Adcetris: Package Insert / Prescribing Info
Package insert / product label
Generic name: brentuximab vedotin
Dosage form: injection, powder, lyophilized, for solution
Drug class: CD30 monoclonal antibodies
J Code (medical billing code): J9042 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Feb 23, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ADCETRIS® (brentuximab vedotin) for injection, for intravenous use
Initial U.S. approval: 2011
Recent Major Changes
Indications and Usage for Adcetris
ADCETRIS is a CD30-directed antibody and microtubule inhibitor conjugate indicated for treatment of:
- •
- Adult patients with previously untreated Stage III or IV classical Hodgkin lymphoma (cHL), in combination with doxorubicin, vinblastine, and dacarbazine (1.1).
- •
- Pediatric patients 2 years and older with previously untreated high risk classical Hodgkin lymphoma (cHL), in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide (1.2).
- •
- Adult patients with classical Hodgkin lymphoma (cHL) at high risk of relapse or progression as post-autologous hematopoietic stem cell transplantation (auto-HSCT) consolidation (1.3).
- •
- Adult patients with classical Hodgkin lymphoma (cHL) after failure of auto-HSCT or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not auto-HSCT candidates (1.4).
- •
- Adult patients with previously untreated systemic anaplastic large cell lymphoma (sALCL) or other CD30-expressing peripheral T-cell lymphomas (PTCL), including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified (NOS), in combination with cyclophosphamide, doxorubicin, and prednisone (1.5).
- •
- Adult patients with systemic anaplastic large cell lymphoma (sALCL) after failure of at least one prior multi-agent chemotherapy regimen (1.6).
- •
- Adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or CD30-expressing mycosis fungoides (MF) who have received prior systemic therapy (1.7).
- •
- Adult patients with relapsed or refractory large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) NOS, DLBCL arising from indolent lymphoma, or high-grade B-cell lymphoma (HGBL), after two or more lines of systemic therapy who are not eligible for auto-HSCT or CAR T-cell therapy, in combination with lenalidomide and a rituximab product (1.8).
Adcetris Dosage and Administration
- •
- Administer only as an intravenous infusion over 30 minutes (2.1).
- •
- The recommended dosage as monotherapy for adult patients is 1.8 mg/kg up to a maximum of 180 mg every 3 weeks (2.1).
- •
- The recommended dosage in combination with chemotherapy for adult patients with previously untreated Stage III or IV cHL is 1.2 mg/kg up to a maximum of 120 mg every 2 weeks for a maximum of 12 doses (2.1).
- •
- The recommended dosage in combination with chemotherapy for pediatric patients 2 years and older with previously untreated high risk cHL is 1.8 mg/kg up to a maximum of 180 mg every 3 weeks for a maximum of 5 doses (2.1).
- •
- The recommended dosage in combination with chemotherapy for adult patients with previously untreated PTCL is 1.8 mg/kg up to a maximum of 180 mg every 3 weeks for 6 to 8 doses (2.1).
- •
- The recommended dosage in combination with lenalidomide and a rituximab product for adult patients with relapsed or refractory LBCL is 1.2 mg/kg up to a maximum of 120 mg every 3 weeks (2.1).
- •
- Avoid use in patients with severe renal impairment (2.2)
- •
- Reduce dose in patients with mild hepatic impairment; avoid use in patients with moderate or severe hepatic impairment (2.3).
Dosage Forms and Strengths
For injection: 50 mg lyophilized powder in a single-dose vial (3).
Contraindications
Concomitant use with bleomycin due to pulmonary toxicity (4).
Warnings and Precautions
- •
- Peripheral neuropathy: Monitor patients for neuropathy and institute dose modifications accordingly (5.1).
- •
- Anaphylaxis and infusion reactions: If an infusion reaction occurs, interrupt the infusion. If anaphylaxis occurs, immediately discontinue the infusion (5.2).
- •
- Hematologic toxicities: Monitor complete blood counts. Monitor for signs of infection. Manage using dose delays and growth factor support (5.3).
- •
- Serious infections and opportunistic infections: Closely monitor patients for the emergence of bacterial, fungal or viral infections (5.4).
- •
- Tumor lysis syndrome: Closely monitor patients with rapidly proliferating tumor or high tumor burden (5.5).
- •
- Hepatotoxicity: Monitor liver enzymes and bilirubin (5.8).
- •
- Pulmonary toxicity: Monitor patients for new or worsening symptoms (5.10).
- •
- Serious dermatologic reactions: Discontinue if Stevens-Johnson syndrome or toxic epidermal necrolysis occurs (5.11).
- •
- Gastrointestinal complications: Monitor patients for new or worsening symptoms (5.12).
- •
- Hyperglycemia: Monitor patients for new or worsening hyperglycemia. Manage with anti-hyperglycemic medications as clinically indicated (5.13).
- •
- Embryo-Fetal toxicity: Can cause fetal harm. Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus and to use effective contraception (5.14, 8.1, 8.3).
Adverse Reactions/Side Effects
The most common adverse reactions (≥20%) are peripheral neuropathy, nausea, fatigue, musculoskeletal pain, constipation, diarrhea, vomiting, pyrexia, upper respiratory tract infection, mucositis, abdominal pain, and rash.
The most common laboratory abnormalities (≥20%) are decreased neutrophils, increased creatinine, decreased hemoglobin, decreased lymphocytes, increased glucose, increased alanine aminotransferase (ALT), and increased aspartate aminotransferase (AST) (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Seagen Inc. at 1-855-473-2436 or FDA at 1-800-FDA-1088 or www.fda.gov/Safety/MedWatch.
Drug Interactions
Concomitant use of strong CYP3A4 inhibitors or inducers has the potential to affect the exposure to monomethyl auristatin E (MMAE) (7.1).
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2025
Full Prescribing Information
WARNING: PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY (PML)
JC virus infection resulting in PML and death can occur in patients receiving ADCETRIS [see Warnings and Precautions (5.9), Adverse Reactions (6.1)].
1. Indications and Usage for Adcetris
1.1 Previously Untreated Stage III or IV Classical Hodgkin Lymphoma (cHL), in Combination with Chemotherapy
ADCETRIS is indicated for the treatment of adult patients with previously untreated Stage III or IV cHL, in combination with doxorubicin, vinblastine, and dacarbazine.
1.2 Previously Untreated High Risk Classical Hodgkin Lymphoma (cHL), in Combination with Chemotherapy
ADCETRIS is indicated for the treatment of pediatric patients 2 years and older with previously untreated high risk cHL, in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide.
1.3 Classical Hodgkin Lymphoma (cHL) Consolidation
ADCETRIS is indicated for the treatment of adult patients with cHL at high risk of relapse or progression as post-autologous hematopoietic stem cell transplantation (auto-HSCT) consolidation.
1.4 Relapsed Classical Hodgkin Lymphoma (cHL)
ADCETRIS is indicated for the treatment of adult patients with cHL after failure of auto-HSCT or after failure of at least two prior multi-agent chemotherapy regimens in patients who are not auto-HSCT candidates.
1.5 Previously Untreated Systemic Anaplastic Large Cell Lymphoma (sALCL) or Other CD30-Expressing Peripheral T-cell Lymphomas (PTCL), in Combination with Chemotherapy
ADCETRIS is indicated for the treatment of adult patients with previously untreated sALCL or other CD30-expressing PTCL, including angioimmunoblastic T-cell lymphoma and PTCL not otherwise specified (NOS), in combination with cyclophosphamide, doxorubicin, and prednisone.
1.6 Relapsed Systemic Anaplastic Large Cell Lymphoma (sALCL)
ADCETRIS is indicated for the treatment of adult patients with sALCL after failure of at least one prior multi-agent chemotherapy regimen.
1.7 Relapsed Primary Cutaneous Anaplastic Large Cell Lymphoma (pcALCL) or CD30-Expressing Mycosis Fungoides (MF)
ADCETRIS is indicated for the treatment of adult patients with pcALCL or CD30-expressing MF who have received prior systemic therapy.
1.8 Relapsed or Refractory Large B-Cell Lymphoma (LBCL)
ADCETRIS in combination with lenalidomide and a rituximab product is indicated for the treatment of adult patients with relapsed or refractory LBCL, including diffuse large B-cell lymphoma (DLBCL) NOS, DLBCL arising from indolent lymphoma, or high-grade B-cell lymphoma (HGBL), after two or more lines of systemic therapy who are not eligible for auto-HSCT or chimeric antigen receptor (CAR) T-cell therapy.
2. Adcetris Dosage and Administration
2.1 Recommended Dosage
The recommended ADCETRIS dosage is provided in Table 1. Administer ADCETRIS as a 30-minute intravenous infusion.
For recommended dosage for patients with renal or hepatic impairment, see Dosage and Administration (2.2 and 2.3).
For dosing instructions of combination agents administered with ADCETRIS, see Clinical Studies (14.1, 14.2, and 14.5) and the manufacturer’s prescribing information.
|
Indication |
Recommended Dose* |
Frequency and Duration |
|
Adult patients with previously untreated Stage III or IV classical Hodgkin lymphoma |
1.2 mg/kg up to a maximum of 120 mg in combination with chemotherapy |
Administer every 2 weeks until a maximum of 12 doses, disease progression, or unacceptable toxicity |
|
Pediatric patients with previously untreated high risk classical Hodgkin lymphoma |
1.8 mg/kg up to a maximum of 180 mg in combination with chemotherapy |
Administer every 3 weeks with each cycle of chemotherapy for a maximum of 5 doses |
|
Adult patients with classical Hodgkin lymphoma consolidation |
1.8 mg/kg up to a maximum of 180 mg |
Initiate ADCETRIS treatment within 4‑6 weeks post-auto-HSCT or upon recovery from auto-HSCT |
|
Adult patients with relapsed classical Hodgkin lymphoma |
1.8 mg/kg up to a maximum of 180 mg |
Administer every 3 weeks until disease progression or unacceptable toxicity |
|
Adult patients with previously untreated systemic ALCL or other CD30-expressing peripheral T-cell lymphomas |
1.8 mg/kg up to a maximum of 180 mg in combination with chemotherapy |
Administer every 3 weeks with each cycle of chemotherapy for 6 to 8 doses |
|
Adult patients with relapsed Systemic ALCL |
1.8 mg/kg up to a maximum of 180 mg |
Administer every 3 weeks until disease progression or unacceptable toxicity |
|
Adult patients with relapsed primary cutaneous ALCL or CD30-expressing mycosis fungoides |
1.8 mg/kg up to a maximum of 180 mg |
Administer every 3 weeks until a maximum of 16 cycles, disease progression, or unacceptable toxicity |
|
Adult patients with relapsed or refractory LBCL |
1.2 mg/kg up to a maximum of 120 mg in combination with lenalidomide and rituximab† |
Administer every 3 weeks until disease progression, or unacceptable toxicity |
2.2 Recommended Dosage in Patients with Renal Impairment
No dosage adjustment is required for mild renal impairment (CrCL greater than 50‑80 mL/min) and moderate renal impairment (CrCL 30-50 mL/min).
Avoid use in patients with severe (CrCL less than 30 mL/min) renal impairment [see Warnings and Precautions (5.6)].
2.3 Recommended Dosage in Patients with Hepatic Impairment
Adult patients with previously untreated Stage III or IV classical Hodgkin lymphoma
Reduce the dosage of ADCETRIS to 0.9 mg/kg up to a maximum of 90 mg every 2 weeks for patients with mild hepatic impairment (Child-Pugh A).
Avoid use in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment [see Warnings and Precautions (5.7)].
Adult patients with relapsed or refractory LBCL
Reduce the dosage of ADCETRIS to 0.9 mg/kg up to a maximum of 90 mg every 3 weeks for patients with mild hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] and aspartate transaminase [AST] > ULN, or total bilirubin >1 to 1.5 × ULN and any AST).
Avoid use in patients with moderate and severe hepatic impairment (total bilirubin >1.5 × ULN) [see Warnings and Precautions (5.7)].
Hepatic impairment is defined per the National Cancer Institute Organ Dysfunction Working Group.
All other indications
Reduce the dosage of ADCETRIS to 1.2 mg/kg up to a maximum of 120 mg every 3 weeks for patients with mild hepatic impairment (Child-Pugh A).
Avoid use in patients with moderate (Child-Pugh B) and severe (Child-Pugh C) hepatic impairment [see Warnings and Precautions (5.7)].
2.4 Recommended Prophylactic Medications
In adult patients with previously untreated Stage III or IV cHL who are treated with ADCETRIS + doxorubicin, vinblastine, and dacarbazine (AVD), administer G‑CSF beginning with Cycle 1.
In pediatric patients with previously untreated high risk cHL who are treated with ADCETRIS + doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide (AVEPC), administer G-CSF beginning with Cycle 1.
In adult patients with previously untreated PTCL who are treated with ADCETRIS + cyclophosphamide, doxorubicin, and prednisone (CHP), administer G-CSF beginning with Cycle 1.
In adult patients with relapsed or refractory LBCL who are treated with ADCETRIS + lenalidomide + rituximab, administer G-CSF beginning with Cycle 1.
2.5 Dosage Modifications for Adverse Reactions
|
Monotherapy |
Severity† |
Dosage Modification |
|
|
Peripheral Neuropathy |
|||
|
1.2 mg/kg up to a maximum of 120 mg every 2 weeks |
In combination with chemotherapy |
Grade 2 |
Reduce dose to 0.9 mg/kg up to a maximum of 90 mg every 2 weeks. |
|
Grade 3 |
Hold ADCETRIS dosing until improvement to Grade 2 or lower. |
||
|
Grade 4 |
Discontinue dosing. |
||
|
1.2 mg/kg up to a maximum of 120 mg every 3 weeks |
In combination with lenalidomide and rituximab |
Grade 2 |
Sensory neuropathy: If resolves to Grade 1 or lower before the next scheduled dose, resume at the same dose level. If Grade 2 persists at the next scheduled dose, reduce one dose level. |
|
Grade 3 |
Sensory neuropathy: Hold ADCETRIS dosing until improvement to Grade 2 or lower, then restart treatment at a reduced dosage of 0.9 mg/kg up to a maximum of 90 mg every 3 weeks. |
||
|
Grade 4 |
Discontinue dosing. |
||
|
1.8 mg/kg up to a maximum of 180 mg every 3 weeks |
As monotherapy |
New or |
Hold dosing until improvement to baseline or Grade 1. |
|
Grade 4 |
Discontinue dosing. |
||
|
In combination with chemotherapy |
Grade 2 |
Sensory neuropathy: Continue treatment at same dose. |
|
|
Grade 3 |
Sensory neuropathy: Reduce dose to 1.2 mg/kg, up to a maximum of 120 mg every 3 weeks. |
||
|
Grade 4 |
Discontinue dosing. |
||
|
Neutropenia |
|||
|
1.2 mg/kg up to a maximum of 120 mg every 2 weeks |
In combination with chemotherapy |
Grade 3 or 4 |
Administer G‑CSF prophylaxis for subsequent cycles for patients not receiving primary G‑CSF prophylaxis. |
|
1.2 mg/kg up to a maximum of 120 mg every 3 weeks |
In combination with lenalidomide and rituximab |
Grade 3 or 4 |
Hold dosing until improvement to baseline or Grade 2 or lower. |
|
1.8 mg/kg up to a maximum of 180 mg every 3 weeks |
In combination with chemotherapy |
Grade 3 or 4 |
Administer G-CSF prophylaxis in subsequent cycles for patients not receiving primary G-CSF prophylaxis. |
|
1.8 mg/kg up to a maximum of 180 mg every 3 weeks |
As monotherapy |
Grade 3 or 4 |
Hold dosing until improvement to baseline or Grade 2 or lower. |
|
Recurrent Grade 4 despite G‑CSF prophylaxis |
Consider discontinuation or dose reduction to 1.2 mg/kg up to a maximum of 120 mg every 3 weeks. |
||
|
Severity |
Dosage Modification |
|
|
Peripheral Neuropathy† |
||
|
1.8 mg/kg up to a maximum of 180 mg every 3 weeks |
Grade 2† |
Reduce dose of vincristine per prescribing information. |
|
Grade 3† |
Discontinue vincristine. |
|
|
Grade 4† |
Discontinue ADCETRIS and vincristine. |
|
|
Neutropenia |
||
|
1.8 mg/kg up to a maximum of 180 mg every 3 weeks |
Grade 3 or 4 |
Reduce dose to 1.2 mg/kg up to a maximum of 120 mg every 3 weeks in patients who are unable to start a cycle >5 weeks after the start of the previous cycle (>2-week delay) due to neutropenia. |
2.6 Instructions for Preparation and Administration
Administration
- •
- Administer ADCETRIS as an intravenous infusion only.
- •
- Do not mix ADCETRIS with, or administer as an infusion with, other medicinal products.
Reconstitution
- •
- Follow procedures for proper handling and disposal of hazardous drugs1.
- •
- Use appropriate aseptic technique for reconstitution and preparation of dosing solutions.
- •
- Determine the number of 50 mg vials needed based on the patient’s weight and the prescribed dose [see Dosage and Administration (2.1)].
- •
- Reconstitute each 50 mg vial of ADCETRIS with 10.5 mL of Sterile Water for Injection to yield a single-dose solution containing 5 mg/mL brentuximab vedotin.
- •
- Direct the stream toward the wall of vial and not directly at the cake or powder.
- •
- Gently swirl the vial to aid dissolution. DO NOT SHAKE.
- •
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The reconstituted solution should be clear to slightly opalescent, colorless, and free of visible particulates.
- •
- Following reconstitution, dilute immediately into an infusion bag. If not diluted immediately, store the solution refrigerated at 2°C to 8°C (36°F to 46°F) and use within 24 hours of reconstitution. DO NOT FREEZE.
- •
- Discard any unused portion left in the vial.
Dilution
- •
- Calculate the required volume of 5 mg/mL reconstituted ADCETRIS solution needed.
- •
- Withdraw this amount from the vial and immediately add it to an infusion bag containing 0.9% Sodium Chloride Injection, USP, 5% Dextrose Injection or Lactated Ringer's Injection to achieve a final concentration of 0.4 mg/mL to 1.8 mg/mL brentuximab vedotin.
- •
- Gently invert the bag to mix the solution.
- •
- Following dilution, infuse the ADCETRIS solution immediately. If not used immediately, store the solution refrigerated at 2°C to 8°C (36°F to 46°F) and use within 24 hours of reconstitution. DO NOT FREEZE.
3. Dosage Forms and Strengths
For injection: 50 mg of brentuximab vedotin as a sterile, white to off-white lyophilized, preservative-free cake or powder in a single-dose vial for reconstitution.
4. Contraindications
ADCETRIS is contraindicated with concomitant bleomycin due to pulmonary toxicity (e.g., interstitial infiltration and/or inflammation) [see Adverse Reactions (6.1)].
5. Warnings and Precautions
5.1 Peripheral Neuropathy
ADCETRIS treatment causes a peripheral neuropathy that is predominantly sensory. Cases of peripheral motor neuropathy have also been reported. ADCETRIS-induced peripheral neuropathy is cumulative.
In studies of ADCETRIS as monotherapy, 62% of patients experienced any grade of peripheral neuropathy. The median time to onset was 3 months (range, 0–12). Of the patients who experienced neuropathy, 62% had complete resolution, 24% had partial improvement, and 14% had no improvement at their last evaluation. The median time from onset to resolution or improvement was 5 months (range, 0–45). Of the patients with ongoing neuropathy (38%), 71% had Grade 1, 24% had Grade 2, and 4% had Grade 3.
In ECHELON-1 (Study 5), 67% of patients treated with ADCETRIS + AVD experienced any grade of peripheral neuropathy. The median time to onset of any grade was 2 months (range, 0–7), of Grade 2 was 3 months (range, 0–6) and of Grade 3 was 4 months (range, <1–7). By the time of the primary analysis, 43% of affected patients had complete resolution, 24% had partial improvement, and 33% had no improvement at their last evaluation. The median time from onset to resolution or improvement of any grade was 2 months (range, 0–32).
At the updated analysis of ECHELON-1, 72% of the patients who experienced peripheral neuropathy had complete resolution, 14% had partial improvement, and 14% had no improvement. The median time to partial improvement was 2.9 months (range, <1–50), and the median time to complete resolution was 6.6 months (range, <1–67). Of the patients with ongoing neuropathy (28%), 57% had Grade 1, 30% had Grade 2, 12% had Grade 3, and <1% had Grade 4.
In ECHELON-2 (Study 6), 52% of patients treated with ADCETRIS + CHP experienced new or worsening peripheral neuropathy of any grade (by maximum grade, 34% Grade 1, 15% Grade 2, 3% Grade 3, <1% Grade 4). The peripheral neuropathy was predominantly sensory (94% sensory, 16% motor) and had a median onset time of 2 months (range, <1–5). At last evaluation, 50% had complete resolution of neuropathy, 12% had partial improvement, and 38% had no improvement. The median time to resolution or improvement overall was 4 months (range, 0–45). Of patients with ongoing neuropathy (50%), 72% had Grade 1, 25% had Grade 2, and 3% had Grade 3.
In AHOD1331 (Study 7), 20% of pediatric patients treated with ADCETRIS + AVEPC experienced peripheral neuropathy of any grade (7% Grade 3, <1% Grade 4). Peripheral neuropathy was predominantly sensory. Of the patients who experienced peripheral neuropathy, 81% experienced sensory neuropathy and 29% experienced motor neuropathy.
In ECHELON-3 (Study 8), 27% of patients treated with ADCETRIS + lenalidomide + a rituximab product experienced peripheral neuropathy of any grade (by maximum grade, 14% Grade 1, 7% Grade 2, 5% Grade 3). The peripheral neuropathy was predominantly sensory and had a median onset time of 3 months (range, <1-10). Peripheral neuropathy resulted in ADCETRIS dose reduction in 6% of treated patients, and permanent discontinuation in 4.5%. At last evaluation, 7% of the patients who experienced peripheral neuropathy had complete resolution of neuropathy, 10% had partial improvement, and 83% had no improvement. The median time to resolution was 2 months (range <1-3). The median time to improvement was 4 months (range, 3-4). Of patients who experienced peripheral neuropathy, 93% had ongoing peripheral neuropathy (47% had Grade 1, 33% had Grade 2, and 13% had Grade 3).
Monitor patients for symptoms of neuropathy, such as hypoesthesia, hyperesthesia, paresthesia, discomfort, a burning sensation, neuropathic pain, or weakness. Patients experiencing new or worsening peripheral neuropathy may require a delay, change in dose, or discontinuation of ADCETRIS [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
5.2 Anaphylaxis and Infusion Reactions
Infusion-related reactions, including anaphylaxis, have occurred with ADCETRIS. Monitor patients during infusion. If anaphylaxis occurs, immediately and permanently discontinue administration of ADCETRIS and administer appropriate medical therapy. If an infusion-related reaction occurs, interrupt the infusion and institute appropriate medical management. Patients who have experienced a prior infusion-related reaction should be premedicated for subsequent infusions. Premedication may include acetaminophen, an antihistamine, and a corticosteroid.
5.3 Hematologic Toxicities
Fatal and serious cases of febrile neutropenia have been reported with ADCETRIS. Prolonged (≥1 week) severe neutropenia and Grade 3 or Grade 4 thrombocytopenia or anemia can occur with ADCETRIS.
Start primary prophylaxis with G‑CSF beginning with Cycle 1 for adult patients who receive ADCETRIS in combination for previously untreated Stage III or IV cHL or previously untreated PTCL or relapsed or refractory LBCL, and pediatric patients who receive ADCETRIS in combination with chemotherapy for previously untreated high risk cHL [see Dosage and Administration (2.3) and Adverse Reactions (6.1)].
Monitor complete blood counts prior to each dose of ADCETRIS. Monitor more frequently for patients with Grade 3 or 4 neutropenia. Monitor patients for fever. If Grade 3 or 4 neutropenia develops, consider dose delays, reductions, discontinuation, or G-CSF prophylaxis with subsequent ADCETRIS doses [see Dosage and Administration (2.2, 2.3)].
5.4 Serious Infections and Opportunistic Infections
Serious infections and opportunistic infections such as pneumonia, bacteremia, and sepsis or septic shock (including fatal outcomes) have been reported in patients treated with ADCETRIS. Monitor patients closely during treatment for the emergence of possible bacterial, fungal, or viral infections.
5.5 Tumor Lysis Syndrome
Patients with rapidly proliferating tumor and high tumor burden may be at increased risk of tumor lysis syndrome. Monitor closely and take appropriate measures.
5.6 Increased Toxicity in the Presence of Severe Renal Impairment
The frequency of ≥ Grade 3 adverse reactions and deaths was greater in patients with severe renal impairment compared to patients with normal renal function. Due to higher MMAE exposure, ≥Grade 3 adverse reactions may be more frequent in patients with severe renal impairment compared to patients with normal renal function. Avoid the use of ADCETRIS in patients with severe renal impairment [creatinine clearance (CrCL) <30 mL/min] [see Use in Specific Populations (8.6)].
5.7 Increased Toxicity in the Presence of Moderate or Severe Hepatic Impairment
The frequency of ≥ Grade 3 adverse reactions and deaths was greater in patients with moderate and severe hepatic impairment compared to patients with normal hepatic function. Avoid the use of ADCETRIS in patients with moderate (Child-Pugh B) or severe (Child-Pugh C) hepatic impairment [see Use in Specific Populations (8.7)].
5.8 Hepatotoxicity
Fatal and serious cases of hepatotoxicity have occurred in patients receiving ADCETRIS. Cases were consistent with hepatocellular injury, including elevations of transaminases and/or bilirubin. Cases have occurred after the first dose of ADCETRIS or after ADCETRIS rechallenge. Preexisting liver disease, elevated baseline liver enzymes, and concomitant medications may also increase the risk. Monitor liver enzymes and bilirubin. Patients experiencing new, worsening, or recurrent hepatotoxicity may require a delay, change in dose, or discontinuation of ADCETRIS.
5.9 Progressive Multifocal Leukoencephalopathy
Fatal cases of JC virus infection resulting in PML have been reported in ADCETRIS-treated patients. First onset of symptoms occurred at various times from initiation of ADCETRIS therapy, with some cases occurring within 3 months of initial exposure. In addition to ADCETRIS therapy, other possible contributory factors include prior therapies and underlying disease that may cause immunosuppression. Consider the diagnosis of PML in any patient presenting with new-onset signs and symptoms of central nervous system abnormalities. Hold ADCETRIS dosing for any suspected case of PML and discontinue ADCETRIS dosing if a diagnosis of PML is confirmed.
5.10 Pulmonary Toxicity
Fatal and serious events of noninfectious pulmonary toxicity including pneumonitis, interstitial lung disease, and acute respiratory distress syndrome (ARDS), have been reported. Monitor patients for signs and symptoms of pulmonary toxicity, including cough and dyspnea. In the event of new or worsening pulmonary symptoms, hold ADCETRIS dosing during evaluation and until symptomatic improvement.
5.11 Serious Dermatologic Reactions
Fatal and serious cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported with ADCETRIS. If SJS or TEN occurs, discontinue ADCETRIS and administer appropriate medical therapy.
5.12 Gastrointestinal Complications
Fatal and serious events of acute pancreatitis have been reported. Other fatal and serious gastrointestinal (GI) complications include perforation, hemorrhage, erosion, ulcer, intestinal obstruction, enterocolitis, neutropenic colitis, and ileus. Lymphoma with preexisting GI involvement may increase the risk of perforation. In the event of new or worsening GI symptoms, including severe abdominal pain, perform a prompt diagnostic evaluation and treat appropriately.
5.13 Hyperglycemia
Serious events of hyperglycemia, such as new-onset hyperglycemia, exacerbation of pre-existing diabetes mellitus, and ketoacidosis (including fatal outcomes) have been reported in ADCETRIS-treated patients. In studies of ADCETRIS monotherapy, 8% of patients experienced any grade hyperglycemia, with 6% experiencing Grade 3 or 4 hyperglycemia. The median time to onset for any grade or Grade 3 or 4 was 1 month (range, 0-10). Hyperglycemia occurred more frequently in patients with high body mass index or diabetes. Monitor serum glucose and if hyperglycemia develops, administer anti-hyperglycemic medications as clinically indicated.
5.14 Embryo-Fetal Toxicity
Based on the mechanism of action and findings in animals, ADCETRIS can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of ADCETRIS in pregnant women. In animal reproduction studies, brentuximab vedotin caused embryo-fetal toxicities, including significantly decreased embryo viability, and fetal malformations at maternal exposures that were similar to the clinical dose of 1.8 mg/kg every three weeks.
Advise females of reproductive potential to use effective contraception during ADCETRIS treatment and for 2 months after the last dose of ADCETRIS. Advise male patients with female partners of reproductive potential to use effective contraception during ADCETRIS treatment and for 4 months after the last dose of ADCETRIS. Advise a pregnant woman of the potential risk to the fetus [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- •
- Peripheral Neuropathy [see Warnings and Precautions (5.1)]
- •
- Anaphylaxis and Infusion Reactions [see Warnings and Precautions (5.2)]
- •
- Hematologic Toxicities [see Warnings and Precautions (5.3)]
- •
- Serious Infections and Opportunistic Infections [see Warnings and Precautions (5.4)]
- •
- Tumor Lysis Syndrome [see Warnings and Precautions (5.5)]
- o
- Increased Toxicity in the Presence of Severe Renal Impairment [see Warnings and Precautions (5.6)]
- •
- Increased Toxicity in the Presence of Moderate or Severe Hepatic Impairment [see Warnings and Precautions (5.7)]
- •
- Hepatotoxicity [see Warnings and Precautions (5.8)]
- •
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.9)]
- •
- Pulmonary Toxicity [see Warnings and Precautions (5.10)]
- •
- Serious Dermatologic Reactions [see Warnings and Precautions (5.11)]
- •
- Gastrointestinal Complications [see Warnings and Precautions (5.12)]
- •
- Hyperglycemia [see Warnings and Precautions (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data below reflect exposure to ADCETRIS in 931 adult patients with cHL including 662 patients who received ADCETRIS in combination with chemotherapy in a randomized controlled trial, 269 who received ADCETRIS as monotherapy (167 in a randomized controlled trial and 102 in a single arm trial), and 296 pediatric patients with high risk cHL who received ADCETRIS in combination with chemotherapy. Data summarizing ADCETRIS exposure are also provided for 347 patients with T-cell lymphoma, including 223 patients with PTCL who received ADCETRIS in combination with chemotherapy in a randomized, double-blind, controlled trial; 58 patients with sALCL who received ADCETRIS monotherapy in a single-arm trial; and 66 patients with pcALCL or CD30-expressing MF who received ADCETRIS monotherapy in a randomized, controlled trial. ADCETRIS was administered intravenously at a dose of either 1.2 mg/kg every 2 weeks in combination with AVD, 1.8 mg/kg every 3 weeks in combination with AVEPC in pediatric patients, 1.8 mg/kg every 3 weeks in combination with CHP, or 1.8 mg/kg every 3 weeks as monotherapy.
The most common adverse reactions (≥20%) with monotherapy in adult patients were peripheral neuropathy, fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, diarrhea, pyrexia, rash, and cough.
The most common laboratory abnormalities (≥20%) with monotherapy in adult patients were decreased neutrophils, increased creatinine, increased glucose, increased aspartate aminotransferase (AST), increased alanine aminotransferase (ALT), decreased lymphocytes, decreased hemoglobin, and decreased platelets.
The most common adverse reactions (≥20%) with combination therapy in adult patients were peripheral neuropathy, nausea, fatigue, musculoskeletal pain, constipation, diarrhea, mucositis, vomiting, abdominal pain, pyrexia, alopecia, upper respiratory tract infection, and rash.
The most common laboratory abnormalities (≥20%) with combination therapy in adult patients were decreased neutrophils, increased creatinine, decreased hemoglobin, decreased lymphocytes, increased ALT, increased AST, increased glucose, and increased uric acid.
The most common Grade ≥3 adverse reactions (≥5%) in combination with AVEPC in pediatric patients were neutropenia, anemia, thrombocytopenia, febrile neutropenia, stomatitis, and infection.
Previously Untreated Stage III or IV Classical Hodgkin Lymphoma (Study 5: ECHELON-1)
ADCETRIS in combination with AVD was evaluated for the treatment of previously untreated patients with Stage III or IV cHL in a randomized, open-label, multicenter clinical trial of 1334 patients. Patients were randomized to receive up to 6 cycles of ADCETRIS + AVD or ABVD on Days 1 and 15 of each 28‑day cycle. The recommended starting dose of ADCETRIS was 1.2 mg/kg intravenously over 30 minutes, administered approximately 1 hour after completion of AVD therapy. A total of 1321 patients received at least one dose of study treatment (662 ADCETRIS + AVD, 659 ABVD). The median number of treatment cycles in each study arm was 6 (range, 1–6); 76% of patients on the ADCETRIS + AVD arm received 12 doses of ADCETRIS [see Clinical Studies (14.1)].
After 75% of patients had started study treatment, the use of prophylactic G‑CSF was recommended with the initiation of treatment for all ADCETRIS + AVD treated patients, based on the observed rates of neutropenia and febrile neutropenia [see Dosage and Administration (2.2)]. Among 579 patients on the ADCETRIS + AVD arm who did not receive G‑CSF primary prophylaxis beginning with Cycle 1, 96% experienced neutropenia (21% with Grade 3; 67% with Grade 4), and 21% had febrile neutropenia (14% with Grade 3; 6% with Grade 4). Among 83 patients on the ADCETRIS + AVD arm who received G-CSF primary prophylaxis beginning with Cycle 1, 61% experienced neutropenia (13% with Grade 3; 27% with Grade 4), and 11% experienced febrile neutropenia (8% with Grade 3; 2% with Grade 4).
Serious adverse reactions were reported in 43% of ADCETRIS + AVD-treated patients and 27% of ABVD-treated patients. The most common serious adverse reactions in ADCETRIS + AVD-treated patients were febrile neutropenia (17%), pyrexia (7%), neutropenia and pneumonia (3% each).
Adverse reactions that led to dose delays of one or more drugs in more than 5% of ADCETRIS + AVD-treated patients were neutropenia (21%) and febrile neutropenia (8%) [see Dosage and Administration (2.2)]. Adverse reactions led to treatment discontinuation of one or more drugs in 13% of ADCETRIS + AVD-treated patients. Seven percent of patients treated with ADCETRIS + AVD discontinued due to peripheral neuropathy.
There were 9 on-study deaths among ADCETRIS + AVD-treated patients; 7 were associated with neutropenia, and none of these patients had received G-CSF prior to developing neutropenia.
| AVD = doxorubicin, vinblastine, and dacarbazine ABVD = doxorubicin, bleomycin, vinblastine, and dacarbazine Events were graded using the NCI CTCAE Version 4.03 Events listed are those having a ≥5% difference in rate between treatment arms |
||||||
|
|
ADCETRIS + AVD
|
ABVD
|
||||
|
Body System
|
Any Grade |
Grade 3 |
Grade 4 |
Any Grade |
Grade 3 |
Grade 4 |
|
Blood and lymphatic system disorders |
||||||
|
Anemia* |
98 |
11 |
<1 |
92 |
6 |
<1 |
|
Neutropenia* |
91 |
20 |
62 |
89 |
31 |
42 |
|
Febrile neutropenia |
19 |
13 |
6 |
8 |
6 |
2 |
|
Gastrointestinal disorders |
||||||
|
Constipation |
42 |
2 |
- |
37 |
<1 |
<1 |
|
Vomiting |
33 |
3 |
- |
28 |
1 |
- |
|
Diarrhea |
27 |
3 |
<1 |
18 |
<1 |
- |
|
Stomatitis |
21 |
2 |
- |
16 |
<1 |
- |
|
Abdominal pain |
21 |
3 |
- |
10 |
<1 |
- |
|
Nervous system disorders |
||||||
|
Peripheral sensory neuropathy |
65 |
10 |
<1 |
41 |
2 |
- |
|
Peripheral motor neuropathy |
11 |
2 |
- |
4 |
<1 |
- |
|
General disorders and administration site conditions |
||||||
|
Pyrexia |
27 |
3 |
<1 |
22 |
2 |
- |
|
Musculoskeletal and connective tissue disorders |
||||||
|
Bone pain |
19 |
<1 |
- |
10 |
<1 |
- |
|
Back pain |
13 |
<1 |
- |
7 |
- |
- |
|
Skin and subcutaneous tissue disorders |
||||||
|
Rashes, eruptions and exanthems† |
13 |
<1 |
<1 |
8 |
<1 |
- |
|
Respiratory, thoracic and mediastinal disorders |
||||||
|
Dyspnea |
12 |
1 |
- |
19 |
2 |
- |
|
Investigations |
||||||
|
Decreased weight |
22 |
<1 |
- |
6 |
<1 |
- |
|
Increased alanine aminotransferase |
10 |
3 |
- |
4 |
<1 |
- |
|
Metabolism and nutrition disorders |
||||||
|
Decreased appetite |
18 |
<1 |
- |
12 |
<1 |
- |
|
Psychiatric disorders |
||||||
|
Insomnia |
19 |
<1 |
- |
12 |
<1 |
- |
Previously Untreated High Risk Classical Hodgkin Lymphoma (cHL)
Study 7: AHOD1331
The safety of ADCETRIS was evaluated in Study 7: AHOD1331 [see Clinical Studies (14.1)]. The study included pediatric patients with previously untreated high risk cHL. Patients received ADCETRIS plus AVEPC chemotherapy at 1.8 mg/kg intravenously over 30 minutes prior to other chemotherapy in 21-day cycles (n = 296) or ABVE-PC in 21-day cycles (n = 297). Among patients who received ADCETRIS in combination with AVEPC chemotherapy, the median number of treatment cycles was 5 (range, 1-5).
Serious adverse reactions occurred in 22% of patients who received ADCETRIS plus AVEPC chemotherapy. Serious adverse reactions in >2% of patients included hypotension (3%) and febrile neutropenia (3%).
|
||||
|
ADCETRIS + AVEPC
|
ABVE-PC
|
|||
|
System Organ Class
|
Grade 3 |
Grade 4 |
Grade 3 |
Grade 4 |
|
Blood and lymphatic system disorders |
||||
|
Anemia |
35 |
1.7 |
28 |
2 |
|
Febrile neutropenia |
28 |
3.4 |
31 |
1.7 |
|
Lymphopenia |
13 |
11 |
8 |
18 |
|
Thrombocytopenia* |
10 |
22 |
11 |
16 |
|
Neutropenia |
8 |
43 |
4.4 |
36 |
|
Gastrointestinal disorders |
||||
|
Stomatitis |
10 |
- |
7 |
- |
|
Nausea |
3.7 |
- |
2 |
- |
|
Vomiting |
3.7 |
- |
1.3 |
- |
|
Diarrhea |
2.4 |
- |
0.3 |
- |
|
Colitis |
2 |
0.3 |
1 |
- |
|
Infections and infestations |
||||
|
Infections† |
9 |
2.7 |
7 |
3.4 |
|
Nervous system disorders |
||||
|
Peripheral sensory neuropathy |
6 |
- |
4.4 |
- |
|
Metabolism and nutrition disorders |
||||
|
Hypokalemia |
5 |
0.7 |
6 |
1 |
|
Hyponatremia |
3.4 |
- |
3 |
- |
|
Decreased appetite |
2.7 |
- |
1.7 |
- |
|
Dehydration |
2.7 |
- |
1 |
- |
|
Hepatobiliary disorders |
||||
|
Alanine aminotransferase increased |
3.7 |
0.3 |
2.7 |
0.3 |
|
General disorders and administration site conditions |
||||
|
Infusion-related reactions‡ |
3 |
1 |
5 |
1 |
Classical Hodgkin Lymphoma Post-Auto-HSCT Consolidation (Study 3: AETHERA)
ADCETRIS was studied in 329 patients with cHL at high risk of relapse or progression post-auto-HSCT in a randomized, double-blind, placebo-controlled clinical trial in which the recommended starting dose and schedule was 1.8 mg/kg of ADCETRIS administered intravenously over 30 minutes every 3 weeks or placebo for up to 16 cycles. Of the 329 enrolled patients, 327 (167 ADCETRIS, 160 placebo) received at least one dose of study treatment. The median number of treatment cycles in each study arm was 15 (range, 1–16) and 80 patients (48%) in the ADCETRIS-treatment arm received 16 cycles [see Clinical Studies (14.1)].
Standard international guidelines were followed for infection prophylaxis for herpes simplex virus (HSV), varicella-zoster virus (VZV), and Pneumocystis jiroveci pneumonia (PJP) post-auto-HSCT. Overall, 312 patients (95%) received HSV and VZV prophylaxis with a median duration of 11.1 months (range, 0–20) and 319 patients (98%) received PJP prophylaxis with a median duration of 6.5 months (range, 0–20).
Adverse reactions that led to dose delays in more than 5% of ADCETRIS-treated patients were neutropenia (22%), peripheral sensory neuropathy (16%), upper respiratory tract infection (6%), and peripheral motor neuropathy (6%) [see Dosage and Administration (2.3)]. Adverse reactions led to treatment discontinuation in 32% of ADCETRIS-treated patients. Adverse reactions that led to treatment discontinuation in 2 or more patients were peripheral sensory neuropathy (14%), peripheral motor neuropathy (7%), acute respiratory distress syndrome (1%), paresthesia (1%), and vomiting (1%). Serious adverse reactions were reported in 25% of ADCETRIS-treated patients. The most common serious adverse reactions were pneumonia (4%), pyrexia (4%), vomiting (3%), nausea (2%), hepatotoxicity (2%), and peripheral sensory neuropathy (2%).
| Events were graded using the NCI CTCAE Version 4 | ||||||
|
||||||
|
|
ADCETRIS
|
Placebo
|
||||
|
Body System
|
Any Grade |
Grade 3 |
Grade 4 |
Any Grade |
Grade 3 |
Grade 4 |
|
Blood and lymphatic system disorders |
||||||
|
Neutropenia* |
78 |
30 |
9 |
34 |
6 |
4 |
|
Thrombocytopenia* |
41 |
2 |
4 |
20 |
3 |
2 |
|
Anemia* |
27 |
4 |
- |
19 |
2 |
- |
|
Nervous system disorders |
||||||
|
Peripheral sensory neuropathy |
56 |
10 |
- |
16 |
1 |
- |
|
Peripheral motor neuropathy |
23 |
6 |
- |
2 |
1 |
- |
|
Headache |
11 |
2 |
- |
8 |
1 |
- |
|
Infections and infestations |
||||||
|
Upper respiratory tract infection |
26 |
- |
- |
23 |
1 |
- |
|
General disorders and administration site conditions |
||||||
|
Fatigue |
24 |
2 |
- |
18 |
3 |
- |
|
Pyrexia |
19 |
2 |
- |
16 |
- |
- |
|
Chills |
10 |
- |
- |
5 |
- |
- |
|
Gastrointestinal disorders |
||||||
|
Nausea |
22 |
3 |
- |
8 |
- |
- |
|
Diarrhea |
20 |
2 |
- |
10 |
1 |
- |
|
Vomiting |
16 |
2 |
- |
7 |
- |
- |
|
Abdominal pain |
14 |
2 |
- |
3 |
- |
- |
|
Constipation |
13 |
2 |
- |
3 |
- |
- |
|
Respiratory, thoracic and mediastinal disorders |
||||||
|
Cough |
21 |
- |
- |
16 |
- |
- |
|
Dyspnea |
13 |
- |
- |
6 |
- |
1 |
|
Investigations |
||||||
|
Weight decreased |
19 |
1 |
- |
6 |
- |
- |
|
Musculoskeletal and connective tissue disorders |
||||||
|
Arthralgia |
18 |
1 |
- |
9 |
- |
- |
|
Muscle spasms |
11 |
- |
- |
6 |
- |
- |
|
Myalgia |
11 |
1 |
- |
4 |
- |
- |
|
Skin and subcutaneous tissue disorders |
||||||
|
Pruritus |
12 |
1 |
- |
8 |
- |
- |
|
Metabolism and nutrition disorders |
||||||
|
Decreased appetite |
12 |
1 |
- |
6 |
- |
- |
Relapsed Classical Hodgkin Lymphoma (Study 1)
ADCETRIS was studied in 102 patients with cHL in a single arm clinical trial in which the recommended starting dose and schedule was 1.8 mg/kg intravenously every 3 weeks. Median duration of treatment was 9 cycles (range, 1–16) [see Clinical Studies (14.1)].
Adverse reactions that led to dose delays in more than 5% of ADCETRIS-treated patients were neutropenia (16%) and peripheral sensory neuropathy (13%) [see Dosage and Administration (2.3)]. Adverse reactions led to treatment discontinuation in 20% of ADCETRIS-treated patients. Adverse reactions that led to treatment discontinuation in 2 or more patients were peripheral sensory neuropathy (6%) and peripheral motor neuropathy (3%). Serious adverse reactions were reported in 25% of ADCETRIS-treated patients. The most common serious adverse reactions were peripheral motor neuropathy (4%), abdominal pain (3%), pulmonary embolism (2%), pneumonitis (2%), pneumothorax (2%), pyelonephritis (2%), and pyrexia (2%).
| Events were graded using the NCI CTCAE Version 3.0 | |||
|
|||
|
|
cHL
Total N = 102
|
||
|
Body System
|
Any Grade |
Grade 3 |
Grade 4 |
|
Blood and lymphatic system disorders |
|||
|
Neutropenia* |
54 |
15 |
6 |
|
Anemia* |
33 |
8 |
2 |
|
Thrombocytopenia* |
28 |
7 |
2 |
|
Lymphadenopathy |
11 |
- |
- |
|
Nervous system disorders |
|||
|
Peripheral sensory neuropathy |
52 |
8 |
- |
|
Peripheral motor neuropathy |
16 |
4 |
- |
|
Headache |
19 |
- |
- |
|
Dizziness |
11 |
- |
- |
|
General disorders and administration site conditions |
|||
|
Fatigue |
49 |
3 |
- |
|
Pyrexia |
29 |
2 |
- |
|
Chills |
13 |
- |
- |
|
Infections and infestations |
|||
|
Upper respiratory tract infection |
47 |
- |
- |
|
Gastrointestinal disorders |
|||
|
Nausea |
42 |
- |
- |
|
Diarrhea |
36 |
1 |
- |
|
Abdominal pain |
25 |
2 |
1 |
|
Vomiting |
22 |
- |
- |
|
Constipation |
16 |
- |
- |
|
Skin and subcutaneous tissue disorders |
|||
|
Rash |
27 |
- |
- |
|
Pruritus |
17 |
- |
- |
|
Alopecia |
13 |
- |
- |
|
Night sweats |
12 |
- |
- |
|
Respiratory, thoracic and mediastinal disorders |
|||
|
Cough |
25 |
- |
- |
|
Dyspnea |
13 |
1 |
- |
|
Oropharyngeal pain |
11 |
- |
- |
|
Musculoskeletal and connective tissue disorders |
|||
|
Arthralgia |
19 |
- |
- |
|
Myalgia |
17 |
- |
- |
|
Back pain |
14 |
- |
- |
|
Pain in extremity |
10 |
- |
- |
|
Psychiatric disorders |
|||
|
Insomnia |
14 |
- |
- |
|
Anxiety |
11 |
2 |
- |
|
Metabolism and nutrition disorders |
|||
|
Decreased appetite |
11 |
- |
- |
Previously Untreated Systemic Anaplastic Large Cell Lymphoma or Other CD30-Expressing Peripheral T-Cell Lymphomas (Study 6, ECHELON-2)
ADCETRIS in combination with CHP was evaluated in patients with previously untreated, CD30-expressing PTCL in a multicenter randomized, double-blind, double dummy, actively controlled trial. Patients were randomized to receive ADCETRIS + CHP or CHOP for 6 to 8, 21-day cycles. ADCETRIS was administered on Day 1 of each cycle, with a starting dose of 1.8 mg/kg intravenously over 30 minutes, approximately 1 hour after completion of CHP [see Clinical Studies (14.2)]. The trial required hepatic transaminases ≤3 times upper limit of normal (ULN), total bilirubin ≤1.5 times ULN, and serum creatinine ≤2 times ULN and excluded patients with Grade 2 or higher peripheral neuropathy.
A total of 449 patients were treated (223 with ADCETRIS + CHP, 226 with CHOP), with 6 cycles planned in 81%. In the ADCETRIS + CHP arm, 70% of patients received 6 cycles, and 18% received 8 cycles. Primary prophylaxis with G-CSF was administered to 34% of ADCETRIS + CHP-treated patients and 27% of CHOP-treated patients.
Fatal adverse reactions occurred in 3% of patients in the A+CHP arm and in 4% of patients in the CHOP arms, most often from infection. Serious adverse reactions were reported in 38% of ADCETRIS + CHP- treated patients and 35% of CHOP-treated patients. Serious adverse reactions occurring in >2% of ADCETRIS + CHP-treated patients included febrile neutropenia (14%), pneumonia (5%), pyrexia (4%), and sepsis (3%).
The most common adverse reactions observed ≥2% more in recipients of ADCETRIS + CHP were nausea, diarrhea, fatigue or asthenia, mucositis, pyrexia, vomiting, and anemia. Other common (≥10%) adverse reactions observed ≥2% more with ADCETRIS + CHP were febrile neutropenia, abdominal pain, decreased appetite, dyspnea, edema, cough, dizziness, hypokalemia, decreased weight, and myalgia.
In recipients of ADCETRIS + CHP, adverse reactions led to dose delays of ADCETRIS in 25% of patients, dose reduction in 9% (most often for peripheral neuropathy), and discontinuation of ADCETRIS with or without the other components in 7% (most often from peripheral neuropathy and infection).
| The table includes a combination of grouped and ungrouped terms. CHP = cyclophosphamide, doxorubicin, and prednisone; CHOP = cyclophosphamide, doxorubicin, vincristine, and prednisone Events were graded using the NCI CTCAE Version 4.03 |
||||||
|
||||||
|
|
ADCETRIS + CHP
|
CHOP
|
||||
|
Body System
|
Any Grade |
Grade 3 |
Grade 4 |
Any Grade |
Grade 3 |
Grade 4 |
|
Blood and lymphatic system disorders |
||||||
|
Anemia* |
66 |
13 |
<1 |
59 |
12 |
<1 |
|
Neutropenia* |
59 |
17 |
22 |
58 |
14 |
22 |
|
Lymphopenia* |
51 |
18 |
1 |
57 |
19 |
2 |
|
Febrile neutropenia |
19 |
17 |
2 |
16 |
12 |
4 |
|
Thrombocytopenia* |
17 |
3 |
3 |
13 |
3 |
2 |
|
Gastrointestinal disorders |
||||||
|
Nausea |
46 |
2 |
- |
39 |
2 |
- |
|
Diarrhea |
38 |
6 |
- |
20 |
<1 |
- |
|
Mucositis |
30 |
2 |
<1 |
27 |
3 |
- |
|
Constipation |
29 |
<1 |
<1 |
30 |
1 |
- |
|
Vomiting |
26 |
<1 |
- |
17 |
2 |
- |
|
Abdominal pain |
17 |
1 |
- |
13 |
<1 |
- |
|
Nervous system disorders |
||||||
|
Peripheral neuropathy |
52 |
3 |
<1 |
55 |
4 |
- |
|
Headache |
15 |
<1 |
- |
15 |
<1 |
- |
|
Dizziness |
13 |
- |
- |
9 |
<1 |
- |
|
General disorders and administration site conditions |
||||||
|
Fatigue or asthenia |
35 |
2 |
- |
29 |
2 |
- |
|
Pyrexia |
26 |
1 |
<1 |
19 |
- |
- |
|
Edema |
15 |
<1 |
- |
12 |
<1 |
- |
|
Infections and infestations |
||||||
|
Upper respiratory tract infection |
14 |
<1 |
- |
15 |
<1 |
- |
|
Skin and subcutaneous disorders |
||||||
|
Alopecia |
26 |
- |
- |
25 |
1 |
- |
|
Rash |
16 |
1 |
<1 |
14 |
1 |
- |
|
Musculoskeletal and connective tissue disorders |
||||||
|
Myalgia |
11 |
- |
- |
8 |
- |
- |
|
Respiratory, thoracic and mediastinal disorders |
||||||
|
Dyspnea |
15 |
2 |
- |
11 |
2 |
- |
|
Cough |
13 |
<1 |
- |
10 |
- |
- |
|
Metabolism and nutrition disorders |
||||||
|
Decreased appetite |
17 |
1 |
- |
12 |
1 |
- |
|
Hypokalemia |
12 |
4 |
- |
8 |
<1 |
<1 |
|
Investigations |
||||||
|
Weight decreased |
12 |
<1 |
- |
8 |
<1 |
- |
|
Psychiatric disorders |
||||||
|
Insomnia |
11 |
- |
- |
14 |
- |
- |
Relapsed Systemic Anaplastic Large Cell Lymphoma (Study 2)
ADCETRIS was studied in 58 patients with sALCL in a single arm clinical trial in which the recommended starting dose and schedule was 1.8 mg/kg intravenously every 3 weeks. Median duration of treatment was 7 cycles (range, 1–16) [see Clinical Studies (14.2)].
Adverse reactions that led to dose delays in more than 5% of ADCETRIS-treated patients were neutropenia (12%) and peripheral sensory neuropathy (7%) [see Dosage and Administration (2.3)]. Adverse reactions led to treatment discontinuation in 19% of ADCETRIS-treated patients. The adverse reaction that led to treatment discontinuation in 2 or more patients was peripheral sensory neuropathy (5%). Serious adverse reactions were reported in 41% of ADCETRIS-treated patients. The most common serious adverse reactions were septic shock (3%), supraventricular arrhythmia (3%), pain in extremity (3%), and urinary tract infection (3%).
| sALCL Total N = 58 % of patients |
|||
|---|---|---|---|
| Body System
Adverse Reaction | Any Grade | Grade 3 | Grade 4 |
| Events were graded using the NCI CTCAE Version 3.0 | |||
|
|||
|
Blood and lymphatic system disorders |
|||
|
Neutropenia* |
55 |
12 |
9 |
|
Anemia* |
52 |
2 |
- |
|
Thrombocytopenia* |
16 |
5 |
5 |
|
Lymphadenopathy |
10 |
- |
- |
|
Nervous system disorders |
|||
|
Peripheral sensory neuropathy |
53 |
10 |
- |
|
Headache |
16 |
2 |
- |
|
Dizziness |
16 |
- |
- |
|
General disorders and administration site conditions |
|||
|
Fatigue |
41 |
2 |
2 |
|
Pyrexia |
38 |
2 |
- |
|
Chills |
12 |
- |
- |
|
Pain |
28 |
- |
5 |
|
Edema peripheral |
16 |
- |
- |
|
Infections and infestations |
|||
|
Upper respiratory tract infection |
12 |
- |
- |
|
Gastrointestinal disorders |
|||
|
Nausea |
38 |
2 |
- |
|
Diarrhea |
29 |
3 |
- |
|
Vomiting |
17 |
3 |
- |
|
Constipation |
19 |
2 |
- |
|
Skin and subcutaneous tissue disorders |
|||
|
Rash |
31 |
- |
- |
|
Pruritus |
19 |
- |
- |
|
Alopecia |
14 |
- |
- |
|
Dry skin |
10 |
- |
- |
|
Respiratory, thoracic and mediastinal disorders |
|||
|
Cough |
17 |
- |
- |
|
Dyspnea |
19 |
2 |
- |
|
Musculoskeletal and connective tissue disorders |
|||
|
Myalgia |
16 |
2 |
- |
|
Back pain |
10 |
2 |
- |
|
Pain in extremity |
10 |
2 |
2 |
|
Muscle spasms |
10 |
2 |
- |
|
Psychiatric disorders |
|||
|
Insomnia |
16 |
- |
- |
|
Metabolism and nutrition disorders |
|||
|
Decreased appetite |
16 |
2 |
- |
|
Investigations |
|||
|
Weight decreased |
12 |
3 |
- |
Primary Cutaneous Anaplastic Large Cell Lymphoma and CD30-Expressing Mycosis Fungoides (Study 4: ALCANZA)
ADCETRIS was studied in 131 patients with pcALCL or CD30-expressing MF requiring systemic therapy in a randomized, open-label, multicenter clinical trial in which the recommended starting dose and schedule was ADCETRIS 1.8 mg/kg intravenously over 30 minutes every 3 weeks or physician’s choice of either methotrexate 5 to 50 mg orally weekly or bexarotene 300 mg/m2 orally daily.
Of the 131 enrolled patients, 128 (66 brentuximab vedotin, 62 physician’s choice) received at least one dose of study treatment. The median number of treatment cycles in the ADCETRIS treatment arm was 12 (range, 1–16) compared to 3 (range, 1–16) and 6 (range, 1–16) in the methotrexate and bexarotene arms, respectively. Twenty-four (24) patients (36%) in the ADCETRIS-treatment arm received 16 cycles compared to 5 patients (8%) in the physician’s choice arm [see Clinical Studies (14.2)].
Adverse reactions that led to dose delays in more than 5% of ADCETRIS-treated patients were peripheral sensory neuropathy (15%) and neutropenia (6%) [see Dosage and Administration (2.3)]. Adverse reactions led to treatment discontinuation in 24% of ADCETRIS-treated patients. The most common adverse reaction that led to treatment discontinuation was peripheral neuropathy (12%). Serious adverse reactions were reported in 29% of ADCETRIS-treated patients. The most common serious adverse reactions were cellulitis (3%) and pyrexia (3%).
| ADCETRIS Total N = 66 % of patients | Physician’s Choice*
Total N = 62 % of patients |
|||||
|---|---|---|---|---|---|---|
| Body System
Adverse Reaction | Any Grade | Grade 3 | Grade 4 | Any Grade | Grade 3 | Grade 4 |
| Events were graded using the NCI CTCAE Version 4.03 | ||||||
|
Blood and lymphatic system disorders |
||||||
|
Anemia† |
62 |
- |
- |
65 |
5 |
- |
|
Neutropenia† |
21 |
3 |
2 |
24 |
5 |
- |
|
Thrombocytopenia† |
15 |
2 |
2 |
2 |
- |
- |
|
Nervous system disorders |
||||||
|
Peripheral sensory neuropathy |
45 |
5 |
- |
2 |
- |
- |
|
Gastrointestinal disorders | ||||||
|
Nausea |
36 |
2 |
- |
13 |
- |
- |
|
Diarrhea |
29 |
3 |
- |
6 |
- |
- |
|
Vomiting |
17 |
2 |
- |
5 |
- |
- |
|
General disorders and administration site conditions |
||||||
|
Fatigue |
29 |
5 |
- |
27 |
2 |
- |
|
Pyrexia |
17 |
- |
- |
18 |
2 |
- |
|
Edema peripheral |
11 |
- |
- |
10 |
- |
- |
|
Asthenia |
11 |
2 |
- |
8 |
- |
2 |
|
Skin and subcutaneous tissue disorders |
||||||
|
Pruritus |
17 |
2 |
- |
13 |
3 |
- |
|
Alopecia |
15 |
- |
- |
3 |
- |
- |
|
Rash maculo-papular |
11 |
2 |
- |
5 |
- |
- |
|
Pruritus generalized |
11 |
2 |
- |
2 |
- |
- |
|
Metabolism and nutrition disorders |
||||||
|
Decreased appetite |
15 |
- |
- |
5 |
- |
- |
|
Musculoskeletal and connective tissue disorders |
||||||
|
Arthralgia |
12 |
- |
- |
6 |
- |
- |
|
Myalgia |
12 |
- |
- |
3 |
- |
- |
|
Respiratory, thoracic and mediastinal disorders |
||||||
|
Dyspnea |
11 |
- |
- |
- |
- |
- |
Relapsed or Refractory Large B-Cell Lymphoma (Study 8: ECHELON-3)
The safety of ADCETRIS in combination with lenalidomide and a rituximab product was evaluated in ECHELON-3, a randomized, multicenter, double-blind, placebo-controlled trial in patients with relapsed or refractory LBCL who had received at least 2 prior lines of systemic therapy and who were not eligible for HSCT or CAR T-cell therapy [see Clinical Studies (14.5)].
Patients in the treatment arm (n = 112) received ADCETRIS, 1.2 mg/kg via intravenous infusion every 3 weeks, lenalidomide, and a rituximab product. Placebo replaced ADCETRIS in the placebo plus lenalidomide and rituximab arm (n = 116).
The trial required an absolute neutrophil count ≥1,000/µL, platelet count ≥50,000/µL, creatinine clearance (CrCL) ≥45 mL/min, hepatic transaminases ≤3 times the upper limit of normal (ULN), and bilirubin <1.5 times ULN. The trial excluded patients having Eastern Cooperative Oncology Group (ECOG) performance status above 2, active central nervous system (CNS) lymphoma, and Grade 2 or higher peripheral neuropathy. Granulocyte colony-stimulating factor (G-CSF) primary prophylaxis was required and administered to 98% of patients in the ADCETRIS plus lenalidomide and rituximab arm and 91% of patients in the lenalidomide and rituximab arm.
The median age was 71 years (range: 21 to 89 years); 44% of patients were female; 53% were White, 26% were Asian, and 4% were Hispanic or Latino. There were no Black or African American patients enrolled in ECHELON-3. Among patients who received ADCETRIS, the median number of treatment cycles was 5 (range, 1-34).
Serious adverse reactions occurred in 60% of patients who received ADCETRIS in combination with lenalidomide and a rituximab product. Serious adverse reactions that occurred in >2% of patients included pneumonia (21%), COVID-19 (13%, includes COVID-19 pneumonia), sepsis (9%), febrile neutropenia (7%), hemorrhage (3.6%), urinary tract infection (3.6%), thrombocytopenia (2.7%) and upper respiratory tract infection (2.7%). Fatal adverse reactions occurred in 12% of patients who received ADCETRIS in combination with lenalidomide and a rituximab product, including COVID-19 (4.5%, includes COVID-19 pneumonia), pneumonia (3.6%), and sepsis (1.8%).
Adverse reactions led to dose reduction of ADCETRIS in 6% of patients, all due to peripheral neuropathy. Adverse reactions leading to dose delay of ADCETRIS in more than 5% of patients included neutropenia (23%), COVID-19 (13%), pneumonia (8%), and thrombocytopenia (8%).
Adverse reactions led to discontinuation of ADCETRIS in 20% of patients. Adverse reactions that led to treatment discontinuation in 3 or more patients included peripheral neuropathy (4.5%) and pneumonia (2.7%).
| ADCETRIS + Lenalidomide + Rituximab N = 112 | Placebo + Lenalidomide + Rituximab N = 116 |
|||
|---|---|---|---|---|
| Body System
Adverse Reaction | All Grades
% | Grade 3-4
% | All Grades
% | Grade 3-4
% |
|
||||
|
General disorders and Administration Site Conditions |
||||
|
Fatigue* |
46 |
10 |
29 |
5 |
|
Pyrexia |
15 |
1.8 |
15 |
0.9 |
|
Gastrointestinal disorders |
||||
|
Diarrhea* |
31 |
4.5 |
23 |
1.7 |
|
Constipation |
17 |
1.8 |
18 |
0 |
|
Nausea |
15 |
0.9 |
16 |
0.9 |
|
Abdominal pain* |
12 |
1.8 |
12 |
1.7 |
|
Stomatitis* |
11 |
0 |
7 |
0 |
|
Nervous system disorders |
||||
|
Peripheral neuropathy† |
27 |
5 |
21 |
0 |
|
Infections and Infestations* |
||||
|
COVID-19‡ |
27 |
13 |
16 |
8 |
|
Pneumonia§ |
27 |
21 |
10 |
9 |
|
Upper respiratory tract infection* |
12 |
2.7 |
5 |
0 |
|
Skin and subcutaneous tissue disorders |
||||
|
Rash* |
27 |
2.7 |
16 |
0.9 |
|
Pruritus* |
17 |
0 |
6 |
0 |
|
Renal and urinary disorders |
||||
|
Renal insufficiency |
20 |
3.6 |
14 |
4.3 |
|
Respiratory, thoracic and mediastinal disorders |
||||
|
Cough |
17 |
0 |
9 |
0 |
|
Dyspnea* |
12 |
0.9 |
14 |
2.6 |
|
Metabolism and nutrition disorders |
||||
|
Decreased appetite |
17 |
0.9 |
9 |
0 |
|
Investigations |
||||
|
Weight decreased |
13 |
0.9 |
5 |
0.9 |
Clinically relevant adverse reactions in <10% of patients who received ADCETRIS in combination with lenalidomide and a rituximab product include febrile neutropenia, edema, hypotension, urinary tract infection, sepsis, respiratory tract infection, vomiting, back pain, dizziness, arthralgia, herpes virus infection, bone pain, atrial fibrillation or flutter, lower respiratory tract infection, and cardiac failure.
| ADCETRIS + Lenalidomide + Rituximab N = 112* | Placebo + Lenalidomide + Rituximab N = 116* |
|||
|---|---|---|---|---|
| Laboratory Abnormality* | All Grades
(%) | Grade 3-4
(%) | All Grades
(%) | Grade 3-4
(%) |
|
||||
|
Hematology | ||||
|
Neutrophils decreased |
77 |
49 |
63 |
42 |
|
Lymphocytes decreased |
65 |
38 |
53 |
30 |
|
Platelets decreased |
65 |
29 |
54 |
18 |
|
Hemoglobin decreased |
54 |
19 |
49 |
14 |
|
Chemistry | ||||
|
Alanine aminotransferase increased |
31 |
0.9 |
17 |
0 |
|
Potassium decreased |
31 |
7 |
29 |
2.9 |
|
Albumin decreased |
29 |
0.9 |
25 |
1 |
|
Creatinine increased |
26 |
2.8 |
23 |
0 |
|
Calcium decreased |
21 |
0.9 |
7 |
0 |
Additional Important Adverse Reactions
Infusion reactions
In studies of ADCETRIS as monotherapy (Studies 1–4), 13% of ADCETRIS-treated patients experienced infusion-related reactions. The most common adverse reactions in Studies 1–4 (≥3% in any study) associated with infusion-related reactions were chills (4%), nausea (3–4%), dyspnea (2–3%), pruritus (2–5%), pyrexia (2%), and cough (2%). Grade 3 events were reported in 5 of the 51 ADCETRIS-treated patients who experienced infusion-related reactions.
In a study of ADCETRIS in combination with AVD (Study 5, ECHELON-1), infusion-related reactions were reported in 57 patients (9%) in the ADCETRIS + AVD-treated arm. Grade 3 events were reported in 3 of the 57 patients treated with ADCETRIS + AVD who experienced infusion-related reactions. The most common adverse reaction (≥2%) associated with infusion-related reactions was nausea (2%).
In a study of ADCETRIS in combination with CHP (Study 6, ECHELON-2), infusion-related reactions were reported in 10 patients (4%) in the ADCETRIS + CHP-treated arm: 2 (1%) patients with events that were Grade 3 or higher events, and 8 (4%) patients with events that were less than Grade 3.
In a study of ADCETRIS in combination with lenalidomide and rituximab (Study 8, ECHELON-3), Grade 1 or 2 infusion-related reactions were reported in 6 patients (5%) in the ADCETRIS + lenalidomide + rituximab arm.
Pulmonary toxicity
In a trial in patients with cHL that studied ADCETRIS with bleomycin as part of a combination regimen, the rate of non-infectious pulmonary toxicity was higher than the historical incidence reported with ABVD (doxorubicin, bleomycin, vinblastine, dacarbazine). Patients typically reported cough and dyspnea. Interstitial infiltration and/or inflammation were observed on radiographs and computed tomographic imaging of the chest. Most patients responded to corticosteroids. The concomitant use of ADCETRIS with bleomycin is contraindicated [see Contraindications (4)].
In a study of ADCETRIS in combination with AVD (Study 5, ECHELON-1), non-infectious pulmonary toxicity events were reported in 12 patients (2%) in the ADCETRIS + AVD arm. These events included lung infiltration (6 patients) and pneumonitis (6 patients), or interstitial lung disease (1 patient).
In a study of ADCETRIS in combination with CHP (Study 6, ECHELON-2), non-infectious pulmonary toxicity events were reported in 5 patients (2%) in the ADCETRIS + CHP arm; all 5 events were pneumonitis. Cases of pulmonary toxicity have also been reported in patients receiving ADCETRIS monotherapy. In Study 3 (AETHERA), pulmonary toxicity was reported in 8 patients (5%) in the ADCETRIS-treated arm and 5 patients (3%) in the placebo arm.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
During treatment in patients with relapsed or refractory cHL and relapsed or refractory systemic ALCL in Studies 1 and 2, two of the patients (1%) with persistently positive antibodies experienced adverse reactions consistent with infusion reactions that led to discontinuation of treatment [see Warnings and Precautions (5.2)]. Overall, a higher incidence of infusion-related reactions was observed in patients who developed persistently positive antibodies [see Clinical Pharmacology (12.6)].
6.2 Post Marketing Experience
The following adverse reactions have been identified during post-approval use of ADCETRIS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: febrile neutropenia [see Warnings and Precautions (5.3)].
Gastrointestinal disorders: acute pancreatitis and gastrointestinal complications (including fatal outcomes) [see Warnings and Precautions (5.12)].
Hepatobiliary disorders: hepatotoxicity [see Warnings and Precautions (5.8)].
Infections: PML [see Boxed Warning, Warnings and Precautions (5.9)], serious infections and opportunistic infections [see Warnings and Precautions (5.4)].
Metabolism and nutrition disorders: hyperglycemia [see Warnings and Precautions (5.13)].
Respiratory, thoracic and mediastinal disorders: noninfectious pulmonary toxicity including pneumonitis, interstitial lung disease, and ARDS (some with fatal outcomes) [see Warnings and Precautions (5.10) and Adverse Reactions (6.1)].
Skin and subcutaneous tissue disorders: Toxic epidermal necrolysis, including fatal outcomes [see Warnings and Precautions (5.11)].
Related/similar drugs
7. Drug Interactions
7.1 Effect of Other Drugs on ADCETRIS
CYP3A4 Inhibitors: Co-administration of ADCETRIS with ketoconazole, a potent CYP3A4 inhibitor, increased exposure to MMAE [see Clinical Pharmacology (12.3)], which may increase the risk of adverse reaction. Closely monitor adverse reactions when ADCETRIS is given concomitantly with strong CYP3A4 inhibitors.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
ADCETRIS can cause fetal harm based on the findings from animal studies and the drug’s mechanism of action [see Clinical Pharmacology (12.1)]. In animal reproduction studies, administration of brentuximab vedotin to pregnant rats during organogenesis at doses similar to the clinical dose of 1.8 mg/kg every three weeks caused embryo-fetal toxicities, including congenital malformations (see Data). The available data from case reports on ADCETRIS use in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. Advise a pregnant woman of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Animal Data
In an embryo-fetal developmental study, pregnant rats received 2 intravenous doses of 0.3, 1, 3, or 10 mg/kg brentuximab vedotin during the period of organogenesis (once each on Pregnancy Days 6 and 13). Drug-induced embryo-fetal toxicities were seen mainly in animals treated with 3 and 10 mg/kg of the drug and included increased early resorption (≥99%), post-implantation loss (≥99%), decreased numbers of live fetuses, and external malformations (i.e., umbilical hernias and malrotated hindlimbs). Systemic exposure in animals at the brentuximab vedotin dose of 3 mg/kg is approximately the same exposure in patients with cHL or sALCL who received the recommended dose of 1.8 mg/kg every three weeks.
8.2 Lactation
Risk Summary
There is no information regarding the presence of brentuximab vedotin in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child from ADCETRIS, including cytopenias and neurologic or gastrointestinal toxicities, advise patients that breastfeeding is not recommended during ADCETRIS treatment.
8.3 Females and Males of Reproductive Potential
ADCETRIS can cause fetal harm based on the findings from animal studies and the drug’s mechanism of action [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating ADCETRIS therapy.
Contraception
Females
Advise females of reproductive potential to use effective contraception during ADCETRIS treatment and for 2 months after the last dose of ADCETRIS. Advise females to immediately report pregnancy [see Use in Specific Populations (8.1)].
Males
ADCETRIS may damage spermatozoa and testicular tissue, resulting in possible genetic abnormalities. Males with female sexual partners of reproductive potential should use effective contraception during ADCETRIS treatment and for 4 months after the last dose of ADCETRIS [see Nonclinical Toxicology (13.1)].
Infertility
Females
Based on findings in animal studies with MMAE-containing antibody-drug conjugates (ADCs), ADCETRIS may impair female fertility. The effect on fertility is reversible [see Nonclinical Toxicology (13.1)].
Males
Based on findings in rats, male fertility may be compromised by treatment with ADCETRIS [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ADCETRIS have been established in pediatric patients age 2 years and older with previously untreated high risk classical Hodgkin lymphoma in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide. The safety and effectiveness of ADCETRIS have not been established for all other indications [see Indications and Usage (1)].
Previously Untreated, High Risk Classical Hodgkin Lymphoma (cHL) in Combination with Doxorubicin, Vincristine, Etoposide, Prednisone, and Cyclophosphamide
The safety and effectiveness of ADCETRIS have been established in pediatric patients 2 years and older with previously untreated high risk cHL in combination with doxorubicin, vincristine, etoposide, prednisone, and cyclophosphamide chemotherapy.
Use of ADCETRIS for this indication is supported by evidence from Study 7: AHOD1331, a randomized study which included pediatric patients with previously untreated high risk cHL, including patients in the following age groups: 9 patients 3 to less than 6 years of age, 81 patients 6 to less than 12 years of age, and 345 patients 12 to less than 17 years of age [see Adverse Reactions (6.1) and Clinical Studies (14.1)].
The safety and efficacy of ADCETRIS have not been established for this indication in patients younger than 2 years.
Previously Untreated High Risk Classical Hodgkin Lymphoma (cHL) in Combination with Etoposide, Prednisone, Doxorubicin, Cyclophosphamide, Prednisone, and Dacarbazine
The safety and effectiveness of ADCETRIS in combination with etoposide (E), prednisone (P), and doxorubicin (A)/cyclophosphamide (C), prednisone (P), and dacarbazine (Dac) (AEPA/CAPDac) were assessed but have not been established based on a single arm, open-label trial (NCT01920932) in 77 patients, which included 48 pediatric patients age 6 to less than 17 with previously untreated high risk (IIB, IIIB, IVA, or IVB) cHL. No new safety signals were identified in this study.
Relapsed or Refractory Classical HL (cHL)
ADCETRIS in Combination with Gemcitabine
The safety and effectiveness of ADCETRIS in combination with gemcitabine were assessed but have not been established based on a study (NCT01780662) in 45 patients, which included 18 pediatric patients age 5 to less than 17 with relapsed or refractory cHL. No new safety signals were identified in this study.
ADCETRIS Monotherapy
The safety and effectiveness of ADCETRIS monotherapy was assessed but have not been established based on a study (NCT01492088) in 36 patients, which included 15 pediatric patients age 8 to less than 17 with relapsed or refractory cHL. No new safety signals were identified in this study.
Relapsed or Refractory Systemic ALCL (sALCL)
ADCETRIS monotherapy
The safety and effectiveness of ADCETRIS monotherapy was assessed but have not been established based on a study (NCT01492088) in 36 patients, which included 16 pediatric patients age 7 to less than 17 with sALCL. No new safety signals were identified in this study.
Newly Diagnosed ALK+ ALCL
The safety and effectiveness of ADCETRIS in combination with alternating chemotherapy Courses A (dexamethasone, ifosfamide, methotrexate, etoposide, cytarabine) and B (dexamethasone, methotrexate, cyclophosphamide, doxorubicin) administered every 21 days for a total of 6 cycles was assessed but have not been established based on a study (NCT01979536) in 67 patients, which included 61 pediatric patients age 2 to less than 17 years with newly diagnosed ALK+ ALCL. No new safety signals were identified in this study.
8.5 Geriatric Use
In the clinical trial of ADCETRIS in combination with AVD for patients with previously untreated Stage III or IV cHL (Study 5: ECHELON-1), 9% of ADCETRIS + AVD-treated patients were age 65 and older. Older age was a risk factor for febrile neutropenia, occurring in 39% of patients who were age 65 and older versus 17% of patients less than age 65, who received ADCETRIS + AVD [see Dosage and Administration (2.3)]. The ECHELON-1 trial did not contain sufficient information on patients age 65 and older to determine whether they respond differently from younger patients [see Clinical Studies (14.1)].
In the clinical trial of ADCETRIS in combination with CHP for patients with previously untreated, CD30-expressing PTCL (Study 6: ECHELON-2), 31% of ADCETRIS + CHP-treated patients were age 65 and older [see Clinical Studies (14.2)]. Among older patients, 74% had adverse reactions ≥ Grade 3 and 49% had serious adverse reactions. Among patients younger than age 65, 62% had adverse reactions ≥ Grade 3 and 33% had serious adverse reactions. Older age was a risk factor for febrile neutropenia, occurring in 29% of patients who were age 65 and older versus 14% of patients less than age 65.
Other clinical trials of ADCETRIS in cHL (Study 1; Study 3: AETHERA) and sALCL (Study 2) did not include sufficient numbers of patients who were age 65 and older to determine whether they respond differently from younger patients [see Clinical Studies (14.1, 14.3)].
In the clinical trial of ADCETRIS in pcALCL or CD30-expressing MF (Study 4: ALCANZA), 42% of ADCETRIS-treated patients were age 65 and older [see Clinical Studies (14.4)]. No meaningful differences in safety or efficacy were observed between these patients and younger patients.
In the clinical trial of ADCETRIS in relapsed or refractory LBCL (Study 8: ECHELON-3) [see Clinical Studies (14.5)], 79 (71%) of ADCETRIS-treated patients were age 65 and older. No meaningful differences in safety or efficacy were observed between these patients and younger patients.
8.6 Renal Impairment
Avoid the use of ADCETRIS in patients with severe renal impairment (CrCL <30 mL/min) [see Warnings and Precautions (5.6) and Clinical Pharmacology (12.3)]. No dosage adjustment is required for mild (CrCL >50–80 mL/min) or moderate (CrCL 30–50 mL/min) renal impairment.
8.7 Hepatic Impairment
Avoid the use of ADCETRIS in patients with moderate (Child-Pugh B) or severe (Child-Pugh C) hepatic impairment [see Warnings and Precautions (5.7) and Clinical Pharmacology (12.3)]. Dosage reduction is required in patients with mild (Child-Pugh A) hepatic impairment [see Dosage and Administration (2.3)].
Hepatic impairment for patients with relapsed or refractory large B-cell lymphoma is defined per the National Cancer Institute Organ Dysfunction Working Group.
10. Overdosage
There is no known antidote for overdosage of ADCETRIS. In case of overdosage, the patient should be closely monitored for adverse reactions, particularly neutropenia, and supportive treatment should be administered.
11. Adcetris Description
ADCETRIS (brentuximab vedotin) is a CD30-directed antibody and microtubule inhibitor conjugate consisting of three components: 1) the chimeric IgG1 antibody cAC10, specific for human CD30, 2) the microtubule disrupting agent MMAE, and 3) a protease-cleavable linker that covalently attaches MMAE to cAC10.
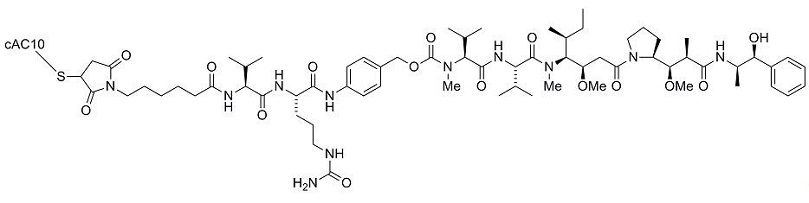
Brentuximab vedotin has an approximate molecular weight of 153 kDa. Approximately 4 molecules of MMAE are attached to each antibody molecule. Brentuximab vedotin is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ADCETRIS (brentuximab vedotin) for injection is supplied as a sterile, white to off-white, preservative-free lyophilized cake or powder in single-dose vials. Following reconstitution with 10.5 mL Sterile Water for Injection, USP, a solution containing 5 mg/mL brentuximab vedotin is produced. The reconstituted product contains 70 mg/mL trehalose dihydrate, 5.6 mg/mL sodium citrate dihydrate, 0.21 mg/mL citric acid monohydrate, and 0.20 mg/mL polysorbate 80 and water for injection. The pH is approximately 6.6.
12. Adcetris - Clinical Pharmacology
12.1 Mechanism of Action
CD30 is a member of the tumor necrosis factor receptor family and is expressed on the surface of sALCL cells and on Hodgkin Reed-Sternberg (HRS) cells in cHL. CD30 is variably expressed in other T-cell lymphomas. Expression of CD30 on healthy tissue and cells is limited. In vitro data suggest that signaling through CD30-CD30L binding may affect cell survival and proliferation.
Brentuximab vedotin is an antibody-drug conjugate (ADC). The antibody is a chimeric IgG1 directed against CD30. The small molecule, MMAE, is a microtubule-disrupting agent. MMAE is covalently attached to the antibody via a linker. Nonclinical data suggest that the anticancer activity of ADCETRIS is due to the binding of the ADC to CD30-expressing cells, followed by internalization of the ADC‑CD30 complex, and the release of MMAE via proteolytic cleavage. Binding of MMAE to tubulin disrupts the microtubule network within the cell, subsequently inducing cell cycle arrest and apoptotic death of the cells. Additionally, in vitro data provide evidence for antibody-dependent cellular phagocytosis (ADCP).
12.2 Pharmacodynamics
Cardiac Electrophysiology
At the recommended dose of 1.8 mg/kg, brentuximab vedotin had no large QTc prolongation (>10ms).
12.3 Pharmacokinetics
The pharmacokinetics of brentuximab vedotin were evaluated in monotherapy and combination chemotherapy in patients with hematological malignancies. The pharmacokinetics of brentuximab vedotin in combination therapy were similar to those in monotherapy. Total antibody and ADC had similar pharmacokinetic profiles. The pharmacokinetics of the ADC and MMAE are presented.
ADC
Maximum concentrations of ADC were observed near the end of infusion. Exposures were approximately dose proportional from 1.2 to 2.7 mg/kg (1.5 times the highest approved recommended dosage).
- •
- 1.8 mg/kg Q3W: Steady state was achieved within 21 days, and minimal to no accumulation of ADC was observed.
- •
- 1.2 mg/kg Q2W: Steady state was achieved within 56 days, 1.27-fold accumulation (14‑day AUC) was observed.
- •
- 1.2 mg/kg Q3W: Steady state was achieved within 21 days, and minimal to no accumulation of ADC was observed.
MMAE
Maximum concentrations of MMAE were observed approximately 1 to 3 days after end of infusion. Exposures decreased with continued administration of ADCETRIS with approximately 50% to 80% of the exposure of the first dose observed at subsequent doses.
- •
- 1.8 mg/kg Q3W: Steady state was achieved within 21 days.
- •
- 1.2 mg/kg Q2W: Steady state was achieved within 56 days.
- •
- 1.2 mg/kg Q3W: Steady state was achieved within 21 days.
Distribution
In humans, the mean steady state volume of distribution was approximately 6–10 L for ADC.
In vitro, the binding of MMAE to human plasma proteins ranged from 68–82%. MMAE is not likely to displace or to be displaced by highly protein-bound drugs.
Elimination
ADC elimination exhibited a multi-exponential decline with a t1/2 of approximately 4 to 6 days.
MMAE elimination exhibited a mono-exponential decline with a t1/2 of approximately 3 to 4 days. Elimination of MMAE appeared to be limited by its rate of release from ADC.
Metabolism
A small fraction of MMAE released from brentuximab vedotin is metabolized. In vitro data indicate that the MMAE metabolism that occurs is primarily via oxidation by CYP3A4/5.
Excretion
After a single dose of 1.8 mg/kg of ADCETRIS in patients, approximately 24% of the total MMAE administered was recovered in both urine and feces over a 1-week period, approximately 72% of which was recovered in the feces, and the majority was excreted unchanged.
Specific Populations
Sex and race do not have a meaningful effect on the pharmacokinetics of brentuximab vedotin.
Pediatric Patients
The pharmacokinetics of brentuximab vedotin and MMAE were evaluated in 65 pediatric patients aged 3 to <6 years (N=3), 6 to <12 years (N=30) and 12 to <17 years (N=32). Following the recommended dosage of brentuximab vedotin 1.8 mg/kg Q3W, the dose-normalized steady state Cavg of brentuximab vedotin in patients 12 to <17 years of age were generally consistent with those in adult patients administered brentuximab vedotin 1.2 mg/kg Q2W. The median AUC of ADC was 22% lower in patients 6 to <12 years of age (median [range] body weight = 28.8 kg [16.2, 80.8 kg]), and 37% lower in patients 3 to <6 years of age (median [range] body weight = 17.0 kg [10.7, 31.1 kg]), respectively, compared to that in patients 12 to <17 years of age (median [range] body weight = 52.7 kg [28.5, 123.9 kg]). The AUC of MMAE was 25% lower in patients 6 to <12 years of age, and 41% lower in patients 3 to <6 years of age, respectively, compared to that in patients 12 to <17 years of age. After accounting for body weight, other factors such as age, sex, race, and baseline albumin had no clinically significant effect on the PK of ADC and MMAE in pediatric patients 3 to <17 years of age.
Renal Impairment
The pharmacokinetics of brentuximab vedotin and MMAE were evaluated after the administration of 1.2 mg/kg of ADCETRIS to patients with mild (CrCL >50–80 mL/min; n=4), moderate (CrCL 30–50 mL/min; n=3) and severe (CrCL <30 mL/min; n=3) renal impairment. The AUC of MMAE was approximately 2-fold higher in patients with severe renal impairment compared to patients with normal renal function and not meaningfully altered in patients with mild or moderate renal impairment.
Hepatic Impairment
The pharmacokinetics of brentuximab vedotin and MMAE were evaluated after the administration of 1.2 mg/kg of ADCETRIS to patients with mild (Child-Pugh A; n=1), moderate (Child-Pugh B; n=5) and severe (Child-Pugh C; n=1) hepatic impairment. The AUC of MMAE was approximately 2.3-fold higher in patients with hepatic impairment compared to patients with normal hepatic function.
Drug Interaction Studies
Effects of Other Drugs on ADCETRIS
Co-administration of ADCETRIS with ketoconazole, a potent CYP3A4 inhibitor, increased exposure to MMAE by approximately 34%.
Co-administration of ADCETRIS with rifampin, a potent CYP3A4 inducer, reduced exposure to MMAE by approximately 46%.
Effects of ADCETRIS on Other Drugs
Co-administration of ADCETRIS did not affect exposure to midazolam, a CYP3A4 substrate.
In vitro studies using human liver microsomes indicate that MMAE inhibits CYP3A4/5 but not other CYP450 isoforms. MMAE did not induce any major CYP450 enzymes in human hepatocytes.
In vitro studies indicate that MMAE is a substrate and not an inhibitor of the efflux transporter P‑glycoprotein (P-gp).
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies (ADA) in the studies described below with incidence of ADA in other studies, including those of ADCETRIS or of other brentuximab vedotin products.
Among adult patients with relapsed or refractory cHL and relapsed or refractory systemic ALCL in Studies 1 and 2 [see Clinical Studies (14.1) and (14.3)], treatment-emergent ADA (or anti-brentuximab vedotin antibodies) developed in 37% (58/156) of patients who were tested for anti-brentuximab vedotin antibodies. Approximately 7% of patients in these trials developed persistently positive antibodies (positive test at more than 2 time points) and 30% developed transiently positive antibodies (positive at 1 or 2 post-baseline time points). Two of the patients (1%) with persistently positive antibodies experienced adverse reactions consistent with infusion reactions that led to discontinuation of treatment. Overall, a higher incidence of infusion-related reactions was observed in patients who developed persistently positive antibodies. The incidence of treatment-emergent neutralizing antibodies against brentuximab vedotin was 62% (36/58). The effect of anti-brentuximab vedotin antibodies on efficacy is not known.
Among pediatric patients with previously untreated high risk cHL in Study 7 [see Clinical Studies (14.1)], of the 26 patients tested, none of the patients tested positive for anti-brentuximab vedotin antibodies.
Among adult patients with LBCL in Study 8 [see Clinical Studies (14.5)], treatment-emergent anti-brentuximab vedotin antibodies developed in 8% (8/97) of patients who were tested. The effect of anti-brentuximab vedotin antibodies on efficacy is not known.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with brentuximab vedotin or the small molecule (MMAE) have not been conducted.
MMAE was genotoxic in the rat bone marrow micronucleus study through an aneugenic mechanism. This effect is consistent with the pharmacological effect of MMAE as a microtubule-disrupting agent. MMAE was not mutagenic in the bacterial reverse mutation assay (Ames test) or the L5178Y mouse lymphoma forward mutation assay.
Fertility studies with brentuximab vedotin or MMAE have not been conducted. However, results of repeat-dose toxicity studies indicate the potential for brentuximab vedotin to impair female and male reproductive function and fertility. In a 4-week repeat-dose toxicity study in rats with weekly dosing at 0.5, 5, or 10 mg/kg brentuximab vedotin, seminiferous tubule degeneration, Sertoli cell vacuolation, reduced spermatogenesis, and aspermia were observed. Effects in animals were seen mainly at 5 and 10 mg/kg of brentuximab vedotin. These doses are approximately 3 and 6-fold the human recommended dose of 1.8 mg/kg, respectively, based on body weight.
MMAE-containing ADCs have been associated with adverse ovarian effects when administered to sexually immature animals. Adverse effects included decrease in, or absence of, secondary and tertiary ovarian follicles after weekly administration to cynomolgus monkeys in studies of 4-week duration. These effects showed a trend towards recovery 6 weeks after the end of dosing; no changes were observed in primordial follicles.
14. Clinical Studies
14.1 Classical Hodgkin Lymphoma
Randomized Clinical Trial in Previously Untreated Stage III or IV Classical Hodgkin Lymphoma (Study 5: ECHELON-1, NCT01712490)
The efficacy of ADCETRIS in combination with chemotherapy for the treatment of patients with previously untreated Stage III or IV cHL was evaluated in a randomized, open-label, 2-arm, multicenter trial. Of the 1334 total patients, 664 patients were randomized to the ADCETRIS + doxorubicin [A], vinblastine [V] and dacarbazine [D] (ADCETRIS + AVD) arm and 670 patients were randomized to the A+ bleomycin [B] + V + D (ABVD) arm. Patients in both treatment arms were treated intravenously on Days 1 and 15 of each 28-day cycle for up to 6 cycles. Dosing in each treatment arm was administered according to the following:
- •
- ADCETRIS + AVD arm: ADCETRIS 1.2 mg/kg over 30 minutes, doxorubicin 25 mg/m2, vinblastine 6 mg/m2, and dacarbazine 375 mg/m2
- •
- ABVD arm: doxorubicin 25 mg/m2, bleomycin 10 units/m2, vinblastine 6 mg/m2, and dacarbazine 375 mg/m2
Efficacy was established based on modified progression-free survival (modified PFS) per independent review facility (IRF). A modified PFS event is defined as progression, death, or receipt of additional anticancer therapy for patients who are not in a complete response (CR) after completion of frontline therapy.
Patients had Stage III (36%) or IV disease (64%), and 62% had extranodal involvement at diagnosis. Most patients were male (58%) and white (84%). The median age was 36 years (range, 18‑83); 186 patients (14%) were 60 years or older.
The efficacy results are summarized in Table 13 and Figure 1.
|
||
|
Modified Progression-Free Survival per IRF* |
ADCETRIS + AVD
|
ABVD
|
|
Number of events (%) |
117 (18%) |
146 (22%) |
|
Median months (95% CI) |
NE† |
NE† |
|
Hazard ratio (95% CI)‡ |
0.77 (0.60, 0.98) |
|
|
P-value§ |
0.035 |
|
|
Reason leading to a modified PFS event | ||
|
Progressive disease |
90 (14) |
102 (15) |
|
Death due to any cause |
18 (3) |
22 (3) |
|
Receipt of additional anticancer therapy for |
9 (1) |
22 (3) |
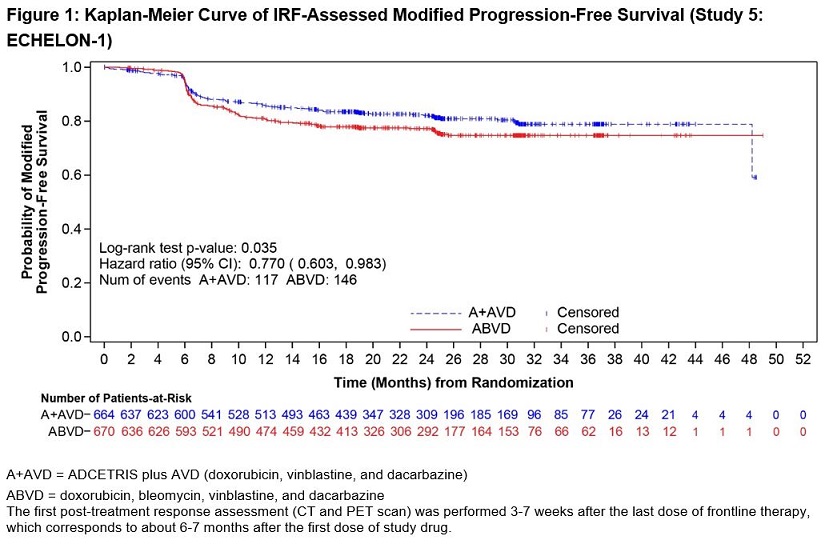
At the time of the modified PFS analysis, the prespecified interim OS analysis did not demonstrate a significant difference. The CR rate per IRF assessment at the end of the randomized regimen was 73% on the ADCETRIS + AVD arm and 70% on the ABVD arm.
A prespecified second interim analysis showed a statistically significant improvement in OS in the ADCETRIS + AVD arm (39 deaths) compared to the ABVD arm (64 deaths). With an estimated median follow-up of 6.1 years, the stratified hazard ratio was 0.59 (95% CI, 0.396; 0.879), with a 2-sided p-value of 0.009 (significance level, 0.0365). Median OS was not reached in either treatment arm (Figure 2).
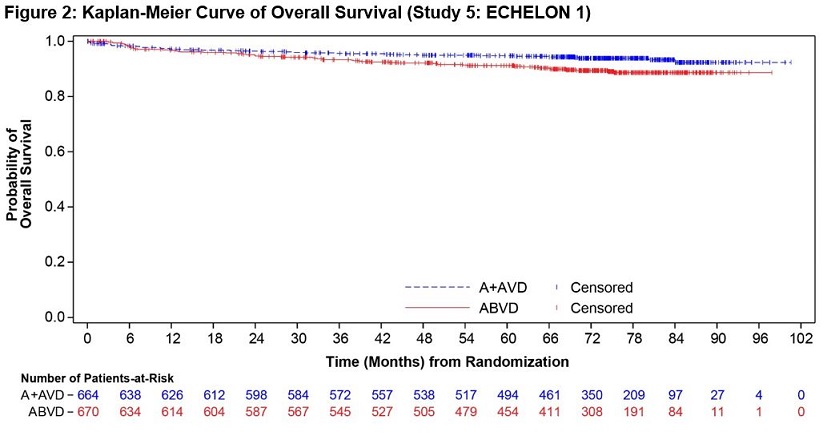
Randomized Clinical Trial in Previously Untreated High Risk Classical Hodgkin Lymphoma (Study 7, AHOD1331, NCT02166463)
The efficacy of ADCETRIS in combination with chemotherapy for the treatment of pediatric patients (2 to <22 years of age) with previously untreated, high risk cHL was evaluated in a randomized, open-label, actively controlled trial. High risk was defined as Ann Arbor Stage IIB with bulk disease, Stage IIIB, Stage IVA, and Stage IVB. Of the 600 total patients randomized, 300 were randomized to ADCETRIS + Doxorubicin [A], Vincristine [V], Etoposide [E], Prednisone [P], Cyclophosphamide [C] (ADCETRIS + AVEPC) arm and 300 patients were randomized to A+ Bleomycin [B]+V+E+P+C (ABVE-PC) arm. Patients in each treatment arm received up to 5 cycles of the following:
- •
- ADCETRIS + AVEPC arm: ADCETRIS 1.8 mg/kg over 30 minutes (day 1), doxorubicin 25 mg/m2 (days 1 and 2), vincristine 1.4 mg/m2 (day 8), etoposide 125 mg/m2 (days 1-3), prednisone 20 mg/m2 BID (days 1-7), cyclophosphamide 600 mg/m2 (days 1 and 2)
- •
- ABVE-PC arm: doxorubicin 25 mg/m2 (days 1 and 2), bleomycin 5 units/m2 (day1) and 10 units/m2 (day 8), vincristine 1.4 mg/m2 (days 1 and 8), etoposide 125 mg/m2 (days 1-3), prednisone 20 mg/m2 BID (days 1-7), cyclophosphamide 600 mg/m2 (days 1 and 2)
The median age was 15 years (range: 3-21 years); 53% were male, 74% were White, 11% Black, and 3% Asian. Nine patients were <6 years, 81 patients were 6 to <12 years, 448 patients were 12 to <18 years, and 62 patients were ≥18 years. Of the 600 enrolled patients, 20% had disease stage of IIB with bulk disease, 19% had IIIB, 29% had IVA, and 31% had IVB.
Efficacy was established based on event-free-survival (EFS), defined as the time from randomization to the earliest of disease progression or relapse, second malignancy, or death due to any cause. Efficacy results are summarized in Table 14.
| NR = Not reached. | ||
|
||
|
Event-Free Survival |
ADCETRIS + AVEPC
|
ABVE-PC
|
|
Number of Events (%) |
23 (8) |
52 (17) |
|
Median (95% CI) |
NR |
NR |
|
Hazard Ratio (95% CI)* |
0.41 (0.25, 0.67) |
|
|
P-value (log-rank test)† |
0.0002 |
|
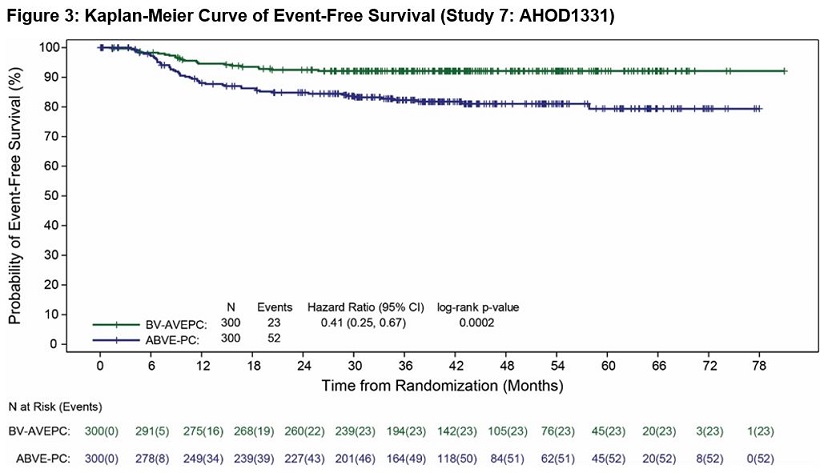
Randomized Placebo-Controlled Clinical Trial in Classical Hodgkin Lymphoma Post-Auto-HSCT Consolidation (Study 3: AETHERA, NCT01100502)
The efficacy of ADCETRIS in patients with cHL at high risk of relapse or disease progression post-auto-HSCT was studied in a randomized, double-blind, placebo-controlled clinical trial. Three hundred twenty-nine (329) patients were randomized 1:1 to receive placebo or ADCETRIS 1.8 mg/kg intravenously over 30 minutes every 3 weeks for up to 16 cycles, beginning 30–45 days post-auto-HSCT. Patients in the placebo arm with progressive disease per investigator could receive ADCETRIS as part of a separate trial. The primary endpoint was progression-free survival (PFS) determined by independent review facility (IRF). Standard international guidelines were followed for infection prophylaxis for HSV, VZV, and PJP post-auto-HSCT [see Clinical Trial Experience (6.1)].
High risk of post-auto-HSCT relapse or progression was defined according to status following frontline therapy: refractory, relapse within 12 months, or relapse ≥12 months with extranodal disease. Patients were required to have obtained a complete response (CR), partial response (PR), or stable disease (SD) to most recent pre-auto-HSCT salvage therapy.
A total of 329 patients were enrolled and randomized (165 ADCETRIS, 164 placebo); 327 patients received study treatment. Patient demographics and baseline characteristics were generally balanced between treatment arms. The 329 patients ranged in age from 18–76 years (median, 32 years) and most were male (53%) and white (94%). Patients had received a median of 2 prior systemic therapies (range, 2–8) excluding autologous hematopoietic stem cell transplantation.
The efficacy results are summarized in Table 15. PFS is calculated from randomization to date of disease progression or death (due to any cause). The median PFS follow-up time from randomization was 22 months (range, 0–49). Study 3 (AETHERA) demonstrated a statistically significant improvement in IRF-assessed PFS and increase in median PFS in the ADCETRIS arm compared with the placebo arm. At the time of the PFS analysis, an interim overall survival analysis demonstrated no difference.
|
Progression-Free Survival per IRF |
ADCETRIS
|
Placebo
|
|
Number of events (%) |
60 (36) |
75 (46) |
|
Median months (95% CI) |
24.1 (11.5, NE†) |
|
|
Stratified Hazard Ratio (95% CI) |
0.57 (0.40, 0.81) |
|
|
Stratified Log-Rank Test P-value |
0.001 |
|
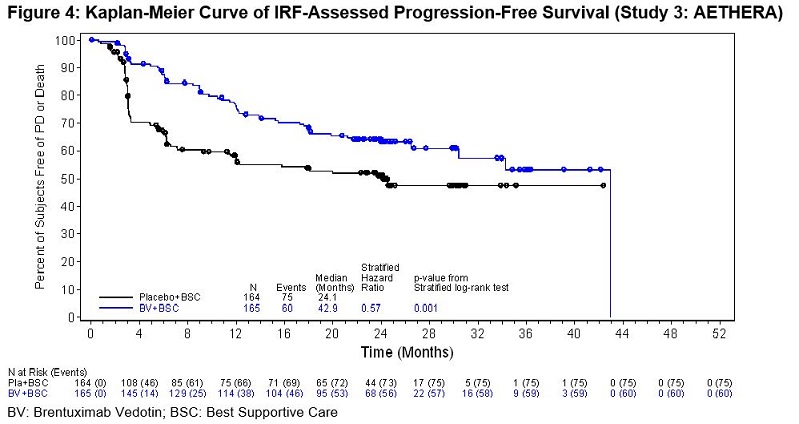
Clinical Trial in Relapsed Classical Hodgkin Lymphoma (Study 1, NCT00848926)
The efficacy of ADCETRIS in patients with cHL who relapsed after autologous hematopoietic stem cell transplantation was evaluated in one open-label, single-arm, multicenter trial. One hundred two (102) patients were treated with 1.8 mg/kg of ADCETRIS intravenously over 30 minutes every 3 weeks. An independent review facility (IRF) performed efficacy evaluations which included overall response rate (ORR = complete response [CR] + partial response [PR]) and duration of response as defined by clinical and radiographic measures including computed tomography (CT) and positron-emission tomography (PET) as defined in the 2007 Revised Response Criteria for Malignant Lymphoma (modified).
The 102 patients ranged in age from 15–77 years (median, 31 years) and most were female (53%) and white (87%). Patients had received a median of 5 prior therapies including autologous hematopoietic stem cell transplantation.
The efficacy results are summarized in Table 16. Duration of response is calculated from date of first response to date of progression or data cutoff date.
14.2 Systemic Anaplastic Large Cell Lymphoma and Other CD30-Expressing Peripheral T-Cell Lymphomas
Randomized Clinical Trial in Previously Untreated Systemic Anaplastic Large Cell Lymphoma or Other CD30-Expressing Peripheral T-Cell Lymphomas (Study 6: ECHELON-2, NCT01777152)
The efficacy of ADCETRIS in combination with chemotherapy for the treatment of adult patients with previously untreated, CD30-expressing PTCL was evaluated in a multicenter, randomized, double-blind, double-dummy, actively controlled trial. For enrollment, the trial required CD30 expression ≥10% per immunohistochemistry. The trial excluded patients with primary cutaneous CD30-positive T-cell lymphoproliferative disorders and lymphomas. The trial required hepatic transaminases ≤3 times ULN, total bilirubin ≤1.5 times ULN, and serum creatinine ≤2 times ULN.
Of the 452 total patients, 226 patients were randomized to the ADCETRIS + CHP arm and 226 patients were randomized to the CHOP arm. Patients in both treatment arms were treated intravenously on Day 1 of each 21-day cycle for 6 to 8 cycles; prednisone was administered orally on Days 1‑5. Dosing in each treatment arm was administered according to the following:
- •
- ADCETRIS + CHP arm: ADCETRIS 1.8 mg/kg over 30 minutes, cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, and prednisone 100 mg orally
- •
- CHOP arm: cyclophosphamide 750 mg/m2, doxorubicin 50 mg/m2, vincristine 1.4 mg/m2, and prednisone 100 mg orally
The median age was 58 years (range: 18 to 85), 63% were male, 62% were White, 22% were Asian, and 78% had an ECOG performance status of 0-1. Of the 452 patients enrolled, the disease subtypes included patients with systemic ALCL [70%; 48% anaplastic lymphoma kinase (ALK) negative and 22% ALK positive], PTCL NOS (16%), angioimmunoblastic T-cell lymphoma (12%), adult T-cell leukemia/lymphoma (2%), and enteropathy-associated T-cell lymphoma (<1%). Most patients had Stage III or IV disease (81%) and a baseline international prognostic index of 2 or 3 (63%).
During randomized treatment, on the ADCETRIS + CHP arm, 70% of patients received 6 cycles and 18% of patients received 8 cycles. On the CHOP arm, 62% of patients received 6 cycles and 19% received 8 cycles.
Efficacy was based on IRF-assessed PFS, which was defined as time from randomization to progression, death due to any cause, or receipt of subsequent anticancer chemotherapy to treat residual or progressive disease. Other efficacy endpoints included PFS in patients with systemic ALCL, overall survival, complete response rate, and overall response rate. Efficacy results are summarized in Table 17. Kaplan-Meier curves for PFS and overall survival are presented in Figure 5 and Figure 6, respectively.
| NE = Not estimable | ||
|
||
|
Outcomes per IRF* |
ADCETRIS + CHP |
CHOP |
|
PFS |
||
|
Number of events, n (%) |
95 (42) |
124 (55) |
|
Median PFS, months (95% CI) |
48.2 (35.2, NE) |
20.8 (12.7, 47.6) |
|
Hazard ratio (95% CI)† |
0.71 (0.54, 0.93) |
|
|
P-value‡ |
0.011 |
|
|
Reason leading to a PFS event, n (%) | ||
|
Progressive disease |
71 (31) |
86 (38) |
|
Death |
13 (6) |
17 (8) |
|
Receipt of subsequent anticancer chemotherapy to treat residual or progressive disease |
11 (5) |
21 (9) |
|
PFS for patients with sALCL |
||
|
N |
163 |
151 |
|
Number of patients with a PFS event, n (%) |
56 (34) |
73 (48) |
|
Median PFS, months (95% CI) |
55.7 (48.2, NE) |
54.2 (13.4, NE) |
|
Hazard ratio (95% CI)† |
0.59 (0.42, 0.84) |
|
|
P-value‡ |
0.003 |
|
|
OS§ |
||
|
Number of deaths |
51 (23) |
73 (32) |
|
Median OS, months (95% CI) |
NE (NE, NE) |
NE (54.2, NE) |
|
Hazard ratio (95% CI)† |
0.66 (0.46, 0.95) |
|
|
P-value‡ |
0.024 |
|
|
CR Rate¶ |
||
|
% (95% CI) |
68 (61, 74) |
56 (49, 62) |
|
P-value# |
0.007 |
|
|
ORR¶ |
||
|
% (95% CI) |
83 (78, 88) |
72 (66, 78) |
|
P-value# |
0.003 |
|
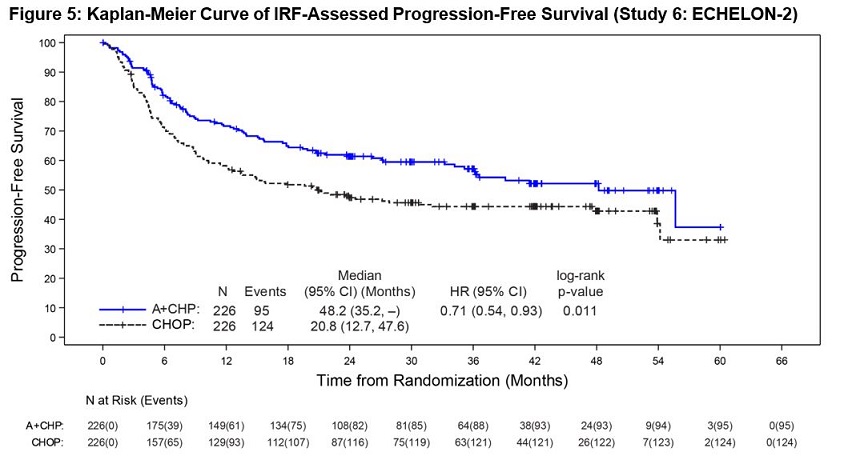
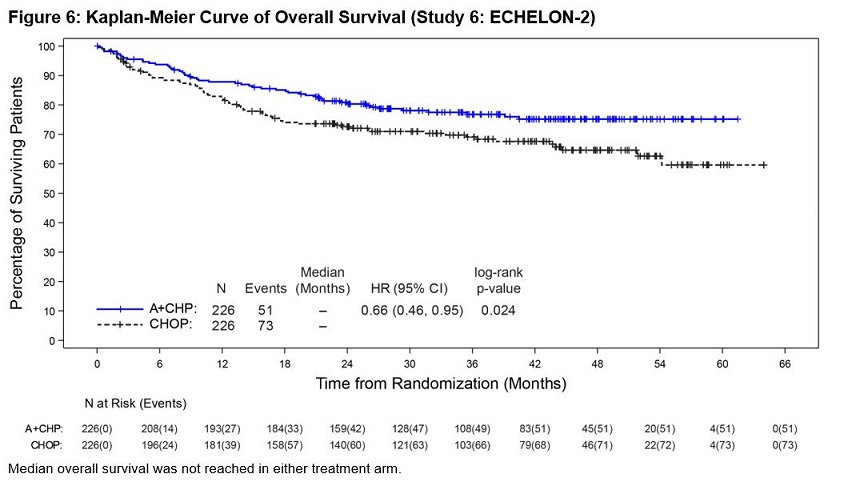
14.3 Systemic Anaplastic Large Cell Lymphoma
Clinical Trial in Relapsed sALCL (Study 2, NCT00866047)
The efficacy of ADCETRIS in patients with relapsed sALCL was evaluated in one open-label, single-arm, multicenter trial. This trial included patients who had sALCL that was relapsed after prior therapy. Fifty-eight (58) patients were treated with 1.8 mg/kg of ADCETRIS administered intravenously over 30 minutes every 3 weeks. An IRF performed efficacy evaluations which included overall response rate (ORR = complete response [CR] + partial response [PR]) and duration of response as defined by clinical and radiographic measures including computed tomography (CT) and positron-emission tomography (PET) as defined in the 2007 Revised Response Criteria for Malignant Lymphoma (modified).
The 58 patients ranged in age from 14–76 years (median, 52 years) and most were male (57%) and white (83%). Patients had received a median of 2 prior therapies; 26% of patients had received prior autologous hematopoietic stem cell transplantation. Fifty percent (50%) of patients were relapsed, and 50% of patients were refractory to their most recent prior therapy. Seventy-two percent (72%) were anaplastic lymphoma kinase (ALK)-negative.
The efficacy results are summarized in Table 18. Duration of response is calculated from date of first response to date of progression or data cutoff date.
|
|
N = 58 |
||
|
Percent (95% CI) |
Duration of Response, in months |
||
|
Median (95% CI) |
Range |
||
|
CR |
57 (44, 70) |
13.2 (10.8, NE*) |
0.7 to 15.9† |
|
PR |
29 (18, 41) |
2.1 (1.3, 5.7) |
0.1 to 15.8† |
|
ORR |
86 (77, 95) |
12.6 (5.7, NE*) |
0.1 to 15.9† |
14.4 Primary Cutaneous Anaplastic Large Cell Lymphoma and CD30-Expressing Mycosis Fungoides
Randomized Clinical Trial in Primary Cutaneous Anaplastic Large Cell Lymphoma and CD30-expressing Mycosis Fungoides (Study 4: ALCANZA, NCT01578499)
The efficacy of ADCETRIS in patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) or mycosis fungoides (MF) requiring systemic therapy was studied in ALCANZA, a randomized, open-label, multicenter clinical trial. In ALCANZA, one hundred thirty-one (131) patients were randomized 1:1 to receive ADCETRIS 1.8 mg/kg intravenously over 30 minutes every 3 weeks or physician’s choice of methotrexate (5 to 50 mg orally weekly) or bexarotene (300 mg/m2 orally daily). The randomization was stratified by baseline disease diagnosis (MF or pcALCL). Patients could receive a maximum of 16 cycles (21-day cycle) of therapy every 3 weeks for those receiving brentuximab vedotin or 48 weeks of therapy for those in the control arm.
Patients with pcALCL must have received prior radiation or systemic therapy, and must have at least 1 biopsy with CD30-expression of ≥10%. Patients with MF must have received prior systemic therapy and have had skin biopsies from at least 2 separate lesions, with CD30-expression of ≥10% in at least 1 biopsy.
A total of 131 patients were randomized (66 ADCETRIS, 65 physician’s choice). The efficacy results were based on 128 patients (64 patients in each arm with CD30-expression of ≥10% in at least one biopsy). Among 128 patients, the patients’ age ranged from 22–83 years (median, 60 years), and 55% of them were male and 85% of them were white. Patients had received a median of 4 prior therapies (range, 0–15), including a median of 1 prior skin-directed therapy (range, 0–9) and 2 systemic therapies (range, 0–11). At study entry, patients were diagnosed as Stage 1 (25%), Stage 2 (38%), Stage 3 (5%), or Stage 4 (13%).
Efficacy was established based on the proportion of patients achieving an objective response (CR+PR) that lasts at least 4 months (ORR4). ORR4 was determined by independent review facility (IRF) using the global response score (GRS), consisting of skin evaluations per modified severity-weighted assessment tool (mSWAT), nodal and visceral radiographic assessment, and detection of circulating Sézary cells (MF patients only). Additional efficacy outcome measures included proportion of patients achieving a complete response (CR) per IRF, and progression-free survival (PFS) per IRF.
The efficacy results are summarized in Table 19 below and the Kaplan-Meier curves of IRF-assessed PFS are shown in Figure 7.
|
||
|
ADCETRIS |
Physician’s Choice*
|
|
|
ORR4† |
||
|
Percent (95% CI‡) |
56.3 (44.1, 68.4) |
12.5 (4.4, 20.6) |
|
P-value§ |
<0.001 |
|
|
ORR |
67.2 (55.7, 78.7) |
20.3 (10.5, 30.2) |
|
CR |
||
|
Percent (95% CI‡) |
15.6 (7.8, 26.9) |
1.6 (0, 8.4) |
|
0.0066 |
||
|
PR |
51.6 (39.3, 63.8) |
18.8 (9.2, 28.3) |
|
PFS |
||
|
Number of events (%) |
36 (56.3) |
50 (78.1) |
|
Median months (95% CI‡) |
16.7 (14.9, 22.8) |
3.5 (2.4, 4.6) |
|
Hazard Ratio (95% CI‡) |
0.27 (0.17, 0.43) |
|
|
<0.001 |
||
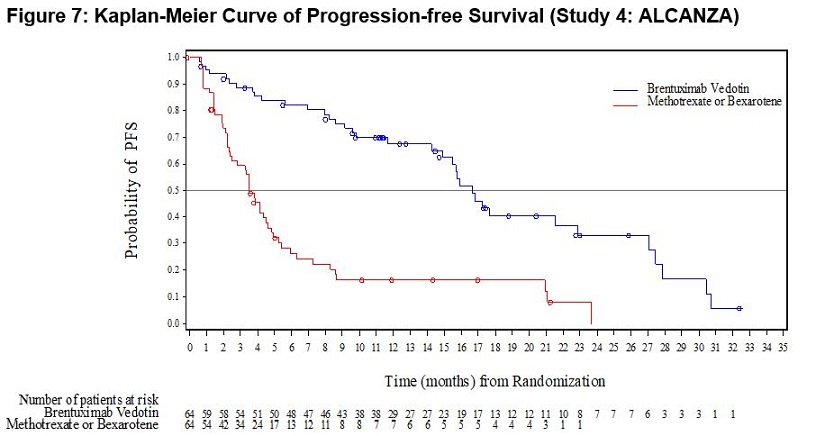
Supportive trials include 2 single-arm trials, which enrolled patients with MF who were treated with ADCETRIS 1.8 mg/kg intravenously over 30 minutes every 3 weeks. Out of 73 patients with MF from the 2 pooled supportive trials, 34% (25/73) achieved ORR4. Among these 73 patients, 35 had 1% to 9% CD30-expression and 31% (11/35) achieved ORR4.
14.5 Relapsed or Refractory Large B-Cell Lymphoma (LBCL)
Randomized Clinical Trial in Relapsed or Refractory Large B-Cell Lymphoma (Study 8: ECHELON-3)
The efficacy of ADCETRIS in combination with lenalidomide and a rituximab product for the treatment of adult patients with relapsed or refractory LBCL after two or more lines of systemic therapy was evaluated in ECHELON-3 (NCT04404283), a randomized, multicenter, double-blind, placebo-controlled trial. Eligible patients were 18 years of age and older with relapsed or refractory LBCL, including DLBCL NOS, DLBCL arising from indolent lymphoma, or HGBL, an ECOG performance status of 0–2, and were not eligible to receive an auto-HSCT or CAR T-cell therapy. The study excluded patients with active CNS lymphoma or Grade 2 or higher peripheral neuropathy.
Patients were randomized 1:1 to receive ADCETRIS plus lenalidomide and a rituximab product or to receive placebo plus lenalidomide and a rituximab product until disease progression or unacceptable toxicity. Randomization was stratified by CD30 expression (≥1% versus <1%), prior allogenic or autologous HSCT therapy, prior CAR T-cell therapy and cell of origin (germinal-center B-cell like [GCB] or non-GCB). Dosing in each treatment arms as follows:
- •
- ADCETRIS plus lenalidomide and rituximab arm: ADCETRIS 1.2 mg/kg via intravenous infusion every 3 weeks, lenalidomide 20 mg orally daily, and rituximab 375 mg/m2 via intravenous infusion every 3 weeks. Starting with Cycle 2, rituximab intravenous treatment could be substituted with rituximab 1,400 mg and hyaluronidase human 23,400 Units via subcutaneous injection every 3 weeks.
- •
- Placebo plus lenalidomide and rituximab arm: lenalidomide 20 mg orally daily, and rituximab 375 mg/m2 via intravenous infusion every 3 weeks. Starting with Cycle 2, rituximab intravenous treatment could be substituted with rituximab 1,400 mg and hyaluronidase human 23,400 Units via subcutaneous injection every 3 weeks.
Prophylaxis with granulocyte-colony stimulating factor (G-CSF) was mandated for both arms.
Of the 230 patients randomized (112 to ADCETRIS plus lenalidomide and rituximab, 118 to placebo plus lenalidomide and rituximab), the median age was 71 years (range 21 to 89 years); 57% were male, 53% were White, 26% were Asian and 4% were Hispanic or Latino. Race and ethnicity was not reported in 46 (20%) patients. There were no Black or African American patients enrolled. Of the total 230 patients, 71% had DLBCL NOS, 26% had transformed disease from prior indolent lymphoma, 16% had HGBL with MYC and BCL2 and/or BCL6 rearrangements or HGBL NOS. Of the patients with DLBCL NOS, 73 (45%) and 91 (55%) had GCB and non-GCB subtypes, respectively. Twenty-nine percent of patients had received prior CAR T-cell therapy and 12% had received prior HSCT. The median number of prior systemic therapies was 3 (range 2-8) with 41% receiving 2 prior therapies and 50% receiving 3 or more prior therapies. Eighty four percent had refractory disease to last therapy.
The major efficacy outcome was overall survival (OS). Additional efficacy outcome measures included progression free survival (PFS) and Objective Response Rate (ORR) according to investigator assessment per the Lugano Criteria for Response Assessment 2014. The results are presented in Table 20 and Figures 8 and 9.
| CI = confidence interval; CR = complete response; HR = hazard ratio; PFS = progression free survival; OS = overall survival. Stratified analyses were based on the stratification factors of cell of origin (GCB, non-GCB) and CD30 status (≥1%, <1%) at randomization. | ||
|
||
|
ADCETRIS + Lenalidomide + Rituximab (N=112) |
Placebo + Lenalidomide + Rituximab (N=118) |
|
|
Overall Survival |
||
|
Deaths, n (%) |
58 (51.8) |
76 (64.4) |
|
Median, months (95% CI) |
13.8 (10.3, 18.8) |
8.5 (5.4, 11.7) |
|
HR (95% CI)* |
0.63 (0.45, 0.89) |
|
|
0.0085 |
||
|
PFS per Investigator |
||
|
Number (%) of patients with event |
71 (63.4) |
85 (72.0) |
|
Progressive disease, n (%) |
56 (50.0) |
71 (60.2) |
|
Death, n (%) |
15 (13.4) |
14 (11.9) |
|
Median, months (95% CI) |
4.2 (2.9, 7.1) |
2.6 (1.4, 3.1) |
|
HR (95% CI)* |
0.53 (0.38, 0.73) |
|
|
p-value† |
<0.0001 |
|
|
Objective Response Rate per Investigator |
||
|
Objective response rate (CR or PR), n (%) |
72 (64.3) |
49 (41.5) |
|
95% CI for ORR§ |
(54.7, 73.1) |
(32.5, 51.0) |
|
p-value¶ |
0.0006 |
|
|
Complete response (CR), n (%) |
45 (40.2) |
22 (18.6) |
Figure 8: Kaplan-Meier Curve of Overall Survival (Study 8: ECHELON-3)
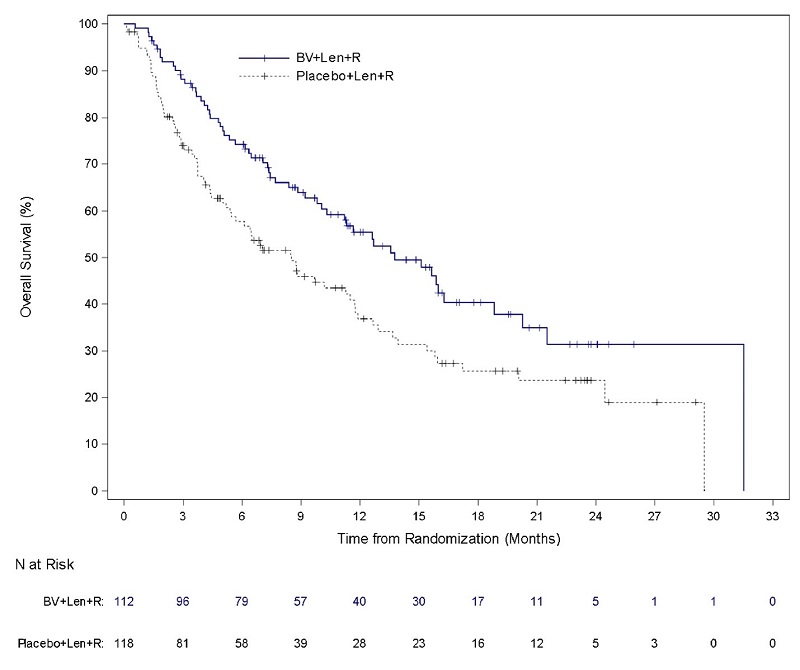
Figure 9: Kaplan-Meier Curve of Progression-Free Survival (Study 8: ECHELON-3)
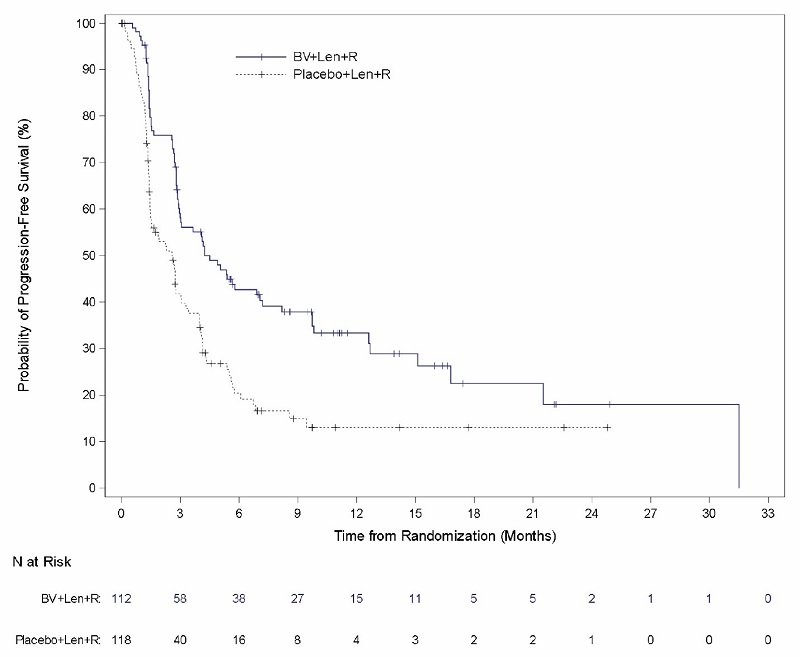
15. References
- 1.
- OSHA Hazardous Drugs. OSHA. [Accessed on 30 July 2013, from http://www.osha.gov/SLTC/hazardousdrugs/index.html]
16. How is Adcetris supplied
How Supplied
ADCETRIS (brentuximab vedotin) for Injection is supplied as a sterile, white to off-white preservative-free lyophilized cake or powder in individually-boxed single-dose vials:
- •
- NDC (51144-050-01), 50 mg brentuximab vedotin
Storage
Store vial refrigerated at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light.
Special Handling
ADCETRIS is a hazardous product. Follow special handling and disposal procedures1.
17. Patient Counseling Information
Peripheral Neuropathy
Advise patients that ADCETRIS can cause a peripheral neuropathy. They should be advised to report to their health care provider any numbness or tingling of the hands or feet or any muscle weakness [see Warnings and Precautions (5.1)].
Fever/Neutropenia
Advise patients to contact their health care provider if a fever of 100.5°F or greater or other evidence of potential infection such as chills, cough, or pain on urination develops [see Warnings and Precautions (5.3)].
Infusion Reactions
Advise patients to contact their health care provider if they experience signs and symptoms of infusion reactions including fever, chills, rash, or breathing problems within 24 hours of infusion [see Warnings and Precautions (5.2)].
Hepatotoxicity
Advise patients to report symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice [see Warnings and Precautions (5.8)].
Progressive Multifocal Leukoencephalopathy
Instruct patients receiving ADCETRIS to immediately report if they have any of the following neurological, cognitive, or behavioral signs and symptoms or if anyone close to them notices these signs and symptoms [see Boxed Warning, Warnings and Precautions (5.9)]:
- •
- changes in mood or usual behavior
- •
- confusion, thinking problems, loss of memory
- •
- changes in vision, speech, or walking
- •
- decreased strength or weakness on one side of the body
Pulmonary Toxicity
Instruct patients to report symptoms that may indicate pulmonary toxicity, including cough or shortness of breath [see Warnings and Precautions (5.10)].
Acute Pancreatitis
Advise patients to contact their health care provider if they develop severe abdominal pain [see Warnings and Precautions (5.12)].
Gastrointestinal Complications
Advise patients to contact their health care provider if they develop severe abdominal pain, chills, fever, nausea, vomiting, or diarrhea [see Warnings and Precautions (5.12)].
Hyperglycemia
Educate patients about the risk of hyperglycemia and how to recognize associated symptoms [see Warnings and Precautions (5.13)].
Females and Males of Reproductive Potential
ADCETRIS can cause fetal harm. Advise women receiving ADCETRIS to use effective contraception during ADCETRIS treatment and for 2 months after the last dose of ADCETRIS.
Advise males with female sexual partners of reproductive potential to use effective contraception during ADCETRIS treatment and for 4 months after the last dose of ADCETRIS [see Use in Specific Populations (8.3)].
Advise patients to report pregnancy immediately [see Warnings and Precautions (5.14)].
Lactation
Advise patients to avoid breastfeeding while receiving ADCETRIS [see Use in Specific Populations (8.2)].

Manufactured by:
Seagen Inc.
Bothell, WA 98021
1-855-473-2436
U.S. License 2257
ADCETRIS, Seagen and  are US registered trademarks of Seagen Inc.
are US registered trademarks of Seagen Inc.
© 2025 Seagen Inc., Bothell, WA 98021. All rights reserved.
LAB-1598-1.0
PACKAGE LABEL
NDC 51144-050-01
ADCETRIS®
(brentuximab vedotin)
FOR INJECTION
50 mg/vial
Single-dose vial. Discard unused portion.
Reconstitution and dilution required
For intravenous use only
Rx Only
Recommended Storage:
Store vial at 2°C to 8°C (36°F to 46°F) in original carton to protect from light.
See Prescribing Information for dosage and dilution.
Manufactured by Seagen Inc., Bothell, WA 98021
U.S. License No. 2257
Seagen®
123456
MMMYYYY

NDC 51144-050-01
ADCETRIS®
(brentuximab vedotin)
FOR INJECTION
50 mg/vial
Single-dose vial. Discard unused portion.
Reconstitution and dilution required
For intravenous use only
Seagen®
Rx Only
Each vial contains 50 mg of Brentuximab vedotin.
No Preservative.
After reconstitution with 10.5 mL of Sterile Water for Injection, USP, the concentration of ADCETRIS (brentuximab vedotin) is 5 mg/mL.
Recommended Dosage:
See Prescribing Information.
Manufactured by:
Seagen Inc.
Bothell, WA 98021
U.S. License No. 2257
For more information:
1-855-4SEAGEN
Recommended Storage:
Store vial at 2°C to 8°C (36°F to 46°F) in original carton to protect from light.
No U.S. Standard of Potency.
ADCETRIS, SEAGEN AND ![]() are US registered trademarks of Seagen Inc.
are US registered trademarks of Seagen Inc.
© 2021 Seagen Inc.
All rights reserved. Printed in USA.
LOT 123456
EXP MMMYYYY
SN 12345678901234
GTIN 12345678901234

| ADCETRIS
brentuximab vedotin injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - SEAGEN INC. (028484371) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| SEAGEN INC. | 028484371 | ANALYSIS(51144-050) | |
Biological Products Related to Adcetris
Find detailed information on biosimilars for this medication.
Frequently asked questions
More about Adcetris (brentuximab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: CD30 monoclonal antibodies
- Breastfeeding
- En español