Lemtrada: Package Insert / Prescribing Info
Package insert / product label
Generic name: alemtuzumab
Dosage form: injection, solution, concentrate
Drug class: CD52 monoclonal antibodies
J Code (medical billing code): J0202 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on May 26, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
LEMTRADA® (alemtuzumab) injection, for intravenous use
Initial U.S. Approval: 2001
WARNING: AUTOIMMUNITY, INFUSION REACTIONS, STROKE, AND MALIGNANCIES
See full prescribing information for complete boxed warning.
- LEMTRADA causes serious, sometimes fatal, autoimmune conditions such as immune thrombocytopenia and anti-glomerular basement membrane disease. Monitor complete blood counts with differential, serum creatinine levels, and urinalysis with urine cell counts monthly until 48 months after the last dose. (5.1)
- LEMTRADA causes serious and life-threatening infusion reactions. LEMTRADA must be administered in a setting with appropriate equipment and personnel to manage anaphylaxis or serious infusion reactions. Monitor patients for two hours after each infusion. Make patients aware that serious infusion reactions can also occur after the 2-hour monitoring period. (5.2)
- Serious and life-threatening stroke has been reported within 3 days of LEMTRADA administration. Instruct patients to seek immediate medical attention if symptoms of stroke occur. (5.3)
- LEMTRADA may cause an increased risk of malignancies, including thyroid cancer, melanoma, and lymphoproliferative disorders. Perform baseline and yearly skin exams. (5.4)
- LEMTRADA is available only through a restricted distribution program. (5.5)
Indications and Usage for Lemtrada
- LEMTRADA is a CD52-directed cytolytic monoclonal antibody indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include relapsing-remitting disease and active secondary progressive disease, in adults. Because of its safety profile, the use of LEMTRADA should generally be reserved for patients who have had an inadequate response to two or more drugs indicated for the treatment of MS. (1.5)
Limitations of Use:
LEMTRADA is not recommended for use in patients with clinically isolated syndrome (CIS) because of its safety profile. (1.5)
Lemtrada Dosage and Administration
- Baseline laboratory tests are required prior to treatment. (2.1)
- Administer LEMTRADA by intravenous infusion over 4 hours for 2 or more treatment courses:
Initial treatment of 2 courses:- First course: 12 mg/day on 5 consecutive days. (2.3)
- Second course: 12 mg/day on 3 consecutive days 12 months after first treatment course. (2.3)
- Premedicate with corticosteroids prior to LEMTRADA infusion for the first 3 days of each treatment course. (2.2)
- Administer antiviral agents for herpetic prophylaxis starting on the first day of LEMTRADA dosing and continuing for a minimum of two months after completion of LEMTRADA dosing or until CD4+ lymphocyte count is more than 200 cells per microliter, whichever occurs later. (2.2)
- Must be diluted prior to administration. (2.4)
Dosage Forms and Strengths
Injection: 12 mg/1.2 mL (10 mg/mL) in a single-dose vial. (3)
Contraindications
Warnings and Precautions
- Immune Thrombocytopenia: Obtain complete blood counts (CBCs) with differential prior to initiation of treatment and at monthly intervals thereafter until 48 months after the last infusion. (5.6)
- Glomerular Nephropathies: Obtain serum creatinine levels, urinalysis with cell counts and urine protein to creatinine ratio prior to initiation of treatment. Monitor serum creatinine levels and urinalysis with cell counts at monthly intervals thereafter until 48 months after the last infusion. (5.7)
- Thyroid Disorders: Obtain thyroid function tests prior to initiation of treatment and every 3 months until 48 months after the last infusion. (5.8)
- Other Autoimmune Cytopenias: Monitor CBCs monthly until 48 months after the last infusion. (2.6, 5.9)
- Autoimmune Hepatitis: If signs of hepatic dysfunction occur, promptly measure serum transaminases and total bilirubin and interrupt or discontinue treatment. (5.10)
- Hemophagocytic Lymphohistiocytosis: Consider this diagnosis and evaluate patients immediately if they develop signs or symptoms of systemic inflammation. Discontinue LEMTRADA if an alternative etiology is not established. (5.11)
- Adult Onset Still's Disease (AOSD): If a patient develops AOSD, they require prompt evaluation and treatment. (5.12)
- Thrombotic Thrombocytopenic Purpura (TTP): Evaluate patients immediately if they develop clinical symptoms or laboratory findings consistent with TTP. Discontinue LEMTRADA if TTP is confirmed or if an alternative etiology is not established. (5.13)
- Autoimmune Encephalitis (AIE): Evaluate patients if they develop signs and symptoms suggestive of AIE, such as subacute onset of memory impairment, altered mental status, psychiatric symptoms, neurological findings, and seizures. (5.14)
- Acquired Hemophilia A: Obtain a coagulopathy panel including aPTT in patients who present with signs such as spontaneous subcutaneous hematomas, extensive bruising, hematuria, epistaxis, or gastrointestinal or other types of bleeding. (5.15)
- Immune-Mediated Colitis: Immune-mediated colitis has been reported in the postmarketing setting. Monitor patients for new or persistent diarrhea or other gastrointestinal symptoms, and evaluate promptly if colitis is suspected. (5.16)
- Infections: Administration is contraindicated in patients with active infection. Do not administer live viral vaccines following a course of LEMTRADA. (4, 5.17)
- Progressive Multifocal Leukoencephalopathy (PML): Withhold LEMTRADA at the first sign or symptom suggestive of PML. (5.18)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥10% and > interferon beta-1a): rash, headache, pyrexia, nasopharyngitis, nausea, urinary tract infection, fatigue, insomnia, upper respiratory tract infection, herpes viral infection, urticaria, pruritus, thyroid gland disorders, fungal infection, arthralgia, pain in extremity, back pain, diarrhea, sinusitis, oropharyngeal pain, paresthesia, dizziness, abdominal pain, flushing, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genzyme Corporation at 1-800-745-4447, option 2 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
Pregnancy: May cause fetal harm. (8.1)
Women of childbearing potential should use effective contraception during and for 4 months after a course of treatment with LEMTRADA. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2024
Full Prescribing Information
WARNING: AUTOIMMUNITY, INFUSION REACTIONS, STROKE, AND MALIGNANCIES
- LEMTRADA causes serious, sometimes fatal, autoimmune conditions such as immune thrombocytopenia and anti-glomerular basement membrane disease. Monitor complete blood counts with differential, serum creatinine levels, and urinalysis with urine cell counts before starting treatment and then at monthly intervals until 48 months after the last dose of LEMTRADA [see Warnings and Precautions (5.1)].
- LEMTRADA causes serious and life-threatening infusion reactions. LEMTRADA must be administered in a setting with appropriate equipment and personnel to manage anaphylaxis or serious infusion reactions. Monitor patients for two hours after each infusion. Make patients aware that serious infusion reactions can also occur after the 2-hour monitoring period [see Warnings and Precautions (5.2)].
- Serious and life-threatening stroke (including ischemic and hemorrhagic stroke) has been reported within 3 days of LEMTRADA administration. Instruct patients to seek immediate medical attention if symptoms of stroke occur [see Warnings and Precautions (5.3)].
- LEMTRADA may cause an increased risk of malignancies, including thyroid cancer, melanoma, and lymphoproliferative disorders. Perform baseline and yearly skin exams [see Warnings and Precautions (5.4)].
- Because of the risk of autoimmunity, infusion reactions, and malignancies, LEMTRADA is available only through restricted distribution under a Risk Evaluation Mitigation Strategy (REMS) Program. Call 1-855-676-6326 to enroll in the LEMTRADA REMS program [see Warnings and Precautions (5.5)].
1. Indications and Usage for Lemtrada
LEMTRADA is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include relapsing-remitting disease and active secondary progressive disease, in adults. Because of its safety profile, the use of LEMTRADA should generally be reserved for patients who have had an inadequate response to two or more drugs indicated for the treatment of MS [see Warnings and Precautions (5)].
Limitations of Use
LEMTRADA is not recommended for use in patients with clinically isolated syndrome (CIS) because of its safety profile [see Warnings and Precautions (5)].
2. Lemtrada Dosage and Administration
2.1 Testing and Procedures Prior to Treatment
Baseline laboratory tests are required prior to treatment with LEMTRADA [see Dosage and Administration (2.6)]. In addition, prior to starting treatment with LEMTRADA [see Warnings and Precautions (5.15)]:
- complete any necessary immunizations at least 6 weeks prior to treatment.
- determine whether patients have a history of varicella or have been vaccinated for varicella zoster virus (VZV). If not, test the patient for antibodies to VZV and consider vaccination for those who are antibody-negative. Postpone treatment with LEMTRADA until 6 weeks after VZV vaccination.
- perform tuberculosis screening according to local guidelines.
- instruct patients to avoid potential sources of Listeria monocytogenes.
2.2 Recommended Premedication and Concomitant Medication
Corticosteroids
Premedicate patients with high dose corticosteroids (1,000 mg methylprednisolone or equivalent) immediately prior to LEMTRADA infusion and for the first 3 days of each treatment course [see Warnings and Precautions (5.2)].
Herpes Prophylaxis
Administer antiviral prophylaxis for herpetic viral infections starting on the first day of each treatment course and continue for a minimum of two months following treatment with LEMTRADA or until the CD4+ lymphocyte count is at least 200 cells per microliter, whichever occurs later [see Warnings and Precautions (5.15)].
2.3 Recommended Dosage
- The recommended dosage of LEMTRADA is 12 mg/day administered by intravenous infusion for 2 treatment courses: First Treatment Course: 12 mg/day on 5 consecutive days (60 mg total dose).
- Second Treatment Course: 12 mg/day on 3 consecutive days (36 mg total dose) administered 12 months after the first treatment course.
Following the second treatment course, subsequent treatment courses of 12 mg per day on 3 consecutive days (36 mg total dose) may be administered, as needed, at least 12 months after the last dose of any prior treatment courses.
2.4 Preparation Instructions
Follow the steps below to prepare the diluted solution of LEMTRADA for intravenous infusion:
- Inspect LEMTRADA visually for particulate matter and discoloration prior to administration. Do not use if particulate matter is present or the solution is discolored. Do not freeze or shake vials prior to use.
- Withdraw 1.2 mL of LEMTRADA from the vial into a syringe using aseptic technique and inject into a 100 mL bag of sterile 0.9% Sodium Chloride, USP or 5% Dextrose in Water, USP.
- Gently invert the bag to mix the solution. Ensure the sterility of the prepared solution because it contains no antimicrobial preservatives. Each vial is for single use only.
Prior to administration, protect diluted LEMTRADA solution from light and store for as long as 8 hours either at room temperature 15°C to 25°C (59°F to 77°F) or keep refrigerated at conditions 2°C to 8°C (36°F to 46°F).
2.5 Infusion Instructions
Infuse LEMTRADA over 4 hours starting within 8 hours after dilution. Extend the duration of the infusion if clinically indicated.
Administer LEMTRADA in a setting in which equipment and personnel to appropriately manage anaphylaxis, serious infusion reactions, myocardial ischemia, myocardial infarction, and cerebrovascular and respiratory adverse reactions are available [see Warnings and Precautions (5.2)].
Do not add or simultaneously infuse other drug substances through the same intravenous line. Do not administer as an intravenous push or bolus.
Obtain a baseline ECG. Monitor vital signs before the infusion and periodically during the infusion. Provide appropriate symptomatic treatment for infusion reactions as needed. Consider immediate discontinuation of the intravenous infusion if severe infusion reactions occur.
Observe patients for infusion reactions during and for at least 2 hours after each LEMTRADA infusion. Consider longer periods of observation if clinically indicated. Inform patients that they should report symptoms that occur during and after each infusion because they may indicate a need for prompt medical intervention [see Warnings and Precautions (5.2)].
2.6 Laboratory Testing and Monitoring to Assess Safety
Measure the urine protein to creatinine ratio prior to initiation of treatment. Conduct the following laboratory tests at baseline and at periodic intervals until 48 months after the last treatment course of LEMTRADA in order to monitor for early signs of potentially serious adverse effects:
- Complete blood count (CBC) with differential (prior to treatment initiation and at monthly intervals thereafter)
- Serum creatinine levels (prior to treatment initiation and at monthly intervals thereafter)
- Urinalysis with urine cell counts (prior to treatment initiation and at monthly intervals thereafter)
- A test of thyroid function, such as thyroid stimulating hormone (TSH) level (prior to treatment initiation and every 3 months thereafter)
- Serum transaminases (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) and total bilirubin levels (prior to treatment initiation and periodically thereafter)
Conduct baseline and yearly skin exams to monitor for melanoma [see Warnings and Precautions (5.4)].
3. Dosage Forms and Strengths
Injection: 12 mg/1.2 mL (10 mg/mL) in a single-dose vial. LEMTRADA is a clear and colorless to slightly yellow solution that requires dilution prior to intravenous infusion.
4. Contraindications
LEMTRADA is contraindicated in patients:
- with known hypersensitivity or anaphylactic reactions to alemtuzumab or any of the excipients in LEMTRADA
- who are infected with human immunodeficiency virus (HIV) because LEMTRADA causes prolonged reductions of CD4+ lymphocyte counts
- with active infection
5. Warnings and Precautions
5.1 Autoimmunity
Treatment with LEMTRADA can result in the formation of autoantibodies and increase the risk of serious autoimmune conditions, which may be life threatening.
In clinical studies (controlled and open-label extension), the following serious autoimmune conditions were reported in LEMTRADA-treated patients, which are described in separate warnings:
- thyroid disorders (36.8%) [see Warnings and Precautions (5.8)]
- immune thrombocytopenia (2%) [see Warnings and Precautions (5.6)]
- glomerular nephropathies (0.3%) [see Warnings and Precautions 5.7]
- autoimmune hemolytic anemia (0.3%), autoimmune pancytopenia (0.2%), and autoimmune neutropenia (0.1%) [see Warnings and Precautions (5.9)]
- acquired hemophilia A (anti-Factor VIII antibodies) (0.1%) [see Warnings and Precautions (5.15)]
In clinical studies (controlled and open-label extension), vitiligo (0.3%), undifferentiated connective tissue disorders (0.2%), type 1 diabetes (0.2%), rheumatoid arthritis (0.1%), and retinal pigment epitheliopathy (0.1%) were also reported in LEMTRADA-treated patients.
In the postmarketing setting, cases of autoimmune hepatitis [see Warnings and Precautions (5.10)], hemophagocytic lymphohistiocytosis [see Warnings and Precautions (5.11)], Adult Onset Still's Disease [see Warnings and Precautions (5.12)], thrombotic thrombocytopenic purpura [see Warnings and Precautions (5.13)], autoimmune encephalitis [see Warnings and Precautions (5.14)], immune-mediated colitis [see Warnings and Precautions (5.16)], Guillain-Barré syndrome, and vasculitis have been reported.
Chronic inflammatory demyelinating polyradiculoneuropathy has been reported in the treatment of patients with B-cell chronic lymphocytic leukemia (B-CLL), as well as other autoimmune disorders, generally at higher and more frequent doses than recommended in MS.
Autoantibodies may be transferred from the mother to the fetus during pregnancy. A case of transplacental transfer of anti-thyrotropin receptor antibodies resulting in neonatal Graves' disease occurred after alemtuzumab treatment in the mother [see Use in Specific Populations (8.1)].
LEMTRADA may increase the risk of other autoimmune conditions because of the broad range of autoantibody formation with LEMTRADA.
Measure the urine protein to creatinine ratio prior to initiation of treatment. Monitor complete blood counts with differential, serum creatinine levels, and urinalysis with urine cell counts before starting treatment and then at monthly intervals until 48 months after the last dose of LEMTRADA to allow for early detection and treatment of autoimmune adverse reactions [see Dosage and Administration (2.6)]. After 48 months, testing should be performed based on clinical findings suggestive of autoimmunity.
LEMTRADA is available only through a restricted program under a REMS [see Warnings and Precautions (5.5)].
5.2 Infusion Reactions
LEMTRADA causes cytokine release syndrome resulting in infusion reactions, some of which may be serious and life threatening. In clinical studies, 92% of LEMTRADA-treated patients experienced infusion reactions. In some patients, infusion reactions were reported more than 24 hours after LEMTRADA infusion. Serious reactions occurred in 3% of patients and included anaphylaxis in 2 patients (including anaphylactic shock), angioedema, bronchospasm, hypotension, chest pain, bradycardia, tachycardia (including atrial fibrillation), transient neurologic symptoms, hypertension, headache, pyrexia, and rash. Other infusion reactions included nausea, urticaria, pruritus, insomnia, chills, flushing, fatigue, dyspnea, pulmonary infiltrates, dysgeusia, dyspepsia, dizziness, and pain. In clinical studies, 0.6% of patients with infusion reactions received epinephrine or atropine.
During postmarketing use, cases of pulmonary alveolar hemorrhage, myocardial ischemia, myocardial infarction, stroke (including ischemic and hemorrhagic stroke), and cervicocephalic (e.g., vertebral, carotid) arterial dissection have been reported. Reactions may occur following any of the doses during the treatment course. In the majority of cases, time to onset was within 1 to 3 days of LEMTRADA infusion. Patients should be informed about the signs and symptoms and advised to seek immediate medical attention if any of these symptoms occur. Cases of severe (including fatal) neutropenia have been reported within 2 months of LEMTRADA infusion; some cases resolved with receiving granulocyte-colony stimulating factor treatment. Mild to moderate decreases in platelet counts, starting at the time of alemtuzumab infusion and often resolving without treatment, have been reported. Other serious and sometimes fatal infusion reactions (e.g., hypoxia, syncope, acute respiratory distress syndrome, respiratory arrest, myocardial infarction, acute cardiac insufficiency, cardiac arrest) have been reported in the treatment of patients with B-CLL, as well as other disorders, generally at higher and more frequent doses than recommended in MS.
Premedicate patients with corticosteroids immediately prior to LEMTRADA infusion for the first 3 days of each treatment course. In clinical studies, patients received 1,000 mg of methylprednisolone for the first 3 days of each LEMTRADA treatment course. Consider pretreatment with antihistamines and/or antipyretics prior to LEMTRADA administration. Infusion reactions may occur despite pretreatment.
Consider additional monitoring in patients with medical conditions which predispose them to cardiovascular or pulmonary compromise. Physicians should alert patients that an infusion reaction could occur within 48 hours of infusion.
LEMTRADA can only be administered in certified healthcare settings that have on-site access to equipment and personnel trained to manage infusion reactions (including anaphylaxis, cerebrovascular, cardiac and respiratory emergencies) [see Dosage and Administration (2.5)].
LEMTRADA is available only through a restricted program under a REMS [see Warnings and Precautions (5.5)].
5.3 Stroke and Cervicocephalic Arterial Dissection
Stroke
In the postmarketing setting, serious and life-threatening stroke (including ischemic and hemorrhagic stroke) has been reported within 3 days of LEMTRADA administration, with most cases occurring within 1 day.
Cervicocephalic Arterial Dissection
In the postmarketing setting, cases of cervicocephalic (e.g., vertebral, carotid) arterial dissection involving multiple arteries have been reported within 3 days of LEMTRADA administration. Ischemic stroke was reported in one of these cases.
Educate patients on the symptoms of stroke and cervicocephalic (e.g., carotid, vertebral) arterial dissection. Instruct patients to seek immediate medical attention if symptoms of stroke or cervicocephalic arterial dissection occur.
5.4 Malignancies
Thyroid Cancer
LEMTRADA may increase the risk of thyroid cancer. In controlled clinical studies, 3 of 919 (0.3%) LEMTRADA-treated patients developed thyroid cancer, compared to none in the interferon beta-1a–treated group. However, screening for thyroid cancer was performed more frequently in the LEMTRADA-treated group, because of the higher incidence of autoimmune thyroid disorders in those patients. Two additional cases of thyroid cancer in LEMTRADA-treated patients occurred in uncontrolled studies.
Patients and healthcare providers should monitor for symptoms of thyroid cancer including a new lump or swelling in the neck, pain in the front of the neck, persistent hoarseness or other voice changes, trouble swallowing or breathing, or a constant cough not due to an upper respiratory tract infection.
Melanoma
LEMTRADA may increase the risk of melanoma. In MS clinical studies (controlled and open-label extension), 5 of 1486 (0.3%) LEMTRADA-treated patients developed melanoma or melanoma in situ. One of those patients had evidence of locally advanced disease.
Perform baseline and yearly skin examinations to monitor for melanoma in patients receiving LEMTRADA.
Lymphoproliferative Disorders and Lymphoma
Cases of lymphoproliferative disorders and lymphoma have occurred in LEMTRADA-treated patients with MS, including a MALT lymphoma, Castleman's Disease, and a fatality following treatment of non-Epstein Barr Virus–associated Burkitt's lymphoma. There are postmarketing reports of Epstein Barr Virus–associated lymphoproliferative disorders in non-MS patients.
Because LEMTRADA is an immunomodulatory therapy, caution should also be exercised in initiating LEMTRADA in patients with preexisting or ongoing malignancies.
LEMTRADA is available only through a restricted program under a REMS [see Warnings and Precautions (5.5)].
5.5 LEMTRADA REMS Program
LEMTRADA is available only through a restricted program under a REMS called the LEMTRADA REMS Program because of the risks of autoimmunity, infusion reactions, and malignancies [see Warnings and Precautions (5.1, 5.2, 5.4)].
Notable requirements of the LEMTRADA REMS Program include the following:
- Prescribers must be certified with the program by enrolling and completing training.
- Patients must enroll in the program and comply with ongoing monitoring requirements [see Dosage and Administration (2.6)].
- Pharmacies must be certified with the program and must only dispense to certified healthcare facilities that are authorized to receive LEMTRADA.
- Healthcare facilities must enroll in the program and verify that patients are authorized before infusing LEMTRADA. Healthcare facilities must have on-site access to equipment and personnel trained to manage infusion reactions.
Further information, including a list of qualified healthcare facilities, is available at 1-855-676-6326.
5.6 Immune Thrombocytopenia
Immune thrombocytopenia (ITP) occurred in 2% of LEMTRADA-treated patients in MS clinical studies (controlled and open-label extension).
In a controlled clinical study in patients with MS, one LEMTRADA-treated patient developed ITP that went unrecognized prior to the implementation of monthly blood monitoring requirements, and died from intracerebral hemorrhage. Nadir platelet counts ≤20,000 cells per microliter as a result of ITP occurred in 2% of all LEMTRADA-treated patients in clinical studies in MS. Anti-platelet antibodies did not precede ITP onset. ITP has been diagnosed more than 3 years after the last LEMTRADA dose.
Symptoms of ITP include easy bruising, petechiae, spontaneous mucocutaneous bleeding (e.g., epistaxis, hemoptysis), and heavier than normal or irregular menstrual bleeding. Hemoptysis may also be indicative of anti-glomerular basement membrane (GBM) disease [see Warnings and Precautions (5.7)], and an appropriate differential diagnosis has to be undertaken. Remind the patient to remain vigilant for symptoms they may experience and to seek immediate medical help if they have any concerns.
Obtain complete blood counts (CBCs) with differential prior to initiation of treatment and at monthly intervals thereafter until 48 months after the last infusion [see Dosage and Administration (2.6)]. After this period of time, testing should be performed based on clinical findings suggestive of ITP. If ITP is suspected, a complete blood count should be obtained immediately. If ITP onset is confirmed, promptly initiate appropriate medical intervention.
5.7 Glomerular Nephropathies Including Anti-glomerular Basement Membrane Disease
Glomerular nephropathies occurred in 0.3% of LEMTRADA-treated patients in MS clinical studies. There were 3 cases of membranous glomerulonephritis and 2 cases of anti-glomerular basement membrane (anti-GBM) disease.
In postmarketing cases, some LEMTRADA-treated patients with anti-GBM disease developed end-stage renal disease requiring dialysis or renal transplantation. Urgent evaluation and treatment are required because early treatment can improve the preservation of renal function. Anti-GBM disease can be life-threatening if left untreated. Alveolar hemorrhage, manifested as hemoptysis, is a common component of anti-GBM disease and has been reported in postmarketing cases. Cases of anti-GBM disease have been diagnosed up to 40 months after the last dose of LEMTRADA.
Symptoms of nephropathy may include edema, hematuria, change in urine color, decreased urine output, fatigue, dyspnea, and hemoptysis. Patients and caregivers should be instructed to seek medical advice if they have concerns.
Obtain serum creatinine levels, urinalysis with cell counts, and urine protein to creatinine ratio prior to initiation of treatment. Obtain serum creatinine levels and urinalysis with cell counts at monthly intervals thereafter until 48 months after the last infusion. After this period of time, testing should be performed based on clinical findings suggestive of nephropathies.
For urine dipstick results of 1+ protein or greater, measure the urine protein to creatinine ratio. For urine protein to creatinine ratio greater than 200 mg/g, increase in serum creatinine greater than 30%, or unexplained hematuria, perform further evaluation for nephropathies. Increased serum creatinine with hematuria or signs of pulmonary involvement of anti-GBM disease (e.g., hemoptysis, exertional dyspnea) warrant immediate evaluation. Early detection and treatment of nephropathies may decrease the risk of poor outcomes.
5.8 Thyroid Disorders
Thyroid endocrine disorders, including autoimmune thyroid disorders, occurred in 36.8% of LEMTRADA-treated patients in MS clinical studies (controlled and open-label extension). Newly diagnosed thyroid disorders occurred throughout the uncontrolled clinical study follow-up period, more than 7 years after the first LEMTRADA dose. Autoimmune thyroid disorders included Graves' disease, hyperthyroidism, hypothyroidism, autoimmune thyroiditis, and goiter. Graves' ophthalmopathy with decreased vision, eye pain, and exophthalmos occurred in 2% of LEMTRADA-treated patients. Seven patients required surgical orbital decompression. Serious thyroid events occurred in about 5.2% of LEMTRADA-treated patients in clinical studies and included cardiac and psychiatric events associated with thyroid disease. Of all LEMTRADA-treated patients, 3.8% underwent thyroidectomy.
Thyroid disease poses special risks in women who are pregnant [see Use in Specific Populations (8.1)].
Obtain thyroid function tests, such as TSH levels, prior to initiation of treatment and every 3 months thereafter until 48 months after the last infusion. Continue to test thyroid function after 48 months if clinically indicated or in case of pregnancy.
In patients with ongoing thyroid disorder, LEMTRADA should be administered only if the potential benefit justifies the potential risks.
5.9 Other Autoimmune Cytopenias
Autoimmune cytopenias such as neutropenia (0.1%), hemolytic anemia (0.3%), and pancytopenia (0.2%) occurred in LEMTRADA-treated patients in MS clinical studies (controlled and open-label extension). In cases of autoimmune hemolytic anemia, patients tested positive for direct antiglobulin antibodies, and nadir hemoglobin levels ranged from 2.9–8.6 g/dL. Symptoms of autoimmune hemolytic anemia include weakness, chest pain, jaundice, dark urine, and tachycardia. One LEMTRADA-treated patient with autoimmune pancytopenia died from sepsis.
During postmarketing use, additional autoimmune cytopenias, including fatal autoimmune hemolytic anemia and aplastic anemia, have been reported in the treatment of patients with B-CLL, as well as other disorders, generally at higher and more frequent doses of alemtuzumab than recommended in MS.
Use CBC results to monitor for cytopenias. Prompt medical intervention is indicated if a cytopenia is confirmed.
5.10 Autoimmune Hepatitis
Autoimmune hepatitis causing clinically significant liver injury, including acute liver failure requiring transplant, has been reported in patients treated with LEMTRADA in the postmarketing setting. If a patient develops clinical signs, including unexplained liver enzyme elevations or symptoms suggestive of hepatic dysfunction (e.g., unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine), promptly measure serum transaminases and total bilirubin and interrupt or discontinue treatment with LEMTRADA, as appropriate.
Prior to starting treatment with LEMTRADA, obtain serum transaminases (ALT and AST) and total bilirubin levels. Obtain transaminase levels and total bilirubin levels at periodic intervals until 48 months after the last dose.
5.11 Hemophagocytic Lymphohistiocytosis
Hemophagocytic lymphohistiocytosis (HLH) has occurred in patients taking LEMTRADA. HLH is a life-threatening syndrome of pathologic immune activation characterized by clinical signs and symptoms of extreme systemic inflammation. It is associated with high mortality rates if not recognized early and treated. Common findings include fever, hepatosplenomegaly, rash, lymphadenopathy, neurologic symptoms (e.g., mental status changes, ataxia, or seizures), cytopenias, high serum ferritin, hypertriglyceridemia, and liver function and coagulation abnormalities. Hemophagocytosis may be seen on histologic examination of bone marrow, spleen, or lymph nodes. In cases of HLH reported with LEMTRADA, most patients presented with fever, elevated ferritin, transaminitis, hypertriglyceridemia, and all patients required hospitalization. Although the small number of cases limits the ability to draw conclusions pertaining to mean or range of latency for HLH, symptoms have been reported to occur within approximately thirteen months to thirty-three months following the initiation of treatment. Patients who develop early manifestations of pathologic immune activation should be evaluated immediately, and a diagnosis of HLH should be considered. LEMTRADA should be discontinued if an alternate etiology for the signs or symptoms cannot be established.
5.12 Adult Onset Still's Disease (AOSD)
During postmarketing use, Adult Onset Still's Disease (AOSD) has been reported in patients treated with LEMTRADA. AOSD is a rare inflammatory condition that requires urgent evaluation and treatment. Patients with AOSD may have a combination of the following signs and symptoms: fever, arthritis, rash, and leukocytosis in the absence of infections, malignancies, and other rheumatic conditions. Patients with manifestations of AOSD should be evaluated immediately and LEMTRADA should be discontinued if an alternate etiology for the signs or symptoms cannot be established.
5.13 Thrombotic Thrombocytopenic Purpura (TTP)
TTP has been reported in patients treated with LEMTRADA. TTP is characterized by thrombocytopenia, microangiopathic hemolytic anemia, neurological sequelae, fever, and renal impairment. TTP is associated with high morbidity and mortality rates if not recognized and treated early. LEMTRADA should be discontinued if TTP is confirmed or an alternate etiology for the signs or symptoms cannot be established.
5.14 Autoimmune Encephalitis (AIE)
During postmarketing use, cases of AIE have been reported in patients treated with LEMTRADA. AIE can present with a variety of clinical manifestations, including subacute onset of memory impairment, altered mental status, psychiatric symptoms, neurological findings, and seizures. LEMTRADA should be discontinued if AIE is confirmed by the presence of neural autoantibodies or an alternate etiology cannot be established.
5.15 Acquired Hemophilia A
Cases of acquired hemophilia A (anti-Factor VIII antibodies) have been reported in both the clinical trial and postmarketing settings. Patients typically present with spontaneous subcutaneous hematomas and extensive bruising, although hematuria, epistaxis, gastrointestinal, or other types of bleeding may occur. Obtain a coagulopathy panel including aPTT in patients who present with such symptoms. Patients should be informed about the signs and symptoms of acquired hemophilia A and advised to seek immediate medical attention if any of these symptoms occur.
5.16 Immune-Mediated Colitis
Immune-mediated colitis, which can present as a severe and acute-onset form of colitis, has been reported in patients receiving LEMTRADA in the postmarketing setting. Some cases of colitis were serious, requiring hospitalization. Systemic corticosteroids were required in some of these patients. The time from treatment initiation to onset of symptoms in these cases ranged from a few months to years.
Monitor patients for immune-mediated colitis during and after LEMTRADA treatment, and evaluate promptly if signs and symptoms that may indicate immune-mediated colitis, such as new or persistent diarrhea or other gastrointestinal signs and symptoms, occur.
It is unknown whether immune-mediated colitis in patients treated with LEMTRADA is a monophasic disorder, or whether recurrent flares (as are seen in patients with inflammatory bowel disease) will occur.
5.17 Infections
Infections occurred in 71% of LEMTRADA-treated patients compared to 53% of patients treated with interferon beta-1a in controlled clinical studies in MS up to 2 years in duration. Infections that occurred more often in LEMTRADA-treated patients than interferon beta-1a patients included nasopharyngitis, urinary tract infection, upper respiratory tract infection, sinusitis, herpetic infections, influenza, and bronchitis. Serious infections occurred in 3% of patients treated with LEMTRADA as compared to 1% of patients treated with interferon beta-1a. Serious infections in the LEMTRADA group included: appendicitis, gastroenteritis, pneumonia, herpes zoster, and tooth infection.
Do not administer live viral vaccines following a course of LEMTRADA. Patients treated with LEMTRADA have altered immunity and may be at increased risk of infection following administration of live viral vaccines.
LEMTRADA administration is contraindicated in patients with active infection [see Contraindications (4)].
Concomitant use of LEMTRADA with antineoplastic or immunosuppressive therapies could increase the risk of immunosuppression.
Opportunistic Infections
In the postmarketing setting, serious, sometimes fatal, opportunistic infections have been reported in patients taking LEMTRADA, including aspergillosis, coccidioidomycosis, histoplasmosis, Pneumocystis jirovecii pneumonia, nocardiosis, Epstein-Barr virus, and cytomegalovirus infections.
Listeria Monocytogenes Infections
Listeria monocytogenes infections (e.g., meningitis, encephalitis, sepsis, and gastroenteritis), including fatal cases of Listeria meningoencephalitis, have occurred in LEMTRADA-treated patients. Listeria infections have occurred as early as 3 days after treatment and up to 8 months after the last LEMTRADA dose. The duration of increased risk for Listeria infection after LEMTRADA treatment is unknown.
Advise patients to avoid or adequately heat foods that are potential sources of Listeria monocytogenes (e.g., deli meat, dairy products made with unpasteurized milk, soft cheeses, or undercooked meat, seafood, or poultry). Initiate these Listeria precautions prior to starting LEMTRADA treatment. The incubation period for Listeria monocytogenes ranges from 3 to 70 days. In most cases, signs and symptoms of invasive listeriosis start within 1 month of exposure to Listeria monocytogenes. Symptoms of Listeria infection include fever, chills, diarrhea, nausea, vomiting, headache, pains in joints and muscles, neck stiffness, difficulty walking, mental status changes, coma, and other neurologic changes. As is the case with many infections, treatment cannot always prevent mortality and morbidity related to Listeria infections. Therefore, advise patients to watch for symptoms of Listeria infection and seek prompt medical help if symptoms occur.
Herpes Viral Infections
In controlled clinical studies, 16% of LEMTRADA-treated patients developed a herpes viral infection compared to 3% of interferon beta-1a patients. These events included oral herpes (8.8%), herpes zoster (4.2%), herpes simplex (1.8%), and genital herpes (1.3%). Serious herpetic infections in LEMTRADA-treated patients included primary varicella (0.1%), herpes zoster (0.2%), and herpes meningitis (0.1%). Administer antiviral agents for herpetic prophylaxis at appropriate suppressive dosing regimens. Administer antiviral prophylaxis for herpetic viral infections starting on the first day of each treatment course and continue for a minimum of two months following treatment with LEMTRADA or until the CD4+ lymphocyte count is ≥200 cells per microliter, whichever occurs later [see Dosage and Administration (2.2)].
Human Papilloma Virus
Cervical human papilloma virus (HPV) infection, including cervical dysplasia, occurred in 2% of LEMTRADA-treated patients. Annual HPV screening is recommended for female patients.
Tuberculosis
Tuberculosis occurred in patients treated with LEMTRADA and interferon beta-1a in controlled clinical studies. Active and latent tuberculosis cases occurred in 0.3% of LEMTRADA-treated patients, most often in endemic regions. Perform tuberculosis screening according to local guidelines prior to initiation of LEMTRADA. For patients testing positive in tuberculosis screening, treat by standard medical practice prior to therapy with LEMTRADA.
Fungal Infections
Fungal infections, especially oral and vaginal candidiasis, occurred more commonly in LEMTRADA-treated patients (12%) than in patients treated with interferon beta-1a (3%) in controlled clinical studies in MS.
Infections in Non-MS Patients
During postmarketing use, serious and sometimes fatal viral, bacterial, protozoan, and fungal infections, including some due to reactivation of latent infections, have been reported in the treatment of patients with B-CLL, as well as other disorders, generally at higher and more frequent doses than recommended in MS.
Hepatitis
No data are available on the association of LEMTRADA with Hepatitis B virus (HBV) or Hepatitis C virus (HCV) reactivation because patients with evidence of active or chronic infections were excluded from the clinical studies. Consider screening patients at high risk of HBV and/or HCV infection before initiation of LEMTRADA and exercise caution in prescribing LEMTRADA to patients identified as carriers of HBV and/or HCV as these patients may be at risk of irreversible liver damage relative to a potential virus reactivation as a consequence of their pre-existing status.
5.18 Progressive Multifocal Leukoencephalopathy (PML)
Progressive multifocal leukoencephalopathy (PML) has occurred in a patient with MS treated with LEMTRADA. PML is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability. PML was diagnosed two months after the second course of LEMTRADA. The patient had previously received multiple MS therapies, but had not received other drugs for treatment of MS for more than one year. The patient had no other identified systemic medical conditions resulting in compromised immune system function and had not previously been treated with natalizumab, which has a known association with PML. The patient was not taking any immunosuppressive or immunomodulatory medications concomitantly. After the diagnosis of PML, the patient developed immune reconstitution inflammatory syndrome (IRIS). The patient's condition improved, but mild residual neurologic sequelae remained at last follow-up.
At the first sign or symptom suggestive of PML, withhold LEMTRADA and perform an appropriate diagnostic evaluation. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
MRI findings may be apparent before clinical signs or symptoms. Cases of PML, diagnosed based on MRI findings and the detection of JCV DNA in the cerebrospinal fluid in the absence of clinical signs or symptoms specific to PML, have been reported in patients treated with other MS medications associated with PML. Many of these patients subsequently became symptomatic with PML. Therefore, monitoring with MRI for signs that may be consistent with PML may be useful, and any suspicious findings should lead to further investigation to allow for an early diagnosis of PML, if present. Following discontinuation of another MS medication associated with PML, lower PML-related mortality and morbidity have been reported in patients who were initially asymptomatic at diagnosis compared to patients who had characteristic clinical signs and symptoms at diagnosis. It is not known whether these differences are due to early detection and discontinuation of MS treatment or due to differences in disease in these patients.
5.19 Acute Acalculous Cholecystitis
LEMTRADA may increase the risk of acute acalculous cholecystitis. In controlled clinical studies, 0.2% of LEMTRADA-treated MS patients developed acute acalculous cholecystitis, compared to 0% of patients treated with interferon beta-1a. During postmarketing use, additional cases of acute acalculous cholecystitis have been reported in LEMTRADA-treated patients. Time to onset of symptoms ranged from less than 24 hours to 2 months after LEMTRADA infusion. Typical risk or predisposing factors such as concurrent critical illness was often not reported. Abnormal ultrasound or computed tomography was used to support the diagnosis of acute acalculous cholecystitis in some cases. Some patients were treated conservatively with antibiotics and recovered without surgical intervention, whereas others underwent cholecystectomy.
Symptoms of acute acalculous cholecystitis include abdominal pain, abdominal tenderness, fever, nausea, and vomiting. Leukocytosis and abnormal liver enzymes are also commonly observed. Acute acalculous cholecystitis is a condition that is associated with high morbidity and mortality rates if not diagnosed early and treated. If acute acalculous cholecystitis is suspected, evaluate and treat promptly.
5.20 Pneumonitis
In clinical studies, 6 of 1217 (0.5%) LEMTRADA-treated patients had pneumonitis of varying severity. Cases of hypersensitivity pneumonitis and pneumonitis with fibrosis occurred in clinical studies. Patients should be advised to report symptoms of pneumonitis, which may include shortness of breath, cough, wheezing, chest pain or tightness, and hemoptysis.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described below and elsewhere in the labeling:
- Autoimmunity [see Boxed Warning and Warnings and Precautions (5.1)]
- Infusion Reactions [see Boxed Warning and Warnings and Precautions (5.2)]
- Stroke and Cervicocephalic Arterial Dissection [see Warnings and Precautions (5.3)]
- Malignancies [see Warnings and Precautions (5.4)]
- Immune Thrombocytopenia [see Warnings and Precautions (5.6)]
- Glomerular Nephropathies Including Anti-glomerular Basement Membrane Disease [see Warnings and Precautions (5.7)]
- Thyroid Disorders [see Warnings and Precautions (5.8)]
- Other Autoimmune Cytopenias [see Warnings and Precautions (5.9)]
- Autoimmune Hepatitis [see Warnings and Precautions (5.10)]
- Hemophagocytic Lymphohistiocytosis [see Warnings and Precautions (5.11)]
- Adult Onset Still's Disease [see Warnings and Precautions (5.12)]
- Thrombotic Thrombocytopenic Purpura (TTP) [see Warnings and Precautions (5.13)]
- Autoimmune Encephalitis (AIE) [see Warnings and Precautions (5.14)]
- Acquired Hemophilia A [see Warnings and Precautions (5.15)]
- Immune-Mediated Colitis [see Warnings and Precautions (5.16)]
- Infections [see Warnings and Precautions (5.17)]
- Progressive Multifocal Leukoencephalopathy (PML) [see Warnings and Precautions (5.18)]
- Acute Acalculous Cholecystitis [see Warnings and Precautions (5.19)]
- Pneumonitis [see Warnings and Precautions (5.20)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In controlled clinical trials (Study 1 and Study 2), a total of 811 patients with relapsing forms of MS received LEMTRADA. The population was 18 to 55 years of age, 65% were female, and 92% were Caucasian. A total of 811 patients received 1 course of therapy, and 789 patients received a second course of therapy at 12 months. The overall follow-up in the controlled trials was equivalent to 1622 patient years.
In MS clinical studies (controlled and open-label extension), overall, a total of 1217 patients received LEMTRADA. Approximately 60% of patients received a total of 2 treatment courses and approximately 24% of patients received a total of 3 treatment courses; others received a total of 4 or more treatment courses, although data beyond 3 treatment courses are limited. The overall follow-up was 6858 person-years. Patients had a median of 6 years of follow-up from the first LEMTRADA dose, with approximately 14% having at least 7 years of follow-up.
Most Common Adverse Reactions
In controlled clinical trials, the most common adverse reactions with LEMTRADA (in at least 10% of patients and more frequently than in interferon beta-1a) were rash, headache, pyrexia, nasopharyngitis, nausea, urinary tract infection, fatigue, insomnia, upper respiratory tract infection, herpes viral infection, urticaria, pruritus, thyroid gland disorders, fungal infection, arthralgia, pain in extremity, back pain, diarrhea, sinusitis, oropharyngeal pain, paresthesia, dizziness, abdominal pain, flushing, and vomiting.
Table 1 lists adverse reactions occurring in ≥5% of LEMTRADA-treated patients in Study 1 and 2 and at the same or at a higher rate than interferon beta-1a.
| LEMTRADA (N=811) % | interferon beta-1a 44 mcg (N=389) % |
|
|---|---|---|
| Rash | 53 | 6 |
| Headache | 52 | 23 |
| Pyrexia | 29 | 9 |
| Nasopharyngitis | 25 | 19 |
| Nausea | 21 | 9 |
| Urinary tract infection | 19 | 8 |
| Fatigue | 18 | 13 |
| Insomnia | 16 | 15 |
| Upper respiratory tract infection | 16 | 13 |
| Herpes viral infection | 16 | 3 |
| Urticaria | 16 | 2 |
| Pruritus | 14 | 2 |
| Thyroid gland disorders | 13 | 3 |
| Fungal infection | 13 | 4 |
| Arthralgia | 12 | 9 |
| Pain in extremity | 12 | 9 |
| Back pain | 12 | 8 |
| Diarrhea | 12 | 6 |
| Sinusitis | 11 | 8 |
| Oropharyngeal pain | 11 | 5 |
| Paresthesia | 10 | 8 |
| Dizziness | 10 | 5 |
| Abdominal pain | 10 | 5 |
| Flushing | 10 | 4 |
| Vomiting | 10 | 3 |
| Cough | 9 | 4 |
| Chills | 9 | 3 |
| Dysgeusia | 8 | 7 |
| Influenza | 8 | 6 |
| Dermatitis | 8 | 5 |
| Dyspepsia | 8 | 4 |
| Blood in urine | 8 | 3 |
| Dyspnea | 8 | 1 |
| Tachycardia | 8 | 1 |
| Anxiety | 7 | 6 |
| Muscular weakness | 7 | 6 |
| Bronchitis | 7 | 4 |
| Chest discomfort | 7 | 2 |
| Muscle spasms | 6 | 5 |
| Myalgia | 6 | 5 |
| Decrease in CD4 lymphocytes | 6 | 2 |
| Decrease in CD8 lymphocytes | 6 | 2 |
| Asthenia | 5 | 4 |
| Decrease in T-lymphocyte count | 5 | 3 |
| Erythema | 5 | 2 |
| Peripheral edema | 5 | 2 |
| Epistaxis | 5 | 2 |
| Neck pain | 5 | 2 |
| Abnormal uterine bleeding | 5 | 1 |
6.2 Lymphopenia
Nearly all (99.9%) patients treated with LEMTRADA in MS clinical trials experienced lymphopenia. The lowest lymphocyte counts occurred approximately by 1 month after each course of treatment. The mean lymphocyte count at 1 month after LEMTRADA treatment was 0.25 × 109 L (range 0.02–2.30 × 109 L) and 0.32 (0.02–1.81 × 109 L) for treatment courses 1 and 2, respectively. Total lymphocyte counts increased to reach the lower limit of normal in approximately 40% of patients by 6 months after each LEMTRADA treatment course and approximately 80% of patients by 12 months after each course [see Clinical Pharmacology (12.2)].
6.3 Suicidal Behavior or Ideation
In clinical studies, 0.6% of patients in both the LEMTRADA and interferon beta-1a groups had events of attempted suicide or suicidal ideation. There were no completed suicides in either clinical study treatment group. Suicidal behavior or ideation occurred in patients with or without a history of a psychiatric or thyroid disorder. Advise patients to report immediately any symptoms of depression or suicidal ideation to the prescribing physician.
6.4 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The incidence of antibodies is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including inhibitory antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to LEMTRADA with the incidence of antibodies to other products may be misleading.
Using an enzyme-linked immunosorbent assay (ELISA) and a competitive binding assay, anti-alemtuzumab binding antibodies were detected in 62%, 67%, and 29% of LEMTRADA-treated patients, at months 1, 3, and 12 (Course 1) as well as 83%, 83%, and 75% of LEMTRADA-treated patients at months 13, 15, and 24 (Course 2). Samples that tested positive for binding antibodies were further evaluated for evidence of in vitro inhibition using a flow cytometry assay. Neutralizing antibodies were detected in 87%, 46%, and 5% of positive binding antibody patients at months 1, 3, and 12 (Course 1) as well as 94%, 88%, and 42% of positive binding antibody patients at months 13, 15, and 24 (Course 2). Anti-alemtuzumab antibodies were associated with decreased alemtuzumab concentration during Course 2, but not Course 1. Through 2 treatment courses, there was no evidence from clinical trials that the presence of binding or inhibitory anti-alemtuzumab antibodies had a significant effect on clinical outcomes, total lymphocyte count, or adverse events. High titer anti-alemtuzumab antibodies, which were observed in 13 patients, were associated with incomplete lymphocyte depletion following a third or fourth treatment course, but there was no clear effect of anti-alemtuzumab antibodies on the clinical efficacy or safety profile of LEMTRADA.
6.5 Postmarketing Experience
The following adverse reactions have been identified during post approval use of alemtuzumab. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Postmarketing Experience with LEMTRADA
Blood and Lymphatic System Disorders: Acquired hemophilia A [see Warnings and Precautions (5.15)], neutropenia, thrombocytopenia [see Warnings and Precautions (5.2)], thrombotic thrombocytopenic purpura [see Warnings and Precautions (5.13)]
Cerebrovascular Disorders: Stroke, including hemorrhagic and ischemic stroke and cervicocephalic arterial dissection [see Warnings and Precautions (5.3)]
Gastrointestinal System Disorders: Cholecystitis, including acalculous cholecystitis and acute acalculous cholecystitis [see Warnings and Precautions (5.19)], immune-mediated colitis [see Warnings and Precautions (5.16)]
Hepatobiliary Disorders: Autoimmune hepatitis [see Warnings and Precautions (5.10)], viral hepatitis [see Warnings and Precautions (5.17)]
Infections and Infestations: Opportunistic infections [see Warnings and Precautions (5.17)], Progressive multifocal leukoencephalopathy [see Warnings and Precautions (5.18)]
Immune System Disorders: Autoimmune hepatitis, vasculitis, Guillain-Barré syndrome [see Warnings and Precautions (5.1)], hemophagocytic lymphohistiocytosis [see Warnings and Precautions (5.11)], sarcoidosis
Musculoskeletal and Connective Tissue Disorders: Adult Onset Still's Disease [see Warnings and Precautions (5.12)]
Nervous System Disorders: Autoimmune encephalitis [see Warnings and Precautions (5.14)], myasthenia gravis, Lambert-Eaton myasthenic syndrome
Pulmonary System Disorders: Pulmonary alveolar hemorrhage [see Warnings and Precautions (5.2)]
Skin and Subcutaneous Tissue Disorders: Alopecia
Postmarketing Experience with CAMPATH
CAMPATH is approved for the treatment of B-cell chronic lymphocytic leukemia (B-CLL) and is generally administered at higher and more frequent doses (e.g., 30 mg) than recommended in the treatment of MS.
Cardiac Disorders: Congestive heart failure, cardiomyopathy, and decreased ejection fraction in non-MS patients previously treated with potentially cardiotoxic agents.
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of LEMTRADA in pregnant women. LEMTRADA was embryolethal in pregnant huCD52 transgenic mice when administered during organogenesis [see Animal data]. Auto-antibodies may develop after administration of LEMTRADA. Placental transfer of anti-thyroid antibodies resulting in neonatal Graves' disease has been reported.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
There is a pregnancy surveillance program for LEMTRADA. If LEMTRADA exposure occurs during pregnancy, healthcare providers and patients are encouraged to report pregnancies by calling 1-800-745-4447, option 2.
Clinical Considerations
LEMTRADA induces persistent thyroid disorders [see Warnings and Precautions (5.8)]. Untreated hypothyroidism in pregnant women increases the risk for miscarriage and may have effects on the fetus including mental retardation and dwarfism. In mothers with Graves' disease, maternal thyroid stimulating hormone receptor antibodies can be transferred to a developing fetus and can cause neonatal Graves' disease. In a patient who developed Graves' disease after treatment with alemtuzumab, placental transfer of anti-thyrotropin receptor antibodies resulted in neonatal Graves' disease with thyroid storm in her infant who was born 1 year after alemtuzumab dosing [see Warnings and Precautions (5.1)].
Data
Animal data
When LEMTRADA was administered to pregnant huCD52 transgenic mice during organogenesis (gestation days [GD] 6–10 or GD 11–15) at doses of 3 or 10 mg/kg IV, no teratogenic effects were observed. However, there was an increase in embryolethality (increased postimplantation loss and the number of dams with all fetuses dead or resorbed) in pregnant animals dosed during GD 11–15. In a separate study in pregnant huCD52 transgenic mice, administration of LEMTRADA during organogenesis (GD 6–10 or GD 11–15) at doses of 3 or 10 mg/kg IV, decreases in B- and T-lymphocyte populations were observed in the offspring at both doses tested.
In pregnant huCD52 transgenic mice administered LEMTRADA at doses of 3 or 10 mg/kg/day IV throughout gestation and lactation, there was an increase in pup deaths during the lactation period at 10 mg/kg. Decreases in T- and B-lymphocyte populations and in antibody response were observed in offspring at both doses tested.
8.2 Lactation
Risk Summary
There are no data on the presence of alemtuzumab in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Alemtuzumab was detected in the milk of lactating huCD52 transgenic mice administered LEMTRADA [see Animal data].
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for LEMTRADA and any potential adverse effects on the breastfed child from LEMTRADA or from the underlying maternal conditions.
Data
Animal data
Alemtuzumab was detected in the milk of lactating huCD52 transgenic mice following intravenous administration of LEMTRADA at a dose of 10 mg/kg on postpartum days 8–12. Serum levels of alemtuzumab were similar in lactating mice and offspring on postpartum Day 13 and were associated with evidence of pharmacological activity (decrease in lymphocyte counts) in the offspring.
8.3 Females and Males of Reproductive Potential
Contraception
Before initiation of LEMTRADA treatment, women of childbearing potential should be counselled on the potential for a serious risk to the fetus. To avoid in utero exposure to LEMTRADA, women of childbearing potential should use effective contraceptive measures when receiving a course of treatment with LEMTRADA and for 4 months following that course of treatment [see Use in Specific Populations (8.1)].
Infertility
In huCD52 transgenic mice, administration of LEMTRADA prior to and during the mating period resulted in adverse effects on sperm parameters in males and reduced number of corpora lutea and implantations in females [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients less than 17 years of age have not been established. Use of LEMTRADA is not recommended in pediatric patients due to the risks of autoimmunity, infusion reactions, and stroke, and because it may increase the risk of malignancies (thyroid, melanoma, lymphoproliferative disorders, and lymphoma) [see Warnings and Precautions (5.1, 5.2, 5.3, 5.4)].
10. Overdosage
Two MS patients experienced serious reactions (headache, rash, and either hypotension or sinus tachycardia) after a single accidental infusion up to 60 mg of LEMTRADA. Doses of LEMTRADA greater than those recommended may increase the intensity and/or duration of infusion reactions or its immune effects. There is no known antidote for alemtuzumab overdosage.
11. Lemtrada Description
Alemtuzumab is a recombinant humanized IgG1 kappa monoclonal antibody directed against the cell surface glycoprotein, CD52. Alemtuzumab has an approximate molecular weight of 150 kD. Alemtuzumab is produced in mammalian cell (Chinese hamster ovary) suspension culture in a nutrient medium containing neomycin. Neomycin is not detectable in the final product.
LEMTRADA (alemtuzumab) injection is a sterile, clear and colorless to slightly yellow, solution (pH 7.2 ± 0.2) for intravenous infusion.
Each 1 mL of solution contains 10 mg alemtuzumab, dibasic sodium phosphate (1.15 mg), disodium edetate dihydrate (0.0187 mg), polysorbate 80 (0.1 mg), potassium chloride (0.2 mg), potassium dihydrogen phosphate (0.2 mg), sodium chloride (8 mg), and Water for Injection, USP.
12. Lemtrada - Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism by which alemtuzumab exerts its therapeutic effects in multiple sclerosis is unknown but is presumed to involve binding to CD52, a cell surface antigen present on T and B lymphocytes, and on natural killer cells, monocytes, and macrophages. Following cell surface binding to T and B lymphocytes, alemtuzumab results in antibody-dependent cellular cytolysis and complement-mediated lysis.
12.2 Pharmacodynamics
Effects of LEMTRADA on the Lymphocyte Population
LEMTRADA depletes circulating T and B lymphocytes after each treatment course. In clinical trials, the lowest cell counts occurred 1 month after a course of treatment at the time of the first post-treatment blood count. Lymphocyte counts then increased over time: B cell counts usually recovered within 6 months; T cell counts increased more slowly and usually remained below baseline 12 months after treatment. Approximately 60% of patients had total lymphocyte counts below the lower limit of normal 6 months after each treatment course and 20% had counts below the lower limit of normal after 12 months.
Reconstitution of the lymphocyte population varies for the different lymphocyte subtypes. At Month 1 in clinical trials, the mean CD4+ lymphocyte count was 40 cells per microliter, and, at Month 12, 270 cells per microliter. At 30 months, approximately half of patients had CD4+ lymphocyte counts that remained below the lower limit of normal.
12.3 Pharmacokinetics
The pharmacokinetics of LEMTRADA were evaluated in a total of 148 patients with relapsing forms of MS who received 12 mg/day on 5 consecutive days, followed by 12 mg/day on 3 consecutive days 12 months following the first treatment course.
Absorption
Serum concentrations increased with each consecutive dose within a treatment course, with the highest observed concentrations occurring following the last infusion of a treatment course. The mean maximum concentration was 3014 ng/mL on Day 5 of the first treatment course, and 2276 ng/mL on Day 3 of the second treatment course.
Distribution
LEMTRADA is largely confined to the blood and interstitial space with a central volume of distribution of 14.1 L.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies to assess the carcinogenic or genotoxic potential of LEMTRADA have not been conducted.
When LEMTRADA (3 or 10 mg/kg IV) was administered to huCD52 transgenic male mice on 5 consecutive days prior to cohabitation with untreated wild-type females, no effect on fertility or reproductive performance was observed. However, adverse effects on sperm parameters (including abnormal morphology [detached/no head] and reduced total count and motility) were observed at both doses tested.
When LEMTRADA (3 or 10 mg/kg IV) was administered to huCD52 transgenic female mice for 5 consecutive days prior to cohabitation with untreated wild-type males, there was a decrease in the average number of corpora lutea and implantation sites and an increase in postimplantation loss, resulting in fewer viable embryos at the higher dose tested.
14. Clinical Studies
The efficacy of LEMTRADA was demonstrated in two studies (Study 1 and 2) that evaluated LEMTRADA 12 mg in patients with relapsing-remitting multiple sclerosis (RRMS). LEMTRADA was administered by intravenous infusion once daily over a 5-day course, followed one year later by intravenous infusion once daily over a 3-day course. Both studies included patients who had experienced at least 2 relapses during the 2 years prior to trial entry and at least 1 relapse during the year prior to trial entry. Neurological examinations were performed every 12 weeks and at the time of suspected relapse. Magnetic resonance imaging (MRI) evaluations were performed annually.
Study 1
Study 1 was a 2-year randomized, open-label, rater-blinded, active comparator (interferon beta-1a 44 micrograms administered subcutaneously three times a week) controlled study in patients with RRMS. Patients entering Study 1 had Expanded Disability Status Scale (EDSS) scores of 5 or less and had to have experienced at least one relapse while on interferon beta or glatiramer acetate therapy.
Patients were randomized to receive LEMTRADA (n=426) or interferon beta-1a (n=202). At baseline, the mean age was 35 years, the mean disease duration was 4.5 years, and the mean EDSS score was 2.7.
The clinical outcome measures were the annualized relapse rate (ARR) over 2 years and the time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase above baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 6 months. The MRI outcome measure was the change in T2 lesion volume.
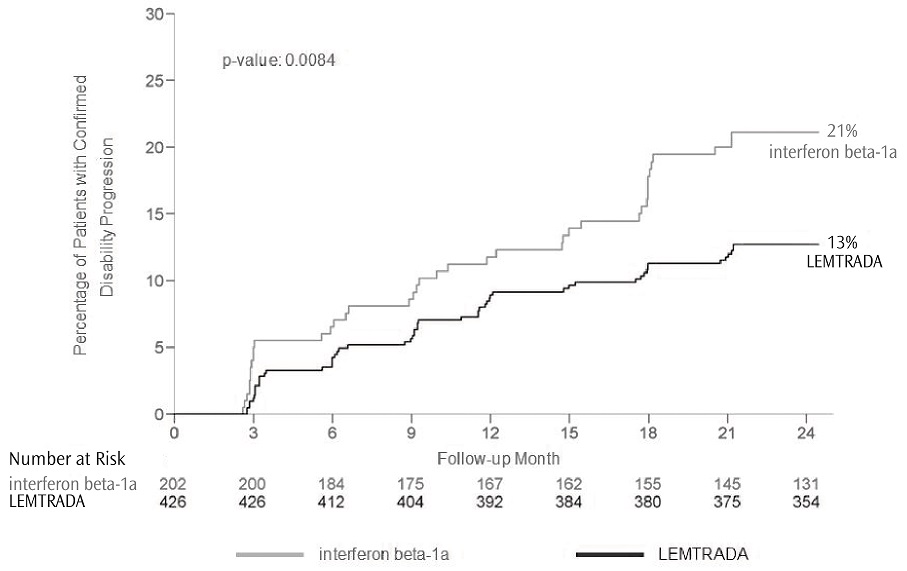
The annualized relapse rate was significantly lower in patients treated with LEMTRADA than in patients who received interferon beta-1a. Time to onset of 6-month confirmed disability progression was significantly delayed with LEMTRADA treatment compared to interferon beta-1a. There was no significant difference between the treatment groups for the change in T2 lesion volume. The results of Study 1 are shown in Table 2 and Figure 1.
| LEMTRADA | interferon beta-1a 44 mcg | p-value | |
|---|---|---|---|
| (N=426) | (N=202) | ||
| Clinical Outcomes | |||
| Annualized relapse rate | 0.26 | 0.52 | <0.0001 |
| Relative reduction | 49% | ||
| Proportion of patients with disability progression at Year 2 | 13% | 21% | 0.0084 |
| Relative risk reduction | 42% | ||
| Percent of patients remaining relapse-free at Year 2 | 65% | 47% | <0.0001 |
| MRI Outcomes | |||
| Percent change in T2 lesion volume from baseline | -1.3 | -1.2 | 0.14 |
Figure 1: Time to 6-month Confirmed Disability Progression (Study 1)
Study 2
Study 2 was a 2-year randomized, open-label, rater-blinded, active comparator (interferon beta-1a 44 micrograms administered subcutaneously three times a week) controlled study in patients with RRMS. Patients entering Study 2 had EDSS scores of 3 or less and no prior treatment for multiple sclerosis.
Patients were randomized to receive LEMTRADA (n=376) or interferon beta-1a (n=187). At baseline, the mean age was 33 years, the mean disease duration was 2 years, and the mean EDSS score was 2.
The clinical outcome measures were the annualized relapse rate (ARR) over 2 years and the time to confirmed disability progression, as defined in Study 1. The MRI outcome measure was the change in T2 lesion volume.
The annualized relapse rate was significantly lower in patients treated with LEMTRADA than in patients who received interferon beta-1a. There was no significant difference between the treatment groups for the time to confirmed disability progression and for the primary MRI endpoint (change in T2 lesion volume). The results for Study 2 are shown in Table 3.
| LEMTRADA | interferon beta-1a 44 mcg | p-value | |
|---|---|---|---|
| (N=376) | (N=187) | ||
| Clinical Outcomes | |||
| Annualized relapse rate Relative reduction | 0.18 55% | 0.39 | <0.0001 |
| Proportion of patients with disability progression at Year 2 Relative risk reduction | 8% 30% | 11% | 0.22 |
| Percent of patients remaining relapse-free at Year 2 | 78% | 59% | <0.0001 |
| MRI Outcomes | |||
| Percent change in T2 lesion volume from baseline | -9.3 | -6.5 | 0.31 |
16. How is Lemtrada supplied
16.1 How Supplied
LEMTRADA (alemtuzumab) injection is a sterile, clear and colorless to slightly yellow solution for intravenous infusion, containing no antimicrobial preservatives.
Each LEMTRADA carton (NDC: 58468-0200-1) contains one single-dose vial that delivers 12 mg/1.2 mL (10 mg/mL). The vial stopper is not made with natural rubber latex.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Autoimmunity
- Advise patients to contact their healthcare provider promptly if they experience any symptoms of potential autoimmune disease. Give examples of important symptoms such as bleeding, easy bruising, petechiae, purpura, hematuria, edema, jaundice, or hemoptysis [see Warnings and Precautions (5.1)].
- Advise patients of the importance of monthly blood and urine tests for 48 months following the last course of LEMTRADA to monitor for signs of autoimmunity because early detection and prompt treatment can help prevent serious and potentially fatal outcomes associated with these events. Advise patients that monitoring may need to continue past 48 months if they have signs or symptoms of autoimmunity.
- Advise patients that LEMTRADA may cause hyperthyroid or hypothyroid disorders.
- Advise patients to contact their healthcare provider if they experience symptoms reflective of a potential thyroid disorder such as unexplained weight loss or gain, fast heartbeat or palpitations, nervousness, worsening tiredness, eye swelling, constipation, or feeling cold.
- Advise women of childbearing potential of the risks of pregnancy with concomitant thyroid disease. Advise women of childbearing potential to discuss pregnancy planning with their doctor.
- Cases of autoimmune hepatitis have been reported in patients treated with LEMTRADA. Advise patients to contact their healthcare provider right away if they develop signs or symptoms suggestive of hepatic dysfunction such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, jaundice and/or dark urine, or bleeding or bruising more easily than normal.
- Advise patients to contact their healthcare provider if they experience symptoms of acquired hemophilia A such as spontaneous bruising, nose bleeds, painful or swollen joints, other types of bleeding, or bleeding from a cut that may take longer than usual to stop.
- Advise patients that cases of autoimmune encephalitis can occur after receiving LEMTRADA. This condition may include symptoms such as behavior and psychiatric changes, movement disorders, short-term memory loss or seizures, as well as other symptoms that may resemble an MS relapse.
Infusion Reactions
- Advise patients that infusion reactions can occur at the time of infusion or after they leave the infusion center [see Warnings and Precautions (5.2)].
- Instruct the patient to remain at the infusion center for at least 2 hours after each LEMTRADA infusion, or longer at the discretion of the healthcare provider. Advise patients that symptoms of infusion reactions may occur after they leave the infusion center and to report these symptoms to their healthcare provider.
- Advise patients that their healthcare provider will monitor vital signs, including blood pressure, before and during the infusion and to contact their healthcare provider promptly if they experience infusion reactions, which include swelling in the mouth or throat, difficulty breathing, weakness, abnormal heart rate (fast, slow, or irregular), chest pain, rash, facial drooping, sudden severe headache, weakness on one side of the body, difficulty with speech, or neck pain.
- Instruct patients that there have also been reports of rare but serious infusion reactions, including bleeding in the lung, chest tightness/pain or discomfort, heart attack, and stroke or tears in blood vessels supplying the brain, which should be reported to your healthcare provider.
- Advise patients that reactions may occur following any of the doses during the treatment course. In the majority of cases, reactions occurred within 1–3 days of the infusion.
Stroke and Cervicocephalic Arterial Dissection
- Educate patients on the symptoms and instruct patients to seek immediate medical attention if symptoms of stroke or cervicocephalic arterial dissection occur (e.g., neck pain, weakness on one side, facial droop, difficulty with speech, sudden severe headache) [see Warnings and Precautions (5.3)].
Malignancies
- Advise patients that LEMTRADA may increase their risk of malignancies including thyroid cancer and melanoma [see Warnings and Precautions (5.4)].
- Advise patients to report symptoms of thyroid cancer, including a new lump or swelling in the neck, pain in the front of the neck, hoarseness or other voice changes that do not go away, trouble swallowing or breathing, or a constant cough not due to a cold.
- Advise patients that they should have baseline and yearly skin examinations.
LEMTRADA REMS Program
- LEMTRADA is available only through a restricted program called the LEMTRADA REMS Program [see Warnings and Precautions (5.5)]. Inform the patient of the following notable requirements:
- Patients and providers must be enrolled in the program.
- Patients must comply with the ongoing monitoring requirements.
- Patients must report any side effects or symptoms to their doctor.
- LEMTRADA is available only at certified infusion centers participating in the program. Therefore, provide patients with information on the LEMTRADA REMS Program in order to locate an infusion center.
- Advise patients to read the LEMTRADA REMS material for patients, LEMTRADA Treatment and Infusion Reactions Patient Guide.
- Instruct patients to carry the LEMTRADA REMS Patient Safety Information Card with them in case of an emergency.
Hemophagocytic Lymphohistiocytosis
- Inform patients that treatment with LEMTRADA may increase the risk of a type of excessive immune activation (hemophagocytic lymphohistiocytosis), which can be fatal, particularly if not diagnosed and treated early.
- Advise patients to contact their healthcare provider immediately if they experience symptoms such as fever, swollen glands, skin rash, or new neurologic symptoms such as mental status changes, ataxia, or seizures.
- In cases reported with LEMTRADA, symptoms occurred within approximately thirteen months to thirty-three months following the initiation of treatment.
Adult Onset Still's Disease (AOSD)
- Inform patients that AOSD is a rare condition that has the potential to cause multi-organ inflammation with several symptoms such as fever >39°C or 102.2°F lasting more than 1 week, pain, stiffness with or without swelling in multiple joints, and/or a skin rash [see Warnings and Precautions (5.12)].
- Instruct patients if they experience a combination of these symptoms to contact their healthcare provider immediately.
Thrombotic Thrombocytopenic Purpura
- Inform patients that there have been reports of TTP in patients treated with LEMTRADA and that this is a potentially life-threatening condition [see Warnings and Precautions (5.13)].
- Instruct patients to get prompt medical attention if they experience symptoms of TTP such as fever, fatigue, pallor, purpura, jaundice, tachycardia, dyspnea, hematuria, dark-colored urine, decreased urine volume, abdominal pain, nausea, vomiting, or new neurological symptoms such as confusion, altered mental status, vision or speech changes, or seizures.
Immune-Mediated Colitis
- Advise patients to promptly contact their healthcare provider if they experience any signs and symptoms of colitis, including diarrhea, abdominal pain, and blood in stool [see Warnings and Precautions (5.16)].
Infections
- Advise patients to contact their healthcare provider if they develop symptoms of serious infection such as fatigue, fever, or swollen glands [see Warnings and Precautions (5.17)].
- Advise patients to complete any necessary immunizations at least 6 weeks prior to treatment with LEMTRADA [see Dosage and Administration (2.1)]. Advise patients that they should talk to their healthcare provider before taking any vaccine after recent treatment with LEMTRADA [see Warnings and Precautions (5.17)].
- Advise patients to avoid or adequately heat foods that are potential sources of Listeria monocytogenes prior to receiving LEMTRADA and if they have had a recent course of LEMTRADA. The duration of increased risk for Listeria infection after LEMTRADA administration is not known. Inform patients that Listeria infection can lead to significant complications or death [see Warnings and Precautions (5.17)].
- Advise patients to take their prescribed medication for herpes prophylaxis as directed by their healthcare provider [see Warnings and Precautions (5.17)].
- Advise patients that yearly HPV screening is recommended [see Warnings and Precautions (5.17)].
Progressive Multifocal Leukoencephalopathy
- Inform patients that progressive multifocal leukoencephalopathy (PML) has occurred in a patient who received LEMTRADA. Inform the patient that PML is characterized by a progression of deficits and usually leads to death or severe disability over weeks or months. Instruct the patient of the importance of contacting their doctor if they develop any symptoms suggestive of PML. Inform the patient that typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes [see Warnings and Precautions (5.18)].
Acute Acalculous Cholecystitis
- Advise patients to report symptoms of acute acalculous cholecystitis. These include abdominal pain, abdominal tenderness, fever, nausea, and vomiting [see Warnings and Precautions (5.19)].
Pneumonitis
- Advise patients that pneumonitis has been reported in patients treated with LEMTRADA [see Warnings and Precautions (5.20)]. Advise patients to report symptoms of lung disease such as shortness of breath, cough, wheezing, chest pain or tightness, and hemoptysis.
Concomitant Use of CAMPATH
- Advise patients that alemtuzumab is the same drug as CAMPATH for use in B-CLL. Patients should inform their healthcare provider if they have taken CAMPATH [see Warnings and Precautions (5.21)].
Fetal Risk
- Inform patients that LEMTRADA may cause fetal harm. Discuss with women of childbearing age whether they are pregnant, might be pregnant, or are trying to become pregnant. Advise women of childbearing age of the need for effective contraception during LEMTRADA treatment and for 4 months after a treatment course of LEMTRADA. Advise the patient that if she should nevertheless become pregnant, she should immediately inform her physician.
- Advise patients exposed to LEMTRADA during pregnancy that there is a pregnancy safety surveillance program that monitors pregnancy outcomes [see Use in Specific Populations (8.1)]. If exposure occurs during pregnancy, healthcare providers and patients are encouraged to report pregnancies by calling 1-800-745-4447, option 2.
Manufactured and distributed by:
Genzyme Corporation
Cambridge, MA 02141
A SANOFI COMPANY
US License Number: 1596
LEMTRADA and CAMPATH are registered trademarks of Genzyme Corporation. ©2024 Genzyme Corporation.
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: May 2024 | ||
|
MEDICATION GUIDE
|
|||
|
Read this Medication Guide before you start receiving LEMTRADA and before you begin each treatment course. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. |
|||
|
What is the most important information I should know about LEMTRADA? LEMTRADA can cause serious side effects, including:
|
|||
|
|
||
|
|||
|
|
||
|
Side effects may happen while you receive LEMTRADA and for 4 years after you stop receiving LEMTRADA. Your healthcare provider will order blood and urine tests before you receive, while you are receiving, and every month for 4 years after you receive your last LEMTRADA infusion. You may need to continue these blood and urine tests after 4 years if you have any autoimmune signs or symptoms. The blood and urine tests will help your healthcare provider watch for signs and symptoms of serious autoimmune problems. It is important to have your blood and urine tested, even if you are feeling well and do not have any symptoms from LEMTRADA and your multiple sclerosis. This may help your healthcare provider find any problems early.
|
|||
|
|
||
|
To lower your chances of getting a serious infusion reaction, your healthcare provider will give you a medicine called corticosteroids before your first 3 infusions of a treatment course. You may also be given other medicines before or after the infusion to try to reduce your chances of these reactions or to treat them after they happen.
|
|||
|
|
||
|
|||
|
|
||
|
You should have your skin checked before you start receiving LEMTRADA and each year while you are receiving treatment to monitor symptoms of skin cancer. Because of your risk of autoimmunity, infusion reactions, and the risk of some kinds of cancers, LEMTRADA is only available through a restricted program called the LEMTRADA Risk Evaluation and Mitigation Strategy (REMS) Program. Call 1-855-676-6326 to enroll in the LEMTRADA REMS Program.
|
|||
|
What is LEMTRADA? LEMTRADA is a prescription medicine used to treat relapsing forms of multiple sclerosis (MS), to include relapsing-remitting disease and active secondary progressive disease, in adults. Since treatment with LEMTRADA can increase your risk of getting certain conditions and diseases, LEMTRADA is generally prescribed for people who have tried 2 or more MS medicines that have not worked well enough. LEMTRADA is not recommended for use in patients with clinically isolated syndrome (CIS). It is not known if LEMTRADA is safe and effective for use in children under 17 years of age. |
|||
|
Who should not receive LEMTRADA? Do not receive LEMTRADA if you:
|
|||
|
What should I tell my healthcare provider before receiving LEMTRADA? Before receiving LEMTRADA, tell your healthcare provider if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. LEMTRADA and other medicines may affect each other causing side effects. Especially tell your healthcare provider if you take medicines that increase your chance of getting infections, including medicines used to treat cancer or to control your immune system. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||
|
How will I receive LEMTRADA?
|
|||
|
What are the possible side effects of LEMTRADA? LEMTRADA may cause serious side effects including:
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
||
|
Your healthcare provider will do blood tests to check for cytopenias. Call your healthcare provider right away if you have symptoms listed above.
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
||
|
|||
Call your healthcare provider right away if you have symptoms of a serious infection, such as fever or swollen glands. You may need to go to the hospital for treatment if you get a serious infection. It is important to tell the healthcare providers that you have received LEMTRADA. Talk to your healthcare provider before you get vaccinations after receiving LEMTRADA. Certain vaccinations may increase your chances of getting infections. |
|||
|
|||
|
|
||
|
|||
|
|
||
|
|||
|
|
||
|
The most common side effects of LEMTRADA include: |
|||
|
|
||
|
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of LEMTRADA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088. |
|||
|
General information about the safe and effective use of LEMTRADA. This Medication Guide summarizes the most important information about LEMTRADA. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about LEMTRADA that is written for health professionals. For more information, go to www.LemtradaREMS.com or call Genzyme at 1-855-676-6326. |
|||
|
What are the ingredients in LEMTRADA? Active ingredient: alemtuzumab Inactive ingredients: dibasic sodium phosphate, disodium edetate dihydrate, polysorbate 80, potassium chloride, potassium dihydrogen phosphate, sodium chloride, and Water for Injection, USP. |
|||
|
Manufactured and distributed by: LEMTRADA and CAMPATH are registered trademarks of Genzyme Corporation. |
|||
| LEMTRADA
alemtuzumab injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Boehringer Ingelheim Pharma GmbH & Co. KG | 340700520 | ANALYSIS(58468-0200) , API MANUFACTURE(58468-0200) , MANUFACTURE(58468-0200) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi-Aventis Deutschland GmbH | 313218430 | ANALYSIS(58468-0200) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| EUROAPI UK LIMITED | 229522842 | PACK(58468-0200) , LABEL(58468-0200) | |
Biological Products Related to Lemtrada
Find detailed information on biosimilars for this medication.
More about Lemtrada (alemtuzumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (21)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: CD52 monoclonal antibodies
- Breastfeeding