Tanovea
This treatment applies to the following species: Company: Elanco US
Company: Elanco US
(rabacfosadine for injection)
16.4 mg rabacfosadine per vial
Antineoplastic
For intravenous use in dogs only
Approved by FDA under NADA# 141-545
Tanovea Caution
Federal law restricts this drug to use by or on the order of a licensed veterinarian. Use only as directed.
Description
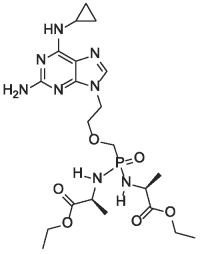
TANOVEA (rabacfosadine for injection) is an acyclic nucleotide phosphonate. The molecular weight of rabacfosadine is 526.5 g/mole. The empirical formula is C21H35N8O6P. The structural formula is:
TANOVEA is supplied as a sterile, white to off-white lyophilized powder in the form of a cake contained in a 3 mL amber glass vial. Each single-use vial contains 16.4 mg of rabacfosadine, present as the succinate salt, along with 20 mg of mannitol and 1.6 mg of citrate as excipients.
INDICATION:
TANOVEA is indicated for the treatment of lymphoma in dogs.
Dosage and Administration
Always provide the Client Information Sheet to the dog owner with each dose administration.
Administer TANOVEA at 1 mg/kg body weight as a 30-minute intravenous infusion, once every three weeks, for up to five doses.
Stepwise dose reductions to 0.8 mg/kg and 0.66 mg/kg or dose delays may be used to manage adverse reactions.
TANOVEA is supplied as a sterile lyophilized powder for reconstitution before use. After reconstitution with 2 mL of 0.9% Sodium Chloride Injection, USP, the reconstituted solution contains 8.2 mg/mL of rabacfosadine.
Reconstitution and administration of TANOVEA
Wear chemotherapy-resistant gloves, goggles, and protective clothing in the preparation and administration of TANOVEA. Use aseptic technique in the preparation and administration of TANOVEA.
Reconstitution instructions
1. Obtain the desired number of vials from the refrigerator.
2. Add 2 mL of 0.9% Sodium Chloride Injection, USP to the single-use vial.
3. Gently invert the vial several times until the drug has completely dissolved and the solution is particle free.
4. The solution should be clear without visible particulates. If particulates are observed, the solution should be discarded.
Dilution for infusion and administration instructions
1. TANOVEA should be diluted for infusion within 4 hours of reconstitution.
2. Add the calculated volume of reconstituted TANOVEA (8.2 mg/mL) to 0.9% Sodium Chloride Injection, USP in a polyvinyl chloride (PVC) infusion bag or polypropylene infusion syringe to yield a total infusion volume of 2 mL/kg.
3. The volume of reconstituted TANOVEA should be calculated based on the exact weight of the dog. (See Table 1. for example doses and administration volumes).
4. The infusion solution should be used within 24 hours of being added to the infusion bag or syringe and within 4 hours of being added to an intravenous transfer set. Protection from light is not needed.
5. Administer TANOVEA as a 30-minute intravenous infusion.
Table 1. Example doses and volumes at 1 mg/kg.
|
Dog weight (kg) |
Dog weight (lb) |
Dose (mg) |
Volume of reconstituted TANOVEA solution (mL) |
Volume 0.9% NaCl (mL) |
Total infusion volume (mL) |
Rate of infusion (mL/minute) |
|
5a |
11 |
5 |
0.6 |
9.4 |
10 |
0.3 |
|
10 |
22 |
10 |
1.2 |
18.8 |
20 |
0.7 |
|
15 |
33 |
15 |
1.8 |
28.2 |
30 |
1.0 |
|
20 |
44 |
20 |
2.4 |
37.6 |
40 |
1.3 |
|
25 |
55 |
25 |
3.0 |
47.0 |
50 |
1.7 |
|
30 |
66 |
30 |
3.7 |
56.3 |
60 |
2.0 |
|
35 |
77 |
35 |
4.3 |
65.7 |
70 |
2.3 |
|
40a |
88 |
40 |
4.9 |
75.1 |
80 |
2.7 |
a TANOVEA can be administered to dogs less than 5 kg and greater than 40 kg. Always calculate the dose based on the exact weight of the dog.
Compatibility of administration sets containing ethyl vinyl acetate (EVA) has not been evaluated.
Contraindications
Do not use in dogs with pulmonary fibrosis or a history of chronic pulmonary disease that could lead to fibrosis, such as chronic bronchitis (see WARNINGS). Do not use in West Highland White Terriers due to a genetic predisposition for development of pulmonary fibrosis. Use with caution in other terrier breeds due to potential genetic predisposition for development of pulmonary fibrosis.
Do not use in dogs that are pregnant, lactating, or intended for breeding. Rabacfosadine is cytotoxic and may cause birth defects and affect female and male fertility. TANOVEA has not been evaluated in pregnant, lactating, or breeding dogs.
Warnings
USER SAFETY WARNINGS:
NOT FOR USE IN HUMANS. KEEP THIS AND ALL MEDICATIONS OUT OF THE REACH OF CHILDREN. Do not store near food or with medications intended for use in humans. Do not eat, drink, or smoke while handling the product.
CHILDREN SHOULD NOT COME INTO CONTACT WITH TANOVEA. Children should not come into contact with feces, urine, vomit, and saliva of treated dogs for 5 days after each treatment.
Drug Handling and Administration
Pregnant women, women who may become pregnant, and nursing women should not handle, prepare, or administer TANOVEA. Rabacfosadine is cytotoxic and may cause birth defects and affect female and male fertility.
Use standard measures for the safe handling of all chemotherapeutic drugs. Refer to Occupational Safety and Health Administration (OSHA) for appropriate guidelines, recommendations, and regulations for handling antineoplastic agents.
Do not come into direct contact with TANOVEA. Wear chemotherapy-resistant gloves, goggles, and protective clothing when handling or administering TANOVEA. After removing and disposal of gloves, wash hands immediately and thoroughly with soap and water.
Accidental Exposure to TANOVEA
In the case of accidental sett-injection:
● Remove glove.
● Let the wound bleed a few drops of blood.
● Rinse the wound thoroughly with tap water.
● Seek medical advice immediately and show the package insert, label, or client information sheet to the physician.
In case of accidental skin contact:
● Wash the affected area immediately and thoroughly with soap and water.
In the case of accidental eye exposure:
● Remove contact lenses.
● Rinse the eyes with large amounts of tap water (use eyewash station if present) for 10 minutes while holding back the eyelid.
● Seek medical advice immediately and show the package insert, label, or client information sheet to the physician.
In the case of accidental ingestion:
● Seek medical advice immediately and show the package insert, label, or client information sheet to the physician.
Handling of Excreta and Soiled Items
Do not come into direct contact with the treated dog’s feces, urine, vomit, and saliva for 5 days after each treatment with TANOVEA.
When cleaning up feces, urine, vomit, and saliva, wear disposable chemotherapy-resistant gloves to collect the contaminated substances with disposable absorptive material (such as paper towels) and place them into a plastic bag. Carefully remove the gloves and place them in the bag, and tie or fasten it securely before general disposal. Wash hands immediately and thoroughly with soap and water afterwards.
Do not wash any items soiled with feces, urine, vomit, and saliva from the dog for 5 days after each treatment with other laundry.
Wear disposable chemotherapy-resistant gloves when handling the dog’s toys, food bowl, and water bowl. Wash food and water bowls separately from other items for 5 days after each treatment.
Accidental Exposure to Excreta
In the case of direct skin contact with feces, urine, vomit, and saliva of dogs for 5 days after each treatment:
● Wash the affected skin immediately and thoroughly with soap and water.
ANIMAL SAFETY WARNINGS:
TANOVEA is associated with life-threatening or fatal pulmonary fibrosis. The pulmonary fibrosis may be an idiosyncratic toxicity. Monitoring for signs of pulmonary dysfunction is recommended (see ADVERSE REACTIONS).
Precautions
TANOVEA is associated with dermatopathies (e.g., otitis, alopecia, dermatitis, erythema, pruritus, hyperpigmentation, skin ulcerations, and bacterial skin infections) which can worsen with subsequent treatment (see ADVERSE REACTIONS). Careful monitoring for changes in the skin and ears is recommended. Dose reductions and/or dose delays should be considered to mitigate dermatologic adverse reactions.
TANOVEA can cause neutropenia with a nadir around seven days post-treatment. Dogs should be frequently monitored for evidence of neutropenia during treatment with TANOVEA (see ADVERSE REACTIONS and TARGET ANIMAL SAFETY).
Extravasation of TANOVEA may cause pain and/or tissue injury. If extravasation is suspected, discontinue infusion immediately. Monitor the site for evidence of injury; consider local and systemic treatment measures as needed.
The safety and effectiveness of TANOVEA has not been evaluated in conjunction with other chemotherapeutic agents or other modalities for the treatment of lymphoma.
The effect of concomitant medications on the metabolism of TANOVEA has not been evaluated.
Adverse Reactions
A randomized, placebo-controlled, masked, multicenter clinical field study evaluated the effectiveness and safety of TANOVEA for the treatment of lymphoma.
One-hundred and twenty dogs received TANOVEA at a dose of 1 mg/kg, administered as a 30-minute intravenous infusion once every three weeks for one to five doses. Thirty-eight dogs received a placebo (saline) infusion.
The Veterinary Cooperative Oncology Group - common terminology criteria for adverse events (VCOG-CTCAE)1 definitions were used to grade the adverse reactions observed: Grade 1 (mild), Grade 2 (moderate), Grade 3 (severe), Grade 4 (life-threatening), and Grade 5 (death or euthanasia). Most adverse reactions were Grade 1 or 2. The adverse reactions observed in the study and number of dogs experiencing each adverse reaction is summarized in Table 2 below.
The most common adverse reactions included diarrhea, decreased appetite, vomiting, lethargy, weight loss, and neutropenia. Though diarrhea, decreased appetite, vomiting, lethargy, and weight loss were observed in both groups, the incidence was higher in the TANOVEA group and there were more Grade 2 and 3 adverse reactions compared to the Placebo group.
Table 2. Adverse Reactions Reported During the Field Study
|
|
TANOVEA (n=120) |
Placebo (n=38) |
||
|
n |
% |
n |
% |
|
|
Diarrhea |
105 |
87.5 |
19 |
50 |
|
Decreased appetite |
82 |
68.3 |
15 |
39.5 |
|
Emesis |
82 |
68.3 |
9 |
23.7 |
|
Lethargy |
76 |
63.3 |
24 |
63.2 |
|
Weight loss |
58 |
48.3 |
4 |
10.5 |
|
Neutropeniaa |
55 |
45.8 |
0 |
|
|
Polydipsia |
40 |
33.3 |
6 |
15.8 |
|
Anorexia |
34 |
28.3 |
7 |
18.4 |
|
Otitisb |
31 |
25.8 |
0 |
|
|
Alopecia |
30 |
25 |
2 |
5.3 |
|
Adipsia |
29 |
24.2 |
5 |
13.2 |
|
Polyuria |
29 |
24.2 |
2 |
5.3 |
|
Dermatitis |
25 |
20.8 |
0 |
|
|
Increased appetite |
24 |
20 |
6 |
15.8 |
|
Hypoalbuminemia |
24 |
20 |
5 |
13.2 |
|
Anemia |
20 |
16.7 |
3 |
7.9 |
|
Hematochezia |
20 |
16.7 |
0 |
|
|
Dehydration |
17 |
14.2 |
1 |
2.6 |
|
Nausea |
15 |
12.5 |
2 |
5.3 |
|
Erythema |
15 |
12.5 |
1 |
2.6 |
|
Pruritus |
15 |
12.5 |
1 |
2.6 |
|
Hyperpigmentation |
14 |
11.7 |
0 |
|
|
Leukopenia |
14 |
11.7 |
0 |
|
|
Monocytosis |
13 |
10.8 |
3 |
7.9 |
|
Elevated alanine aminotransferase (ALT) |
13 |
10.8 |
2 |
5.3 |
|
Proteinuria |
13 |
10.8 |
2 |
5.3 |
|
Pulmonary disorderc |
13 |
10.8 |
0 |
|
|
Elevated creatine-kinase (CK) |
12 |
10 |
1 |
2.6 |
|
Oliguria |
12 |
10 |
1 |
2.6 |
|
Urinary tract infection |
12 |
10 |
1 |
2.6 |
|
Cough |
11 |
9.2 |
3 |
7.9 |
|
Increased blood urea nitrogen (BUN) or creatinine |
11 |
9.2 |
2 |
5.3 |
|
Elevated aspartate aminotransferase (AST) |
11 |
9.2 |
0 |
|
|
Neutrophilia |
10 |
8.3 |
3 |
7.9 |
|
Hematuria |
10 |
8.3 |
1 |
2.6 |
|
Massd |
10 |
8.3 |
0 |
|
|
Hyperthermia |
9 |
7.5 |
4 |
10.5 |
|
Thrombocytopenia |
9 |
7.5 |
3 |
7.9 |
|
Elevated symmetrical dimethylarginine (SDMA) |
9 |
7.5 |
2 |
5.3 |
|
Hypocalcemia |
9 |
7.5 |
2 |
5.3 |
|
Glucosuriae |
9 |
7.5 |
0 |
|
|
Skin ulceration |
9 |
7.5 |
0 |
|
|
Tachypnea |
8 |
6.7 |
5 |
13.2 |
|
Dyspnea |
7 |
5.8 |
5 |
13.2 |
|
Elevated total bilirubin |
7 |
5.8 |
1 |
2.6 |
|
Hypokalemia |
7 |
5.8 |
1 |
2.6 |
|
Bacterial skin infection |
7 |
5.8 |
0 |
|
|
Desquamation |
7 |
5.8 |
0 |
|
|
Leukocytosis |
6 |
5 |
5 |
13.2 |
|
Pinnal irritation |
6 |
5 |
1 |
2.6 |
|
Digestive tract disorders NOSf |
6 |
5 |
0 |
|
|
Hypoproteinemia |
6 |
5 |
0 |
|
aMost neutropenias were Grade 1 or 2. However, eight instances of Grade 3 and three instances of Grade 4 neutropenia were reported. Neutropenia was reported seven days after treatment and returned to the normal range in all but two instances by the next cycle.
bRepresents combined terms of Otitis Externa and Otitis Not Otherwise Specified (NOS)
cPulmonary disorders included pulmonary fibrosis or possible pulmonary fibrosis (five dogs), pulmonary interstitial pattern (three dogs), pneumonia (three dogs), alveolar pattern on radiographs (two dogs), pneumomediastinum (one dog), pneumonitis (one dog), pulmonary infiltrates on radiographs (one dog). Some dogs were reported with more than one abnormality.
dMasses included skin and subcutaneous masses
eGlucosuria ranges from 1+ to 4+. In one dog, a 1+ ketonuria was reported at the same time as a 4+ glucosuria.
fDigestive tract disorders NOS included regurgitation (three dogs) and abnormal stool color (three dogs).
The type and frequency of reported adverse reactions did not vary greatly by cycle of TANOVEA treatment. A single cycle was the day of treatment and the 20 days after treatment (total of 21 days). However, during Cycles 1 and 2 (after the first or second dose), the majority of adverse reactions were primarily related to gastrointestinal and constitutional signs (i.e. lethargy, weight loss), while dermatopathies began to occur more frequently in Cycles 3 through 5 (after the third through fifth dose).
Serious Adverse Reactions
Serious adverse events (SAEs) were reported in 20% of dogs in the TANOVEA group (24 of 120 dogs) and 13% of dogs in the placebo group (five of 38 dogs). In the 24 TANOVEA dogs, SAEs included pulmonary fibrosis (five dogs) and dermatopathy (six dogs) as described below. Other SAEs included three cases of progressive disease (Grade 5), three cases of unrelated neoplasia/comorbidities, and one case each of hepatopathy (Grade 3), renal insufficiency (Grade 3), neutropenia (Grade 4), nausea (Grade 3), lymph node abscess (Grade 3), colitis (Grade 3), and hematochezia (Grade 3).
Pulmonary Fibrosis
Five dogs in the TANOVEA group developed severe adverse reactions associated with pulmonary fibrosis. The clinical signs included dyspnea, tachypnea, and orthopnea. Four of these dogs also had pneumomediastinum, pneumothorax, and/or emphysema diagnosed radiographically. All five dogs had radiographic findings that could be associated with pulmonary fibrosis along with accompanying pulmonary clinical signs. All five dogs were euthanized (four dogs) or died (one dog) due to the pulmonary fibrosis (either during the study or after removal from the study). Two of these dogs had a histologic confirmation of fibrosis on necropsy. The median time from randomization to first detection of clinical signs was 87 days (range 84 to 140) and the median time from randomization to death was 127 days (range 112 to 172). At the time of withdrawal (due to adverse reactions), all five dogs had a complete response to treatment. Pulmonary fibrosis may be an idiosyncratic adverse reaction with an unknown mechanism of action.
Dermatopathy
Over half of the patients (67/120 dogs) treated with TANOVEA experienced one or more dermatologic adverse reactions during their treatment. These adverse reactions were predominantly Grade 1 and primarily included otitis, alopecia, dermatitis, erythema, pruritus, hyperpigmentation, skin ulcerations, and bacterial skin infections.
Dermatologic adverse reactions typically presented by Cycle 3 (after the third dose), suggesting a cumulative effect of TANOVEA-associated dermatopathy. Dose reductions and dose delays were implemented to mitigate dermal adverse reactions. Six dogs in the TANOVEA group had Grade 3 or Grade 4 dermal adverse reactions which appeared in Cycle 2 (two dogs), Cycle 3 (three dogs), and Cycle 5 (one dog). Grade 3 adverse reactions included bacterial skin infections, skin ulcerations, desquamation, and dermatitis. Grade 4 adverse reactions included skin ulceration (severe erythema and ulceration of skin on limbs, ventrum, and foot pads) and desquamation (moist desquamation along the inguinal region and encompassing perivulvar area).
Euthanasia
Nine dogs in the TANOVEA group (8%) and two dogs in the placebo group (5%) were euthanized while on study. Reasons for euthanasia included four cases of progressive lymphoma (one with multiple comorbidities and pre-existing dermatopathy), two cases of pulmonary fibrosis, one case of probable hemangiosarcoma, one case of pyelonephritis/renal failure, and one case was unspecified. An additional three dogs died/were euthanized after withdrawal from the study due to suspected pulmonary fibrosis. One dog was euthanized after withdrawal from the study due to sepsis secondary to dermatopathy.
Dose Reductions and Dose Delays
Twenty-nine dogs in the TANOVEA group dogs received dose reductions (from 1 to 0.8 mg/kg), which were typically employed on the second to fourth dose of TANOVEA. Four dogs received a second stepwise reduction (from 0.8 to 0.66 mg/kg), which were employed on the third to fifth dose. The primary reason for dose reduction was gastrointestinal toxicity (diarrhea, vomiting, nausea), followed by dermatopathies, weight loss, anorexia/hyporexia, neutropenia, thrombocytopenia, and hypoalbuminemia. All dogs continued on the reduced dose(s) for the remainder of the study or until withdrawn.
Fourteen dogs in the TANOVEA group had a dose delay, which typically was one week beyond the intended treatment day. The primary reason cited in all cases was dermatopathy, and one case also cited weight loss. Eight dogs had both a dose reduction and delay, primarily attributable to dermatopathies.
Infusion Site Observations
On the day of treatment, two incidences of bruising were reported in dogs in the TANOVEA group. Infusion site observations seven days after treatment in dogs in the TANOVEA group included pigmentation (seven incidences), scaling (five incidences), erythema (three incidences), and swelling (two incidences).
Conditional Approval Experience:
Rabacfosadine for injection was conditionally approved and marketed from 2016-2021 under the proprietary name TANOVEA-CA1. The adverse events voluntarily reported for TANOVEA-CA1 (i.e., reports not associated with the clinical study) were similar to those described in the ADVERSE REACTIONS section above.
CONTACT INFORMATION:
To report suspected adverse events, for technical assistance, or to obtain a copy of the Safety Data Sheet, contact Elanco US Inc. at 1-877-468-3832.
For additional information about adverse drug experience reporting for animal drugs, contact FDA at 1-888-FDA-VETS or http://www.fda.gov/reportanimalae.
INFORMATION FOR DOG OWNERS:
Always provide the Client Information Sheet and review it with the dog owner or person responsible for care of the dog. Advise dog owners about possible adverse reactions, when to contact a veterinarian, and how to clean up any feces, urine, vomit, or saliva from dogs treated with TANOVEA. The Client Information Sheet also contains warnings for humans and what to do in case of accidental human exposure to TANOVEA.
Clinical Pharmacology
Rabacfosadine is a prodrug of 9-(2-phosphonylmethoxyethyl)-N6-cyclopropyl-2,6-diaminopurine (cPrPMEDAP) and 9-(2-phosphonylmethoxyethyl) guanine (PMEG). Rabacfosadine is hydrolyzed intracellularly to cPrPMEDAP and subsequently deaminated to PMEG. PMEG is then converted to its active phosphorylated form, PMEG diphosphate (PMEGpp), which is a potent, chain-terminating inhibitor of the major nuclear, replicative deoxyribonucleic acid (DNA) polymerases. The two prodrug moieties present in rabacfosadine increase the permeability and accumulation of metabolites in various cell types including peripheral blood mononuclear cells (PBMCs).
In vitro rabacfosadine has been demonstrated to inhibit DNA synthesis, resulting in S phase arrest and induction of apoptosis. Also in vitro rabacfosadine inhibits the proliferation of mitogen-stimulated lymphocytes and lymphoma/leukemia cell lines.
The in vivo data from studies in healthy dogs and dogs with lymphoma illustrated rapid clearance of rabacfosadine following a 30-minute intravenous infusion resulting in plasma exposure to the metabolite cPrPMEDAP and undetectable levels of the generally more cytotoxic agent PMEG. The exposure of rabacfosadine and cPrPMEDAP was linear with no gender differences. The mean peak plasma concentrations (Cmax) for rabacfosadine and cPrPMEDAP occurred at approximately 30 minutes (Tmax) and 1-2 hours, respectively. In dogs with lymphoma, rabacfosadine and cPrPMEDAP had a plasma half-life of <0.5 hour and 6 hours, respectively. In contrast to plasma, high concentrations of PMEG were readily detectable in PBMCs. Following rabacfosadine administration at 0.82 mg/kg over 30 minutes by intravenous infusion (once every two or three weeks), cPrPMEDAP and PMEGpp concentrations in lymphoid cells and tissues (represented by PBMCs) were 131 and 1,420 nM, respectively. In lymphoid cells, high concentrations of the active metabolite PMEGpp accumulated and persisted for greater than 24 hours. In PBMCs, the metabolites cPrPMEDAP and PMEGpp showed a terminal half-life of 25 hours and 68.7 hours, respectively. The observed PMEGpp concentrations in lymphoid cells and tissues are sufficient to allow for the desired antiproliferative effect following a single intravenous infusion administered once every three weeks.
Following intravenous administration of [14C]-rabacfosadine to dogs, the excretion of radiolabeled material was similar in both feces and urine accounting for 42.4% and 32.5% of the dose, respectively, at 120 hours.
By 24 hours, radioactivity was widely distributed throughout the body. Some of the highest concentrations in tissues observed, with the exception of excretory organs, were found in lymphoid tissues. The mean overall elimination of radioactivity after intravenous dosing was 79.7% over 120 hours.
Approximately 10% of the administered dose was retained in tissues. The overall mass balance including radioactivity retained in the tissues would be estimated to be a mean of 88.8%. No central nervous system penetration was observed.
While the cytotoxic agent PMEG has been observed in PBMCs, PMEG was not observed in the liver, kidney, bile, or urine following administration of [14C]-rabacfosadine to dogs. Because the effective clinical plasma concentration is anticipated to be <2 µM and rabacfosadine is a prodrug that has a short half-life in dogs, rabacfosadine is unlikely to be a clinically relevant inhibitor or inducer of the major CYP450 pathways.
Effectiveness
The effectiveness and safety of TANOVEA (rabacfosadine for injection) for the treatment of lymphoma was evaluated in a randomized, placebo-controlled, masked, multicenter clinical field study. TANOVEA was compared to placebo (saline) using progression free survival (PFS) as the primary endpoint. PFS was defined as the interval between the date of randomization and the first date that criteria for progressive disease were met, or the date of death, whichever occurred first. Response assessments were made according to the Veterinary Cooperative Oncology Group Response Evaluation Criteria for Peripheral Nodal Lymphoma in Dogs.2 The study enrolled dogs of any breed, except West Highland White Terrier, or any sex diagnosed with multicentric lymphoma with at least one measurable peripheral lymph node.
One hundred and fifty-eight dogs were randomly assigned to treatment with either 1 mg/kg TANOVEA (n=120) or placebo (n=38) as a 30-minute intravenous infusion, once every three weeks for five doses, or until disease progression or withdrawal from the study for another cause. Dose delays of up to 14 days or stepwise dose reductions to 0.8 mg/kg and 0.66 mg/kg were allowed to manage adverse reactions. Effectiveness was evaluated in 148 dogs (112 in the TANOVEA group and 36 in the placebo control group).
The effectiveness analysis demonstrated a statistically significantly longer median PFS (P <0.0001) in dogs administered TANOVEA compared to placebo (82 days vs. 21 days, respectively).
The median PFS was 151 days in dogs in the TANOVEA group that exhibited a partial response (PR) or complete response (CR) and 168 days for dogs with a CR. The best overall response rate (BORR; the percent of dogs with a PR or CR as their highest response on study) was 73.2% (82/112 dogs) for dogs in the TANOVEA group (50.9% CR; 57/112 dogs) vs. 5.6% (2/36 dogs) for dogs in the placebo group (0% CR; 0/36 dogs).
For dogs with B-cell lymphoma, the median PFS was greater for dogs in the TANOVEA group compared to dogs in the placebo group (126 vs. 21 days, respectively). A smaller trend was observed for dogs with T-cell lymphoma (29 vs. 17 days, respectively). In the TANOVEA group, the BORR was 80.4% (74/92 dogs) in dogs with B-cell lymphoma (58.7% CR; 54/92 dogs), and 40.0% (8/20 dogs) in dogs with T-cell lymphoma (15.0% CR; 3/20 dogs).
For dogs naïve to prior chemotherapy regimens, the median PFS was greater for dogs in the TANOVEA group compared to dogs in the placebo group (143 vs. 19 days, respectively), with a shorter duration of PFS observed for dogs that had received prior chemotherapy (63 vs. 21 days, respectively). Dogs in the TANOVEA group with one prior chemotherapy had a median PFS of 82 days (127 days in dogs with a response to treatment). Dogs with B-cell lymphoma in the TANOVEA group with one prior chemotherapy regimen had a median PFS of 107 days (172 days in dogs with a response to treatment). Dogs with T-cell lymphoma in the TANOVEA group with one prior chemotherapy regimen had a median PFS of 21 days.
The BORR in dogs in the TANOVEA group were impacted by the number of prior chemotherapy regimens, with a BORR of 88.7% (47/53 dogs) in naïve dogs (62.3% CR; 33/53 dogs), 70.3% (26/37 dogs) in dogs with one prior chemotherapy regimen (54.1% CR; 20/37 dogs), and 40.9% (9/22 dogs) in dogs with more than one prior chemotherapy regimen (18.2% CR; 4/22 dogs).
See Table 3 below for a response summary in TANOVEA treated dogs.
Table 3. Response Summary in TANOVEA-Treated Dogs
|
|
Overall |
B-cell |
T-cell |
Naive |
1 priora |
>1 priora |
1 prior (B-cell)a |
1 prior (T-cell)a |
>1 prior (B-cell)a |
>1 prior (T-cell)a |
|
n |
112 |
92 |
20 |
53 |
37 |
22 |
30 |
7 |
18 |
4 |
|
Median PFS (days) |
82 |
126 |
29 |
143 |
82 |
41 |
107 |
21 |
39 |
60 |
|
Median PFS (responding dogs) |
151 |
161 |
55 |
160 |
127 |
64 |
172 |
N/Ab |
64 |
N/Ac |
|
Median PFS (CR only) |
168 |
168 |
63 |
168 |
172 |
NEd |
172 |
N/Ab |
NEd |
NOe |
|
% BORR |
73.2 |
80.4 |
40 |
88.7 |
70.3 |
40.9 |
83.3 |
4.3 |
44.4 |
25 |
|
% CR (anytime) |
50.9 |
58.7 |
15 |
62.3 |
54.1 |
18.2 |
63.3 |
14.3 |
22.2 |
0 |
|
% PR (anytime) |
22.3 |
21.7 |
25 |
26.4 |
16.2 |
22.7 |
20 |
0 |
22.2 |
25 |
a Refers to number of prior chemotherapy regimens (e.g., 0, 1, >1)
b N/A: Not applicable. Includes one dog with a PFS of 43 days
c N/A: Not applicable. Includes one dog with a PFS of 60 days
d NE: Not estimable. Includes four dogs with PFS of 50, 63, 161, and 360 days
e NO: No observations
At the time of the Day 112 visit (one month after the last treatment), 33.0% (37/112 dogs) of dogs in the TANOVEA group were progression free compared with 0 (0/36 dogs) placebo dogs. Forty-four TANOVEA dogs completed five cycles of treatment, and five dogs had a CR at Day 365 (study end). Only one placebo dog completed five cycles and no placebo dogs achieved a CR. The primary reason for early withdrawal from the study for all dogs in both groups was disease progression, followed by death and adverse reactions.
Concomitant medications were used primarily for diarrhea, decreased appetite, and dermatopathy (bacterial skin infections). The most commonly used concomitant medications included antiemetics, antibiotics, electrolyte solutions, sedatives, appetite stimulants, analgesics, and topical corticosteroids with anti-infectives. No other chemotherapies or systemic corticosteroids were used in the study.
TARGET ANIMAL SAFETY:
The margin of safety and toxicity of rabacfosadine (not commercial formulation) was evaluated in three laboratory safety studies and one laboratory cardiovascular study. For each laboratory safety study, there were six male and six female, seven to eight month old Beagle dogs per treatment group. Three dogs/sex/group were necropsied following the last dose and three dogs/sex/group were necropsied following a 21-day recovery period. The VCOG-CTCAE1 definitions were used to grade the adverse reactions observed.
In the first study, a single dose of rabacfosadine was administered by a 30-minute intravenous infusion at doses of 0 (5% dextrose for injection), 0.25, 0.82, 2.5, and 8.2 mg/kg. Doses up to 2.5 mg/kg were tolerated. There were no adverse signs in the dogs administered 0.25 mg/kg. One dog administered 0.82 mg/kg vomited once. Dogs administered 2.5 mg/kg had abnormal feces (black, green, liquid, red, soft, mucoid) beginning on Day 4 and resolving by Day 10, and vomiting. There was a dose dependent weight loss and decreased food consumption in dogs administered ≥0.82 mg/kg. In all groups, including control, there were dermatologic changes (fur loss, thin fur, dry skin, red skin, skin lesions, scabs); there appeared to be a higher incidence in the dogs administered 2.5 mg/kg. Hematological changes included dose dependent leukopenia and Grade 1 to 4 neutropenia in dogs administered ≥0.82 mg/kg, with the nadir occurring between Days 6 and 9 and recovery occurring by Day 12. The 8.2 mg/kg dose was not tolerated. Dogs administered 8.2 mg/kg had clinical signs of abnormal feces, dehydration, vomiting, salivation, fever, tremors, decreased activity, and weakness resulting in a moribund state, and were either preterminally found dead or euthanized by Day 7. On necropsy at Day 3, pathology changes included dose-dependent effects on the gastrointestinal tract, lymphoid tissue, bone marrow, prostate, adrenal cortex, and kidneys (see below for more details). Following a 21-day recovery period, the majority of the changes in the intestines and lymphoid tissues reversed. Dose-dependent effects on the kidney were still observed.
In the second study, rabacfosadine was administered by a 30-minute intravenous infusion at doses of 0 (5% dextrose for injection), 0.082, 0.25, and 0.82 mg/kg once daily for 5 consecutive days and was tolerated at all dose levels. Dogs administered 0.82 mg/kg/day had suspected dehydration, vomiting, abnormal feces (soft, liquid, green, red), anorexia/hyporexia, decreased activity, and fever. Dogs with suspected dehydration received Lactated Ringers Solution subcutaneously. Treatment-related body weight loss and decreased food consumption were observed in dogs administered ≥0.25 mg/kg/day. In all groups, including control, there were dermatologic changes (fur loss, thin fur, dry skin, red skin, skin lesions, and scabs); there appeared to be a higher incidence in the groups administered drug. Hematological changes included dose dependent leukopenia in all treated groups and dose dependent Grade 1 to 4 neutropenia in dogs administered ≥0.25 mg/kg/day with nadirs occurring between Days 6 and 9 with recovery by Day 12. On necropsy at Day 6, pathology changes included dose-dependent effects on the gastrointestinal tract, lymphoid tissue, bone marrow, and male reproductive system (see below for more details). Following a 21-day recovery period, dose-dependent changes were present in the gastrointestinal tract, salivary gland, kidney, and testes in all treated dogs, in the pancreas and thymus in dogs administered ≥0.25 mg/kg/day, and in the bone marrow in dogs administered 0.82 mg/kg/day.
In the third study, rabacfosadine was administered by a 30-minute intravenous infusion at doses of 0 (5% dextrose for injection), 0.25, 0.50, and 1.0 mg/kg once every 7 days for 3 doses was tolerated at all dose levels. Treatment-related body weight loss and decreased food consumption were observed in dogs administered 1.0 mg/kg. In all groups, including control, there were dermatologic changes (fur loss, thin fur, dry skin, red skin, skin lesions, scabs); there appeared to be a higher incidence in the groups administered drug. Hematological changes included Grade 1 neutropenia predominantly in dogs administered 1.0 mg/kg. On necropsy on Day 16, pathology changes included dose-dependent effects on the gastrointestinal tract and lymphoid tissue, and non-dose dependent effects on the salivary gland, male reproductive tract, and kidney (see below for more details). These changes had only partial reversibility after a 21-day recovery period.
The pathology changes observed in the three studies included, in the gastrointestinal tract, necrosis of the stomach and combinations of mucosal hemorrhage, dilatation of mucosal glands/crypts, necrosis of crypt epithelial cells, atrophy of the mucosa/villi, edema, and inflammation of the intestinal wall. In the lymphoid tissue, observations included atrophy and necrosis of the thymus, spleen, lymph nodes, and gut associated lymphoid tissue (GALT). In the bone marrow, observations included hematopoietic hypocellularity. In the mandibular salivary gland, observations included glandular cell necrosis, atrophy, and inflammation. In the male reproductive tract, observations included small testes and epididymides, degeneration/atrophy of the seminiferous epithelium, and necrosis of the prostate. In the kidneys, observations included renal tubular vacuolation, dilatation, basophilia, degeneration, necrosis, and fibrosis. Tubular regeneration was observed following the recovery period. In the adrenal gland, observations included increased mitotic figures/necrosis in the adrenal cortex and adrenal fibrosis. In the pancreas, observations included acinar cell necrosis.
In the cardiovascular study, four telemeterized male, seven to eight month old Beagle dogs were administered 0 (5% dextrose for injection), 0.25, and 2.5 mg/kg rabacfosadine intravenously in an escalating dose regimen with a washout between doses. Dogs were monitored for 24 hours following dosing. There were no treatment-related effects on arterial blood pressure (mean, systolic, diastolic), heart rate, or electrocardiographic parameters.
STORAGE CONDITIONS:
Unopened vials: Store the vials refrigerated at 2 °C to 8 °C (36 °F to 46 °F). Retain in the original package to protect from light.
After reconstitution: Store at 20 °C to 25 °C (68 °F to 77 °F). TANOVEA should be diluted for infusion within 4 hours of reconstitution. The infusion solution should be used within 24 hours of being added to the infusion bag or syringe and within 4 hours of attachment to an intravenous administration set. Protection from light is not needed.
Disposal
Dispose of any unused product or waste materials in accordance with proper procedures for cytotoxic drugs.How Supplied
TANOVEA is supplied in a 3 mL amber Type I glass vial with rubber stopper, aluminum over-seal, and plastic flip-off cap, packaged in a carton. Each vial contains 16.4 mg of rabacfosadine, as succinate salt.
Pack size: 4 vials and 10 vials
References
1. Veterinary Cooperative Oncology Group. Veterinary cooperative oncology group - common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1.1. Veterinary and Comparative Oncology 2016;14(4):417-446.
2. Vail DM, et al. Response evaluation criteria for peripheral nodal lymphoma in dogs (v1.0)-a veterinary cooperative oncology group (VCOG) consensus document. Veterinary and Comparative Oncology 2009;8(1):28-37.
Manufactured for:
Elanco US Inc., Greenfield, IN 46140
Product of Canada
Revision date: January 2022
Approved by FDA under NADA # 141-545
Tanovea, Elanco and the diagonal bar logo are trademarks of Elanco or its affiliates.
© 2022 Elanco or its affiliates
PA103455X
CPN: 1131092.2
2500 INNOVATION WAY, GREENFIELD, IN, 46140
| Customer Service: | 317-276-1262 | |
| Technical Service: | 800-428-4441 | |
| Website: | www.elanco.us | |
| Email: | elanco@elanco.com |
 |
THIS SERVICE AND DATA ARE PROVIDED "AS IS". DVMetrics assumes no liability, and each user assumes full risk, responsibility, and liability, related to its use of the DVMetrics service and data. See the Terms of Use for further details. |
![]()
Copyright © 2025 Animalytix LLC. Updated: 2025-08-27