Dofetilide: Package Insert / Prescribing Info
Package insert / product label
Dosage form: capsule
Drug class: Group III antiarrhythmics
Medically reviewed by Drugs.com. Last updated on Sep 16, 2025.
On This Page
- Description
- Clinical Pharmacology
- Drug Interactions
- Clinical Studies
- Indications and Usage
- Contraindications
- Warnings
- Precautions
- Patient Counseling Information
- Adverse Reactions/Side Effects
- Overdosage
- Dosage and Administration
- How Supplied/Storage and Handling
- Storage and Handling
- Medication Guide
|
To minimize the risk of induced arrhythmia, patients initiated or re-initiated on dofetilide capsules should be placed for a minimum of 3 days in a facility that can provide calculations of creatinine clearance, continuous electrocardiographic monitoring, and cardiac resuscitation. For detailed instructions regarding dose selection, see DOSAGE AND ADMINISTRATION. |
Dofetilide Description
Dofetilide capsules are an antiarrhythmic drug with Class III (cardiac action potential duration prolonging) properties. Its empirical formula is C19H27N3O5S2 and it has a molecular weight of 441.6. The structural formula is
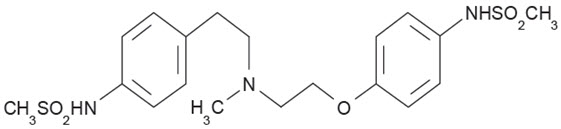
The chemical name for dofetilide is:
N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamide.
Dofetilide is a white to off-white powder. It is very slightly soluble in water and propan-2-ol and is soluble in 0.1M aqueous sodium hydroxide, acetone, and aqueous 0.1M hydrochloric acid.
Dofetilide capsules contain the following inactive ingredients: microcrystalline cellulose, corn starch, colloidal silicon dioxide and magnesium stearate. The capsule shells contain gelatin, titanium dioxide, red iron oxide, yellow iron oxide, and black ink. The imprint ink contains iron oxide black, shellac, ethanol, n-butyl alcohol, isopropyl alcohol, propylene glycol, and ammonium hydroxide. Dofetilide capsules are supplied for oral administration in three dosage strengths: 125 mcg (0.125 mg) dark caramel and white capsules, 250 mcg (0.25 mg) light orange capsules, and 500 mcg (0.5 mg) light orange and white capsules.
Dofetilide - Clinical Pharmacology
Mechanism of Action
Dofetilide shows Vaughan Williams Class III antiarrhythmic activity. The mechanism of action is blockade of the cardiac ion channel carrying the rapid component of the delayed rectifier potassium current, IKr. At concentrations covering several orders of magnitude, dofetilide blocks only IKr with no relevant block of the other repolarizing potassium currents (e.g., IKs, IK1). At clinically relevant concentrations, dofetilide has no effect on sodium channels (associated with Class I effect), adrenergic alpha-receptors, or adrenergic beta-receptors.
Electrophysiology
Dofetilide increases the monophasic action potential duration in a predictable, concentration-dependent manner, primarily due to delayed repolarization. This effect, and the related increase in effective refractory period, is observed in the atria and ventricles in both resting and paced electrophysiology studies. The increase in QT interval observed on the surface ECG is a result of prolongation of both effective and functional refractory periods in the His-Purkinje system and the ventricles.
Dofetilide did not influence cardiac conduction velocity and sinus node function in a variety of studies in patients with or without structural heart disease. This is consistent with a lack of effect of dofetilide on the PR interval and QRS width in patients with pre-existing heart block and/or sick sinus syndrome.
In patients, dofetilide terminates induced re-entrant tachyarrhythmias (e.g., atrial fibrillation/flutter and ventricular tachycardia) and prevents their re-induction. Dofetilide does not increase the electrical energy required to convert electrically induced ventricular fibrillation, and it significantly reduces the defibrillation threshold in patients with ventricular tachycardia and ventricular fibrillation undergoing implantation of a cardioverter-defibrillator device.
Hemodynamic Effects
In hemodynamic studies, dofetilide had no effect on cardiac output, cardiac index, stroke volume index, or systemic vascular resistance in patients with ventricular tachycardia, mild to moderate congestive heart failure or angina, and either normal or low left ventricular ejection fraction. There was no evidence of a negative inotropic effect related to dofetilide therapy in patients with atrial fibrillation. There was no increase in heart failure in patients with significant left ventricular dysfunction (see CLINICAL STUDIES, Safety in Patients with Structural Heart Disease, DIAMOND Studies). In the overall clinical program, dofetilide did not affect blood pressure. Heart rate was decreased by 4-6 bpm in studies in patients.
Pharmacokinetics, General
Absorption and Distribution
The oral bioavailability of dofetilide is >90%, with maximal plasma concentrations occurring at about 2-3 hours in the fasted state. Oral bioavailability is unaffected by food or antacid. The terminal half-life of dofetilide is approximately 10 hours; steady state plasma concentrations are attained within 2-3 days, with an accumulation index of 1.5 to 2.0. Plasma concentrations are dose proportional. Plasma protein binding of dofetilide is 60-70%, is independent of plasma concentration, and is unaffected by renal impairment. Volume of distribution is 3 L/kg.
Metabolism and Excretion
Approximately 80% of a single dose of dofetilide is excreted in urine, of which approximately 80% is excreted as unchanged dofetilide with the remaining 20% consisting of inactive or minimally active metabolites. Renal elimination involves both glomerular filtration and active tubular secretion (via the cation transport system, a process that can be inhibited by cimetidine, trimethoprim, prochlorperazine, megestrol, ketoconazole and dolutegravir). In vitro studies with human liver microsomes show that dofetilide can be metabolized by CYP3A4, but it has a low affinity for this isoenzyme. Metabolites are formed by N-dealkylation and N-oxidation. There are no quantifiable metabolites circulating in plasma, but 5 metabolites have been identified in urine.
Pharmacokinetics in Special Populations
Renal Impairment
In volunteers with varying degrees of renal impairment and patients with arrhythmias, the clearance of dofetilide decreases with decreasing creatinine clearance. As a result, and as seen in clinical studies, the half-life of dofetilide is longer in patients with lower creatinine clearances. Because increase in QT interval and the risk of ventricular arrhythmias are directly related to plasma concentrations of dofetilide, dosage adjustment based on calculated creatinine clearance is critically important (see DOSAGE AND ADMINISTRATION). Patients with severe renal impairment (creatinine clearance <20 mL/min) were not included in clinical or pharmacokinetic studies (see CONTRAINDICATIONS).
Hepatic Impairment
There was no clinically significant alteration in the pharmacokinetics of dofetilide in volunteers with mild to moderate hepatic impairment (Child-Pugh Class A and B) compared to age- and weight-matched healthy volunteers. Patients with severe hepatic impairment were not studied.
Patients with Heart Disease
Population pharmacokinetic analyses indicate that the plasma concentration of dofetilide in patients with supraventricular and ventricular arrhythmias, ischemic heart disease, or congestive heart failure are similar to those of healthy volunteers, after adjusting for renal function.
Women
A population pharmacokinetic analysis showed that women have approximately 12-18% lower dofetilide oral clearances than men (14-22% greater plasma dofetilide levels), after correction for weight and creatinine clearance. In females, as in males, renal function was the single most important factor influencing dofetilide clearance. In normal female volunteers, hormone replacement therapy (a combination of conjugated estrogens and medroxyprogesterone) did not increase dofetilide exposure.
Dose-Response and Concentration Response for Increase in QT Interval
Increase in QT interval is directly related to dofetilide dose and plasma concentration. Figure 1 shows that the relationship in normal volunteers between dofetilide plasma concentrations and change in QTc is linear, with a positive slope of approximately 15-25 msec/(ng/mL) after the first dose and approximately 10-15 msec/(ng/mL) at Day 23 (reflecting a steady state of dosing). A linear relationship between mean QTc increase and dofetilide dose was also seen in patients with renal impairment, in patients with ischemic heart disease, and in patients with supraventricular and ventricular arrhythmias.
Figure 1: Mean QTc-Concentration Relationship in Young Volunteers Over 24 Days
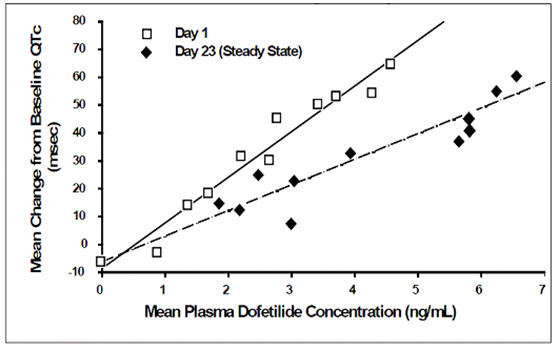
Note: The range of dofetilide plasma concentrations achieved with the 500 mcg BID dose adjusted for creatinine clearance is 1-3.5 ng/mL.
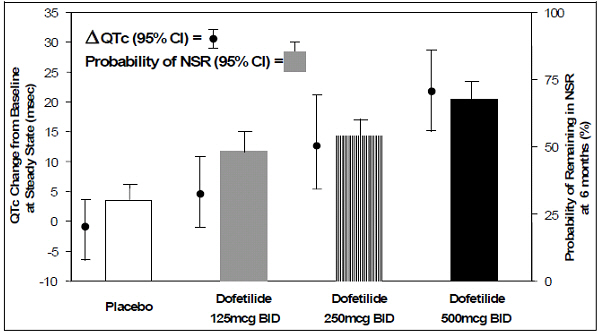
The relationship between dose, efficacy, and the increase in QTc from baseline at steady state for the two randomized, placebo-controlled studies (described further below) is shown in Figure 2. The studies examined the effectiveness of dofetilide capsules in conversion to sinus rhythm and maintenance of normal sinus rhythm after conversion in patients with atrial fibrillation/flutter of >1 week duration. As shown, both the probability of a patient's remaining in sinus rhythm at six months and the change in QTc from baseline at steady state of dosing increased in an approximately linear fashion with increasing dose of dofetilide capsules. Note that in these studies, doses were modified by results of creatinine clearance measurement and in-hospital QTc prolongation.
Clinical Studies
Chronic Atrial Fibrillation and/or Atrial Flutter
Two randomized, parallel, double-blind, placebo-controlled, dose-response trials evaluated the ability of dofetilide capsules 1) to convert patients with atrial fibrillation or atrial flutter (AF/AFl) of more than 1 week duration to normal sinus rhythm (NSR) and 2) to maintain NSR (delay time to recurrence of AF/AFl) after drug-induced or electrical cardioversion. A total of 996 patients with a one week to two year history of atrial fibrillation/atrial flutter were enrolled. Both studies randomized patients to placebo or to doses of dofetilide capsules 125 mcg, 250 mcg, 500 mcg, or in one study a comparator drug, given twice a day (these doses were lowered based on calculated creatinine clearance and, in one of the studies, for QT interval or QTc). All patients were started on therapy in a hospital where their ECG was monitored (see DOSAGE AND ADMINISTRATION).
Patients were excluded from participation if they had had syncope within the past 6 months, AV block greater than first degree, MI or unstable angina within 1 month, cardiac surgery within 2 months, history of QT interval prolongation or polymorphic ventricular tachycardia associated with use of antiarrhythmic drugs, QT interval or QTc >440 msec, serum creatinine >2.5 mg/mL, significant diseases of other organ systems; used cimetidine; or used drugs known to prolong the QT interval.
Both studies enrolled mostly Caucasians (over 90%), males (over 70%), and patients ≥65 years of age (over 50%). Most (>90%) were NYHA Functional Class I or II. Approximately one-half had structural heart disease (including ischemic heart disease, cardiomyopathies, and valvular disease) and about one-half were hypertensive. A substantial proportion of patients were on concomitant therapy, including digoxin (over 60%), diuretics (over 20%), and ACE inhibitors (over 30%). About 90% were on anticoagulants.
Acute conversion rates are shown in Table 1 for randomized doses (doses were adjusted for calculated creatinine clearance and, in Study 1, for QT interval or QTc). Of patients who converted pharmacologically, approximately 70% converted within 24-36 hours.
| Dofetilide Capsules Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg
BID | 250 mcg
BID | 500 mcg
BID |
||
|
Study 1 |
5/82 (6%) |
8/82 (10%) |
23/77 (30%) |
1/84 (1%) |
|
Study 2 |
8/135 (6%) |
14/133 (11%) |
38/129 (29%) |
2/137 (1%) |
Patients who did not convert to NSR with randomized therapy within 48-72 hours had electrical cardioversion. Those patients remaining in NSR after conversion in hospital were continued on randomized therapy as outpatients (maintenance period) for up to one year unless they experienced a recurrence of atrial fibrillation/atrial flutter or withdrew for other reasons.
Table 2 shows, by randomized dose, the percentage of patients at 6 and 12 months in both studies who remained on treatment in NSR and the percentage of patients who withdrew because of recurrence of AF/AFl or adverse events.
| Dofetilide Capsules Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
| Note that columns do not add up to 100% due to discontinuations for "other" reasons. | ||||
|
Study 1 | ||||
|
Randomized |
82 |
82 |
77 |
84 |
|
Achieved NSR |
60 |
61 |
61 |
68 |
|
6 months | ||||
|
Still on treatment in NSR |
38% |
44% |
52% |
32% |
|
D/C for recurrence |
55% |
49% |
33% |
63% |
|
D/C for AEs |
3% |
3% |
8% |
4% |
|
12 months | ||||
|
Still on treatment in NSR |
32% |
26% |
46% |
22% |
|
D/C for recurrence |
58% |
57% |
36% |
72% |
|
D/C for AEs |
7% |
11% |
8% |
6% |
|
Study 2 | ||||
|
Randomized |
135 |
133 |
129 |
137 |
|
Achieved NSR |
103 |
118 |
100 |
106 |
|
6 months | ||||
|
Still on treatment in NSR |
41% |
49% |
57% |
22% |
|
D/C for recurrence |
48% |
42% |
27% |
72% |
|
D/C for AEs |
9% |
6% |
10% |
4% |
|
12 months | ||||
|
Still on treatment in NSR |
25% |
42% |
49% |
16% |
|
D/C for recurrence |
59% |
47% |
32% |
76% |
|
D/C for AEs |
11% |
6% |
12% |
5% |
Table 3 and Figures 3 and 4 show, by randomized dose, the effectiveness of dofetilide capsules in maintaining NSR using Kaplan Meier analysis, which shows patients remaining on treatment.
| Dofetilide Capsules Dose | Placebo | |||
|---|---|---|---|---|
| 125 mcg BID | 250 mcg BID | 500 mcg BID | ||
|
Study 1 | ||||
|
p-value vs. placebo |
P=0.21 |
P=0.10 |
P<0.001 | |
|
Median time to recurrence (days) |
31 |
179 |
>365 |
27 |
|
Study 2 | ||||
|
p-value vs. placebo |
P=0.006 |
P<0.001 |
P<0.001 | |
|
Median time to recurrence (days) |
182 |
>365 |
>365 |
34 |
Median time to recurrence of AF/AFl could not be estimated accurately for the 250 mcg BID treatment group in Study 2 and the 500 mcg BID treatment groups in Studies 1 and 2 because dofetilide capsules maintained >50% of patients (51%, 58%, and 66%, respectively) in NSR for the 12 months duration of the studies.
Figure 3: Maintenance of Normal Sinus Rhythm, Dofetilide Capsules Regimen vs. Placebo (Study 1)
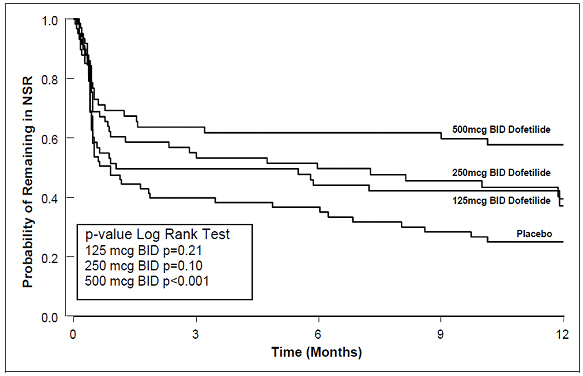
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 62% and 58%, respectively, for dofetilide capsules 500 mcg BID; 50% and 37%, respectively, for dofetilide capsules 250 mcg BID; and 37% and 25%, respectively, for placebo.
Figure 4: Maintenance of Normal Sinus Rhythm, Dofetilide Capsules Regimen vs. Placebo (Study 2)
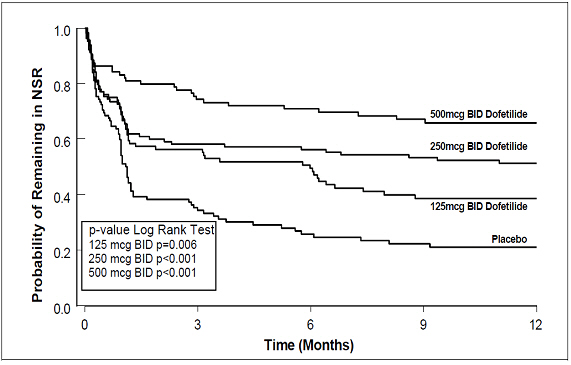
The point estimates of the probabilities of remaining in NSR at 6 and 12 months were 71% and 66%, respectively, for dofetilide capsules 500 mcg BID; 56% and 51%, respectively, for dofetilide capsules 250 mcg BID; and 26% and 21%, respectively, for placebo.
In both studies, dofetilide capsules resulted in a dose-related increase in the number of patients maintained in NSR at all time periods and delayed the time of recurrence of sustained AF. Data pooled from both studies show that there is a positive relationship between the probability of staying in NSR, dofetilide capsules dose, and increase in QTc (see Figure 2 in CLINICAL PHARMACOLOGY, Dose-Response and Concentration Response for Increase in QT Interval).
Analysis of pooled data for patients randomized to a dofetilide capsules dose of 500 mcg twice daily showed that maintenance of NSR was similar in both males and females, in both patients aged <65 years and patients ≥65 years of age, and in both patients with atrial flutter as a primary diagnosis and those with a primary diagnosis of atrial fibrillation.
During the period of in-hospital initiation of dosing, 23% of patients in Studies 1 and 2 had their dose adjusted downward on the basis of their calculated creatinine clearance, and 3% had their dose down-titrated due to increased QT interval or QTc. Increased QT interval or QTc led to discontinuation of therapy in 3% of patients.
Safety in Patients with Structural Heart Disease: DIAMOND Studies (The Danish Investigations of Arrhythmia and Mortality on Dofetilide)
The two DIAMOND studies were 3-year trials comparing the effects of dofetilide capsules and placebo on mortality and morbidity in patients with impaired left ventricular function (ejection fraction ≤35%). Patients were treated for at least one year. One study was in patients with moderate to severe (60% NYHA Class III or IV) congestive heart failure (DIAMOND CHF) and the other was in patients with recent myocardial infarction (DIAMOND MI) (of whom 40% had NYHA Class III or IV heart failure). Both groups were at relatively high risk of sudden death. The DIAMOND trials were intended to determine whether dofetilide capsules could reduce that risk. The trials did not demonstrate a reduction in mortality; however, they provide reassurance that, when initiated carefully, in a hospital or equivalent setting, dofetilide capsules did not increase mortality in patients with structural heart disease, an important finding because other antiarrhythmics [notably the Class IC antiarrhythmics studied in the Cardiac Arrhythmia Suppression Trial (CAST) and a pure Class III antiarrhythmic, d-sotalol (SWORD)] have increased mortality in post-infarction populations. The DIAMOND trials therefore provide evidence of a method of safe use of dofetilide capsules in a population susceptible to ventricular arrhythmias. In addition, the subset of patients with AF in the DIAMOND trials provide further evidence of safety in a population of patients with structural heart disease accompanying the AF. Note, however, that this AF population was given a lower (250 mcg BID) dose (see CLINICAL STUDIES, DIAMOND Patients with Atrial Fibrillation).
In both DIAMOND studies, patients were randomized to 500 mcg BID of dofetilide capsules, but this was reduced to 250 mcg BID if calculated creatinine clearance was 40–60 mL/min, if patients had AF, or if QT interval prolongation (>550 msec or >20% increase from baseline) occurred after dosing. Dose reductions for reduced calculated creatinine clearance occurred in 47% and 45% of DIAMOND CHF and MI patients, respectively. Dose reductions for increased QT interval or QTc occurred in 5% and 7% of DIAMOND CHF and MI patients, respectively. Increased QT interval or QTc (>550 msec or >20% increase from baseline) resulted in discontinuation of 1.8% of patients in DIAMOND CHF and 2.5% of patients in DIAMOND MI.
In the DIAMOND studies, all patients were hospitalized for at least 3 days after treatment was initiated and monitored by telemetry. Patients with QTc greater than 460 msec, second or third degree AV block (unless with pacemaker), resting heart rate <50 bpm, or prior history of polymorphic ventricular tachycardia were excluded.
DIAMOND CHF studied 1518 patients hospitalized with severe CHF who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 311 deaths from all causes in patients randomized to dofetilide capsules (n=762) and 317 deaths in patients randomized to placebo (n=756). The probability of survival at one year was 73% (95% CI: 70% – 76%) in the dofetilide capsules group and 72% (95% CI: 69% – 75%) in the placebo group. Similar results were seen for cardiac deaths and arrhythmic deaths. Torsade de Pointes occurred in 25/762 patients (3.3%) receiving dofetilide capsules. The majority of cases (76%) occurred within the first 3 days of dosing. In all, 437/762 (57%) of patients on dofetilide capsules and 459/756 (61%) on placebo required hospitalization. Of these, 229/762 (30%) of patients on dofetilide capsules and 290/756 (38%) on placebo required hospitalization because of worsening heart failure.
DIAMOND MI studied 1510 patients hospitalized with recent myocardial infarction (2-7 days) who had confirmed impaired left ventricular function (ejection fraction ≤35%). Patients received a median duration of therapy of greater than one year. There were 230 deaths in patients randomized to dofetilide capsules (n=749) and 243 deaths in patients randomized to placebo (n=761). The probability of survival at one year was 79% (95% CI: 76% – 82%) in the dofetilide capsules group and 77% (95% CI: 74% – 80%) in the placebo group. Cardiac and arrhythmic mortality showed a similar result. Torsade de Pointes occurred in 7/749 patients (0.9%) receiving dofetilide capsules. Of these, 4 cases occurred within the first 3 days of dosing and 3 cases occurred between Day 4 and the conclusion of the study. In all, 371/749 (50%) of patients on dofetilide capsules and 419/761 (55%) on placebo required hospitalization. Of these, 200/749 (27%) of patients on dofetilide capsules and 205/761 (27%) on placebo required hospitalization because of worsening heart failure.
DIAMOND Patients with Atrial Fibrillation (the DIAMOND AF subpopulation). There were 506 patients in the two DIAMOND studies who had atrial fibrillation (AF) at entry to the studies (249 randomized to dofetilide capsules and 257 randomized to placebo). DIAMOND AF patients randomized to dofetilide capsules received 250 mcg BID; 65% of these patients had impaired renal function, so that 250 mcg BID represents the dose they would have received in the AF trials, which would give drug exposure similar to a person with normal renal function given 500 mcg BID. In the DIAMOND AF subpopulation, there were 111 deaths (45%) in the 249 patients in the dofetilide capsules group and 116 deaths (45%) in the 257 patients in the placebo group. Hospital readmission rates for any reason were 125/249 or 50% on dofetilide capsules and 156/257 or 61% for placebo. Of these, readmission rates for worsening heart failure were 73/249 or 29% on dofetilide capsules and 102/257 or 40% for placebo.
Of the 506 patients in the DIAMOND studies who had atrial fibrillation or flutter at baseline, 12% of patients in the dofetilide capsules group and 2% of patients in the placebo group had converted to normal sinus rhythm after one month. In those patients converted to normal sinus rhythm, 79% of the dofetilide capsules group and 42% of the placebo group remained in normal sinus rhythm for one year.
In the DIAMOND studies, although Torsade de Pointes occurred more frequently in the dofetilide capsules-treated patients (see ADVERSE REACTIONS), dofetilide capsules, given with an initial 3-day hospitalization and with dose modified for reduced creatinine clearance and increased QT interval, was not associated with an excess risk of mortality in these populations with structural heart disease in the individual studies or in an analysis of the combined studies. The presence of atrial fibrillation did not affect outcome.
Indications and Usage for Dofetilide
Maintenance of Normal Sinus Rhythm (Delay in AF/AFl Recurrence)
Dofetilide capsules are indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm. Because dofetilide capsules can cause life threatening ventricular arrhythmias, it should be reserved for patients in whom atrial fibrillation/atrial flutter is highly symptomatic.
In general, antiarrhythmic therapy for atrial fibrillation/atrial flutter aims to prolong the time in normal sinus rhythm. Recurrence is expected in some patients (see CLINICAL STUDIES).
Contraindications
Dofetilide capsules are contraindicated in patients with congenital or acquired long QT syndromes. Dofetilide capsules should not be used in patients with a baseline QT interval or QTc >440 msec (500 msec in patients with ventricular conduction abnormalities). Dofetilide capsules are also contraindicated in patients with severe renal impairment (calculated creatinine clearance <20 mL/min).
The concomitant use of verapamil or the cation transport system inhibitors cimetidine, trimethoprim (alone or in combination with sulfamethoxazole), or ketoconazole with dofetilide capsules is contraindicated (see WARNINGS and PRECAUTIONS, Drug-Drug Interactions), as each of these drugs cause a substantial increase in dofetilide plasma concentrations. In addition, other known inhibitors of the renal cation transport system such as prochlorperazine, dolutegravir and megestrol should not be used in patients on dofetilide capsules.
The concomitant use of hydrochlorothiazide (alone or in combinations such as with triamterene) with dofetilide capsules is contraindicated (see PRECAUTIONS, Drug-Drug Interactions) because this has been shown to significantly increase dofetilide plasma concentrations and QT interval prolongation.
Dofetilide capsules are also contraindicated in patients with a known hypersensitivity to the drug.
Warnings
Ventricular Arrhythmia
Dofetilide capsules can cause serious ventricular arrhythmias, primarily Torsade de Pointes (TdP) type ventricular tachycardia, a polymorphic ventricular tachycardia associated with QT interval prolongation. QT interval prolongation is directly related to dofetilide plasma concentration. Factors such as reduced creatinine clearance or certain dofetilide drug interactions will increase dofetilide plasma concentration. The risk of TdP can be reduced by controlling the plasma concentration through adjustment of the initial dofetilide dose according to creatinine clearance and by monitoring the ECG for excessive increases in the QT interval.
Treatment with dofetilide must therefore be started only in patients placed for a minimum of three days in a facility that can provide electrocardiographic monitoring and in the presence of personnel trained in the management of serious ventricular arrhythmias. Calculation of the creatinine clearance for all patients must precede administration of the first dose of dofetilide. For detailed instructions regarding dose selection, see DOSAGE AND ADMINISTRATION.
The risk of dofetilide induced ventricular arrhythmia was assessed in three ways in clinical studies: 1) by description of the QT interval and its relation to the dose and plasma concentration of dofetilide; 2) by observing the frequency of TdP in dofetilide capsules-treated patients according to dose; 3) by observing the overall mortality rate in patients with atrial fibrillation and in patients with structural heart disease.
Relation of QT Interval to Dose
The QT interval increases linearly with increasing dofetilide capsules dose (see Figures 1 and 2 in CLINICAL PHARMACOLOGY and Dose-Response and Concentration Response for Increase in QT Interval).
Frequency of Torsade de Pointes
In the supraventricular arrhythmia population (patients with AF and other supraventricular arrhythmias), the overall incidence of Torsade de Pointes was 0.8%. The frequency of TdP by dose is shown in Table 4. There were no cases of TdP on placebo.
| Dofetilide Capsules Dose | |||||
|---|---|---|---|---|---|
| <250 mcg
BID | 250 mcg
BID | >250–500 mcg
BID | >500 mcg
BID | All Doses | |
|
Number of Patients |
217 |
388 |
703 |
38 |
1346 |
|
Torsade de Pointes |
0 |
1 (0.3%) |
6 (0.9%) |
4 (10.5%) |
11 (0.8%) |
As shown in Table 5, the rate of TdP was reduced when patients were dosed according to their renal function (see CLINICAL PHARMACOLOGY, Pharmacokinetics in Special Populations, Renal Impairment and DOSAGE AND ADMINISTRATION).
| Population: | Total | Before | After |
|---|---|---|---|
| n/N % | n/N % | n/N % | |
|
Supraventricular Arrhythmias |
11/1346 (0.8%) |
6/193 (3.1%) |
5/1153 (0.4%) |
|
DIAMOND CHF |
25/762 (3.3%) |
7/148 (4.7%) |
18/614 (2.9%) |
|
DIAMOND MI |
7/749 (0.9%) |
3/101 (3.0%) |
4/648 (0.6%) |
|
DIAMOND AF |
4/249 (1.6%) |
0/43 (0%) |
4/206 (1.9%) |
The majority of the episodes of TdP occurred within the first three days of dofetilide capsules therapy (10/11 events in the studies of patients with supraventricular arrhythmias; 19/25 and 4/7 events in DIAMOND CHF and DIAMOND MI, respectively; 2/4 events in the DIAMOND AF subpopulation).
Mortality
In a pooled survival analysis of patients in the supraventricular arrhythmia population (low prevalence of structural heart disease), deaths occurred in 0.9% (12/1346) of patients receiving dofetilide capsules and 0.4% (3/677) in the placebo group. Adjusted for duration of therapy, primary diagnosis, age, gender, and prevalence of structural heart disease, the point estimate of the hazard ratio for the pooled studies (dofetilide capsules/placebo) was 1.1 (95% CI: 0.3, 4.3). The DIAMOND CHF and MI trials examined mortality in patients with structural heart disease (ejection fraction ≤35%). In these large, double-blind studies, deaths occurred in 36% (541/1511) of dofetilide capsules patients and 37% (560/1517) of placebo patients. In an analysis of 506 DIAMOND patients with atrial fibrillation/flutter at baseline, one year mortality on dofetilide capsules was 31% vs. 32% on placebo (see CLINICAL STUDIES).
Because of the small number of events, an excess mortality due to dofetilide capsules cannot be ruled out with confidence in the pooled survival analysis of placebo-controlled trials in patients with supraventricular arrhythmias. However, it is reassuring that in two large placebo-controlled mortality studies in patients with significant heart disease (DIAMOND CHF/MI), there were no more deaths in dofetilide capsules-treated patients than in patients given placebo (see CLINICAL STUDIES).
Drug-Drug Interactions
(see CONTRAINDICATIONS)
Because there is a linear relationship between dofetilide plasma concentration and QTc, concomitant drugs that interfere with the metabolism or renal elimination of dofetilide may increase the risk of arrhythmia (Torsade de Pointes). Dofetilide is metabolized to a small degree by the CYP3A4 isoenzyme of the cytochrome P450 system and an inhibitor of this system could increase systemic dofetilide exposure. More important, dofetilide is eliminated by cationic renal secretion, and three inhibitors of this process have been shown to increase systemic dofetilide exposure. The magnitude of the effect on renal elimination by cimetidine, trimethoprim, and ketoconazole (all contraindicated concomitant uses with dofetilide) suggests that all renal cation transport inhibitors should be contraindicated.
Hypokalemia and Potassium-Depleting Diuretics
Hypokalemia or hypomagnesemia may occur with administration of potassium-depleting diuretics, increasing the potential for Torsade de Pointes. Potassium levels should be within the normal range prior to administration of dofetilide capsules and maintained in the normal range during administration of dofetilide capsules (see DOSAGE AND ADMINISTRATION).
Use with Drugs that Prolong QT Interval and Antiarrhythmic Agents
The use of dofetilide capsules in conjunction with other drugs that prolong the QT interval has not been studied and is not recommended. Such drugs include phenothiazines, cisapride, bepridil, tricyclic antidepressants, certain oral macrolides, and certain fluoroquinolones. Class I or Class III antiarrhythmic agents should be withheld for at least three half-lives prior to dosing with dofetilide capsules. In clinical trials, dofetilide capsules were administered to patients previously treated with oral amiodarone only if serum amiodarone levels were below 0.3 mg/L or amiodarone had been withdrawn for at least three months.
Precautions
Renal Impairment
The overall systemic clearance of dofetilide is decreased and plasma concentration increased with decreasing creatinine clearance. The dose of dofetilide capsules must be adjusted based on creatinine clearance (see DOSAGE AND ADMINISTRATION). Patients undergoing dialysis were not included in clinical studies, and appropriate dosing recommendations for these patients are unknown. There is no information about the effectiveness of hemodialysis in removing dofetilide from plasma.
Hepatic Impairment
After adjustment for creatinine clearance, no additional dose adjustment is required for patients with mild or moderate hepatic impairment. Patients with severe hepatic impairment have not been studied. Dofetilide capsules should be used with particular caution in these patients.
Cardiac Conduction Disturbances
Animal and human studies have not shown any adverse effects of dofetilide on conduction velocity. No effect on AV nodal conduction following dofetilide capsules treatment was noted in normal volunteers and in patients with 1st degree heart block. Patients with sick sinus syndrome or with 2nd or 3rd degree heart block were not included in the Phase 3 clinical trials unless a functioning pacemaker was present. Dofetilide capsules have been used safely in conjunction with pacemakers (53 patients in DIAMOND studies, 136 in trials in patients with ventricular and supraventricular arrhythmias).
Information for Patients
Please refer patient to the Medication Guide.
Prior to initiation of dofetilide capsules therapy, the patient should be advised to read the Medication Guide and reread it each time therapy is renewed in case the patient's status has changed. The patient should be fully instructed on the need for compliance with the recommended dosing of dofetilide capsules and the potential for drug interactions, and the need for periodic monitoring of QTc and renal function to minimize the risk of serious abnormal rhythms.
Medications and Supplements
Assessment of patients' medication history should include all over-the-counter, prescription, and herbal/natural preparations with emphasis on preparations that may affect the pharmacokinetics of dofetilide capsules such as cimetidine (see CONTRAINDICATIONS), trimethoprim alone or in combination with sulfamethoxazole (see WARNINGS, CONTRAINDICATIONS), prochlorperazine (see WARNINGS, CONTRAINDICATIONS), megestrol (see WARNINGS, CONTRAINDICATIONS), ketoconazole (see WARNINGS, CONTRAINDICATIONS), dolutegravir (see CONTRAINDICATIONS), hydrochlorothiazide (alone or in combinations such as with triamterene) (see CONTRAINDICATIONS), other cardiovascular drugs (especially verapamil – see CONTRAINDICATIONS), phenothiazines, and tricyclic antidepressants (see WARNINGS). If a patient is taking dofetilide capsules and requires anti-ulcer therapy, omeprazole, ranitidine, or antacids (aluminum and magnesium hydroxides) should be used as alternatives to cimetidine, as these agents have no effect on the pharmacokinetics of dofetilide capsules. Patients should be instructed to notify their health care providers of any change in over-the-counter, prescription, or supplement use. If a patient is hospitalized or is prescribed a new medication for any condition, the patient must inform the health care provider of ongoing dofetilide capsules therapy. Patients should also check with their health care provider and/or pharmacist prior to taking a new over-the-counter preparation.
Drug-Drug Interactions
Cimetidine
(see WARNINGS, CONTRAINDICATIONS) Concomitant use of cimetidine is contraindicated. Cimetidine at 400 mg BID (the usual prescription dose) co-administered with dofetilide capsules (500 mcg BID) for 7 days has been shown to increase dofetilide plasma levels by 58%. Cimetidine at doses of 100 mg BID (OTC dose) resulted in a 13% increase in dofetilide plasma levels (500 mcg single dose). No studies have been conducted at intermediate doses of cimetidine. If a patient requires dofetilide capsules and anti-ulcer therapy, it is suggested that omeprazole, ranitidine, or antacids (aluminum and magnesium hydroxides) be used as alternatives to cimetidine, as these agents have no effect on the pharmacokinetic profile of dofetilide capsules.
Verapamil
(see CONTRAINDICATIONS) Concomitant use of verapamil is contraindicated. Co-administration of dofetilide capsules with verapamil resulted in increases in dofetilide peak plasma levels of 42%, although overall exposure to dofetilide was not significantly increased. In an analysis of the supraventricular arrhythmia and DIAMOND patient populations, the concomitant administration of verapamil with dofetilide was associated with a higher occurrence of Torsade de Pointes.
Ketoconazole
(see WARNINGS, CONTRAINDICATIONS) Concomitant use of ketoconazole is contraindicated. Ketoconazole at 400 mg daily (the maximum approved prescription dose) co-administered with dofetilide capsules (500 mcg BID) for 7 days has been shown to increase dofetilide Cmax by 53% in males and 97% in females, and AUC by 41% in males and 69% in females.
Trimethoprim Alone or in Combination with Sulfamethoxazole
(see WARNINGS, CONTRAINDICATIONS) Concomitant use of trimethoprim alone or in combination with sulfamethoxazole is contraindicated. Trimethoprim 160 mg in combination with 800 mg sulfamethoxazole co-administered BID with dofetilide capsules (500 mcg BID) for 4 days has been shown to increase dofetilide AUC by 103% and Cmax by 93%.
Hydrochlorothiazide (HCTZ) Alone or in Combination with Triamterene
(see CONTRAINDICATIONS) Concomitant use of HCTZ alone or in combination with triamterene is contraindicated. HCTZ 50 mg QD or HCTZ/triamterene 50/100 mg QD was co-administered with dofetilide capsules (500 mcg BID) for 5 days (following 2 days of diuretic use at half dose). In patients receiving HCTZ alone, dofetilide AUC increased by 27% and Cmax by 21%. However, the pharmacodynamic effect increased by 197% (QTc increase over time) and by 95% (maximum QTc increase). In patients receiving HCTZ in combination with triamterene, dofetilide AUC increased by 30% and Cmax by 16%. However, the pharmacodynamic effect increased by 190% (QTc increase over time) and by 84% (maximum QTc increase). The pharmacodynamic effects can be explained by a combination of the increase in dofetilide exposure and the reductions in serum potassium. In the DIAMOND trials, 1252 patients were treated with dofetilide capsules and diuretics concomitantly, of whom 493 died compared to 508 deaths among the 1248 patients receiving placebo and diuretics. Of the 229 patients who had potassium depleting diuretics added to their concomitant medications in the DIAMOND trials, the patients on dofetilide capsules had a non-significantly reduced relative risk for death of 0.68 (95% CI: 0.376, 1.230).
Potential Drug Interactions
Dofetilide is eliminated in the kidney by cationic secretion. Inhibitors of renal cationic secretion are contraindicated with dofetilide capsules. In addition, drugs that are actively secreted via this route (e.g., triamterene, metformin, and amiloride) should be co-administered with care as they might increase dofetilide levels.
Dofetilide is metabolized to a small extent by the CYP3A4 isoenzyme of the cytochrome P450 system. Inhibitors of the CYP3A4 isoenzyme could increase systemic dofetilide exposure. Inhibitors of this isoenzyme (e.g., macrolide antibiotics, azole antifungal agents, protease inhibitors, serotonin reuptake inhibitors, amiodarone, cannabinoids, diltiazem, grapefruit juice, nefazadone, norfloxacin, quinine, zafirlukast) should be cautiously co-administered with dofetilide capsules as they can potentially increase dofetilide levels. Dofetilide is not an inhibitor of CYP3A4 nor of other cytochrome P450 isoenzymes (e.g., CYP2C9, CYP2D6) and is not expected to increase levels of drugs metabolized by CYP3A4.
Other Drug Interaction Information
Digoxin
Studies in healthy volunteers have shown that dofetilide capsules do not affect the pharmacokinetics of digoxin. In patients, the concomitant administration of digoxin with dofetilide was associated with a higher occurrence of Torsade de Pointes. It is not clear whether this represents an interaction with dofetilide capsules or the presence of more severe structural heart disease in patients on digoxin; structural heart disease is a known risk factor for arrhythmia. No increase in mortality was observed in patients taking digoxin as concomitant medication.
Other Drugs
In healthy volunteers, amlodipine, phenytoin, glyburide, ranitidine, omeprazole, hormone replacement therapy (a combination of conjugated estrogens and medroxyprogesterone), antacid (aluminum and magnesium hydroxides), and theophylline did not affect the pharmacokinetics of dofetilide capsules. In addition, studies in healthy volunteers have shown that dofetilide capsules do not affect the pharmacokinetics or pharmacodynamics of warfarin, or the pharmacokinetics of propranolol (40 mg twice daily), phenytoin, theophylline, or oral contraceptives.
Population pharmacokinetic analyses were conducted on plasma concentration data from 1445 patients in clinical trials to examine the effects of concomitant medications on clearance or volume of distribution of dofetilide. Concomitant medications were grouped as ACE inhibitors, oral anticoagulants, calcium channel blockers, beta blockers, cardiac glycosides, inducers of CYP3A4, substrates and inhibitors of CYP3A4, substrates and inhibitors of P-glycoprotein, nitrates, sulphonylureas, loop diuretics, potassium sparing diuretics, thiazide diuretics, substrates and inhibitors of tubular organic cation transport, and QTc-prolonging drugs. Differences in clearance between patients on these medications (at any occasion in the study) and those off medications varied between -16% and +3%. The mean clearances of dofetilide were 16% and 15% lower in patients on thiazide diuretics and inhibitors of tubular organic cation transport, respectively.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Dofetilide had no genotoxic effects, with or without metabolic activation, based on the bacterial mutation assay and tests of cytogenetic aberrations in vivo in mouse bone marrow and in vitro in human lymphocytes. Rats and mice treated with dofetilide in the diet for two years showed no evidence of an increased incidence of tumors compared to controls. The highest dofetilide dose administered for 24 months was 10 mg/kg/day to rats and 20 mg/kg/day to mice. Mean dofetilide AUCs(0–24hr) at these doses were about 26 and 10 times, respectively, the maximum likely human AUC.
There was no effect on mating or fertility when dofetilide was administered to male and female rats at doses as high as 1.0 mg/kg/day, a dose that would be expected to provide a mean dofetilide AUC(0–24hr) about 3 times the maximum likely human AUC. Increased incidences of testicular atrophy and epididymal oligospermia and a reduction in testicular weight were, however, observed in other studies in rats. Reduced testicular weight and increased incidence of testicular atrophy were also consistent findings in dogs and mice. The no effect doses for these findings in chronic administration studies in these 3 species (3, 0.1, and 6 mg/kg/day) were associated with mean dofetilide AUCs that were about 4, 1.3, and 3 times the maximum likely human AUC, respectively.
Pregnancy
Dofetilide has been shown to adversely affect in utero growth and survival of rats and mice when orally administered during organogenesis at doses of 2 or more mg/kg/day. Other than an increased incidence of non-ossified 5th metacarpal, and the occurrence of hydroureter and hydronephroses at doses as low as 1 mg/kg/day in the rat, structural anomalies associated with drug treatment were not observed in either species at doses below 2 mg/kg/day. The clearest drug-effect associations were for sternebral and vertebral anomalies in both species; cleft palate, adactyly, levocardia, dilation of cerebral ventricles, hydroureter, hydronephroses, and unossified metacarpal in the rat; and increased incidence of unossified calcaneum in the mouse. The "no observed adverse effect dose" in both species was 0.5 mg/kg/day. The mean dofetilide AUCs(0– 24hr) at this dose in the rat and mouse are estimated to be about equal to the maximum likely human AUC and about half the likely human AUC, respectively. There are no adequate and well controlled studies in pregnant women. Therefore, dofetilide should only be administered to pregnant women where the benefit to the patient justifies the potential risk to the fetus.
Nursing Mothers
There is no information on the presence of dofetilide in breast milk. Patients should be advised not to breast-feed an infant if they are taking dofetilide capsules.
Geriatric Use
Of the total number of patients in clinical studies of dofetilide capsules, 46% were 65 to 89 years old. No overall differences in safety, effect on QTc, or effectiveness were observed between elderly and younger patients. Because elderly patients are more likely to have decreased renal function with a reduced creatinine clearance, care must be taken in dose selection (see DOSAGE AND ADMINISTRATION).
Use in Women
Female patients constituted 32% of the patients in the placebo-controlled trials of dofetilide capsules. As with other drugs that cause Torsade de Pointes, dofetilide capsules were associated with a greater risk of Torsade de Pointes in female patients than in male patients. During the dofetilide capsules clinical development program, the risk of Torsade de Pointes in females was approximately 3 times the risk in males. Unlike Torsade de Pointes, the incidence of other ventricular arrhythmias was similar in female patients receiving dofetilide capsules and patients receiving placebo. Although no study specifically investigated this risk, in post-hoc analyses, no increased mortality was observed in females on dofetilide capsules compared to females on placebo.
Adverse Reactions/Side Effects
The dofetilide capsules clinical program involved approximately 8,600 patients in 130 clinical studies of normal volunteers and patients with supraventricular and ventricular arrhythmias. Dofetilide capsules were administered to 5,194 patients, including two large, placebo-controlled mortality trials (DIAMOND CHF and DIAMOND MI) in which 1,511 patients received dofetilide capsules for up to three years.
In the following section, adverse reaction data for cardiac arrhythmias and non-cardiac adverse reactions are presented separately for patients included in the supraventricular arrhythmia development program and for patients included in the DIAMOND CHF and MI mortality trials (see CLINICAL STUDIES, Safety in Patients with Structural Heart Disease, DIAMOND Studies, for a description of these trials).
In studies of patients with supraventricular arrhythmias, a total of 1,346 and 677 patients were exposed to dofetilide capsules and placebo for 551 and 207 patient years, respectively. A total of 8.7% of patients in the dofetilide groups were discontinued from clinical trials due to adverse events compared to 8.0% in the placebo groups. The most frequent reason for discontinuation (>1%) was ventricular tachycardia (2.0% on dofetilide vs. 1.3% on placebo). The most frequent adverse events were headache, chest pain, and dizziness.
Serious Arrhythmias and Conduction Disturbances
Torsade de Pointes is the only arrhythmia that showed a dose-response relationship to dofetilide capsules treatment. It did not occur in placebo treated patients. The incidence of Torsade de Pointes in patients with supraventricular arrhythmias was 0.8% (11/1346) (see WARNINGS). The incidence of Torsade de Pointes in patients who were dosed according to the recommended dosing regimen (see DOSAGE AND ADMINISTRATION) was 0.8% (4/525). Table 6 shows the frequency by randomized dose of serious arrhythmias and conduction disturbances reported as adverse events in patients with supraventricular arrhythmias.
| Dofetilide Capsules Dose | Placebo | ||||
|---|---|---|---|---|---|
| Arrhythmia event: | <250 mcg
BID N=217 | 250 mcg
BID N=388 | >250–500 mcg
BID N=703 | >500 mcg
BID N=38 | N=677 |
|
3.7% |
2.6% |
3.4% |
15.8% |
2.7% |
|
|
Ventricular fibrillation |
0 |
0.3% |
0.4% |
2.6% |
0.1% |
|
Ventricular tachycardia |
3.7% |
2.6% |
3.3% |
13.2% |
2.5% |
|
Torsade de Pointes |
0 |
0.3% |
0.9% |
10.5% |
0 |
|
Various forms of block | |||||
|
AV block |
0.9% |
1.5% |
0.4% |
0 |
0.3% |
|
Bundle branch block |
0 |
0.5% |
0.1% |
0 |
0.1% |
|
Heart block |
0 |
0.5% |
0.1% |
0 |
0.1% |
In the DIAMOND trials, a total of 1,511 patients were exposed to dofetilide capsules for 1757 patient years. The incidence of Torsade de Pointes was 3.3% in CHF patients and 0.9% in patients with a recent MI.
Table 7 shows the incidence of serious arrhythmias and conduction disturbances reported as adverse events in the DIAMOND subpopulation that had AF at entry to these trials.
| Dofetilide Capsules | Placebo | |
|---|---|---|
| N=249 | N=257 | |
|
14.5% |
13.6% |
|
|
Ventricular fibrillation |
4.8% |
3.1% |
|
Ventricular tachycardia |
12.4% |
11.3% |
|
Torsade de Pointes |
1.6% |
0 |
|
Various forms of block | ||
|
AV block |
0.8% |
2.7% |
|
(Left) bundle branch block |
0 |
0.4% |
|
Heart block |
1.2% |
0.8% |
Other Adverse Reactions
Table 8 presents other adverse events reported with a frequency of >2% on dofetilide capsules and reported numerically more frequently on dofetilide capsules than on placebo in the studies of patients with supraventricular arrhythmias.
| Dofetilide Capsules | Placebo | |
|---|---|---|
| Adverse Event | % | % |
|
headache |
11 |
9 |
|
chest pain |
10 |
7 |
|
dizziness |
8 |
6 |
|
respiratory tract infection |
7 |
5 |
|
dyspnea |
6 |
5 |
|
nausea |
5 |
4 |
|
flu syndrome |
4 |
2 |
|
insomnia |
4 |
3 |
|
accidental injury |
3 |
1 |
|
back pain |
3 |
2 |
|
procedure (medical/surgical/health service) |
3 |
2 |
|
diarrhea |
3 |
2 |
|
rash |
3 |
2 |
|
abdominal pain |
3 |
2 |
Adverse events reported at a rate >2% but no more frequently on dofetilide capsules than on placebo were: angina pectoris, anxiety, arthralgia, asthenia, atrial fibrillation, complications (application, injection, incision, insertion, or device), hypertension, pain, palpitation, peripheral edema, supraventricular tachycardia, sweating, urinary tract infection, ventricular tachycardia.
The following adverse events have been reported with a frequency of ≤2% and numerically more frequently with dofetilide capsules than placebo in patients with supraventricular arrhythmias: angioedema, bradycardia, cerebral ischemia, cerebrovascular accident, edema, facial paralysis, flaccid paralysis, heart arrest, increased cough, liver damage, migraine, myocardial infarct, paralysis, paresthesia, sudden death, and syncope.
The incidences of clinically significant laboratory test abnormalities in patients with supraventricular arrhythmias were similar for patients on dofetilide capsules and those on placebo. No clinically relevant effects were noted in serum alkaline phosphatase, serum GGT, LDH, AST, ALT, total bilirubin, total protein, blood urea nitrogen, creatinine, serum electrolytes (calcium, chloride, glucose, magnesium, potassium, sodium), or creatine kinase. Similarly, no clinically relevant effects were observed in hematologic parameters.
In the DIAMOND population, adverse events other than those related to the post-infarction and heart failure patient population were generally similar to those seen in the supraventricular arrhythmia groups.
To report SUSPECTED ADVERSE EVENTS, contact Dr.Reddy’s Laboratories Inc., at 1-888-375-3784 or FDA at 1-800-FDA-1088 or http://www.fda.gov/medwatch for voluntary reporting of adverse reactions.
Related/similar drugs
Overdosage
There is no known antidote to dofetilide capsules; treatment of overdose should therefore be symptomatic and supportive. The most prominent manifestation of overdosage is likely to be excessive prolongation of the QT interval.
In cases of overdose, cardiac monitoring should be initiated. Charcoal slurry may be given soon after overdosing but has been useful only when given within 15 minutes of dofetilide capsules administration. Treatment of Torsade de Pointes or overdose may include administration of isoproterenol infusion, with or without cardiac pacing. Administration of intravenous magnesium sulfate may be effective in the management of Torsade de Pointes. Close medical monitoring and supervision should continue until the QT interval returns to normal levels.
Isoproterenol infusion into anesthetized dogs with cardiac pacing rapidly attenuates the dofetilide-induced prolongation of atrial and ventricular effective refractory periods in a dose-dependent manner. Magnesium sulfate, administered prophylactically either intravenously or orally in a dog model, was effective in the prevention of dofetilide-induced Torsade de Pointes ventricular tachycardia. Similarly, in man, intravenous magnesium sulfate may terminate Torsade de Pointes, irrespective of cause.
Dofetilide capsules overdose was rare in clinical studies; there were two reported cases of dofetilide capsules overdose in the oral clinical program. One patient received very high multiples of the recommended dose (28 capsules), was treated with gastric aspiration 30 minutes later, and experienced no events. One patient inadvertently received two 500 mcg doses one hour apart and experienced ventricular fibrillation and cardiac arrest 2 hours after the second dose.
In the supraventricular arrhythmia population, only 38 patients received doses greater than 500 mcg BID, all of whom received 750 mcg BID irrespective of creatinine clearance. In this very small patient population, the incidence of Torsade de Pointes was 10.5% (4/38 patients), and the incidence of new ventricular fibrillation was 2.6% (1/38 patients).
Dofetilide Dosage and Administration
- •
- Therapy with dofetilide capsules must be initiated (and, if necessary, re-initiated) in a setting that provides continuous electrocardiographic (ECG) monitoring and in the presence of personnel trained in the management of serious ventricular arrhythmias. Patients should continue to be monitored in this way for a minimum of three days. Additionally, patients should not be discharged within 12 hours of electrical or pharmacological conversion to normal sinus rhythm.
- •
- The dose of dofetilide capsules must be individualized according to calculated creatinine clearance and QTc. (QT interval should be used if the heart rate is <60 beats per minute. There are no data on use of dofetilide capsules when the heart rate is <50 beats per minute.) The usual recommended dose of dofetilide capsules is 500 mcg BID, as modified by the dosing algorithm described below. For consideration of a lower dose, see Special Considerations below.
- •
- Serum potassium should be maintained within the normal range before dofetilide capsules treatment is initiated and should be maintained within the normal range while the patient remains on dofetilide capsules therapy. (See WARNINGS, Hypokalemia and Potassium-Depleting Diuretics). In clinical trials, potassium levels were generally maintained above 3.6-4.0 mEq/L.
- •
- Patients with atrial fibrillation should be anticoagulated according to usual medical practice prior to electrical or pharmacological cardioversion. Anticoagulant therapy may be continued after cardioversion according to usual medical practice for the treatment of people with AF. Hypokalemia should be corrected before initiation of dofetilide capsules therapy (see WARNINGS, Ventricular Arrhythmia).
- •
- Patients to be discharged on dofetilide capsules therapy from an inpatient setting as described above must have an adequate supply of dofetilide capsules, at the patient's individualized dose, to allow uninterrupted dosing until the patient can fill a dofetilide capsules prescription.
Instructions for Individualized Dose Initiation
Initiation of Dofetilide Capsules Therapy
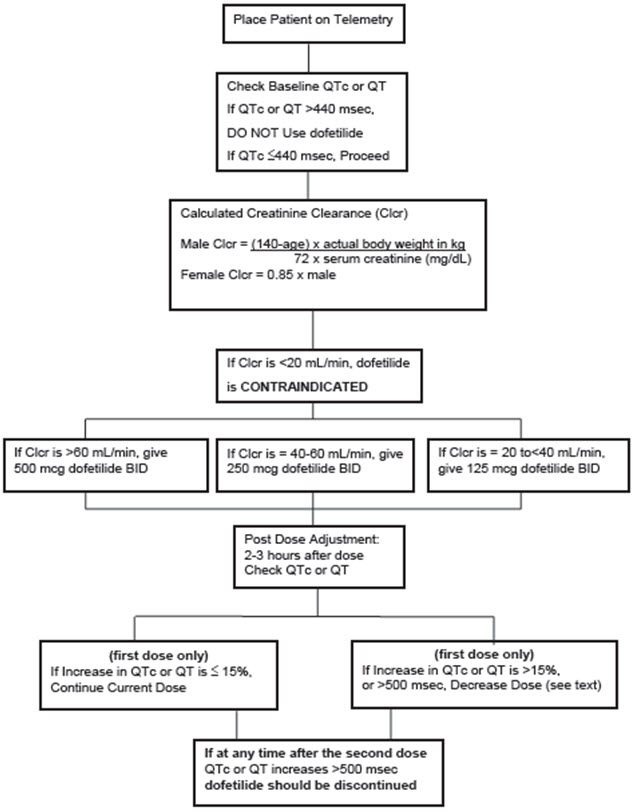
Step 1. Electrocardiographic assessment: Prior to administration of the first dose, the QTc or QT must be checked using an average of 5-10 beats. If the QTc or QT is greater than 440 msec (500 msec in patients with ventricular conduction abnormalities), dofetilide capsules are contraindicated. If heart rate is less than 60 beats per minute, QT interval should be used. Proceed to Step 2 if the QTc or QT is 440 msec. Patients with heart rates <50 beats per minute have not been studied.
Step 2. Calculation of creatinine clearance: Prior to the administration of the first dose, the patient's creatinine clearance must be calculated using the following formula:
|
creatinine clearance (male) = |
(140-age) × actual body weight in kg |
|
72 × serum creatinine (mg/dL) |
|
creatinine clearance (female) = |
(140-age) × actual body weight in kg × 0.85 |
|
72 × serum creatinine (mg/dL) |
When serum creatinine is given in µmol/L, divide the value by 88.4 (1 mg/dL = 88.4 µmol/L).
Step 3. Starting Dose: The starting dose of dofetilide capsules is determined as follows:
| Calculated Creatinine Clearance | Dofetilide Capsules Dose |
|---|---|
|
>60 mL/min |
500 mcg twice daily |
|
40 to 60 mL/min |
250 mcg twice daily |
|
20 to <40 mL/min |
125 mcg twice daily |
|
<20 mL/min |
Dofetilide capsules are contraindicated in these patients |
Step 4. Administer the adjusted dofetilide capsules dose and begin continuous ECG monitoring.
Step 5. At 2-3 hours after administering the first dose of dofetilide capsules, determine the QTc or QT (if heart rate is less than 60 beats per minute). If the QTc or QT has increased by greater than 15% compared to the baseline established in Step 1 OR if the QTc or QT is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), subsequent dosing should be adjusted as follows:
| If the Starting Dose Based on Creatinine Clearance is: | Then the Adjusted Dose (for QTc or QT Prolongation) is: | |
|---|---|---|
|
500 mcg twice daily |
250 mcg twice daily |
|
|
250 mcg twice daily |
125 mcg twice daily |
|
|
125 mcg twice daily |
125 mcg once a day |
Step 6. At 2-3 hours after each subsequent dose of dofetilide capsules, determine the QTc or QT (if heart rate is less than 60 beats per minute) (for in-hospital doses 2-5). No further down titration of dofetilide capsules based on QTc or QT is recommended.
NOTE: If at any time after the second dose of dofetilide capsules is given the QTc or QT is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide capsules should be discontinued.
Step 7. Patients are to be continuously monitored by ECG for a minimum of three days, or for a minimum of 12 hours after electrical or pharmacological conversion to normal sinus rhythm, whichever is greater.
The steps described above are summarized in the following diagram:
Maintenance of Dofetilide Capsules Therapy
Renal function and QTc or QT (if heart rate is less than 60 beats per minute) should be re-evaluated every three months or as medically warranted. If QTc or QT exceeds 500 milliseconds (550 msec in patients with ventricular conduction abnormalities), dofetilide capsules therapy should be discontinued and patients should be carefully monitored until QTc or QT returns to baseline levels. If renal function deteriorates, adjust dose as described in Initiation of Dofetilide Capsules Therapy, Step 3.
Special Considerations
Consideration of a Dose Lower than that Determined by the Algorithm
The dosing algorithm shown above should be used to determine the individualized dose of dofetilide capsules. In clinical trials (see CLINICAL STUDIES), the highest dose of 500 mcg BID of dofetilide capsules as modified by the dosing algorithm led to greater effectiveness than lower doses of 125 or 250 mcg BID as modified by the dosing algorithm. The risk of Torsade de Pointes, however, is related to dose as well as to patient characteristics (see WARNINGS). Physicians, in consultation with their patients, may therefore in some cases choose doses lower than determined by the algorithm. It is critically important that if at any time this lower dose is increased, the patient needs to be rehospitalized for three days. Previous toleration of higher doses does not eliminate the need for rehospitalization.
The maximum recommended dose in patients with a calculated creatinine clearance greater than 60 mL/min is 500 mcg BID; doses greater than 500 mcg BID have been associated with an increased incidence of Torsade de Pointes.
A patient who misses a dose should NOT double the next dose. The next dose should be taken at the usual time.
Cardioversion
If patients do not convert to normal sinus rhythm within 24 hours of initiation of dofetilide capsules therapy, electrical conversion should be considered. Patients continuing on dofetilide capsules after successful electrical cardioversion should continue to be monitored by electrocardiography for 12 hours post cardioversion, or a minimum of 3 days after initiation of dofetilide capsules therapy, whichever is greater.
Switch to Dofetilide Capsules from Class I or other Class III Antiarrhythmic Therapy
Before initiating dofetilide capsules therapy, previous antiarrhythmic therapy should be withdrawn under careful monitoring for a minimum of three (3) plasma half-lives. Because of the unpredictable pharmacokinetics of amiodarone, dofetilide capsules should not be initiated following amiodarone therapy until amiodarone plasma levels are below 0.3 mcg/mL or until amiodarone has been withdrawn for at least three months.
How is Dofetilide supplied
Dofetilide capsules, 125 mcg (0.125 mg) are supplied as No. 4 capsules with a dark caramel cap and white body, imprinted with ML 125 in black ink, and are available in:
Dofetilide capsules, 250 mcg (0.25 mg) are supplied as No. 4 capsules with a light orange cap and light orange body, imprinted with ML 250 in black ink, and are available in:
Dofetilide capsules, 500 mcg (0.5 mg) are supplied as No. 2 capsules with a light orange cap and white body, imprinted with ML 500 in black ink, and are available in:
| 125 mcg
(0.125 mg) | 250 mcg
(0.25 mg) | 500 mcg
(0.5 mg) |
|
|---|---|---|---|
|
Body |
125 |
250 |
500 |
|
Cap |
ML |
ML |
ML |
|
Cartons of 40 capsules (10 capsules each blister pack x 4) |
0904-7522-08 |
0904-7523-08 |
0904-7524-08 |
WARNING: These Unit Dose packages are not child resistant and are Intended for Institutional Use Only. Keep this and all drugs out of the reach of children.
Distributed by:
Dr. Reddy’s Laboratories Inc.
Princeton, NJ 08540
Packaged and Distributed by:
MAJOR® PHARMACEUTICALS
Indianapolis, IN 46268 USA
Refer to package label for Distributor's NDC Number
Revised: August 2023
61013
Medication Guide
Dofetilide Capsules
(doe-FEH-till-ide)
Read the Medication Guide before you start taking dofetilide capsules and each time you get a refill. This information does not take the place of talking with your doctor about your condition or treatment.
What is the most important information I should know about Dofetilide Capsules?
Dofetilide capsules can cause serious side effects, including a type of abnormal heartbeat called Torsade de Pointes, which can lead to death.
To establish the right dose of dofetilide capsules, treatment with dofetilide capsules must be started in a hospital where your heart rate and kidney function will be checked for the first 3 days of treatment. It is important that when you go home, you take the exact dose of dofetilide capsules that your doctor prescribed for you.
While you take dofetilide capsules, always watch for signs of abnormal heartbeat.
Call your doctor and go to the hospital right away if you:
- •
- feel faint
- •
- become dizzy, or
- •
- have a fast heartbeat
What are Dofetilide Capsules?
Dofetilide capsules is a prescription medicine that is used to treat an irregular heartbeat (atrial fibrillation or atrial flutter).
It is not known if dofetilide capsules are safe and effective in children under 18 years of age.
Who should not take Dofetilide Capsules?
Do not take dofetilide capsules if you:
- •
- have an irregular heartbeat called long QT syndrome
- •
- have kidney problems or are on kidney dialysis
- •
- take any of these medicines:
- o
- cimetidine (TAGAMET, TAGAMET HB)1
- o
- verapamil (CALAN, CALAN SR, COVERA-HS, ISOPTIN, ISOPTIN SR, VERELAN, VERELAN PM, TARKA)
- o
- ketoconazole (NIZORAL, XOLEGEL, EXTINA)
- o
- trimethoprim alone (PROLOPRIM, TRIMPEX) or the combination of trimethoprim and sulfamethoxazole (BACTRIM, SEPTRA SULFATRIM)
- o
- prochlorperazine (COMPAZINE, COMPO)
- o
- megestrol (MEGACE)
- o
- dolutegravir (TIVICAY)
- o
- hydrochlorothiazide alone or in combination with other medicines (such as ESIDRIX, EZIDE, HYDRODIURIL, HYDRO-PAR, MICROZIDE, or ORETIC)
- Ask your doctor if you are not sure if any of your medicines are the kind listed above.
- •
- are allergic to dofetilide in dofetilide capsules. See the end of this leaflet for a complete list of ingredients in dofetilide capsules.
What should I tell my doctor before taking Dofetilide Capsules?
Before taking dofetilide capsules, tell your doctor about all of your medical conditions including if you:
- •
- have heart problems
- •
- have kidney or liver problems
- •
- are pregnant or plan to become pregnant. It is not known if dofetilide will harm your unborn baby.
- •
- are breast-feeding or plan to breast-feed. It is not known if dofetilide passes into your breast milk. You and your doctor should decide if you will take dofetilide capsules or breast-feed. You should not do both.
Especially tell your doctor if you take medicines to treat:
- •
- heart problems
- •
- high blood pressure
- •
- depression or other mental problems
- •
- asthma
- •
- allergies, or hay fever
- •
- skin problems
- •
- infections
Ask your doctor if you are not sure about the medicines you take. Tell your doctor about all prescription and non-prescription medicines, vitamins, dietary supplements, and any natural or herbal remedies. Dofetilide capsules and other medicines may affect each other, causing serious side effects. If you take dofetilide capsules with certain medicines, you will be more likely to have a different type of abnormal heartbeat. See ""
Know the medicines you take. Keep a list of your medicines and show it to your doctor and pharmacist when you get a new medicine.
How should I take Dofetilide Capsules?
- •
- Take dofetilide capsules exactly as your doctor tells you.
- •
- Do not change your dofetilide capsules dose unless your doctor tells you to.
- •
- Your doctor will do tests before you start and while you take dofetilide capsules.
- •
- Do not stop taking dofetilide capsules until your doctor tells you to stop. If you miss a dose, just take the next dose at your regular time. Do not take 2 doses of dofetilide capsules at the same time.
- •
- Dofetilide capsules can be taken with or without food.
- •
- If you take too much dofetilide capsules, call your doctor or go to the nearest hospital emergency room right away. Take your dofetilide capsules with you to show to the doctor.
What are the possible side effects of Dofetilide Capsules?
Dofetilide capsules can cause serious side effects, including a type of abnormal heartbeat called Torsade de Pointes, which can lead to death. See "What is the most important information I should know about Dofetilide Capsules?"
The most common side effects of dofetilide capsules include:
- •
- headache
- •
- chest pain
- •
- dizziness
Call your doctor right away if you have signs of electrolyte imbalance:
- •
- severe diarrhea
- •
- unusual sweating
- •
- vomiting
- •
- not hungry (loss of appetite)
- •
- increased thirst (drinking more than normal)
Tell your doctor if you have any side effects that bother you or do not go away.
These are not all the possible side effects of dofetilide capsules. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Dofetilide Capsules?
- •
- Store dofetilide capsules between 59° to 86°F (15° to 30°C).
- •
- Keep dofetilide capsules away from moisture and humidity.
- •
- Keep dofetilide capsules in a tightly closed container.
- •
- Keep dofetilide capsules and all medicines out of the reach of children.
General information about Dofetilide Capsules
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use dofetilide capsules for a condition for which it was not prescribed. Do not give dofetilide capsules to other people, even if they have the same symptoms you have. It may harm them.
This Medication Guide summarizes the most important information about dofetilide capsules. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about dofetilide capsules that is written for health professionals.
For more about dofetilide capsules, call 1-888-375-3784.
What are the ingredients in Dofetilide Capsules?
Active ingredient: dofetilide, USP
Inactive ingredients:
- Capsule fill: microcrystalline cellulose, corn starch, colloidal silicon dioxide, and magnesium stearate
- Capsule shell: gelatin, titanium dioxide, red iron oxide and yellow iron oxide
- Imprinting ink: iron oxide black, shellac, ethanol, n-butyl alcohol, isopropyl alcohol, propylene glycol, and ammonium hydroxide
Rx only
- 1
- Listed trademarks are the property of their respective owners.
Distributed by:
Dr. Reddy’s Laboratories Inc.
Princeton, NJ 08540
Packaged and Distributed by:
MAJOR® PHARMACEUTICALS
Indianapolis, IN 46268 USA
Refer to package label for Distributor's NDC Number
Revised: August 2023
61013
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Package/Label Display Panel
MAJOR®
NDC 0904-7522-08
Unit Dose
Dofetilide
Capsules
125 mcg (0.125 mg)
Pharmacist: Dispense with
Medication Guide
40 CAPSULES (4 x 10)
Rx only



| DOFETILIDE
dofetilide capsule |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| DOFETILIDE
dofetilide capsule |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| DOFETILIDE
dofetilide capsule |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| Labeler - Major Pharmaceuticals (191427277) |
More about dofetilide
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (78)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: group III antiarrhythmics
- En español
