAZEPINES OR OTHER CNS DEPRESSANTS
Risk of Medication Errors
Ensure accuracy when prescribing, dispensing, and administering NUCYNTA oral solution. Dosing errors due to confusion between mg and mL can result in accidental overdose and death [see Dosage and Administration (2.1), Warnings and Precautions (5.1)].
Addiction, Abuse, and Misuse
NUCYNTA oral solution exposes patients and other users to the risks of opioid addiction, abuse, and misuse, which can lead to overdose and death. Assess each patient’s risk prior to prescribing NUCYNTA oral solution, and monitor all patients regularly for the development of these behaviors and conditions [see Warnings and Precautions (5.2)].
Life-Threatening Respiratory Depression
Serious, life-threatening, or fatal respiratory depression may occur with use of NUCYNTA oral solution. Monitor for respiratory depression, especially during initiation of NUCYNTA oral solution or following a dose increase [see Warnings and Precautions (5.3)].
Accidental Ingestion
Accidental ingestion of even one dose of NUCYNTA oral solution, especially by children, can result in a fatal overdose of tapentadol [see Warnings and Precautions (5.3)].
Neonatal Opioid Withdrawal Syndrome
Prolonged use of NUCYNTA oral solution during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. If opioid use is required for a prolonged period in a pregnant woman, advise the patient of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Warnings and Precautions (5.4)].
Risks From Concomitant Use With Benzodiazepines Or Other CNS Depressants
Concomitant use of opioids with benzodiazepines or other central nervous system (CNS) depressants, including alcohol, may result in profound sedation, respiratory depression, coma, and death [see Warnings and Precautions (5.5), Drug Interactions (7)].
- Reserve concomitant prescribing of NUCYNTA oral solution and benzodiazepines or other CNS depressants for use in patients for whom alternative treatment options are inadequate.
- Limit dosages and durations to the minimum required.
- Follow patients for signs and symptoms of respiratory depression and sedation.
1. Indications and Usage for Nucynta Solution
NUCYNTA (tapentadol) oral solution is indicated for the management of acute pain severe enough to require an opioid analgesic and for which alternative treatments are inadequate in adults.
Limitations of Use
Because of the risks of addiction, abuse, and misuse with opioids, even at recommended doses [see Warnings and Precautions (5.2)], reserve NUCYNTA oral solution for use in patients for whom alternative treatment options [e.g., non-opioid analgesics or opioid combination products]:
- Have not been tolerated, or are not expected to be tolerated,
- Have not provided adequate analgesia, or are not expected to provide adequate analgesia
2. Nucynta Solution Dosage and Administration
2.1 Important Dosage and Administration Instructions
Ensure accuracy when prescribing, dispensing, and administering NUCYNTA oral solution to avoid dosing errors due to confusion between mg and mL, which could result in accidental overdose and death. Ensure the proper dose is communicated and dispensed. The oral solution contains 20 mg tapentadol per milliliter (mL), when writing prescriptions, include both the total dose in mg and the total dose in volume.
Always use the enclosed calibrated measuring syringe when administering NUCYNTA oral solution to ensure the dose is measured and administered accurately. An oral syringe is supplied with dose marks corresponding directly to 2.5 mL (equals 50 mg) oral solution, 3.75 mL (equals 75 mg) oral solution, and 5 mL (equals 100 mg) oral solution.
Do not use household teaspoons or tablespoons to measure NUCYNTA oral solution, as using a tablespoon instead of a teaspoon could lead to overdosage.
Inform patients of the availability of FDA-approved patient labeling, Instructions for Use, for step-by-step instructions for patients on how to use the medicine bottle and the oral syringe.
Use the lowest effective dosage for the shortest duration consistent with individual patient treatment goals [see Warnings and Precautions (5)].
Initiate the dosing regimen for each patient individually, taking into account the patient's severity of pain, patient response, prior analgesic treatment experience, and risk factors for addiction, abuse, and misuse [see Warnings and Precautions (5.2)].
Monitor patients closely for respiratory depression, especially within the first 24-72 hours of initiating therapy and following dosage increases with NUCYNTA and adjust the dosage accordingly [see Warnings and Precautions (5.3)]
2.2 Initial Dosage
Initiate treatment with NUCYNTA oral solution in a dosing range of 50 mg (2.5 mL) to 100 mg (5 mL) every 4 to 6 hours as needed for pain.
On the first day of dosing, the second dose may be administered as soon as one hour after the first dose, if adequate pain relief is not attained with the first dose. Subsequent dosing is 2.5 mL (equivalent to 50 mg), 3.75 mL (equivalent to 75 mg), or 5 mL (equivalent to 100 mg) every 4 to 6 hours and should be adjusted to maintain adequate analgesia with acceptable tolerability.
Daily doses greater than 700 mg on the first day of therapy and 600 mg on subsequent days have not been studied and are not recommended.
NUCYNTA oral solution may be given with or without food [see Clinical Pharmacology (12.3)].
Conversion from NUCYNTA oral solution to NUCYNTA ER
Patients can be converted from NUCYNTA oral solution to NUCYNTA ER using the equivalent total daily dose of NUCYNTA oral solution and dividing it into two equal doses of NUCYNTA ER separated by approximately 12-hour intervals. As an example, a patient receiving 50 mg of NUCYNTA oral solution four times per day (200 mg/day) may be converted to 100 mg NUCYNTA ER twice a day.
2.3 Dosage Modifications in Patients with Hepatic Impairment
The safety and efficacy of NUCYNTA oral solution has not been studied in patients with severe hepatic impairment (Child-Pugh Score 10-15) and use in this population is not recommended [see Warnings and Precautions (5.15)].
Initiate treatment of patients with moderate hepatic impairment (Child-Pugh Score 7 to 9) with 50 mg no more frequently than once every 8 hours (maximum of three doses in 24 hours). Further treatment should reflect maintenance of analgesia with acceptable tolerability, to be achieved by either shortening or lengthening the dosing interval. Monitor closely for respiratory and central nervous system depression [see Clinical Pharmacology (12.3)].
No dosage adjustment is recommended in patients with mild hepatic impairment (Child-Pugh Score 5 to 6) [see Clinical Pharmacology (12.3)].
2.4 Titration and Maintenance of Therapy
Continually reevaluate patients receiving NUCYNTA oral solution to assess the maintenance of pain control and the relative incidence of adverse reactions, as well as monitoring for the development of addiction, abuse, or misuse [see Warnings and Precautions (5.2)]. Frequent communication is important among the prescriber, other members of the healthcare team, the patient, and the caregiver/family during periods of changing analgesic requirements, including initial titration.
If the level of pain increases after dosage stabilization, attempt to identify the source of increased pain before increasing the NUCYNTA oral solution dosage. If unacceptable opioid-related adverse reactions are observed, consider reducing the dosage. Adjust the dosage to obtain an appropriate balance between management of pain and opioid-related adverse reactions.
2.5 Discontinuation of NUCYNTA oral solution
When a patient who has been taking NUCYNTA regularly and may be physically dependent no longer requires therapy with NUCYNTA taper the dose gradually, by 25% to 50% every 2 to 4 days, while monitoring carefully for signs and symptoms of withdrawal. If the patient develops these signs or symptoms, raise the dose to the previous level and taper more slowly, either by increasing the interval between decreases, decreasing the amount of change in dose, or both. Do not abruptly discontinue NUCYNTA oral solution in a physically-dependent patient [see Warnings and Precautions (5.13), Drug Abuse and Dependence (9.3)].
3. Dosage Forms and Strengths
Oral solution: 20 mg/mL in 100 mL and 200 mL fill bottles with child-resistant closure [see Description (11) and How Supplied/Storage and Handling (16)].
4. Contraindications
NUCYNTA oral solution is contraindicated in patients with:
- Significant respiratory depression [see Warnings and Precautions (5.3)]
- Acute or severe bronchial asthma in an unmonitored setting or in the absence of resuscitative equipment [see Warnings and Precautions (5.6)]
- Known or suspected gastrointestinal obstruction, including suspected paralytic ileus [see Warnings and Precautions (5.11)]
- Hypersensitivity to tapentadol (e.g. anaphylaxis, angioedema) or to any other ingredients of the product [see Adverse Reactions (6.2)].
- Concurrent use of monoamine oxidase inhibitors (MAOIs) or use of MAOIs within the last 14 days [see Drug Interactions (7)].
5. Warnings and Precautions
5.1 Risk of Accidental Overdose and Death due to Medication Errors
Dosing errors can result in accidental overdose and death. Avoid dosing errors that may result from confusion between mg and mL when prescribing, dispensing, and administering NUCYNTA oral solution. Ensure that the dose is communicated clearly and dispensed accurately. Always use the enclosed calibrated syringe when administering NUCYNTA to ensure the dose is measured and administered accurately. Do not use a teaspoon or a tablespoon to measure a dose. A household teaspoon or tablespoon is not an adequate measuring device. Given the inexactitude of the household spoon measure and the possibility of using a tablespoon instead of a teaspoon, which could lead to overdosage, it is strongly recommended that caregivers obtain and use a calibrated measuring device. Health care providers should recommend a calibrated device that can measure and deliver the prescribed dose accurately, and instruct caregivers to use extreme caution in measuring the dosage.
5.2 Addiction, Abuse, and Misuse
NUCYNTA oral solution contains tapentadol, a Schedule II controlled substance.
As an opioid, NUCYNTA oral solution exposes users to the risks of addiction, abuse, and misuse [see Drug Abuse and Dependence (9)].
Although the risk of addiction in any individual is unknown, it can occur in patients appropriately prescribed NUCYNTA oral solution. Addiction can occur at recommended dosages and if the drug is misused or abused.
Assess each patient’s risk for opioid addiction, abuse, or misuse prior to prescribing NUCYNTA oral solution and monitor all patients receiving NUCYNTA oral solution for the development of these behaviors and conditions. Risks are increased in patients with a personal or family history of substance abuse (including drug or alcohol abuse or addiction) or mental illness (e.g., major depression). The potential for these risks should not, however, prevent the proper management of pain in any given patient. Patients at increased risk may be prescribed opioids such as NUCYNTA oral solution, but use in such patients necessitates intensive counseling about the risks and proper use of NUCYNTA oral solution along with intensive monitoring for signs of addiction, abuse, and misuse.
Opioids are sought by drug abusers and people with addiction disorders and are subject to criminal diversion. Consider these risks when prescribing or dispensing NUCYNTA oral solution. Strategies to reduce these risks include prescribing the drug in the smallest appropriate quantity and advising the patient on the proper disposal of unused drug [see Patient Counseling Information (17)]. Contact local state professional licensing board or state controlled substances authority for information on how to prevent and detect abuse or diversion of this product.
5.3 Life Threatening-Respiratory Depression
Serious, life-threatening, or fatal respiratory depression has been reported with the use of opioids, even when used as recommended. Respiratory depression, if not immediately recognized and treated, may lead to respiratory arrest and death. Management of respiratory depression may include close observation, supportive measures, and use of opioid antagonists, depending on the patient’s clinical status [see Overdosage (10)]. Carbon dioxide (CO2) retention from opioid-induced respiratory depression can exacerbate the sedating effects of opioids.
While serious, life-threatening, or fatal respiratory depression can occur at any time during the use of NUCYNTA oral solution, the risk is greatest during the initiation of therapy or following a dosage increase. Monitor patients closely for respiratory depression, especially within the first 24-72 hours of initiating therapy with and following dosage increases of NUCYNTA oral solution.
To reduce the risk of respiratory depression, proper dosing and titration of NUCYNTA oral solution are essential [see Dosage and Administration (2.2)]. Overestimating the NUCYNTA oral solution dosage when converting patients from another opioid product can result in a fatal overdose with the first dose.
Accidental ingestion of even one dose of NUCYNTA oral solution, especially by children, can result in respiratory depression and death due to an overdose of tapentadol.
5.4 Neonatal Opioid Withdrawal Syndrome
Prolonged use of NUCYNTA oral solution during pregnancy can result in withdrawal in the neonate. Neonatal opioid withdrawal syndrome, unlike opioid withdrawal syndrome in adults, may be life-threatening if not recognized and treated, and requires management according to protocols developed by neonatology experts. Observe newborns for signs of neonatal opioid withdrawal syndrome and manage accordingly. Advise pregnant women using opioids a prolonged period of the risk of neonatal opioid withdrawal syndrome and ensure that appropriate treatment will be available [see Use in Specific Populations (8.1), Patient Counseling Information (17)].
5.5 Risks from Concomitant Use with Benzodiazepines or Other CNS Depressants
Profound sedation, respiratory depression, coma, and death may result from the concomitant use of NUCYNTA oral solution with benzodiazepines or other CNS depressants (e.g., nonbenzodiazepine sedatives/hypnotics, anxiolytics, tranquilizers, muscle relaxants, general anesthetics, antipsychotics, other opioids, alcohol). Because of these risks, reserve concomitant prescribing of these drugs for use in patients for whom alternative treatment options are inadequate.
Observational studies have demonstrated that concomitant use of opioid analgesics and benzodiazepines increases the risk of drug-related mortality compared to use of opioid analgesics alone. Because of similar pharmacological properties, it is reasonable to expect similar risk with the concomitant use of other CNS depressant drugs with opioid analgesics [see Drug Interactions (7)].
If the decision is made to prescribe a benzodiazepine or other CNS depressant concomitantly with an opioid analgesic, prescribe the lowest effective dosages and minimum durations of concomitant use. In patients already receiving an opioid analgesic, prescribe a lower initial dose of the benzodiazepine or other CNS depressant than indicated in the absence of an opioid, and titrate based on clinical response. If an opioid analgesic is initiated in a patient already taking a benzodiazepine or other CNS depressant, prescribe a lower initial dose of the opioid analgesic, and titrate based on clinical response. Follow patients closely for signs and symptoms of respiratory depression and sedation.
Advise both patients and caregivers about the risks of respiratory depression and sedation when NUCYNTA oral solution is used with benzodiazepines or other CNS depressants (including alcohol and illicit drugs). Advise patients not to drive or operate heavy machinery until the effects of concomitant use of the benzodiazepine or other CNS depressant have been determined. Screen patients for risk of substance use disorders, including opioid abuse and misuse, and warn them of the risk for overdose and death associated with the use of additional CNS depressants including alcohol and illicit drugs [see Drug Interactions (7) and Patient Counseling Information (17)].
5.6 Life-Threatening Respiratory Depression in Patients with Chronic Pulmonary Disease or in Elderly, Cachectic, or Debilitated Patients
The use of NUCYNTA oral solution in patients with acute or severe bronchial asthma in an unmonitored setting or in the absence of resuscitative equipment is contraindicated.
Patients with Chronic Pulmonary Disease: NUCYNTA oral solution-treated patients with significant chronic obstructive pulmonary disease or cor pulmonale, and those with a substantially decreased respiratory reserve, hypoxia, hypercapnia, or pre-existing respiratory depression are at increased risk of decreased respiratory drive including apnea, even at recommended dosages of NUCYNTA oral solution [see Warnings and Precautions (5.6)].
Elderly, Cachectic, or Debilitated Patients:Life-threatening respiratory depression is more likely to occur in elderly, cachectic, or debilitated patients because they may have altered pharmacokinetics or altered clearance compared to younger, healthier patients [see Warnings and Precautions (5.6)].
Monitor such patients closely, particularly when initiating and titrating NUCYNTA® and when NUCYNTA oral solution is given concomitantly with other drugs that depress respiration [see Warnings and Precautions (5.6)]. Alternatively, consider the use of non-opioid analgesics in these patients.
5.7 Serotonin Syndrome with Concomitant Use of Serotonergic Drugs
Cases of serotonin syndrome, a potentially life-threatening condition, have been reported during concurrent use of tapentadol with serotonergic drugs. Serotonergic drugs include selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), triptans, 5-HT3 receptor antagonists, drugs that affect the serotonergic neurotransmitter system (e.g. mirtazapine, trazodone, tramadol), and drugs that impair metabolism of serotonin (including MAO inhibitors, both those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue). This may occur within the recommended dosage range.
Serotonin syndrome symptoms may include mental-status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea) and can be fatal [see Drug Interactions (7)] The onset of symptoms generally occurs within several hours to a few days of concomitant use, but may occur later than that. Discontinue NUCYNTA oral solution if serotonin syndrome is suspected.
5.8 Adrenal Insufficiency
Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use. Presentation of adrenal insufficiency may include non-specific symptoms and signs including nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. If adrenal insufficiency is suspected, confirm the diagnosis with diagnostic testing as soon as possible. If adrenal insufficiency is diagnosed, treat with physiologic replacement doses of corticosteroids. Wean the patient off of the opioid to allow adrenal function to recover and continue corticosteroid treatment until adrenal function recovers. Other opioids may be tried as some cases reported use of a different opioid without recurrence of adrenal insufficiency. The information available does not identify any particular opioids as being more likely to be associated with adrenal insufficiency.
5.9 Severe Hypotension
NUCYNTA oral solution may cause severe hypotension including orthostatic hypotension and syncope in ambulatory patients. There is increased risk in patients whose ability to maintain blood pressure has already been compromised by a reduced blood volume or concurrent administration of certain CNS depressant drugs (e.g. phenothiazines or general anesthetics) [see Drug Interactions (7)]. Monitor these patients for signs of hypotension after initiating or titrating the dosage of NUCYNTA oral solution. In patients with circulatory shock, NUCYNTA oral solution may cause vasodilation that can further reduce cardiac output and blood pressure. Avoid the use of NUCYNTA oral solution in patients with circulatory shock.
5.10 Risks of Use in Patients with Increased Intracranial Pressure, Brain Tumors, Head Injury, or Impaired Consciousness
In patients who may be susceptible to the intracranial effects of CO2 retention (e.g., those with evidence of increased intracranial pressure or brain tumors), NUCYNTA oral solution may reduce respiratory drive, and the resultant CO2 retention can further increase intracranial pressure. Monitor such patients for signs of sedation and respiratory depression, particularly when initiating therapy with NUCYNTA oral solution.
Opioids may also obscure the clinical course in a patient with a head injury. Avoid the use of NUCYNTA oral solution in patients with impaired consciousness or coma.
5.11 Risks of Use in Patients with Gastrointestinal Conditions
NUCYNTA oral solution is contraindicated in patients with known or suspected gastrointestinal obstruction, including paralytic ileus.
The tapentadol in NUCYNTA oral solution may cause spasm of the sphincter of Oddi. Opioids may cause increases in serum amylase. Monitor patients with biliary tract disease, including acute pancreatitis for worsening symptoms.
5.12 Increased Risk of Seizures in Patients with Seizure Disorders
The tapentadol in NUCYNTA oral solution may increase the frequency of seizures in patients with seizure disorders, and may increase the risk of seizures occurring in other clinical settings associated with seizures. Monitor patients with a history of seizure disorders for worsened seizure control during NUCYNTA oral solution therapy.
5.13 Withdrawal
Avoid the use of mixed agonist/antagonist (e.g., pentazocine, nalbuphine, and butorphanol) or partial agonist (e.g., buprenorphine) analgesics in patients who are receiving a full opioid agonist analgesic, including NUCYNTA oral solution. In these patients, mixed agonist/antagonist and partial agonist analgesics may reduce the analgesic effect and/or precipitate withdrawal symptoms [see Drug Interactions (7)].
When discontinuing NUCYNTA oral solution in a physically-dependent patient, gradually taper the dosage [see Dosage and Administration (2.5)]. Do not abruptly discontinue NUCYNTA in these patients [see Drug Abuse and Dependence (9.3)].
5.14 Risks of Driving and Operating Machinery
NUCYNTA oral solution may impair the mental or physical abilities needed to perform potentially hazardous activities such as driving a car or operating machinery. Warn patients not to drive or operate dangerous machinery unless they are tolerant to the effects of NUCYNTA oral solution and know how they will react to the medication.
5.15 Interactions with Alcohol, Other Opioids, and Drugs of Abuse
Due to its mu-opioid agonist activity, NUCYNTA oral solution may be expected to have additive effects when used in conjunction with alcohol, other opioids, or illicit drugs that cause central nervous system depression, respiratory depression, hypotension, and profound sedation, coma or death [see Drug Interactions (7)]. Instruct patients not to consume alcoholic beverages or use prescription or non-prescription products containing alcohol, other opioids, or drugs of abuse while on NUCYNTA oral solution therapy [see Drug Interactions (7)].
5.16 Risk of Toxicity in Patients with Hepatic Impairment
A study with NUCYNTA oral solution in subjects with hepatic impairment showed higher serum concentrations of tapentadol than in those with normal hepatic function. Avoid use of NUCYNTA oral solution in patients with severe hepatic impairment. Reduce the dose of NUCYNTA oral solution in patients with moderate hepatic impairment [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)]. Closely monitor patients with moderate hepatic impairment for respiratory and central nervous system depression when receiving NUCYNTA oral solution.
5.17 Risk of Toxicity in Patients with Renal Impairment
Use of NUCYNTA oral solution in patients with severe renal impairment is not recommended due to accumulation of a metabolite formed by glucuronidation of tapentadol. The clinical relevance of the elevated metabolite is not known [see Clinical Pharmacology (12.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed, or described in greater detail in other sections:
- Addiction, Abuse, and Misuse [see Warnings and Precautions (5.2)]
- Life-Threatening Respiratory Depression [see Warnings and Precautions (5.2)]
- Neonatal Opioid Withdrawal Syndrome [see Warnings and Precautions (5.4)]
- Interactions with CNS Benzodiazepine or Other Depressants [see Warnings and Precautions (5.5)]
- Serotonin Syndrome [see Warnings and Precautions (5.7)]
- Adrenal Insufficiency [see Warnings and Precautions (5.8)]
- Severe Hypotension [see Warnings and Precautions (5.9)]
- Gastrointestinal Adverse Reactions [see Warnings and Precautions (5.11)]
- Seizures [see Warnings and Precautions (5.12)]
- Withdrawal [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Based on data from nine Phase 2/3 studies that administered multiple doses (seven placebo- and/or active-controlled, one noncontrolled and one Phase 3 active-controlled safety study) the most common adverse reactions (reported by ≥10% in any NUCYNTA dose group) were: nausea, dizziness, vomiting and somnolence.
The most common reasons for discontinuation due to adverse reactions in the studies described above (reported by ≥1% in any NUCYNTA dose group) were dizziness (2.6% vs. 0.5%), nausea (2.3% vs. 0.6%), vomiting (1.4% vs. 0.2%), somnolence (1.3% vs. 0.2%) and headache (0.9% vs. 0.2%) for NUCYNTA- and placebo-treated patients, respectively. Seventy-six percent of NUCYNTA-treated patients from the nine studies experienced adverse events.
NUCYNTA was studied in multiple-dose, active- or placebo-controlled studies, or noncontrolled studies (n = 2178), in single-dose studies (n = 870), in open-label study extension (n = 483) and in Phase 1 studies (n = 597). Of these, 2034 patients were treated with doses of 50 mg to 100 mg of NUCYNTA dosed every 4 to 6 hours.
The data described below reflect exposure to NUCYNTA in 3161 patients, including 449 exposed for 45 days. NUCYNTA was studied primarily in placebo- and active-controlled studies (n = 2266, and n = 2944, respectively). The population was 18 to 85 years old (mean age 46 years), 68% were female, 75% white and 67% were postoperative. Most patients received NUCYNTA doses of 50 mg, 75 mg, or 100 mg every 4 to 6 hours.
| System/Organ Class MedDRA Preferred Term | NUCYNTA
21 mg – 120 mg (n = 2178) % | Placebo (n = 619) % |
|---|---|---|
| Gastrointestinal disorders | ||
| Nausea | 30 | 13 |
| Vomiting | 18 | 4 |
| Constipation | 8 | 3 |
| Dry mouth | 4 | <1 |
| Dyspepsia | 2 | <1 |
| General disorders and administration site conditions | ||
| Fatigue | 3 | <1 |
| Feeling hot | 1 | <1 |
| Infections and infestations | ||
| Nasopharyngitis | 1 | <1 |
| Upper respiratory tract infection | 1 | <1 |
| Urinary tract infection | 1 | <1 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 2 | 0 |
| Nervous system disorders | ||
| Dizziness | 24 | 8 |
| Somnolence | 15 | 3 |
| Tremor | 1 | <1 |
| Lethargy | 1 | <1 |
| Psychiatric disorders | ||
| Insomnia | 2 | <1 |
| Confusional state | 1 | 0 |
| Abnormal dreams | 1 | <1 |
| Anxiety | 1 | < |
| Skin and subcutaneous tissue disorders | ||
| Pruritus | 5 | 1 |
| Hyperhidrosis | 3 | <1 |
| Pruritus generalized | 3 | <1 |
| Rash | 1 | <1 |
| Vascular disorders | ||
| Hot flush | 1 | <1 |
The following adverse drug reactions occurred in less than 1% of NUCYNTA-treated patients in the pooled safety data from nine Phase 2/3 clinical studies:
Cardiac disorders: heart rate increased, heart rate decreased
Eye disorders: visual disturbance
Gastrointestinal disorders: abdominal discomfort, impaired gastric emptying
General disorders and administration site conditions: irritability, edema, drug withdrawal syndrome, feeling drunk
Immune system disorders: hypersensitivity
Investigations: gamma-glutamyltransferase increased, alanine aminotransferase increased, aspartate aminotransferase increased
Musculoskeletal and connective tissue disorders: involuntary muscle contractions, sensation of heaviness
Nervous system disorders: hypoesthesia, paresthesia, disturbance in attention, sedation, dysarthria, depressed level of consciousness, memory impairment, ataxia, presyncope, syncope, coordination abnormal, seizure
Psychiatric disorders: euphoric mood, disorientation, restlessness, agitation, nervousness, thinking abnormal
Renal and urinary disorders: urinary hesitation, pollakiuria
Respiratory, thoracic and mediastinal disorders: oxygen saturation decreased, cough, dyspnea, respiratory depression
Skin and subcutaneous tissue disorders: urticarial
Vascular disorders: blood pressure decreased
In the pooled safety data, the overall incidence of adverse reactions increased with increased dose of NUCYNTA, as did the percentage of patients with adverse reactions of nausea, dizziness, vomiting, somnolence, and pruritus.
6.2 Post-marketing Experience
The following additional adverse reactions have been identified during post-approval use of tapentadol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal disorders: diarrhea
Nervous system disorders: headache
Psychiatric disorders: hallucination, suicidal ideation, panic attack
Cardiac disorders: palpitations
Serotonin syndrome: Cases of serotonin syndrome, a potentially life-threatening condition, have been reported during concomitant use of opioids with serotonergic drugs.
Adrenal insufficiency: Cases of adrenal insufficiency have been reported with opioid use, more often following greater than one month of use.
Anaphylaxis: Anaphylaxis has been reported with ingredients contained in NUCYNTA oral solution.
Androgen deficiency: Cases of androgen deficiency have occurred with chronic use of opioids [see Clinical Pharmacology (12.2)].
Related/similar drugs
7. Drug Interactions
Table 2 includes clinically significant drug interactions with NUCYNTA oral solution.
| Benzodiazepines and other Central Nervous System (CNS) Depressants | |
| Clinical Impact: | Due to additive pharmacologic effect, the concomitant use of benzodiazepines or other CNS depressants including alcohol, increases the risk of respiratory depression, profound sedation, coma, and death. |
| Intervention: | Reserve concomitant prescribing of these drugs for use in patients for whom alternative treatment options are inadequate. Limit dosages and durations to the minimum required. Follow patients closely for signs of respiratory depression and sedation [see Warnings and Precautions (5.6)]. |
| Examples: | Benzodiazepines and other sedatives/hypnotics, anxiolytics, tranquilizers, muscle relaxants, general anesthetics, antipsychotics, other opioids, alcohol. |
| Serotonergic Drugs | |
| Clinical Impact: | The concomitant use of opioids with other drugs that affect the serotonergic neurotransmitter system has resulted in serotonin syndrome [see Warnings and Precautions 5.7]. |
| Intervention: | If concomitant use is warranted, carefully observe the patient, particularly during treatment initiation and dose adjustment. Discontinue NUCYNTA oral solution if serotonin syndrome is suspected. |
| Examples: | Selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), triptans, 5-HT3 receptor antagonists, drugs that affect the serotonin neurotransmitter system (e.g., mirtazapine, trazodone, tramadol), monoamine oxidase (MAO) inhibitors (those intended to treat psychiatric disorders and also others, such as linezolid and intravenous methylene blue). |
| Monoamine Oxidase Inhibitors (MAOIs) | |
| Clinical Impact: | MAOI interactions with opioids may manifest as serotonin syndrome or opioid toxicity (e.g., respiratory depression, coma) [see Warnings and Precautions (5.3)]. |
| Intervention: | Do not use NUCYNTA oral solution in patients taking MAOIs or within 14 days of stopping such treatment. If urgent use of an opioid is necessary, use test doses and frequent titration of small doses of other opioids (such as oxycodone, hydrocodone, oxymorphone, hydrocodone, or buprenorphine) to treat pain while closely monitoring blood pressure and signs and symptoms of CNS and respiratory depression. |
| Examples: | phenelzine, tranylcypromine, linezolid |
| Mixed Agonist/Antagonist and Partial Agonist Opioid Analgesics | |
| Clinical Impact: | May reduce the analgesic effect of NUCYNTA oral solution and/or precipitate withdrawal symptoms. |
| Intervention: | Avoid concomitant use. |
| Examples: | butorphanol, nalbuphine, pentazocine, buprenorphine |
| Muscle Relaxants | |
| Clinical Impact: | Tapentadol may enhance the neuromuscular blocking action of skeletal muscle relaxants and produce an increased degree of respiratory depression. |
| Intervention: | Monitor patients for signs of respiratory depression that may be greater than otherwise expected and decrease the dosage of NUCYNTA oral solution and/or the muscle relaxant as necessary. |
| Diuretics | |
| Clinical Impact: | Opioids can reduce the efficacy of diuretics by inducing the release of antidiuretic hormone. |
| Intervention: | Monitor patients for signs of diminished diuresis and/or effects on blood pressure and increase the dosage of the diuretic as needed. |
| Anticholinergic Drugs | |
| Clinical Impact: | The concomitant use of anticholinergic drugs may increase risk of urinary retention and/or severe constipation, which may lead to paralytic ileus. |
| Intervention: | Monitor patients for signs of urinary retention or reduced gastric motility when NUCYNTA oral solution is used concomitantly with anticholinergic drugs. |
| Alcohol, Other Opioids, and Drugs of Abuse | |
| Clinical Impact: | Due to its mu-opioid agonist activity, NUCYNTA oral solution may be expected to have additive effects when used in conjunction with alcohol, other opioids, or illicit drugs that cause central nervous system depression, respiratory depression, hypotension, and profound sedation, coma or death [see Warnings and Precautions (5.4)]. |
| Intervention: | Instruct patients not to consume alcoholic beverages or use prescription or non-prescription products containing alcohol, other opioids, or drugs of abuse while on NUCYNTA oral solution therapy. |
| Examples: | Alcohol, other opioids, illicit drugs |
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Category C
Risk Summary
Prolonged use of opioid analgesics during pregnancy may cause neonatal opioid withdrawal syndrome [see Warnings and Precautions (5.4)]. Available data with NUCYNTA oral solution are insufficient to inform a drug-associated risk for major birth defects and miscarriage.
In animal reproduction studies, embryofetal mortality and structural malformations were observed with subcutaneous administration of tapentadol during organogenesis to rabbits and delays in skeletal maturation were observed in rats at exposures equivalent to and less than the maximum recommended human dose (MRHD), respectively. When administered to pregnant rats during organogenesis and through lactation, increased pup mortality was noted following oral tapentadol exposures to doses equivalent to the MRHD [see Data].
Based on animal data, advise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy can occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Prolonged use of opioid analgesics during pregnancy for medical or nonmedical purposes can result in physical dependence in the neonate and neonatal opioid withdrawal syndrome shortly after birth.
Neonatal opioid withdrawal syndrome presents as irritability, hyperactivity and abnormal sleep pattern, high pitched cry, tremor, vomiting, diarrhea and failure to gain weight. The onset, duration, and severity of neonatal opioid withdrawal syndrome vary based on the specific opioid used, duration of use, timing and amount of last maternal use, and rate of elimination of the drug by the newborn. Observe newborns for symptoms of neonatal opioid withdrawal syndrome and manage accordingly [see Warnings and Precautions (5.4)].
Labor or Delivery
Opioids cross the placenta and may produce respiratory depression and psycho-physiologic effects in neonates. An opioid antagonist, such as naloxone, must be available for reversal of opioid-induced respiratory depression in the neonate. NUCYNTA oral solution is not recommended for use in pregnant women during or immediately prior to labor, when other analgesic techniques are more appropriate. Opioid analgesics, including NUCYNTA oral solution, can prolong labor through actions which temporarily reduce the strength, duration, and frequency of uterine contractions. However, this effect is not consistent and may be offset by an increased rate of cervical dilation, which tends to shorten labor. Monitor neonates exposed to opioid analgesics during labor for signs of excess sedation and respiratory depression.
Animal Data
Tapentadol HCl was evaluated for teratogenic effects in pregnant rats and rabbits following subcutaneous exposure during organogenesis. When tapentadol was administered twice daily by the subcutaneous route in rats at dose levels of 10, 20, or 40 mg/kg/day [producing up to 1 times the plasma exposure at the maximum recommended human dose (MRHD) of 700 mg/day based on an area under the time-curve (AUC) comparison], no teratogenic effects were observed. Evidence of embryo fetal toxicity included transient delays in skeletal maturation (i.e. reduced ossification) at the 40 mg/kg/day dose which was associated with significant maternal toxicity.
Administration of tapentadol HCl in rabbits at doses of 4, 10, or 24 mg/kg/day by subcutaneous injection [producing 0.2, 0.6, and 1.85 times the plasma exposure at the MRHD based on an AUC comparison] revealed embryo fetal toxicity at doses ≥10 mg/kg/day. Findings included reduced fetal viability, skeletal delays and other variations. In addition, there were multiple malformations including gastroschisis/thoracogastroschisis, amelia/phocomelia, and cleft palate at doses ≥10 mg/kg/day and above, and ablepharia, encephalopathy, and spina bifida at the high dose of 24 mg/kg/day. Embryofetal toxicity, including malformations, may be secondary to the significant maternal toxicity observed in the study.
In a study of pre- and postnatal development in rats, oral administration of tapentadol at doses of 20, 50, 150, or 300 mg/kg/day to pregnant and lactating rats during the late gestation and early postnatal period [resulting in up to 1.7 times the plasma exposure at the MRHD on an AUC basis] did not influence physical or reflex development, the outcome of neurobehavioral tests or reproductive parameters. Treatment-related developmental delay was observed, including incomplete ossification, and significant reductions in pup body weights and body weight gains at doses associated with maternal toxicity (150 mg/kg/day and above). At maternal tapentadol doses ≥150 mg/kg/day, a dose-related increase in pup mortality was observed through postnatal Day 4.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There is insufficient/limited information on the excretion of tapentadol in human or animal breast milk. Physicochemical and available pharmacodynamic/toxicological data on tapentadol point to excretion in breast milk and risk to the breastfeeding child cannot be excluded. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for NUCYNTA oral solution and any potential adverse effects on the breastfed infant from NUCYNTA oral solution or from the underlying maternal condition.
Clinical Considerations
Infants exposed to NUCYNTA oral solution through breast milk should be monitored for excess sedation and respiratory depression. Withdrawal symptoms can occur in breastfed infants when maternal administration of an opioid analgesic is stopped, or when breast-feeding is stopped.
8.3 Females and Males of Reproductive Potential
Infertility
Chronic use of opioids may cause reduced fertility in females and males of reproductive potential. It is not known whether these effects on fertility are reversible [see Adverse Reactions (6.2)].
8.4 Pediatric Use
The safety and effectiveness of NUCYNTA oral solution in pediatric patients less than 18 years of age have not been established.
8.5 Geriatric Use
Of the total number of patients in Phase 2/3 double-blind, multiple-dose clinical studies of NUCYNTA, 19% were 65 and over, while 5% were 75 and over. No overall differences in effectiveness were observed between these patients and younger patients. The rate of constipation was higher in subjects greater than or equal to 65 years than those less than 65 years (12% vs. 7%).
Elderly patients (aged 65 years or older) may have increased sensitivity to tapentadol. In general, use caution when selecting a dosage for an elderly patient, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
Respiratory depression is the chief risk for elderly patients treated with opioids, and has occurred after large initial doses were administered to patients who were not opioid-tolerant or when opioids were co-administered with other agents that depress respiration. Titrate the dosage of NUCYNTA oral solution slowly in geriatric patients and monitor closely for signs of central nervous system and respiratory depression [see Warnings and Precautions (5.6)].
Tapentadol is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
8.6 Hepatic Impairment
Administration of tapentadol resulted in higher exposures and serum levels of tapentadol in subjects with impaired hepatic function compared to subjects with normal hepatic function [see Clinical Pharmacology (12.3)]. Use of NUCYNTA oral solution is not recommended in patients with severe hepatic impairment (Child- Pugh Score 10 to 15). The dose of NUCYNTA oral solution should be reduced in patients with moderate hepatic impairment (Child-Pugh Score 7 to 9) [see Dosage and Administration (2.4)].No dosage adjustment is recommended in patients with mild hepatic impairment (Child-Pugh Score 5 to 6) [see Warnings and Precautions (5.16), Clinical Pharmacology (12.3)]
8.7 Renal Impairment
Use of NUCYNTA oral solution in patients with severe renal impairment (creatinine clearance less than 30 mL/minute) is not recommended. No dosage adjustment is recommended in patients with mild or moderate renal impairment (creatinine clearance 30-90 mL/minute) [see Warnings and Precautions (5.17), Clinical Pharmacology (12.3)].
9. Drug Abuse and Dependence
9.1 Controlled Substance
NUCYNTA oral solution contains tapentadol, a Schedule II controlled substance.
9.2 Abuse
NUCYNTA oral solution contains tapentadol, a substance with a high potential for abuse similar to other opioids including fentanyl, hydrocodone, hydromorphone, methadone, morphine, oxycodone, and oxymorphone. NUCYNTA can be abused and is subject to misuse, addiction, and criminal diversion [see Warnings and Precautions (5.2)].
All patients treated with opioids require careful monitoring for signs of abuse and addiction, because use of opioid analgesic products carries the risk of addiction even under appropriate medical use.
All patients treated with opioids require careful monitoring for signs of abuse and addiction, since use of opioid analgesic products carries the risk of addiction even under appropriate medical use.
Prescription drug abuse is the intentional non-therapeutic use of a prescription drug, even once, for its rewarding psychological or physiological effects.
Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that develop after repeated substance use and includes: a strong desire to take the drug, difficulties in controlling its use, persisting in its use despite harmful consequences, a higher priority given to drug use than to other activities and obligations, increased tolerance, and sometimes a physical withdrawal.
“Drug seeking” behavior is very common in persons with substance use disorders. Drug-seeking tactics include emergency calls or visits near the end of office hours, refusal to undergo appropriate examination, testing or referral, repeated “loss” of prescriptions, tampering with prescriptions, and reluctance to provide prior medical records or contact information for other treating health care provider (s). “Doctor shopping” (visiting multiple prescribers to obtain additional prescriptions) is common among drug abusers and people suffering from untreated addiction Preoccupation with achieving adequate pain relief can be appropriate behavior in a patient with poor pain control..
Abuse and addiction are separate and distinct from physical dependence and tolerance. Health care providers should be aware that addiction may not be accompanied by concurrent tolerance and symptoms of physical dependence in all addicts. In addition, abuse of opioids can occur in the absence of true addiction.
NUCYNTA oral solution like other opioids, can be diverted for non-medical use into illicit channels of distribution. Careful record-keeping of prescribing information, including quantity, frequency, and renewal requests, as required by law, is strongly advised.
Proper assessment of the patient, proper prescribing practices, periodic re-evaluation of therapy, and proper dispensing and storage are appropriate measures that help to limit abuse of opioid drugs.
Risks Specific to Abuse of NUCYNTA
NUCYNTA oral solution is for oral use only. Abuse of NUCYNTA oral solution poses a risk of overdose and death. The risk is increased with concurrent abuse of NUCYNTA oral solution with alcohol and other central nervous system depressants.
Parenteral drug abuse is commonly associated with transmission of infectious diseases such as hepatitis and HIV.
9.3 Dependence
Both tolerance and physical dependence can develop during chronic opioid therapy. Tolerance is the need for increasing doses of opioids to maintain a defined effect such as analgesia (in the absence of disease progression or other external factors). Tolerance may occur to both the desired and undesired effects of drugs, and may develop at different rates for different effects.
Physical dependence results in withdrawal symptoms after abrupt discontinuation or a significant dosage reduction of a drug. Withdrawal also may be precipitated through the administration of drugs with opioid antagonist activity (e.g., naloxone, nalmefene), or mixed agonist/antagonist analgesics (e.g., pentazocine, butorphanol, nalbuphine) or partial agonists (e.g., buprenorphine). Physical dependence may not occur to a clinically significant degree until after several days to weeks of continued opioid usage.
NUCYNTA oral solution should not be abruptly discontinued in a physically-dependent patient [see Dosage and Administration (2.5)]. If NUCYNTA oral solution is abruptly discontinued in a physically-dependent patient, a withdrawal syndrome may occur. Some or all of the following can characterize this syndrome: restlessness, lacrimation, rhinorrhea, yawning, perspiration, chills, myalgia, and mydriasis Other signs and symptoms also may develop, including: irritability, anxiety, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhea, or increased blood pressure, respiratory rate, or heart rate.
Infants born to mothers physically dependent on opioids will also be physically dependent and may exhibit respiratory difficulties and withdrawal symptoms [see Use in Specific Populations (8.1)].
10. Overdosage
Clinical Presentation
Acute overdosage with NUCYNTA oral solution can be manifested by respiratory depression, somnolence progressing to stupor or coma, skeletal muscle flaccidity, cold and clammy skin, constricted pupils, and in some cases, pulmonary edema, bradycardia, hypotension partial or complete airway obstruction, atypical snoring, and death. Marked mydriasis rather than miosis may be seen due to severe hypoxia in overdose situations [see Clinical Pharmacology (12.2)].
Treatment of Overdose
In case of overdose, priorities are the reestablishment of a patent and protected airway and institution of assisted or controlled ventilation if needed. Employ other supportive measures (including oxygen, and vasopressors) in the management of circulatory shock and pulmonary edema as indicated. Cardiac arrest or arrhythmias will require advanced life support techniques.
The opioid antagonists, naloxone or nalmefene, are specific antidotes to respiratory depression resulting from opioid overdose. For clinically significant respiratory or circulatory depression secondary to tapentadol overdose, administer an opioid antagonist. Opioid antagonists should not be administered in the absence of clinically significant respiratory or circulatory depression secondary to tapentadol overdose.
Because the duration of opioid reversal is expected to be less than the duration of action of tapentadol in NUCYNTA oral solution, carefully monitor the patient until spontaneous respiration is reliably re-established. If the response to an opioid antagonist is suboptimal or only brief in nature, administer additional antagonist as directed in the product’s prescribing information.
In an individual physically dependent on opioids, administration of the recommended usual dosage of the antagonist will precipitate an acute withdrawal. The severity of the withdrawal symptoms experienced will depend on the degree of physical dependence and the dose of the antagonist administered. If a decision is made to treat serious respiratory depression in the physically dependent patient, administration of the antagonist should be begun with care and by titration with smaller than usual doses of the antagonist.
11. Nucynta Solution Description
NUCYNTA (tapentadol) oral solution is a mu-opioid receptor agonist, available in a liquid solution for oral administration. The chemical name is 3-[(1R,2R)-3-(dimethylamino)-1-ethyl-2- methylpropyl]phenol monohydrochloride and it has the following chemical structure:
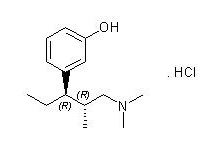
The molecular weight of tapentadol HCl is 257.80, and the molecular formula is C14H23NO·HCl. The n-octanol:water partition coefficient log P value is 2.87. The pKa values are 9.34 and 10.45.
NUCYNTA (tapentadol) oral solution is supplied as a clear, colorless solution and contains 20 mg/mL of tapentadol (corresponding to 23 mg/mL of tapentadol hydrochloride). The inactive ingredients in NUCYNTA oral solution include: citric acid monohydrate, purified water, raspberry flavor, sodium hydroxide, and sucralose.
12. Nucynta Solution - Clinical Pharmacology
12.1 Mechanism of Action
Tapentadol is a mu-opioid receptor (MOR) agonist and a norepinephrine reuptake inhibitor (NRI). Analgesia in animal models is derived from both of these properties.
12.2 Pharmacodynamics
Effects on the Central Nervous System (CNS)
Tapentadol produces respiratory depression by direct effect on the brainstem respiratory centers. The respiratory depression involves a reduction in the responsiveness of the brain stem respiratory centers to both increases in carbon dioxide tension and electrical stimulation.
Tapentadol causes miosis, even in total darkness. Pinpoint pupils are a sign of opioid overdose but are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origin may produce similar findings). Marked mydriasis rather than miosis may be seen with hypoxia in overdose situations
Effects on the Gastrointestinal Tract and on Other Smooth Muscle
Tapentadol causes a reduction in motility associated with an increase in smooth muscle tone in the antrum of the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased, while tone is increased to the point of spasm, resulting in constipation. Other opioid-induced effects may include a reduction in biliary and pancreatic secretions, spasm of sphincter of Oddi, and transient elevations in serum amylase.
Effects on the Cardiovascular System
There was no effect of therapeutic and supratherapeutic doses of tapentadol on the QT interval. In a randomized, double-blind, placebo- and positive-controlled crossover study, healthy subjects were administered five consecutive doses of NUCYNTA 100 mg every 6 hours, NUCYNTA 150 mg every 6 hours, placebo and a single oral dose of moxifloxacin. Similarly, NUCYNTA had no relevant effect on other ECG parameters (heart rate, PR interval, QRS duration, T-wave or U-wave morphology).
Tapentadol produces peripheral vasodilation which may result in orthostatic hypotension or syncope. Manifestations of histamine release and/or peripheral vasodilation may include pruritus, flushing, red eyes, sweating, and/or orthostatic hypotension.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticotropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans [see Adverse Reactions (6.2)]. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon [see Adverse Reactions (6.2)].
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date [see Adverse Reactions (6.2)].
Effects on the Immune System
Opioids have been shown to have a variety of effects on components of the immune system in in vitro and animal models. The clinical significance of these findings is unknown. Overall, the effects of opioids appear to be modestly immunosuppressive.
Concentration-Efficacy Relationships
The minimum effective analgesic concentration will vary widely among patients, especially among patients who have been previously treated with potent agonist opioids. The minimum effective analgesic concentration of tapentadol for any individual patient may increase over time due to an increase in pain, the development of a new pain syndrome and/or the development of analgesic tolerance [see Dosage and Administration (2.1)].
Concentration-Adverse Experience Relationships
There is a relationship between increasing tapentadol plasma concentration and increasing frequency of dose-related adverse reactions such as nausea, vomiting, CNS effects, and respiratory depression. In opioid-tolerant patients, the situation may be altered by the development of tolerance to opioid-related adverse reactions [see Dosage and Administration (2.1, 2.2)].
12.3 Pharmacokinetics
Absorption
The mean absolute bioavailability after single-dose administration (fasting) of NUCYNTA is approximately 32% due to extensive first-pass metabolism. Maximum serum concentrations of tapentadol are typically observed at around 1.25 hours after dosing.
Dose-proportional increases in the Cmax and AUC values of tapentadol have been observed over the 50 to 150 mg dose range.
A multiple (every 6 hour) dose study with doses ranging from 75 to 175 mg tapentadol showed a mean accumulation factor of 1.6 for the parent drug and 1.8 for the major metabolite tapentadol-O-glucuronide, which are primarily determined by the dosing interval and apparent half-life of tapentadol and its metabolite.
Food Effect
The AUC and Cmax increased by 25% and 16%, respectively, when NUCYNTA was administered after a high-fat, high-calorie breakfast. NUCYNTA may be given with or without food.
Distribution
Tapentadol is widely distributed throughout the body. Following intravenous administration, the volume of distribution (Vz) for tapentadol is 540 +/- 98 L. The plasma protein binding is low and amounts to approximately 20%.
Elimination
Metabolism
In humans, about 97% of the parent compound is metabolized. Tapentadol is mainly metabolized via Phase 2 pathways, and only a small amount is metabolized by Phase 1 oxidative pathways. The major pathway of tapentadol metabolism is conjugation with glucuronic acid to produce glucuronides. After oral administration approximately 70% (55% O-glucuronide and 15% sulfate of tapentadol) of the dose is excreted in urine in the conjugated form. A total of 3% of drug was excreted in urine as unchanged drug. Tapentadol is additionally metabolized to N-desmethyl tapentadol (13%) by CYP2C9 and CYP2C19 and to hydroxy tapentadol (2%) by CYP2D6, which are further metabolized by conjugation. Therefore, drug metabolism mediated by cytochrome P450 system is of less importance than phase 2 conjugation.
None of the metabolites contribute to the analgesic activity.
Excretion
Tapentadol and its metabolites are excreted almost exclusively (99%) via the kidneys. The terminal half-life is on average 4 hours after oral administration. The total clearance is 1530 +/- 177 mL/min.
Specific Populations
Age: Geriatric Population
The mean exposure (AUC) to tapentadol was similar in elderly subjects compared to young adults, with a 16% lower mean Cmax observed in the elderly subject group compared to young adult subjects.
Hepatic Impairment
Administration of NUCYNTA resulted in higher exposures and serum levels to tapentadol in subjects with impaired hepatic function compared to subjects with normal hepatic function. The ratio of tapentadol pharmacokinetic parameters for the mild hepatic impairment group (Child- Pugh Score 5 to 6) and moderate hepatic impairment group (Child-Pugh Score 7 to 9) in comparison to the normal hepatic function group were 1.7 and 4.2, respectively, for AUC; 1.4 and 2.5, respectively, for Cmax; and 1.2 and 1.4, respectively, for t1/2. The rate of formation of tapentadol-O-glucuronide was lower in subjects with increased liver impairment.
Renal Impairment
AUC and Cmax of tapentadol were comparable in subjects with varying degrees of renal function (from normal to severely impaired). In contrast, increasing exposure (AUC) to tapentadol-O-glucuronide was observed with increasing degree of renal impairment. In subjects with mild (CLCR = 50 to <80 mL/min), moderate (CLCR = 30 to <50 mL/min), and severe (CLCR = <30 mL/min) renal impairment, the AUC of tapentadol-O-glucuronide was 1.5-, 2.5-, and 5.5- fold higher compared with normal renal function, respectively.
Drug Interaction Studies
Pharmacokinetic Drug Interactions
Tapentadol is mainly metabolized by Phase 2 glucuronidation, a high capacity/low affinity system; therefore, clinically relevant interactions caused by Phase 2 metabolism are unlikely to occur. Naproxen and probenecid increased the AUC of tapentadol by 17% and 57%, respectively. These changes are not considered clinically relevant and no change in dose is required.
No changes in the pharmacokinetic parameters of tapentadol were observed when acetaminophen and acetylsalicylic acid were given concomitantly.
In vitro studies did not reveal any potential of tapentadol to either inhibit or induce cytochrome P450 enzymes. Furthermore, a minor amount of NUCYNTA is metabolized via the oxidative pathway. Thus, clinically relevant interactions mediated by the cytochrome P450 system are unlikely to occur.
The pharmacokinetics of tapentadol were not affected when gastric pH or gastrointestinal motility were increased by omeprazole and metoclopramide, respectively.
Plasma protein binding of tapentadol is low (approximately 20%). Therefore, the likelihood of pharmacokinetic drug-drug interactions by displacement from the protein binding site is low.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Tapentadol was administered to rats (diet) and mice (oral gavage) for two years.
In mice, tapentadol HCl was administered by oral gavage at dosages of 50, 100 and 200 mg/kg/day for 2 years (up to 0.2 times the plasma exposure at the maximum recommended human dose [MRHD] on an area under the time-curve [AUC] basis). No increase in tumor incidence was observed at any dose level.
In rats, tapentadol HCl was administered in diet at dosages of 10, 50, 125 and 250 mg/kg/day for two years (up to 0.2 times in the male rats and 0.6 times in the female rats the MRHD on an AUC basis). No increase in tumor incidence was observed at any dose level.
Mutagenesis
Tapentadol did not induce gene mutations in bacteria, but was clastogenic with metabolic activation in a chromosomal aberration test in V79 cells. The test was repeated and was negative in the presence and absence of metabolic activation. The one positive result for tapentadol was not confirmed in vivo in rats, using the two endpoints of chromosomal aberration and unscheduled DNA synthesis, when tested up to the maximum tolerated dose.
Impairment of Fertility
Tapentadol HCl was administered intravenously to male or female rats at dosages of 3, 6, or 12 mg/kg/day (representing exposures of up to approximately 0.4 times the exposure at the MRHD on an AUC basis, based on extrapolation from toxicokinetic analyses in a separate 4-week intravenous study in rats). Tapentadol did not alter fertility at any dose level. Maternal toxicity and adverse effects on embryonic development, including decreased number of implantations, decreased numbers of live conceptuses, and increased pre- and post-implantation losses occurred at dosages ≥6 mg/kg/day.
13.2 Animal Toxicology and/or Pharmacology
In toxicological studies with tapentadol, the most common systemic effects of tapentadol were related to the mu-opioid receptor agonist and norepinephrine reuptake inhibition pharmacodynamic properties of the compound. Transient, dose-dependent and predominantly CNS-related findings were observed, including impaired respiratory function and convulsions, the latter occurring in the dog at plasma levels (Cmax) which are in the range associated with the maximum recommended human dose (MRHD).
14. Clinical Studies
The efficacy and safety of NUCYNTA in the treatment of acute pain has been established in two randomized, double-blind, placebo- and active-controlled studies of moderate to severe pain from first metatarsal bunionectomy and end-stage degenerative joint disease.
14.1 Orthopedic Surgery – Bunionectomy
A randomized, double-blind, parallel-group, active- and placebo-controlled, multiple-dose study demonstrated the efficacy of 50 mg, 75 mg, and 100 mg NUCYNTA given every 4 to 6 hours for 72 hours in patients aged 18 to 80 years experiencing moderate to severe pain following unilateral, first metatarsal bunionectomy surgery. Patients who qualified for the study with a baseline pain score of ≥4 on an 11-point rating scale ranging from 0 to 10 were randomized to 1 of 5 treatments. Patients were allowed to take a second dose of study medication as soon as 1 hour after the first dose on study Day 1, with subsequent dosing every 4 to 6 hours. If rescue analgesics were required, the patients were discontinued for lack of efficacy. Efficacy was evaluated by comparing the sum of pain intensity difference over the first 48 hours (SPID48) versus placebo. NUCYNTA at each dose provided a greater reduction in pain compared to placebo based on SPID48 values.
For various degrees of improvement from baseline to the 48-hour endpoint, Figure 1 shows the fraction of patients achieving that level of improvement. The figures are cumulative, such that every patient that achieves a 50% reduction in pain from baseline is included in every level of improvement below 50%. Patients who did not complete the 48-hour observation period in the study were assigned 0% improvement.
Figure 1: Percentage of Patients Achieving Various Levels of Pain Relief as Measured by Pain Severity at 48 Hours Compared to Baseline- Post Operative Bunionectomy
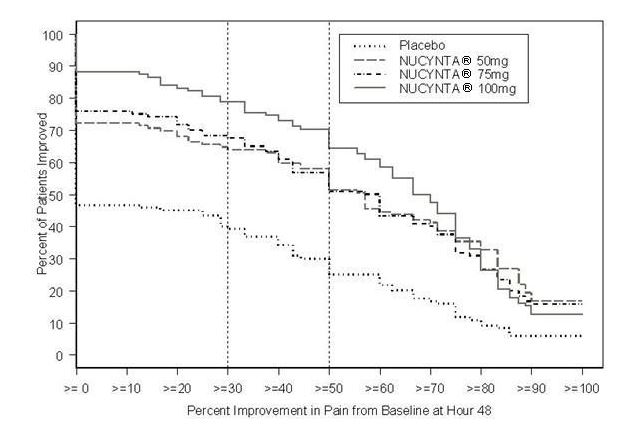
The proportions of patients who showed reduction in pain intensity at 48 hours of 30% or greater, or 50% or greater were significantly higher in patients treated with NUCYNTA at each dose versus placebo.
14.2 End-Stage Degenerative Joint Disease
A randomized, double-blind, parallel-group, active- and placebo-controlled, multiple-dose study evaluated the efficacy and safety of 50 mg and 75 mg NUCYNTA given every 4 to 6 hours during waking hours for 10 days in patients aged 18 to 80 years, experiencing moderate to severe pain from end stage degenerative joint disease of the hip or knee, defined as a 3-day mean pain score of ≥5 on an 11-point pain intensity scale, ranging from 0 to 10. Pain scores were assessed twice daily and assessed the pain the patient had experienced over the previous 12 hours. Patients were allowed to continue non-opioid analgesic therapy for which they had been on a stable regimen before screening throughout the study. Eighty-three percent (83%) of patients in the tapentadol treatment groups and the placebo group took such analgesia during the study. The 75 mg treatment group was dosed at 50 mg for the first day of the study, followed by 75 mg for the remaining nine days. Patients requiring rescue analgesics other than study medication were discontinued for lack of efficacy. Efficacy was evaluated by comparing the sum of pain intensity difference (SPID) versus placebo over the first five days of treatment. NUCYNTA 50 mg and 75 mg provided improvement in pain compared with placebo based on the 5-Day SPID.
For various degrees of improvement from baseline to the Day 5 endpoint, Figure 2 shows the fraction of patients achieving that level of improvement. The figures are cumulative, such that every patient that achieves a 50% reduction in pain from baseline is included in every level of improvement below 50%. Patients who did not complete the 5-day observation period in the study were assigned 0% improvement.
Figure 2: Percentage of Patients Achieving Various Levels of Pain Relief as Measured by Average Pain Severity for the Previous 12 hours, Measured on Study Day 5 Compared to Baseline -- End Stage Degenerative Joint Disease
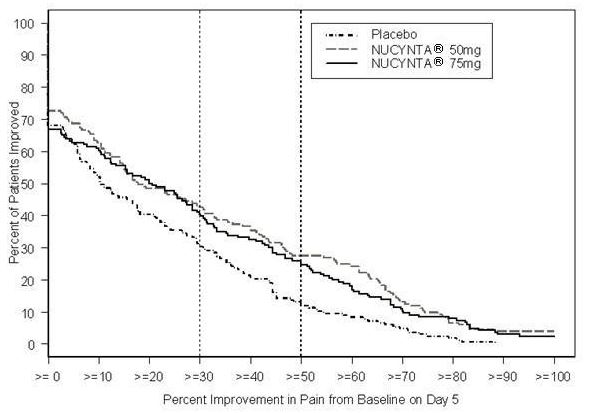
The proportions of patients who showed reduction in pain intensity at 5 days of 30% or greater, or 50% or greater were significantly higher in patients treated with NUCYNTA® at each dose versus placebo.
16. How is Nucynta Solution supplied
NUCYNTA oral solution, 20 mg/mL, is available as a clear, colorless solution. Supplied with calibrated syringe:
Bottles of 100 mL (NDC 50458-817-01)
Bottles of 200 mL (NDC 50458-817-02)
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Medication Errors
Instruct patients how to measure and take the correct dose of NUCYNTA oral solution, and to always use the enclosed syringe when administering NUCNYNTA oral solution to ensure the dose is measured and administered accurately [see Warnings and Precautions (5.1)].
Advise patients that NUCYNTA oral solution is available in 20 mg/mL. Provide detailed instruction on how to measure and take the correct dose of NUCYNTA oral solution, and to always use the enclosed measuring device when administering NUCYNTA oral solution, to ensure that the dose is measured and administered accurately.
If the prescribed concentration is changed, instruct patients on how to correctly measure the new dose to avoid errors which could result in accidental overdose and death.
Addiction, Abuse, and Misuse
Inform patients that the use of NUCYNTA oral solution, even when taken as recommended, can result in addiction, abuse, and misuse, which can lead to overdose and death [see Warnings and Precautions (5.2)]. Instruct patients not to share NUCYNTA oral solution with others and to take steps to protect NUCYNTA oral solution from theft or misuse.
Life-Threatening Respiratory Depression
Inform patients of the risk of life-threatening respiratory depression, including information that the risk is greatest when starting NUCYNTA oral solution or when the dosage is increased, and that it can occur even at recommended dosages [see Warnings and Precautions (5.3)] Advise patients how to recognize respiratory depression and to seek medical attention if they are experiencing breathing difficulties.
Accidental Exposure
Inform patients that accidental ingestion, especially by children, may result in respiratory depression or death [see Warnings and Precautions (5.3)]. Instruct patients to take steps to store NUCYNTA oral solution securely and to dispose of unused NUCYNTA oral solution by flushing the oral solution down the toilet.
Interactions with Benzodiazepines and Other CNS Depressants
Inform patients that potentially fatal serious additive effects may occur if NUCYNTA oral solution is used with benzodiazepines or other CNS depressants, including alcohol, and not to use these concomitantly drugs unless supervised by a health care provider [see Warnings and Precautions (5.5), Drug Interactions (7)].
Serotonin Syndrome
Inform patients that opioids could cause a rare but potentially life-threatening condition resulting from concomitant administration of serotonergic drugs. Warn patients of the symptoms of serotonin syndrome and to seek medical attention right away if symptoms develop. Instruct patients to inform their healthcare provider if they are taking, or plan to take serotonergic medications [see Drug Interactions (7)].
MAOI Interaction
Inform patients not to take NUCYNTA oral solution while using any drugs that inhibit monoamine oxidase. Patients should not start MAOIs while taking NUCYNTA oral solution [see Warnings and Precautions (5.7)]
Adrenal Insufficiency
Inform patients that opioids could cause adrenal insufficiency, a potentially life-threatening condition. Adrenal insufficiency may present with non-specific symptoms and signs such as nausea, vomiting, anorexia, fatigue, weakness, dizziness, and low blood pressure. Advise patients to seek medical attention if they experience a constellation of these symptoms [see Warnings and Precautions (5.8)].
Important Administration Instructions
Instruct patients how to properly take NUCYNTA oral solution, including the following:
- Advise patients to always use the enclosed calibrated oral syringe when administering NUCYNTA oral solution to ensure the dose is measured and administered accurately.
- Advise patients never to use household teaspoons or tablespoons to measure NUCYNTA oral solution.
- Advise patients not to adjust the dose of NUCYNTA oral solution without consulting with a physician or other healthcare professional.
- If patients have been receiving treatment with NUCYNTA oral solution for more than a few weeks and cessation of therapy is indicated, counsel them on the importance of safely tapering the dose as abrupt discontinuation of the medication could precipitate withdrawal symptoms. Provide a dose schedule to accomplish a gradual discontinuation of the medication [see Dosage and Administration (2.4)].
Hypotension
Inform patients that NUCYNTA oral solution may cause orthostatic hypotension and syncope. Instruct patients how to recognize symptoms of low blood pressure and how to reduce the risk of serious consequences should hypotension occur (e.g., sit or lie down, carefully rise from a sitting or lying position). [see Warnings and Precautions (5.9)]
Anaphylaxis
Inform patients that anaphylaxis has been reported with ingredients contained in NUCYNTA oral solution. Advise patients to recognize such a reaction and when to seek medical attention [see Contraindications (4), Adverse Reactions (6)].
Pregnancy
Neonatal Opioid Withdrawal Syndrome
Inform female patients of reproductive potential that prolonged use of NUCYNTA oral solution during pregnancy can result in neonatal opioid withdrawal syndrome, which may be life-threatening if not recognized and treated [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
Embryo-Fetal Toxicity
Inform female patients of reproductive potential that NUCYNTA oral solution can (or may) cause fetal harm and to inform the healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise nursing mothers to monitor infants for increased sleepiness (more than usual), breathing difficulties, or limpness. Instruct nursing mothers to seek immediate medical care if they notice these signs [see Use in Specific Populations (8.2)].
Infertility
Inform patients that chronic use of opioids may cause reduced fertility. It is not known whether these effects on fertility are reversible [see Use in Specific Populations (8.3)]
Driving or Operating Heavy Machinery
Inform patients that NUCYNTA oral solution may impair the ability to perform potentially hazardous activities such as driving a car or operating heavy machinery. Advise patients not to perform such tasks until they know how they will react to the medication [see Warnings and Precautions (5.14)].
Constipation
Advise patients of the potential for severe constipation, including management instructions and when to seek medical attention [see Adverse Reactions (6)].
Disposal of Unused NUCYNTA Oral Solution
Advise patients to dispose of unused NUCYNTA oral solution by flushing down the toilet.
Manufactured for:
Depomed, Inc. (of which Depo NF Sub, LLC is a subsidiary)
Newark, CA 94560
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Issued: 12/2016 | |
|
Medication Guide
NUCYNTA (new-SINN-tah) |
||
|
NUCYNTA oral solution is:
|
||
|
Important information about NUCYNTA oral solution:
|
||
|
Do not take NUCYNTA oral solution if you have:
|
||
|
Before taking NUCYNTA oral solution, tell your healthcare provider if you have a history of: |
||
|
|
|
|
Tell your healthcare provider if you are:
|
||
|
When taking NUCYNTA oral solution:
|
||
|
While taking NUCYNTA oral solution DO NOT:
|
||
|
The possible side effects of NUCYNTA oral solution:
Get emergency medical help if you have:
These are not all the possible side effects of NUCYNTA oral solution. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. For more information go to dailymed.nlm.nih.gov Marketed by: Depomed, Inc (of which Depo NF Sub, LLC is a subsidiary) Newark, CA 94560, www.NUCYNTA.com or call 1-800-526-7736 |
||
Instructions for Use NUCYNTA® (tapentadol), CII oral solution
20 mg/mL
Read the Instructions for Use before you start taking NUCYNTA® oral solution and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment.
Important information about measuring ® oral solution
- Always use the oral syringe provided with your NUCYNTA® oral solution to make sure you measure the right amount.
- You will be provided (See Figure A.):
- 1 bottle of NUCYNTA® oral solution
- 1 oral syringe
- 1 adapter
If you do not receive an oral syringe with your NUCYNTA® oral solution, ask your pharmacist to give you one.
Before you use NUCYNTA® oral solution for the first time:
| 1. | Remove the child-resistant cap and completely remove the foil seal (See Figure B). | 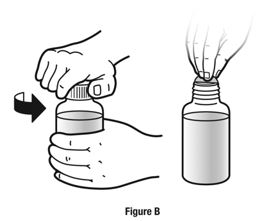 |
| 2. | Push the ribbed end of the adapter into the neck of the bottle until it is firmly in place. The bottom edge of the adapter should fully contact the top rim of the bottle (See Figure C). Do not remove the adapter from the bottle after it is inserted. | 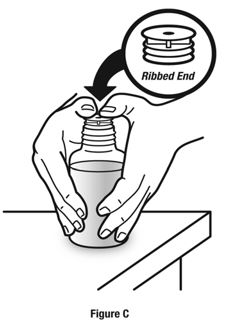 |
| To prepare a dose of NUCYNTA® oral solution: | ||
| 3. | Hold the oral syringe in one hand. With your other hand, fully push down (depress) the plunger (See Figure D). | 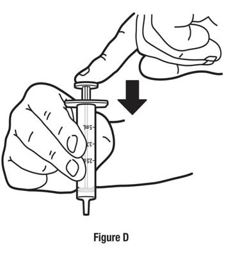 |
| 4. | Insert the tip of the oral syringe into the adapter (See Figure E). | 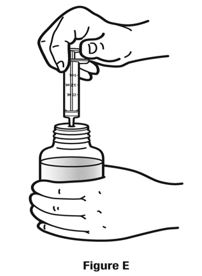 |
| 5. | Turn the bottle upside down. Pull back slowly on the oral syringe plunger to withdraw the dose prescribed by your healthcare provider (2.5 mL, 3.75 mL, or 5 mL). If you see air bubbles in the oral syringe, fully push in the plunger so that the oral solution flows back into the bottle. Then, withdraw your prescribed dose of oral solution (See Figure F). | 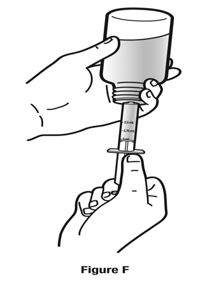 |
| 6. | Leave the oral syringe in the bottle adapter and turn the bottle right-side up. Place the bottle onto a flat surface. Remove the oral syringe from the bottle (See Figure G). | 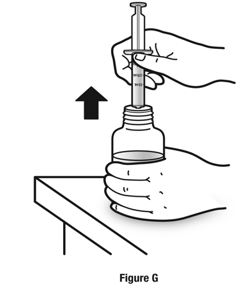 |
| 7. | Place the tip of the oral syringe in your mouth. Squirt the oral solution into your mouth by pushing on the plunger until the oral syringe is empty (See Figure H). | 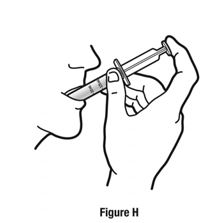 |
| 8. | Leave the adapter in the bottle. Put the child- resistant cap back on the bottle (See Figure I). | 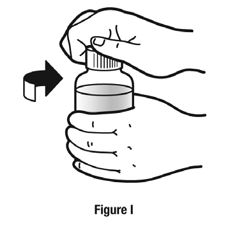 |
| 9. | Remove the plunger from the oral syringe barrel. Rinse the oral syringe with water after each use and let it air dry. When the oral syringe is dry, put the plunger back into the oral syringe barrel for the next use. Do not throw away the oral syringe. | |
|
||
What are the ingredients in NUCYNTA® oral solution?
Active ingredient: tapentadol
Inactive ingredients: citric acid monohydrate, sucralose, raspberry flavor, sodium hydroxide, and purified water.
How should I store NUCYNTA® oral solution?
- Store NUCYNTA® oral solution at room temperature between 68°F to 77°F (20°C to 25°C).
- Store the NUCYNTA® oral solution bottle upright after opening. Keep the oral syringe with your medicine.
- After you stop taking NUCYNTA® oral solution, flush the unused oral solution down the toilet.
Keep NUCYNTA® oral solution out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Depomed, Inc. (of which Depo NF Sub, LLC is a subsidiary)
Newark, CA 94560
Revised: December 2016

| NUCYNTA
tapentadol hydrochloride solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Janssen Pharmaceuticals, Inc. (063137772) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Patheon Pharmaceuticals, Inc. | 005286822 | MANUFACTURE(50458-817) , ANALYSIS(50458-817) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen Pharmaceuticals, Inc. | 080236951 | API MANUFACTURE(50458-817) | |
More about Nucynta (tapentadol)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (280)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Support group
- FDA approval history
- Drug class: Opioids (narcotic analgesics)
- Breastfeeding
- En español
