Tricor: Package Insert / Prescribing Info
Package insert / product label
Generic name: fenofibrate
Dosage form: tablet
Drug class: Fibric acid derivatives
Medically reviewed by Drugs.com. Last updated on Jun 18, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
TRICOR (fenofibrate) tablets, for oral use
Initial U.S. Approval: 1993
Recent Major Changes
Indications and Usage for Tricor
TRICOR is a peroxisome proliferator-activated receptor (PPAR) alpha agonist indicated as an adjunct to diet:
- to reduce triglyceride (TG) levels in adults with severe hypertriglyceridemia (TG greater than or equal to 500 mg/dL) (1).
- to reduce elevated low-density lipoprotein cholesterol (LDL-C) in adults with primary hyperlipidemia when use of recommended LDL-C lowering therapy is not possible (1).
Limitations of Use:
- Markedly elevated levels of serum TG (e.g., >2,000 mg/dL) may increase the risk of developing pancreatitis. The effect of fenofibrate therapy on reducing this risk has not been determined (1).
- Fenofibrate did not reduce coronary heart disease morbidity and mortality in two large, randomized controlled trials of patients with type 2 diabetes mellitus (1).
Tricor Dosage and Administration
-
Severe hypertriglyceridemia: 48 to 145 mg orally once daily; the dosage should be adjusted according to patient response (2.2).
-
Primary hyperlipidemia: 145 mg orally once daily (2.2).
- Administer as a single dose, at any time of day, with or without food (2.2).
- Assess TG when clinically appropriate, as early as 4 to 8 weeks after initiating TRICOR. Discontinue TRICOR in patients who do not have an adequate response after 2 months of treatment (2.2).
-
Renal impairment: Initial dosage of 48 mg orally once daily (2.3).
- Geriatric patients: Select the dosage on the basis of renal function (2.4).
Dosage Forms and Strengths
Tablets: 48 mg and 145 mg (3).
Contraindications
- Severe renal impairment, including those with end-stage renal disease (ESRD) and those receiving dialysis (4).
- Active liver disease including those with unexplained persistent liver function abnormalities (4).
- Pre-existing gallbladder disease (4).
- Hypersensitivity to fenofibrate, fenofibric acid, , or any of the excipients in TRICOR (4).
Warnings and Precautions
-
Hepatotoxicity: Serious drug-induced liver injury, including liver transplantation and death, has been reported with fenofibrates, including TRICOR. Monitor patient’s liver function, including serum ALT, AST, and total bilirubin, at baseline and periodically for the duration of therapy. Discontinue if signs or symptoms of liver injury develop or if elevated enzyme levels persist (5.2).
-
Myopathy and Rhabdomyolysis: Have been reported in patients taking fenofibrates. Risks are increased during co-administration with a statin, in geriatric patients, and in patients with renal impairment or hypothyroidism. Discontinue TRICOR if markedly elevated CK levels occur or if myopathy is either diagnosed or suspected. Temporarily discontinue TRICOR in patients experiencing an acute or serious condition at high risk of developing renal failure secondary to rhabdomyolysis. Inform patients of the risk of myopathy and rhabdomyolysis when starting or increasing the TRICOR dosage. Instruct patients to promptly report any unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever (5.3).
-
Increases in Serum Creatinine: Monitor renal function in patients with renal impairment taking TRICOR. Consider monitoring renal function in patients at risk for renal impairment (5.4).
-
Cholelithiasis: Fenofibrate may increase cholesterol excretion into the bile, leading to cholelithiasis. If cholelithiasis is suspected, gallbladder studies are indicated (5.5).
- Hypersensitivity Reactions: Acute hypersensitivity reactions, including anaphylaxis and angioedema, and delayed hypersensitivity reactions, including severe cutaneous adverse drug reactions have been reported postmarketing. Some cases were life-threatening and required emergency treatment. Discontinue TRICOR and treat appropriately if reactions occur (5.9).
Adverse Reactions/Side Effects
Adverse reactions (≥ 2% and greater than placebo): abnormal liver tests, increased AST, increased ALT, increased CPK, and rhinitis (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact AbbVie Inc. at 1-800-633-9110 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Consider if the benefit of concomitant use of statins or colchicine outweighs the increased risk of myopathy and rhabdomyolysis. Monitor patients for signs and symptoms of myopathy (7).
- Exercise caution in concomitant treatment with coumarin anticoagulants. Reduce the dosage of coumarin to maintain the PT/INR at the desired level to prevent bleeding complications (7).
- Consider the benefits and risks of concomitant use with immunosuppressants and other potentially nephrotoxic agents. Use the lowest effective dosage and monitor renal function (7).
- Administer TRICOR at least 1 hour before or 4 to 6 hours after a bile acid resin to avoid impeding its absorption (7).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Tricor
TRICOR is indicated as adjunctive therapy to diet:
• to reduce triglyceride (TG) levels in adults with severe hypertriglyceridemia (TG greater than or equal to 500 mg/dL).
• to reduce elevated low-density lipoprotein cholesterol (LDL-C) in adults with primary hyperlipidemia when use of recommended LDL-C lowering therapy is not possible.
Limitations of Use
• Markedly elevated levels of serum TG (e.g. > 2,000 mg/dL) may increase the risk of developing pancreatitis. The effect of fenofibrate therapy on reducing this risk has not been determined [see Warnings and Precautions (5.7)].
• Fenofibrate did not reduce coronary heart disease morbidity and mortality in two large, randomized controlled trials of patients with type 2 diabetes mellitus [see Warnings and Precautions (5.1) and Clinical Studies (14.4)].
2. Tricor Dosage and Administration
2.1 Prior to Initiation of TRICOR
• Assess lipid levels before initiating therapy. Identify other causes (e.g., diabetes mellitus, hypothyroidism, or medications) of high TG levels and manage as appropriate.
• Patients should be placed on an appropriate lipid-lowering diet before receiving TRICOR, and should continue this diet during treatment with TRICOR.
• In patients with diabetes and fasting chylomicronemia, improve glycemic control prior to considering starting TRICOR.
2.2 Recommended Dosage and Administration
• Severe hypertriglyceridemia:
○ The recommended dosage of TRICOR is 48 mg or 145 mg orally once daily.
○ Dosage should be individualized according to patient response, and should be adjusted if necessary following repeat lipid determinations at 4 to 8 week intervals.
• Primary hyperlipidemia:
○ The recommended dosage of TRICOR is 145 mg orally once daily.
• Administer TRICOR as a single dose at any time of day, with or without food.
• Advise patients to swallow TRICOR tablets whole. Do not crush, break, dissolve, or chew tablets.
• Assess TG when clinically appropriate, as early as 4 to 8 weeks after initiating TRICOR. Discontinue TRICOR in patients who do not have an adequate response after 2 months of treatment.
• If a dose is missed, advise patients not to take an extra dose. Resume treatment with the next dose.
• Advise patients to take TRICOR at least 1 hour before or 4 hours to 6 hours after a bile acid binding resin to avoid impeding its absorption.
2.3 Recommended Dosage in Patients with Renal Impairment
• Assess renal function prior to initiation of TRICOR and periodically thereafter [see Warnings and Precautions (5.4)].
• Treatment with TRICOR should be initiated at a dosage of 48 mg orally once daily in patients with mild to moderately impaired renal function (eGFR 30 to <60 mL/min/1.73m2), and increased only after evaluation of the effects on renal function and TG levels at this dosage.
• TRICOR is contraindicated in patients with severe renal impairment (eGFR <30 mL/min/1.73m2), including those with end-stage renal disease (ESRD) and those receiving dialysis [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
TRICOR Tablets:
- 48 mg: yellow tablets, imprinted with the code identification letters “FI”.
- 145 mg: white tablets, imprinted with the code identification letters “FO”.
4. Contraindications
TRICOR is contraindicated in patients with:
• Severe renal impairment, including those with end-stage renal disease (ESRD) and those receiving dialysis [see Clinical Pharmacology (12.3)].
• Active liver disease, including those with unexplained persistent liver function abnormalities [see Warnings and Precautions (5.2)].
• Pre-existing gallbladder disease [see Warnings and Precautions (5.5)].
• Hypersensitivity to fenofibrate, fenofibric acid, or any of the excipients in TRICOR. Serious hypersensitivity reactions including anaphylaxis and angioedema have been reported with fenofibrate [see Warnings and Precautions (5.9)].
5. Warnings and Precautions
5.1 Mortality and Coronary Heart Disease Morbidity
Fenofibrate did not reduce cardiovascular disease morbidity or mortality in two large, randomized controlled trials of patients with type 2 diabetes mellitus [see Clinical Studies (14.4)].
Because of chemical, pharmacological, and clinical similarities between fenofibrates, including TRICOR; pemafibrate; clofibrate; and gemfibrozil; the findings in 5 large randomized, placebo-controlled clinical trials with these other fibrate drugs may also apply to TRICOR.
Pemafibrate did not reduce cardiovascular disease morbidity or mortality in a large, randomized, placebo-controlled trial of patients with type 2 diabetes mellitus on background statin therapy [see Clinical Studies (14.4)].
In the Coronary Drug Project, a large trial conducted from 1965 to 1985 in men post myocardial infarction, there was no difference in mortality or nonfatal myocardial infarction between the clofibrate group and the placebo group after 5 years of treatment (NCT00000482).
In a trial conducted by the World Health Organization (WHO) from 1965 to 1976, men without known coronary artery disease were treated with placebo or clofibrate for 5 years and followed for an additional 1 year. There was a statistically significant, higher age-adjusted all-cause mortality in the clofibrate group compared with the placebo group (5.70% vs. 3.96%, p ≤ 0.01). Excess mortality was due to a 33% increase in non-cardiovascular causes, including malignancy, post-cholecystectomy complications, and pancreatitis.
The Helsinki Heart Study, conducted from 1982 to 1987, was a large (n=4,081) trial of middle-aged men without a history of coronary artery disease. Subjects received either placebo or gemfibrozil for 5 years, with a 3.5-year open extension afterward. Total mortality was numerically but not statistically higher in the gemfibrozil randomization group versus placebo [95% confidence interval (CI) of the hazard ratio (HR) 0.91 to 1.64].
A secondary prevention component of the Helsinki Heart Study treated middle-aged men with gemfibrozil or placebo for 5 years. The HR for cardiac deaths was 2.2, 95% CI, 0.94 to 5.05.
5.2 Hepatotoxicity
Serious drug-induced liver injury (DILI), including liver transplantation and death, has been reported with postmarketing use of fenofibrates, including TRICOR. DILI has been reported within the first few weeks of treatment or after several months of therapy and in some cases has reversed with discontinuation of TRICOR treatment. Patients with DILI have experienced signs and symptoms including dark urine, abnormal stool, jaundice, malaise, abdominal pain, myalgia, weight loss, pruritus, and nausea. Many patients had concurrent elevations of total bilirubin, serum alanine transaminase (ALT), and aspartate transaminase (AST). DILI has been characterized as hepatocellular, chronic active, and cholestatic hepatitis, and cirrhosis has occurred in association with chronic active hepatitis.
In clinical trials, an intermediate daily dosage or the maximum recommended daily dosage of fenofibrate have been associated with increases in serum AST or ALT. The incidence of increases in transaminases may be dose related [see Adverse Reactions (6.1)].
TRICOR is contraindicated in patients with active liver disease, including those with unexplained persistent liver function abnormalities. Monitor patient’s liver function, including serum ALT, AST, and total bilirubin, at baseline and periodically for the duration of therapy with TRICOR. Discontinue TRICOR if signs or symptoms of liver injury develop or if elevated enzyme levels persist (ALT or AST > 3 times the upper limit of normal, or if accompanied by elevation of bilirubin). Do not restart TRICOR in these patients if there is no alternative explanation for the liver injury.
5.3 Myopathy and Rhabdomyolysis
TRICOR may cause myopathy [muscle pain, tenderness, or weakness associated with elevated creatine kinase (CK)] and rhabdomyolysis.
Risk Factors for Myopathy
Risk factors for myopathy include age 65 years or older, uncontrolled hypothyroidism, renal impairment, and concomitant use with certain other drugs [see Drug Interactions (7) and Use in Specific Populations (8.6)].
Steps to Prevent or Reduce the Risk of Myopathy and Rhabdomyolysis
Data from observational studies indicate that the risk for rhabdomyolysis is increased when fibrates, including fenofibrates, are co-administered with a statin. Avoid concomitant use unless the benefit of further alterations in TG levels is likely to outweigh the increased risk of this drug combination [see Drug Interactions (7), Clinical Pharmacology (12.3), and Clinical Studies (14.4)].
Cases of myopathy, including rhabdomyolysis, have been reported with TRICOR co-administered with colchicine. Consider whether the benefit of using colchicine concomitantly with TRICOR outweighs the increased risk of myopathy [see Drug Interactions (7)].
Discontinue TRICOR if markedly elevated CK levels occur or if myopathy is either diagnosed or suspected. Muscle symptoms and CK elevations may resolve if TRICOR is discontinued. Temporarily discontinue TRICOR in patients experiencing an acute or serious condition at high risk of developing renal failure secondary to rhabdomyolysis (e.g., sepsis; shock; severe hypovolemia; major surgery; trauma; severe metabolic, endocrine, or electrolyte disorders; or uncontrolled epilepsy).
Inform patients of the risk of myopathy and rhabdomyolysis when starting or increasing the TRICOR dosage. Instruct patients to promptly report any unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever.
5.4 Increases in Serum Creatinine
Increases in serum creatinine have been reported in patients receiving fenofibrates. These increases tend to return to baseline following discontinuation of TRICOR. The clinical significance of this finding is unknown. Monitor renal function in patients with renal impairment taking TRICOR. Renal monitoring should also be considered for patients taking TRICOR at risk for renal insufficiency such as geriatric patients and patients with diabetes. TRICOR is contraindicated in patients with severe renal impairment, including those with end-stage renal disease (ESRD) and those receiving dialysis [see Dosage and Administration (2.3), Use in Specific Populations (8.6), and Clinical Pharmacology (12.3)].
5.5 Cholelithiasis
Fenofibrate may increase cholesterol excretion into the bile, leading to cholelithiasis. If cholelithiasis is suspected, gallbladder studies are indicated. TRICOR therapy should be discontinued if gallstones are found. TRICOR is contraindicated in patients with pre-existing gallbladder disease.
5.6 Increased Bleeding Risk with Coumarin Anticoagulants
Exercise caution when co-administering anticoagulants with TRICOR because of the potentiation of coumarin-type anticoagulant effects in prolonging the prothrombin time/International Normalized Ratio (PT/INR). To prevent bleeding complications, monitor the PT/INR frequently and adjust the dosage of the anticoagulant until the PT/INR has stabilized [see Drug Interactions (7)].
5.7 Pancreatitis
Pancreatitis has been reported in patients taking fenofibrates. This occurrence may represent a failure of efficacy in patients with severe hypertriglyceridemia, a direct drug effect, or a secondary phenomenon mediated through biliary tract stone or sludge formation with obstruction of the common bile duct.
5.8 Hematologic Changes
Mild to moderate hemoglobin, hematocrit, and white blood cell decreases have been observed in patients following initiation of therapy with fenofibrates. However, these levels stabilize during long-term administration. Thrombocytopenia and agranulocytosis have been reported in individuals treated with TRICOR. Periodic monitoring of red and white blood cell counts is recommended during the first 12 months of TRICOR administration.
5.9 Hypersensitivity Reactions
Acute Hypersensitivity
Anaphylaxis and angioedema have been reported with postmarketing use of fenofibrates. In some cases, reactions were life-threatening and required emergency treatment. If a patient develops signs or symptoms of an acute hypersensitivity reaction, advise them to seek immediate medical attention and discontinue TRICOR. TRICOR is contraindicated in patients with a hypersensitivity to fenofibrate, fenofibric acid, or any of the ingredients in TRICOR.
Delayed Hypersensitivity
Severe cutaneous adverse drug reactions (SCAR), including Stevens-Johnson syndrome, Toxic Epidermal Necrolysis, and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), have been reported with postmarketing use of fenofibrates occurring days to weeks after treatment initiation. The cases of DRESS were associated with cutaneous reactions (such as rash or exfoliative dermatitis) and a combination of eosinophilia, fever, systemic organ involvement (renal, hepatic, or respiratory). Discontinue TRICOR and treat patients appropriately if SCAR is suspected.
5.10 Venothromboembolic Disease
In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, pulmonary embolus (PE) and deep vein thrombosis (DVT) were observed at higher rates in the fenofibrate- than the placebo-treated group. Of 9,795 patients enrolled in FIELD, there were 4,900 in the placebo group and 4,895 in the fenofibrate group. For DVT, there were 48 events (1%) in the placebo group and 67 (1.4%) in the fenofibrate group (p = 0.074); and for PE, there were 32 (0.7%) events in the placebo group and 53 (1.1%) in the fenofibrate group (p = 0.022).
In the Coronary Drug Project, a higher proportion of the clofibrate group experienced definite or suspected fatal or nonfatal pulmonary embolism or thrombophlebitis than the placebo group (5.2% vs. 3.3% at five years; p < 0.01).
In the cardiovascular outcome trial with pemafibrate, pulmonary embolism was reported for 37 (0.7%) subjects in the pemafibrate group and 16 (0.3%) subjects in the placebo group. Deep vein thrombosis was reported for 36 (0.7%) subjects in the pemafibrate group and 13 (0.2%) subjects in the placebo group.
5.11 Paradoxical Decreases in HDL Cholesterol Levels
There have been postmarketing and clinical trial reports of severe decreases in high-density lipoprotein cholesterol (HDL-C) levels (as low as 2 mg/dL) occurring in patients with and without diabetes initiated on fibrate therapy, including fenofibrate. The decrease in HDL-C is mirrored by a decrease in apolipoprotein A1. This decrease has been reported to occur within 2 weeks to years after initiation of fibrate therapy. The HDL-C levels remain depressed until fibrate therapy has been withdrawn; the response to withdrawal of fibrate therapy is rapid and sustained. The clinical significance of this decrease in HDL-C is unknown. Check HDL-C levels within the first few months after initiation of TRICOR. If a severely depressed HDL-C level is detected, discontinue TRICOR and monitor HDL-C until it has returned to baseline. TRICOR should not be re-initiated.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described below and elsewhere in the labeling:
- Mortality and coronary heart disease morbidity [see Warnings and Precautions (5.1)]
- Hepatoxicity [see Warnings and Precautions (5.2)]
- Myopathy and Rhabdomyolysis [see Warnings and Precautions (5.3)]
- Increases in Serum Creatinine [see Warnings and Precautions (5.4)]
- Cholelithiasis [see Warnings and Precautions (5.5)]
- Increased Bleeding Risk with Coumarin Anticoagulants [see Warnings and Precautions (5.6)]
- Pancreatitis [see Warnings and Precautions (5.7)]
- Hematologic Changes [see Warnings and Precautions (5.8)]
- Hypersensitivity reactions [see Warnings and Precautions (5.9)]
- Venothromboembolic disease [see Warnings and Precautions (5.10)]
- Paradoxical Decreases in HDL Cholesterol Levels [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of TRICOR has been established in adults with hypertriglyceridemia or primary hyperlipidemia based on adequate and well-controlled trials of other formulations of fenofibrate, referenced below as “fenofibrate” [see Clinical Studies (14)]. Dosages of fenofibrate used in these trials were comparable to TRICOR 145 mg per day [see Clinical Pharmacology (12.3)].
Adverse reactions reported by 2% or more of patients treated with fenofibrate (and greater than placebo) during the double-blind, placebo-controlled trials are listed in Table 1. Adverse reactions led to discontinuation of treatment in 5% of patients treated with fenofibrate and in 3% treated with placebo. Increases in liver function tests were the most frequent events, causing discontinuation of fenofibrate treatment in 1.6% of patients in double-blind trials.
| Adverse Reaction | Placebo (N =365) | Fenofibrate (N = 439) |
| Abnormal Liver Tests | 1% | 8% |
| Abdominal Pain | 4% | 5% |
| Increased ALT | 2% | 3% |
| Increased AST | 1% | 3% |
| Increased Creatine Phosphokinase | 1% | 3% |
| Constipation | 1% | 2% |
| Rhinitis | 1% | 2% |
Other Adverse Reactions
Urticaria
Urticaria was seen in 1.1% vs. 0%, and rash in 1.4% vs. 0.8% of fenofibrate and placebo patients respectively in controlled trials.
Increases in Liver Enzymes
In a pooled analysis of 10 placebo-controlled trials, increases to >3 times the upper limit of normal in ALT occurred in 5.3% of patients taking either an intermediate or the maximum recommended daily dosage of fenofibrate versus 1.1% of patients treated with placebo. In an 8-week trial, the incidence of ALT or AST elevations ≥ 3 times the upper limit of normal was 13% in patients receiving an intermediate daily dosage or the maximum recommended daily dosage of fenofibrate and was 0% in those receiving the lowest recommended daily dosage of fenofibrate or placebo [see Warnings and Precautions (5.2)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of fenofibrate. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood: Anemia, white blood cell decreases
Gastrointestinal: Pancreatitis
General: Asthenia
Hepatobiliary: Increased total bilirubin, hepatitis, cirrhosis
Immune System: Anaphylaxis, angioedema
Lipid Disorders: Severely depressed HDL-cholesterol levels
Musculoskeletal: Myalgia, muscle spasms, rhabdomyolysis, arthralgia
Renal and Urinary: Acute renal failure
Respiratory: Interstitial lung disease
Skin and Subcutaneous Tissue: Photosensitivity reactions days to months after initiation. This may occur in patients who report a prior photosensitivity reaction to ketoprofen.
Related/similar drugs
7. Drug Interactions
Table 2 presents clinically important drug interactions with TRICOR.
| Statins | |
| Clinical Impact: | Fibrates may cause myopathy when given alone. The risk of myopathy and rhabdomyolysis is increased with concomitant use of fibrates with statins. |
| Intervention: | Consider if the benefit of using TRICOR concomitantly with statin therapy outweighs the increased risk of myopathy and rhabdomyolysis. If concomitant use is decided, monitor patients for signs and symptoms of myopathy, particularly during initiation of therapy and during upward dosage titration of statin therapy. |
| Colchicine | |
| Clinical Impact: | Cases of myopathy and rhabdomyolysis have been reported with concomitant use of colchicine with fenofibrates. |
| Intervention: | Consider if the benefit of using colchicine concomitantly with TRICOR outweighs the increased risk of myopathy and rhabdomyolysis. If concomitant use is decided, monitor patients for signs and symptoms of myopathy, particularly during initiation of therapy and during upward dosage titration of colchicine. |
| Coumarin Anticoagulants | |
| Clinical Impact: | Fibrates may cause potentiation of coumarin-type anticoagulant effects with prolongation of the PT/INR. |
| Intervention: | Caution should be exercised when coumarin anticoagulants are given in conjunction with TRICOR. The dosage of the anticoagulants should be reduced to maintain the PT/INR at the desired level to prevent bleeding complications. Frequent PT/INR determinations are advisable until it has been definitely determined that the PT/INR has stabilized |
| Immunosuppressants | |
| Clinical Impact: | Immunosuppressants such as cyclosporine and tacrolimus can produce nephrotoxicity with decreases in creatinine clearance and rises in serum creatinine, and because renal excretion is the primary elimination route of fibrate drugs including TRICOR, there is a risk that an interaction will lead to deterioration of renal function. |
| Intervention: | The benefits and risks of using TRICOR with immunosuppressants and other potentially nephrotoxic agents should be carefully considered, and the lowest effective dosage employed and renal function monitored. |
| Bile-Acid Binding Resins | |
| Clinical Impact: | Bile-acid binding resins may bind other drugs given concurrently. |
| Intervention: | In patients taking a bile acid resin, administer TRICOR at least 1 hour before or 4 to 6 hours after the bile acid resin to avoid impeding its absorption. |
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Limited available data with fenofibrate use in pregnant women are insufficient to determine a drug associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. In animal reproduction studies, no evidence of embryo-fetal toxicity was observed with oral administration of fenofibrate in rats and rabbits during organogenesis at doses less than or comparable to the maximum recommended clinical dosage of 145 mg of TRICOR daily, based on body surface area (mg/m2). Adverse reproductive outcomes occurred at higher doses in the presence of maternal toxicity (see Data). TRICOR should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In pregnant rats given oral dietary doses of 14 mg/kg/day, 127 mg/kg/day, and 361 mg/kg/day from gestation day 6 to 15 during the period of organogenesis, no adverse developmental findings were observed at 14 mg/kg/day (less than the clinical exposure at the maximum recommended human dose [MRHD] of 300 mg fenofibrate daily, comparable to 145 mg of TRICOR daily, based on body surface area comparisons). Increased fetal skeletal malformations were observed at maternally toxic doses (361 mg/kg/day, corresponding to 12 times the clinical exposure at the MRHD) that significantly suppressed maternal body weight gain.
In pregnant rabbits given oral gavage doses of 15 mg/kg/day, 150 mg/kg/day, and 300 mg/kg/day from gestation day 6 to 18 during the period of organogenesis and allowed to deliver, no adverse developmental findings were observed at 15 mg/kg/day (a dose that approximates the clinical exposure at the MRHD, based on body surface area comparisons). Aborted litters were observed at maternally toxic doses (≥ 150 mg/kg/day, corresponding to ≥ 10 times the clinical exposure at the MRHD) that suppressed maternal body weight gain.
In pregnant rats given oral dietary doses of 15 mg/kg/day, 75 mg/kg/day, and 300 mg/kg/day from gestation day 15 through lactation day 21 (weaning), no adverse developmental effects were observed at 15 mg/kg/day (less than the clinical exposure at the MRHD, based on body surface area comparisons), despite maternal toxicity (decreased weight gain). Post-implantation loss was observed at ≥ 75 mg/kg/day (≥ 2 times the clinical exposure at the MRHD) in the presence of maternal toxicity (decreased weight gain). Decreased pup survival was noted at 300 mg/kg/day (10 times the clinical exposure at the MRHD), which was associated with decreased maternal body weight gain/maternal neglect.
8.2 Lactation
Risk Summary
There is no available information on the presence of fenofibrate in human milk, effects of the drug on the breastfed infant, or the effects on milk production. Fenofibrate is present in the milk of rats and is therefore likely to be present in human milk. Because of the potential for serious adverse reactions in breastfed infants, such as disruption of infant lipid metabolism, women should not breastfeed during treatment with TRICOR and for 5 days after the final dose.
8.4 Pediatric Use
The safety and effectiveness of TRICOR have not been established in pediatric patients with severe hypertriglyceridemia or primary hyperlipidemia.
8.5 Geriatric Use
Assess renal function in geriatric patients and follow contraindications and dosing recommendations for patients with renal impairment [see Contraindications (4), Warnings and Precautions (5.3, 5.4), and Use in Specific Populations (8.6)]. While fenofibric acid exposure is not influenced by age, geriatric patients are more likely to have renal impairment, and fenofibric acid is substantially excreted by the kidney [see Clinical Pharmacology (12.3)]. Consider monitoring renal function in geriatric patients taking TRICOR.
8.6 Renal Impairment
TRICOR is contraindicated in patients with severe renal impairment (eGFR <30 mL/min/1.73m2), including those with end-stage renal disease (ESRD) and those receiving dialysis. Dosage reduction is required in patients with mild to moderate renal impairment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)]. Patients with severe renal impairment have 2.7-fold higher exposure of fenofibric acid and increased accumulation of fenofibric acid during chronic dosing compared with healthy volunteers. Renal impairment is a risk factor for myopathy and rhabdomyolysis [see Warnings and Precautions (5.3, 5.4) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
The use of TRICOR has not been evaluated in patients with hepatic impairment. TRICOR is contraindicated in patients with active liver disease, including those with unexplained persistent liver function abnormalities [see Clinical Pharmacology (12.3)].
10. Overdosage
In the event of an overdose of TRICOR, consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdosage management recommendations. There is no specific treatment for overdose with TRICOR. General supportive care of the patient is indicated, including monitoring of vital signs and observation of clinical status, should an overdose occur. If indicated, elimination of unabsorbed drug should be achieved by emesis or gastric lavage; usual precautions should be observed to maintain the airway. Because fenofibric acid is highly bound to plasma proteins, hemodialysis should not be considered.
11. Tricor Description
TRICOR (fenofibrate) tablets is a peroxisome proliferator-activated receptor (PPAR) alpha agonist available as tablets for oral administration. Each tablet contains 48 mg or 145 mg of fenofibrate. The chemical name for fenofibrate is 2-[4-(4-chlorobenzoyl) phenoxy]-2-methyl-propanoic acid, 1-methylethyl ester with the following structural formula:
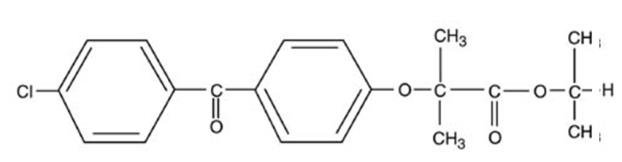
The empirical formula is C20H21O4Cl and the molecular weight is 360.83; fenofibrate is insoluble in water. The melting point is 79 to 82°C. Fenofibrate is a white solid which is stable under ordinary conditions.
Inactive Ingredients
Each tablet contains crospovidone, docusate sodium, hypromellose 2910 (3 cps), lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, sodium lauryl sulfate, and sucrose.
In addition, individual tablets contain:
48 mg tablets
D&C Yellow #10 aluminum lake, FD&C Yellow #6 /sunset yellow FCF aluminum lake, FD&C Blue #2 /indigo carmine aluminum lake, polyvinyl alcohol, soybean lecithin, talc, titanium dioxide, xanthan gum.
145 mg tablets
polyvinyl alcohol, soybean lecithin, talc, titanium dioxide, xanthan gum.
12. Tricor - Clinical Pharmacology
12.1 Mechanism of Action
The active moiety of TRICOR is fenofibric acid. The pharmacological effects of fenofibric acid in both animals and humans have been studied through oral administration of fenofibrate.
The lipid-modifying effects of fenofibric acid seen in clinical practice have been explained in vivo in transgenic mice and in vitro in human hepatocyte cultures by the activation of PPAR alpha receptor. Through this mechanism, fenofibric acid increases lipolysis and elimination of triglyceride-rich particles from plasma by activating lipoprotein lipase and reducing production of apoprotein C-III (an inhibitor of lipoprotein lipase activity).
12.2 Pharmacodynamics
Fenofibric acid, the active metabolite of fenofibrate, produces reductions in total cholesterol (Total-C), total triglycerides, and triglyceride rich lipoprotein (VLDL) in treated patients with severe hypertriglyceridemia.
12.3 Pharmacokinetics
Fenofibrate is a pro-drug of the active chemical moiety fenofibric acid. Fenofibrate is converted by ester hydrolysis in the body to fenofibric acid which is the active constituent measurable in the circulation.
Absorption
The absolute bioavailability of fenofibrate cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection. However, fenofibrate is well absorbed from the gastrointestinal tract. Peak plasma levels of fenofibric acid occur within 6 to 8 hours after administration.
Effect of Food
Exposure to fenofibric acid in plasma, as measured by Cmax and AUC, is not significantly different when a single 145 mg dose of fenofibrate is administered under fasting or nonfasting conditions.
Distribution
Upon multiple dosing of fenofibrate, fenofibric acid steady state is achieved within 9 days. Plasma concentrations of fenofibric acid at steady state are approximately double of those following a single dose. Serum protein binding was approximately 99% in normal and hyperlipidemic subjects.
Elimination
Fenofibric acid is eliminated with a half-life of 20 hours, allowing once daily administration of TRICOR.
Metabolism
Following oral administration, fenofibrate is rapidly hydrolyzed by esterases to the active metabolite, fenofibric acid; no unchanged fenofibrate is detected in plasma.
Fenofibric acid is primarily conjugated with glucuronic acid and then excreted in urine. A small amount of fenofibric acid is reduced at the carbonyl moiety to a benzhydrol metabolite which is, in turn, conjugated with glucuronic acid and excreted in urine.
In vivo metabolism data indicate that neither fenofibrate nor fenofibric acid undergo oxidative metabolism (e.g., cytochrome P450) to a significant extent.
Excretion
After absorption, fenofibrate is mainly excreted in the urine in the form of metabolites, primarily fenofibric acid and fenofibric acid glucuronide. After administration of radiolabeled fenofibrate, approximately 60% of the dose appeared in the urine and 25% was excreted in the feces.
Specific Populations
Geriatric Patients
In geriatric volunteers 77 to 87 years of age, the oral clearance of fenofibric acid following a single oral dose of fenofibrate was 1.2 L/h, which compares to 1.1 L/h in young adults. This indicates that a similar dosage regimen can be used in geriatric patients with normal renal function, without increasing accumulation of the drug or metabolites [see Dosage and Administration (2.4) and Use in Specific Populations (8.5)].
Pediatric Patients
The pharmacokinetics of TRICOR has not been studied in pediatric populations.
Male and Female Patients
No pharmacokinetic difference between males and females has been observed for fenofibrate.
Racial and Ethnic Groups
The influence of race on the pharmacokinetics of fenofibrate has not been studied, however fenofibrate is not metabolized by enzymes known for exhibiting inter-ethnic variability.
Patients with Renal Impairment
The pharmacokinetics of fenofibric acid were examined in patients with mild, moderate, and severe renal impairment. Patients with severe renal impairment (creatinine clearance [CrCl ≤30 mL/min] or estimated glomerular filtration rate [eGFR] < 30 mL/min/1.73m2) showed a 2.7-fold increase in exposure for fenofibric acid and increased accumulation of fenofibric acid during chronic dosing compared to that of healthy subjects. Patients with mild to moderate renal impairment (CrCl 30 mL/min to 80 mL/min or eGFR 30 to 59 mL/min/1.73m2) had similar exposure but an increase in the half-life for fenofibric acid compared to that of healthy subjects [see Dosage and Administration (2.3)].
Patients with Hepatic Impairment
No pharmacokinetic studies have been conducted in patients with hepatic impairment.
Drug Interaction Studies
In vitro studies using human liver microsomes indicate that fenofibrate and fenofibric acid are not inhibitors of cytochrome (CYP) P450 isoforms CYP3A4, CYP2D6, CYP2E1, or CYP1A2. They are weak inhibitors of CYP2C8, CYP2C19 and CYP2A6, and mild-to-moderate inhibitors of CYP2C9 at therapeutic concentrations.
Table 3 describes the effects of co-administered drugs on fenofibric acid systemic exposure. Table 4 describes the effects of co-administered fenofibrate or fenofibric acid on systemic exposure of other drugs.
| Co-
Administered Drug | Dosage Regimen of
Co-Administered Drug | Dosage Regimen of
Fenofibrate | Changes in Fenofibric
Acid Exposure |
|
| AUC | Cmax | |||
| Lipid-lowering medications | ||||
| Atorvastatin | 20 mg once daily for 10 days | Fenofibrate 160 mg1 once daily for 10 days | ↓2% | ↓4% |
| Pravastatin | 40 mg as a single dose | Fenofibrate 3 x 67 mg2 as a single dose | ↓1% | ↓2% |
| Fluvastatin | 40 mg as a single dose | Fenofibrate 160 mg1 as a single dose | ↓2% | ↓10% |
| Anti-diabetic medications | ||||
| Glimepiride | 1 mg as a single dose | Fenofibrate 145 mg1 once daily for 10 days | ↑1% | ↓1% |
| Metformin | 850 mg three times daily for 10 days | Fenofibrate 54 mg1 three times daily for 10 days | ↓9% | ↓6% |
| Rosiglitazone | 8 mg once daily for 5 days | Fenofibrate 145 mg1 once daily for 14 days | ↑10% | ↑3% |
| 1 TriCor (fenofibrate) oral tablet 2 TriCor (fenofibrate) oral micronized capsule |
||||
| Dosage Regimen of
Fenofibrate | Dosage Regimen of Co-
Administered Drug | Changes in Co-Administered
Drug Exposure |
||
| Analyte | AUC | Cmax | ||
| Lipid-lowering medications | ||||
| Fenofibrate 160 mg1 once daily for 10 days | Atorvastatin, 20 mg once daily for 10 days | Atorvastatin | ↓17% | 0% |
| Fenofibrate 3 x 67 mg2 as a single dose | Pravastatin, 40 mg as a single dose | Pravastatin | ↑13% | ↑13% |
| 3α-Hydroxyl-iso- pravastatin | ↑26% | ↑29% | ||
| Fenofibrate 160 mg1 as a single dose | Fluvastatin, 40 mg as a single dose | (+)-3R, 5S- Fluvastatin | ↑15% | ↑16% |
| Anti-diabetic medications | ||||
| Fenofibrate 145 mg1 once daily for 10 days | Glimepiride, 1 mg as a single dose | Glimepiride | ↑35% | ↑18% |
| Fenofibrate 54 mg1 three times daily for 10 days | Metformin, 850 mg three times daily for 10 days | Metformin | ↑3% | ↑6% |
| Fenofibrate 145 mg1 once daily for 14 days | Rosiglitazone, 8 mg once daily for 5 days | Rosiglitazone | ↑6% | ↓1% |
| 1 TriCor (fenofibrate) oral tablet 2 TriCor (fenofibrate) oral micronized capsule |
||||
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, and Impairment of Fertility
Carcinogenesis
Two dietary carcinogenicity studies have been conducted in rats with fenofibrate. In the first 24-month study, Wistar rats were dosed with fenofibrate at 10 mg/kg/day, 45 mg/kg/day, and 200 mg/kg/day, approximately 0.3 times, 1 time, and 6 times the maximum recommended human dose (MRHD) based on body surface area comparisons (mg/m2). At a dose of 200 mg/kg/day (at 6 times the MRHD), the incidence of liver carcinomas was significantly increased in both sexes. A statistically significant increase in pancreatic carcinomas was observed in males at 1 and 6 times the MRHD; an increase in pancreatic adenomas and benign testicular interstitial cell tumors was observed at 6 times the MRHD in males. In a second 24-month study in a different strain of rats (Sprague-Dawley), doses of 10 mg/kg/day and 60 mg/kg/day (0.3 and 2 times the MRHD) produced significant increases in the incidence of pancreatic acinar adenomas in both sexes and increases in testicular interstitial cell tumors in males at 2 times the MRHD.
A 117-week carcinogenicity study was conducted in rats comparing three drugs: fenofibrate 10 mg/kg/day and 60 mg/kg/day (0.3 and 2 times the MRHD), clofibrate (400 mg/kg/day; 2 times the human dose), and gemfibrozil (250 mg/kg/day; 2 times the human dose) based on mg/m2 surface area. Fenofibrate increased pancreatic acinar adenomas in both sexes. Clofibrate increased hepatocellular carcinoma and pancreatic acinar adenomas in males and hepatic neoplastic nodules in females. Gemfibrozil increased hepatic neoplastic nodules in males and females, while all three drugs increased testicular interstitial cell tumors in males.
In a 21-month study in CF-1 mice, fenofibrate 10 mg/kg/day, 45 mg/kg/day, and 200 mg/kg/day (approximately 0.2 times, 1 time, and 3 times the MRHD on the basis of mg/m2 surface area) significantly increased the liver carcinomas in both sexes at 3 times the MRHD. In a second 18-month study at 10 mg/kg/day, 60 mg/kg/day, and 200 mg/kg/day, fenofibrate significantly increased the liver carcinomas in male mice and liver adenomas in female mice at 3 times the MRHD.
Electron microscopy studies have demonstrated peroxisomal proliferation following fenofibrate administration to the rat. An adequate study to test for peroxisome proliferation in humans has not been done, but changes in peroxisome morphology and numbers have been observed in humans after treatment with other members of the fibrate class when liver biopsies were compared before and after treatment in the same individual.
Mutagenesis
Fenofibrate has been demonstrated to be devoid of mutagenic potential in the following tests: Ames, mouse lymphoma, chromosomal aberration and unscheduled DNA synthesis in primary rat hepatocytes.
Impairment of Fertility
In fertility studies rats were given oral dietary doses of fenofibrate, males received 61 days prior to mating and females 15 days prior to mating through weaning which resulted in no adverse effect on fertility at doses up to 300 mg/kg/day (approximately 10 times the MRHD, based on mg/m2 surface area comparisons).
14. Clinical Studies
14.1 Overview of Clinical Trials
The effectiveness of TRICOR has been established in adults with hypertriglyceridemia or primary hyperlipidemia based on adequate and well-controlled trials of other formulations of fenofibrate, referenced below as “fenofibrate.” Dosages of fenofibrate used in these trials were comparable to TRICOR 145 mg per day [see Clinical Pharmacology (12.3)].
14.2 Clinical Trials in Adults with Hypertriglyceridemia
The effects of fenofibrate on serum TG were studied in two randomized, double-blind, placebo-controlled clinical trials of 147 patients with hypertriglyceridemia. Patients were treated for 8 weeks under protocols that differed only in that one entered patients with baseline TG levels of 500 to 1,500 mg/dL, and the other TG levels of 350 to 499 mg/dL. In patients with hypertriglyceridemia and normal cholesterolemia with or without hyperchylomicronemia, treatment with fenofibrate decreased primarily very low density lipoprotein (VLDL) TG and VLDL cholesterol (VLDL-C). Treatment of patients with elevated TG often results in an increase of LDL-C (see Table 5).
| Trial 1 | Placebo | TRICOR | ||||||
| Baseline TG levels
350 to 499 mg/dL | N | Baseline
Mean (mg/dL) | Endpoint
Mean (mg/dL) | Mean %
Change | N | Baseline
Mean (mg/dL) | Endpoint
Mean (mg/dL) | Mean %
Change |
| TG | 28 | 449 | 450 | -0.5 | 27 | 432 | 223 | -46.2* |
| VLDL-TG | 19 | 367 | 350 | 2.7 | 19 | 350 | 178 | -44.1* |
| Total-C | 28 | 255 | 261 | 2.8 | 27 | 252 | 227 | -9.1* |
| HDL-C | 28 | 35 | 36 | 4 | 27 | 34 | 40 | 19.6* |
| LDL-C | 28 | 120 | 129 | 12 | 27 | 128 | 137 | 14.5 |
| VLDL-C | 27 | 99 | 99 | 5.8 | 27 | 92 | 46 | -44.7* |
| Trial 2 | Placebo | TRICOR | ||||||
| Baseline TG levels
500 to 1500 mg/dL | N | Baseline
Mean (mg/dL) | Endpoint
Mean (mg/dL) | Mean %
Change | N | Baseline
Mean (mg/dL) | Endpoint
Mean (mg/dL) | Mean %
Change |
| TG | 44 | 710 | 750 | 7.2 | 48 | 726 | 308 | -54.5* |
| VLDL-TG | 29 | 537 | 571 | 18.7 | 33 | 543 | 205 | -50.6* |
| Total-C | 44 | 272 | 271 | 0.4 | 48 | 261 | 223 | -13.8* |
| HDL-C | 44 | 27 | 28 | 5.0 | 48 | 30 | 36 | 22.9* |
| LDL-C | 42 | 100 | 90 | -4.2 | 45 | 103 | 131 | 45.0* |
| VLDL-C | 42 | 137 | 142 | 11.0 | 45 | 126 | 54 | -49.4* |
| * =p < 0.05 vs. Placebo | ||||||||
14.3 Clinical Trials in Adults with Primary Hyperlipidemia
The effects of fenofibrate were assessed in four randomized, placebo-controlled, double-blind, parallel-group trials in patients with hyperlipidemia and mixed dyslipidemia. TRICOR therapy reduced LDL-C, Total-C, and TG, and increased HDL-C (see Table 6).
Table 6. Mean Percent Change in Lipid Parameters at End of Treatment†
| Treatment Group | Total-C | LDL-C | HDL-C | TG |
| Mean baseline lipid values (n=646) | 306.9 mg/dL | 213.8 mg/dL | 52.3 mg/dL | 191.0 mg/dL |
| All fenofibrate (n=361) | -18.7%* | -20.6%* | +11.0%* | -28.9%* |
| Placebo (n=285) | -0.4% | -2.2% | +0.7% | +7.7% |
| † Duration of study treatment was 3 to 6 months. * p = < 0.05 vs. Placebo |
||||
14.4 Lack of Efficacy in Cardiovascular Outcomes Trials
Fenofibrate did not reduce cardiovascular disease morbidity or mortality in two large, randomized controlled trials of patients with type 2 diabetes mellitus.
The Action to Control Cardiovascular Risk in Diabetes Lipid (ACCORD Lipid) (NCT00000620) trial was a randomized placebo-controlled trial of 5,518 patients (2,765 assigned to receive fenofibrate) with type 2 diabetes mellitus on background statin therapy treated with fenofibrate. The mean age at baseline was 62 years and 31% were female. Overall, 68% were White, 15% were Black or African American; 7% identified as Hispanic or Latino ethnicity. The mean duration of follow-up was 4.7 years. The primary outcome of major adverse cardiovascular events (MACE), a composite of non-fatal myocardial infarction, non-fatal stroke, and cardiovascular disease death was a HR of 0.92 (95% CI, 0.79 to 1.08) for fenofibrate plus statin combination therapy as compared to statin monotherapy.
The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial was a 5-year randomized, placebo-controlled trial of 9,795 patients (4,895 assigned to receive fenofibrate) with type 2 diabetes mellitus treated with fenofibrate. The mean age at baseline was 62 years, 37% were female, and 93% were White. The primary outcome of coronary heart disease events was a HR of 0.89 (95% CI, 0.75 to 1.05) with fenofibrate compared to placebo. The HR for total and coronary heart disease mortality, respectively, was 1.11 (95% CI, 0.95 to 1.29) and 1.19 (95% CI, 0.90 to 1.57) with fenofibrate as compared to placebo.
Because of chemical, pharmacological, and clinical similarities between fenofibrate and pemafibrate, findings in a large randomized, placebo-controlled clinical trial with pemafibrate are relevant for TRICOR.
Pemafibrate did not reduce cardiovascular disease morbidity or mortality in a large, randomized, placebo-controlled trial of patients with type 2 diabetes mellitus (TG levels of 200 to 499 mg per deciliter and HDL-C levels of 40 mg per deciliter or lower), on background statin therapy (NCT03071692). The trial was a randomized placebo-controlled trial of 10,497 patients (5,240 assigned to receive pemafibrate) with type 2 diabetes mellitus on background lipid-lowering therapy. The median age at baseline was 64 years and 28% were female. Overall, 86% were White, 5% were Asian, 3% were Black or African American; 19% identified as Hispanic or Latino ethnicity. The median duration of follow-up was 3.4 years. The primary outcome of major adverse cardiovascular events (MACE), a composite of non-fatal myocardial infarction, non-fatal ischemic stroke, coronary revascularization, and death from cardiovascular causes, was a HR of 1.03 (95% CI, 0.91 to 1.15) for pemafibrate plus statin combination therapy as compared to statin monotherapy.
16. How is Tricor supplied
TRICOR® (fenofibrate tablets) is available in two strengths:
48 mg
Yellow tablets, imprinted with the code identification letters “FI”, available in bottles of 90 (NDC 0074-3173-90).
145 mg
White tablets, imprinted with the code identification letters “FO”, available in bottles of 90 (NDC 0074-3189-90).
Storage
Store at 25°C (77°F); excursions permitted to 15-30°C (59-86°F).
[See USP Controlled Room Temperature]. Keep out of the reach of children. Protect from moisture.
17. Patient Counseling Information
Hepatotoxicity
Inform patients that TRICOR may cause liver enzyme elevations and possibly liver failure. Advise patients to promptly report fatigue, anorexia, right upper abdominal discomfort, dark urine or jaundice [see Contraindications (4) and Warnings and Precautions (5.2)].
Myopathy and Rhabdomyolysis
Advise patients that TRICOR may cause myopathy and rhabdomyolysis. Inform patients that the risk is also increased when taking certain types of medication and they should discuss all medication, both prescription and over the counter, with their healthcare provider. Instruct patients to inform other healthcare providers prescribing a new medication or increasing the dosage of an existing medication that they are taking TRICOR. Instruct patients to promptly report any unexplained muscle pain, tenderness, or weakness particularly if accompanied by malaise or fever [see Warnings and Precautions (5.3) and Drug Interactions (7)].
Increased Bleeding Risk with Coumarin Anticoagulants
Inform patients that the concomitant use of TRICOR with coumarin-type anticoagulants may increase the risk of bleeding. Advise patients if they are taking or planning to take coumarin-type anticoagulants to inform their healthcare providers and that increased monitoring may be necessary [see Warnings and Precautions (5.6) and Drug Interactions (7)].
Hypersensitivity Reactions
Inform patients that serious hypersensitivity reactions, such as anaphylaxis and angioedema, have been reported with fenofibrates. Advise patients to report immediately any signs or symptoms suggesting allergic reaction, and to discontinue drug until they have consulted prescribing physicians [see Warnings and Precautions (5.9)].
Pregnancy
Advise patients to inform their healthcare provider of a known or suspected pregnancy to discuss if TRICOR should be discontinued [see Use in Specific Populations (8.1)].
Lactation
Advise patients that breastfeeding during treatment with TRICOR is not recommended [see Use in Specific Populations (8.2)].
Missed Doses
If a dose is missed, advise patients to not take an extra dose and to resume treatment with the next dose.
Manufactured for AbbVie Inc., North Chicago, IL 60064, U.S.A.
by Fournier Laboratories Ireland Limited, Anngrove, Carrigtwohill Co. Cork, Ireland.
© 2025 AbbVie. All rights reserved.
TRICOR and its design are trademarks of AbbVie Inc.
| TRICOR
fenofibrate tablet |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
| TRICOR
fenofibrate tablet |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - AbbVie Inc. (078458370) |
More about TriCor (fenofibrate)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (16)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: fibric acid derivatives
- Breastfeeding
- En español
Patient resources
Professional resources
Other brands
Lofibra, Antara, Triglide, Fenoglide, Lipofen
