Lapatinib: Package Insert / Prescribing Info
Package insert / product label
Dosage form: tablet
Drug classes: EGFR inhibitors, HER2 inhibitors
Medically reviewed by Drugs.com. Last updated on Jun 18, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
LAPATINIB tablets, for oral use
Initial U.S. Approval: 2007
WARNING: HEPATOTOXICITY
See full prescribing information for complete boxed warning.
Hepatotoxicity has been observed in clinical trials and postmarketing experience. The hepatotoxicity may be severe and deaths have been reported. Causality of the deaths is uncertain.(5.2)
Indications and Usage for Lapatinib
Lapatinib tablet is a kinase inhibitor indicated in combination with: (1)
- capecitabine for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress human epidermal growth factor receptor 2 (HER2) and who have received prior therapy, including an anthracycline, a taxane, and trastuzumab.
Limitations of Use: Patients should have disease progression on trastuzumab prior to initiation of treatment with lapatinib tablets in combination with capecitabine.
- letrozole for the treatment of postmenopausal women with hormone receptor-positive metastatic breast cancer that overexpresses the HER2 receptor for whom hormonal therapy is indicated.
Lapatinib tablets in combination with an aromatase inhibitor have not been compared to a trastuzumab-containing chemotherapy regimen for the treatment of metastatic breast cancer.
Lapatinib Dosage and Administration
The recommended dosage of lapatinib tablets for advanced or metastatic breast cancer is 1,250 mg (5 tablets) given orally once daily on Days 1 to 21 continuously in combination with capecitabine 2,000 mg/m2/day (administered orally in 2 doses approximately 12 hours apart) on Days 1 to 14 in a repeating 21- day cycle. (2.1)
The recommended dose of lapatinib tablets for hormone receptor-positive, HER2-positive metastatic breast cancer is 1,500 mg (6 tablets) given orally once daily continuously in combination with letrozole. When lapatinib tablets are coadministered with letrozole, the recommended dose of letrozole is 2.5 mg once daily. (2.1)
- Lapatinib tablets should be taken at least one hour before or one hour after a meal. However, capecitabine should be taken with food or within 30 minutes after food. (2.1)
- Lapatinib tablets should be taken once daily. Do not divide daily doses of lapatinib tablets. (2.1,12.3)
- Modify dose for cardiac and other toxicities, severe hepatic impairment, diarrhea, and CYP3A4 drug interactions. (2.2)
Dosage Forms and Strengths
250 mg tablets (3)
Contraindications
Known severe hypersensitivity (e.g., anaphylaxis) to this product or any of its components. (4)
Warnings and Precautions
- Decreases in left ventricular ejection fraction (LVEF) have been reported. Confirm normal LVEF before starting lapatinib tablets and continue evaluations during treatment. (5.1)
- Lapatinib has been associated with hepatotoxicity. Monitor liver function tests before initiation of treatment, every 4 to 6 weeks during treatment, and as clinically indicated. Discontinue and do not restart lapatinib tablets if patients experience severe changes in liver function tests. (5.2)
- Dose reduction in patients with severe hepatic impairment should be considered. (2.2, 5.3, 8.7)
- Diarrhea, including severe diarrhea, has been reported during treatment. Manage with antidiarrheal agents, and replace fluids and electrolytes if severe. (5.4)
- Lapatinib has been associated with interstitial lung disease and pneumonitis. Discontinue lapatinib tablets if patients experience severe pulmonary symptoms. (5.5)
- Lapatinib may prolong the QT interval in some patients. Consider electrocardiogram (ECG) and electrolyte monitoring. (5.6,12.2)
- Severe cutaneous reactions have been reported. Discontinue lapatinib tablets if life-threatening reactions are suspected. (5.7)
- Lapatinib tablets can cause fetal harm. Advise patients of the potential risk to the fetus and to use effective contraception. (5.8,8.1, 8.3).
Adverse Reactions/Side Effects
The most common (greater than 20%) adverse reactions during treatment with lapatinib tablets plus capecitabine were diarrhea, palmar-plantar erythrodysesthesia, nausea, rash, vomiting, and fatigue. The most common (greater than or equal to 20%) adverse reactions during treatment with lapatinib tablets plus letrozole were diarrhea, rash, nausea, and fatigue. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Lupin Pharmaceuticals, Inc. at 1-800-399-2561 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Lapatinib tablets are likely to increase exposure to concomitantly administered drugs which are substrates of CYP3A4, CYP2C8, or P-glycoprotein (ABCB1). (7.1)
- Avoid strong CYP3A4 inhibitors. If unavoidable, consider dose reduction of lapatinib tablets in patients coadministered a strong CYP3A4 inhibitor. (2.2, 7.2)
- Avoid strong CYP3A4 inducers. If unavoidable, consider gradual dose increase of lapatinib tablets in patients coadministered a strong CYP3A4 inducer. (2.2, 7.2)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2025
Full Prescribing Information
WARNING: HEPATOTOXICITY
Hepatotoxicity has been observed in clinical trials and postmarketing experience. The hepatotoxicity may be severe and deaths have been reported. Causality of the deaths is uncertain [see Warnings and Precautions (5.2)].
1. Indications and Usage for Lapatinib
Lapatinib tablets are kinase inhibitor indicated in combination with: (1)
- capecitabine for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpress human epidermal growth factor receptor 2 (HER2) and who have received prior therapy, including an anthracycline, a taxane, and trastuzumab.
Limitations of Use: Patients should have disease progression on trastuzumab prior to initiation of treatment with lapatinib tablets in combination with capecetabine.
- letrozole for the treatment of postmenopausal women with hormone receptor-positive metastatic breast cancer that overexpresses the HER2 receptor for whom hormonal therapy is indicated.
Lapatinib tablets in combination with an aromatase inhibitor has not been compared to a trastuzumab-containing chemotherapy regimen for the treatment of metastatic breast cancer.
2. Lapatinib Dosage and Administration
2.1 Recommended Dosing
HER2-Positive Metastatic Breast Cancer: The recommended dose of lapatinib tablets is 1,250 mg given orally once daily on Days 1 to 21 continuously in combination with capecitabine 2,000 mg/m2/day (administered orally in 2 doses approximately 12 hours apart) on Days 1 to 14 in a repeating 21-day cycle. Lapatinib tablets should be taken at least one hour before or one hour after a meal. The dose of lapatinib tablets should be once daily (5 tablets administered all at once); dividing the daily dose is not recommended [see Clinical Pharmacology (12.3)]. Capecitabine should be taken with food or within 30 minutes after food. If a day’s dose is missed, the patient should not double the dose the next day. Treatment should be continued until disease progression or unacceptable toxicity occurs.
Hormone Receptor-Positive, HER2-Positive Metastatic Breast Cancer: The recommended dose of lapatinib tablets is 1,500 mg given orally once daily continuously in combination with letrozole. When coadministered with lapatinib tablets, the recommended dose of letrozole is 2.5 mg once daily. Lapatinib tablets should be taken at least one hour before or one hour after a meal. The dose of lapatinib tablets should be once daily (6 tablets administered all at once); dividing the daily dose is not recommended [ see Clinical Pharmacology (12.3)].
2.2 Dose Modification Guidelines
Cardiac Events: Lapatinib tablets should be discontinued in patients with a decreased left ventricular ejection fraction (LVEF) that is Grade 2 or greater by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE v3.0), and in patients with an LVEF that drops below the institution’s lower limit of normal (LLN) [see Warnings and Precautions (5.1)and Adverse Reactions (6.1)]. Lapatinib tablets in combination with capecitabine may be restarted at a reduced dose (1,000 mg/day) and in combination with letrozole may be restarted at a reduced dose of 1,250 mg/day after a minimum of 2 weeks if the LVEF recovers to normal and the patient is asymptomatic.
Hepatic Impairment: Patients with severe hepatic impairment (Child-Pugh Class C) should have their dose of lapatinib tablets reduced. A dose reduction from 1,250 mg/day to 750 mg/day (HER2-positive metastatic breast cancer indication) or from 1,500 mg/day to 1,000 mg/day (hormone receptor- positive, HER2-positive breast cancer indication) in patients with severe hepatic impairment is predicted to adjust the area under the curve (AUC) to the normal range and should be considered. However, there are no clinical data with this dose adjustment in patients with severe hepatic impairment.
Diarrhea: Lapatinib should be interrupted in patients with diarrhea which is NCI CTCAE Grade 3 or Grade 1 or 2 with complicating features (moderate to severe abdominal cramping, nausea or vomiting greater than or equal to NCI CTCAE Grade 2, decreased performance status, fever, sepsis, neutropenia, frank bleeding, or dehydration). Lapatinib may be reintroduced at a lower dose (reduced from 1,250 mg/day to 1,000 mg/day or from 1,500 mg/day to 1,250 mg/day) when diarrhea resolves to Grade 1 or less. Lapatinib should be permanently discontinued in patients with diarrhea, which is NCI CTCAE Grade 4 [see Warnings and Precautions (5.4) and Adverse Reactions (6.1) ].
Concomitant Strong CYP3A4 Inhibitors: The concomitant use of strong CYP3A4 inhibitors should be avoided (e.g., ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, voriconazole). Grapefruit may also increase plasma concentrations of lapatinib and should be avoided. If patients must be coadministered a strong CYP3A4 inhibitor, based on pharmacokinetic studies, a dose reduction to 500 mg/day of lapatinib is predicted to adjust the lapatinib AUC to the range observed without inhibitors and should be considered. However, there are no clinical data with this dose adjustment in patients receiving strong CYP3A4 inhibitors. If the strong inhibitor is discontinued, a washout period of approximately 1 week should be allowed before the lapatinib dose is adjusted upward to the indicated dose. [see Drug Interactions (7.2)] .
Concomitant Strong CYP3A4 Inducers :The concomitant use of strong CYP3A4 inducers should be avoided (e.g., dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital, St. John's wort). If patients must be coadministered a strong CYP3A4 inducer, based on pharmacokinetic studies, the dose of lapatinib should be titrated gradually from 1,250 mg/day up to 4,500 mg/day (HER2-positive metastatic breast cancer indication) or from 1,500 mg/day up to 5,500 mg/day (hormone receptor-positive, HER2-positive breast cancer indication) based on tolerability. This dose of lapatinib is predicted to adjust the lapatinib AUC to the range observed without inducers and should be considered. However, there are no clinical data with this dose adjustment in patients receiving strong CYP3A4 inducers. If the strong inducer is discontinued, the lapatinib dose should be reduced to the indicated dose. [see Drug Interactions (7.2)].
Other Toxicities: Discontinuation or interruption of dosing with lapatinib tablets may be considered when patients develop greater than or equal to Grade 2 NCI CTCAE toxicity, and can be restarted at the standard dose of 1,250 or 1,500 mg/day when the toxicity improves to Grade 1 or less. If the toxicity recurs, then lapatinib tablets in combination with capecitabine should be restarted at a lower dose (1,000 mg/day) and in combination with letrozole should be restarted at a lower dose of 1,250 mg/day.
See manufacturer's prescribing information for the coadministered product dosage adjustment guidelines in the event of toxicity and other relevant safety information or contraindications.
3. Dosage Forms and Strengths
250 mg tablets — orange colored, oval shaped, film-coated tablets, debossed “NTL” on one side and plain on another side.
4. Contraindications
Lapatinib tablets are contraindicated in patients with known severe hypersensitivity (e.g., anaphylaxis) to this product or any of its components.
5. Warnings and Precautions
5.1 Decreased Left Ventricular Ejection Fraction
Lapatinib tablets has been reported to decrease LVEF [see Adverse Reactions (6.1) ]. In clinical trials, the majority (greater than 57%) of LVEF decreases occurred within the first 12 weeks of treatment; however, data on long-term exposure are limited. Caution should be taken if lapatinib tablets are to be administered to patients with conditions that could impair left ventricular function. LVEF should be evaluated in all patients prior to initiation of treatment with lapatinib tablets to ensure that the patient has a baseline LVEF that is within the institution's normal limits. LVEF should continue to be evaluated during treatment with lapatinib tablets to ensure that LVEF does not decline below the institution's normal limits [see Dosage and Administration (2.2)].
5.2 Hepatotoxicity
Hepatotoxicity [alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than 3 times the upper limit of normal (ULN) and total bilirubin greater than 2 times the ULN] has been observed in clinical trials (less than 1% of patients) and postmarketing experience. The hepatotoxicity may be severe and deaths have been reported. Causality of the deaths is uncertain. The hepatotoxicity may occur days to several months after initiation of treatment. Liver function tests (transaminases, bilirubin, and alkaline phosphatase) should be monitored before initiation of treatment, every 4 to 6 weeks during treatment, and as clinically indicated. If changes in liver function are severe, therapy with lapatinib tablets should be discontinued and patients should not be retreated with lapatinib tablets [see Adverse Reactions (6.1)].
5.3 Patients with Severe Hepatic Impairment
If lapatinib tablets are to be administered to patients with severe pre existing hepatic impairment, dose reduction should be considered [see Dosage and Administration (2.2) and Use in Specific Populations (8.7)]. In patients who develop severe hepatotoxicity while on therapy, lapatinib tablets should be discontinued and patients should not be retreated with lapatinib tablets [see Warnings and Precautions (5.2)].
5.4 Diarrhea
Diarrhea has been reported during treatment with lapatinib [see Adverse Reactions(6.1)].The diarrhea may be severe, and deaths have been reported. Diarrhea generally occurs early during treatment with lapatinib with almost half of those patients with diarrhea first experiencing it within 6 days. This usually lasts 4 to 5 days. Lapatinib-induced diarrhea is usually low-grade, with severe diarrhea of NCI CTCAE Grades 3 and 4 occurring in less than 10% and less than 1% of patients, respectively. Early identification and intervention is critical for the optimal management of diarrhea. Patients should be instructed to report any change in bowel patterns immediately. Prompt treatment of diarrhea with anti-diarrheal agents (such as loperamide) after the first unformed stool is recommended. Severe cases of diarrhea may require administration of oral or intravenous electrolytes and fluids, use of antibiotics, such as fluoroquinolones (especially if diarrhea is persistent beyond 24 hours, there is fever, or Grade 3 or 4 neutropenia), and interruption or discontinuation of therapy with lapatinib [see Dosage and Administration (2.2)].
5.5 Interstitial Lung Disease/Pneumonitis
Lapatinib has been associated with interstitial lung disease and pneumonitis in monotherapy or in combination with other chemotherapies [see Adverse Reactions (6.1)]. Patients should be monitored for pulmonary symptoms indicative of interstitial lung disease or pneumonitis. Lapatinib tablets should be discontinued in patients who experience pulmonary symptoms indicative of interstitial lung disease/pneumonitis, which are greater than or equal to Grade 3 (NCI CTCAE v 3.0).
5.6 QT Prolongation
A concentration-dependent QT prolongation has been associated with lapatinib tablets [see Clinical Pharmacology (12.2)]. Monitor patients who have or may develop prolongation of QTc during treatment with lapatinib tablets. These conditions include patients with hypokalemia or hypomagnesemia, with congenital long QT syndrome, patients taking antiarrhythmic medicines or other medicinal products with known risk for QT prolongation/Torsades de Pointes (TdP), and cumulative high-dose anthracycline therapy. Correct hypokalemia or hypomagnesemia prior to lapatinib tablets administration.
5.7 Severe Cutaneous Reactions
Severe cutaneous reactions have been reported with lapatinib tablets. If life-threatening reactions, such as erythema multiforme, Stevens-Johnson syndrome, or toxic epidermal necrolysis (e.g., progressive skin rash often with blisters or mucosal lesions) are suspected, discontinue treatment with lapatinib tablets.
5.8 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animal studies, lapatinib tablets can cause fetal harm when administered to a pregnant woman. In animal reproductive studies, administration of lapatinib to pregnant rats during the period of organogenesis and through lactation led to death of offspring within the first 4 days after birth at maternal exposures that were ≥ 3.3 times the human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine. When administered to pregnant animals during the period of organogenesis, lapatinib caused fetal anomalies (rats) or abortions (rabbits) at maternally toxic doses (with maternal exposures approximately 6.4 and 0.2 times, respectively, the human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine).
Advise pregnant women and females of reproductive potential of the potential risk to the fetus [see Use in Specific Populations (8.1) and Clinical Pharmacology (12.1)]. Verify the pregnancy status of females of reproductive potential prior to initiation of lapatinib tablets. Advise females of reproductive potential to use effective contraception during treatment with lapatinib tablets and for 1 week after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with lapatinib tablets and for 1 week after the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
HER2-Positive Metastatic Breast Cancer: The safety of lapatinib tablets has been evaluated in more than 12,000 patients in clinical trials. The efficacy and safety of lapatinib tablets in combination with capecitabine in breast cancer was evaluated in 198 patients in a randomized, Phase 3 trial [see Clinical Studies (14.1)]. Adverse reactions, which occurred in at least 10% of patients in either treatment arm and were higher in the combination arm, are shown in Table 1.
The most common adverse reactions (greater than or equal to 20%) during therapy with lapatinib tablets plus capecitabine were gastrointestinal (diarrhea, nausea, and vomiting), dermatologic (palmar-plantar erythrodysesthesia and rash), and fatigue. Diarrhea was the most common adverse reaction resulting in discontinuation of study medication.
The most common Grades 3 and 4 adverse reactions (NCI CTCAE v3.0) were diarrhea and palmar-plantar erythrodysesthesia. Selected laboratory abnormalities are shown in Table 2.
Table 1. Adverse Reactions Occurring in Greater Than or Equal to 10% of Patients
| a NCI CTCAE v3.0. b Grade 3 dermatitis acneiform was reported in less than 1% of patients in the group receiving lapatinib tablets plus capecitabine. |
||||||
|
|
Lapatinib Tablets 1,250 mg/day + Capecitabine 2,000 mg/m2/day (N = 198) |
Capecitabine 2,500 mg/m2/day (N=191) |
||||
|
Reactions |
All Gradesa |
Grade 3 |
Grade 4 |
All Gradesa |
Grade 3 |
Grade 4 |
|
% |
% |
% |
% |
% |
% |
|
|
Gastrointestinal disorders |
|
|
|
|
|
|
|
Diarrhea |
65 |
13 |
1 |
40 |
10 |
0 |
|
Nausea |
44 |
2 |
0 |
43 |
2 |
0 |
|
Vomiting |
26 |
2 |
0 |
21 |
2 |
0 |
|
Stomatitis |
14 |
0 |
0 |
11 |
<1 |
0 |
|
Dyspepsia |
11 |
<1 |
0 |
3 |
0 |
0 |
|
Skin and subcutaneous tissue disorders |
|
|
|
|
|
|
|
Palmar-plantar erythrodysesthesia |
53 |
12 |
0 |
51 |
14 |
0 |
|
Rashb |
28 |
2 |
0 |
14 |
1 |
0 |
|
Dry skin |
10 |
0 |
0 |
6 |
0 |
0 |
|
General disorders and administration site conditions |
|
|
|
|
|
|
|
Mucosal inflammation |
15 |
0 |
0 |
12 |
2 |
0 |
|
Musculoskeletal and connective tissue disorders |
|
|
|
|
|
|
|
Pain in extremity |
12 |
1 |
0 |
7 |
<1 |
0 |
|
Back pain |
11 |
1 |
0 |
6 |
<1 |
0 |
|
Respiratory, thoracic, and mediastinal disorders |
|
|
|
|
|
|
|
Dyspnea |
12 |
3 |
0 |
8 |
2 |
0 |
|
Psychiatric disorders |
|
|
|
|
|
|
|
Insomnia |
10 |
<1 |
0 |
6 |
0 |
0 |
Table 2: Selected Laboratory Abnormalities
| Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase. a NCI CTCAE v3.0. |
||||||
| | Lapatinib Tablets 1,250 mg/day +
Capecitabine 2,000 mg/m2/day | Capecitabine 2,500 mg/m2/day
|
||||
| All Gradesa
| Grade 3
| Grade 4
| All Gradesa
| Grade 3
| Grade 4
|
|
| Parameters
| %
| %
| %
| %
| %
| %
|
| Hematologic
| | | | | | |
| Hemoglobin | 56 | <1 | 0 | 53 | 1 | 0 |
| Platelets | 18 | <1 | 0 | 17 | <1 | <1 |
| Neutrophils | 22 | 3 | <1 | 31 | 2 | 1 |
| Hepatic
| | | | | | |
| Total Bilirubin | 45 | 4 | 0 | 30 | 3 | 0 |
| AST | 49 | 2 | <1 | 43 | 2 | 0 |
| ALT | 37 | 2 | 0 | 33 | 1 | 0 |
Hormone Receptor-Positive, Metastatic Breast Cancer: In a randomized, Phase 3 clinical trial of patients (N = 1286) with hormone receptor-positive, metastatic breast cancer, who had not received chemotherapy for their metastatic disease, patients received letrozole with or without lapatinib tablets. In this trial, the safety profile of lapatinib tablets was consistent with previously reported results from trials of lapatinib tablets in the advanced or metastatic breast cancer population. Adverse reactions, which occurred in at least 10% of patients in either treatment arm and were higher in the combination arm are shown in Table 3. Selected laboratory abnormalities are shown in Table 4.
Table 3. Adverse Reactions Occurring in Greater Than or Equal to 10% of Patients
|
a NCI CTCAE, v3.0. b In addition to the rash reported under “Skin and subcutaneous tissue disorders”, 3 additional subjects in each treatment arm had rash under “Infections and infestations”; none were Grade 3 or 4. |
||||||
| | Lapatinib Tablets 1,500 mg/day +
Letrozole 2.5 mg/day (N = 654) | Letrozole 2.5 mg/day
(N = 624) |
||||
| | All Gradesa
| Grade
3 | Grade
4 | All
Gradesa | Grade 3
| Grade 4
|
|
Reactions | %
| %
| %
| %
| %
| %
|
|
Gastrointestinal disorders | | | | | | |
|
Diarrhea | 64 | 9 | <1 | 20 | <1 | 0 |
|
Nausea | 31 | <1 | 0 | 21 | <1 | 0 |
|
Vomiting | 17 | 1 | <1 | 11 | <1 | <1 |
|
Anorexia | 11 | <1 | 0 | 9 | <1 | 0 |
|
Skin and subcutaneous tissue disorders | | | | | | |
|
Rashb | 44 | 1 | 0 | 13 | 0 | 0 |
|
Dry skin | 13 | <1 | 0 | 4 | 0 | 0 |
|
Alopecia | 13 | <1 | 0 | 7 | 0 | 0 |
|
Pruritus | 12 | <1 | 0 | 9 | <1 | 0 |
|
Nail disorder | 11 | <1 | 0 | <1 | 0 | 0 |
|
General disorders and administration site conditions | | | | | | |
|
Fatigue | 20 | 2 | 0 | 17 | <1 | 0 |
|
Asthenia | 12 | <1 | 0 | 11 | <1 | 0 |
|
Nervous system disorders |
|
|
|
|
|
|
|
Headache | 14 | <1 | 0 | 13 | <1 | 0 |
|
Respiratory, thoracic, and mediastinal disorders |
|
|
|
|
|
|
|
Epistaxis | 11 | <1 | 0 | 2 | <1 | 0 |
Table 4: Selected Laboratory Abnormalities
|
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase. a NCI CTCAE, v3.0. |
||||||
| | Lapatinib Tablets 1,500 mg/day +
Letrozole 2.5 mg/day | Letrozole 2.5 mg/day
|
||||
| All Gradesa
| Grade 3
| Grade 4
| All Gradesa
| Grade 3
| Grade 4
|
|
|
Hepatic Parameters | %
| %
| %
| %
| %
| %
|
|
AST | 53 | 6 | 0 | 36 | 2 | <1 |
|
ALT | 46 | 5 | <1 | 35 | 1 | 0 |
|
Total Bilirubin | 22 | <1 | <1 | 11 | 1 | <1 |
Hormone Receptor-Positive, HER2+ Metastatic Breast Cancer: In another randomized, Phase 3 clinical trial of postmenopausal patients (N = 355) with hormone receptor positive (HR+), HER2-positive metastatic breast cancer (MBC) which had progressed after prior trastuzumab-containing chemotherapy and endocrine therapies, patients received lapatinib tablets with trastuzumab and an aromatase inhibitor (AI) (letrozole, exemestane, or anastrozole), lapatinib tablets with an AI, or trastuzumab with an AI. In this trial, the safety profile of the treatment groups was consistent with the known safety of these agents. The most frequent study treatment-related AEs (> 10%) in each of the lapatinib tablets -containing treatment arms were diarrhea, rash, paronychia, nausea, stomatitis, dermatitis acneiform, and decreased appetite, which were infrequent to absent in the trastuzumab treatment arm. The frequency of cardiac AEs (mostly decrease in ejection fraction) was 7% in the lapatinib tablets + trastuzumab + AI group, 2% in the lapatinib tablets + AI group and 3% in the trastuzumab + AI group. Adverse reactions which occurred in at least 10% of patients in the treatment arms are shown in Table 5.
Table 5: Adverse Reactions Occurring in Greater Than or Equal to 10% of Patients
|
a NCI CTCAE v3.0 b Includes multiple adverse reaction terms for rash. |
||||||||||
| | Lapatinib Tablets
(1,000 mg) + Trastuzumab + AI (N = 118) |
Lapatinib Tablets (1,500 mg) +AI (N = 119) |
Trastuzumab + AI (N = 116) |
|||||||
| | All Gradesa
| Grade
3 | Grade
4 | All Gradesa
| Grade
3 | Grade
4 | All
Gradesa | Grade 3
| Grade 4
|
|
|
Reactions | %
| %
| %
| %
| %
| %
| %
| %
| %
|
|
|
Gastrointestinal disorders |
|
|
|
| ||||||
|
Diarrhea | 69 | 13 | 0 | 51 | 6 | 0 | 9 | 0 | 0 |
|
|
Nausea | 22 | 0 | 0 | 22 | 2 | 0 | 9 | 0 | 0 |
|
|
Stomatitis | 17 | 0 | 0 | 13 | <1 | 0 | 3 | 0 | 0 |
|
|
Vomiting | 10 | 0 | 0 | 14 | 0 | 0 | <1 | <1 | 0 |
|
|
Skin and subcutaneous tissue disorders | | | | | | | | | |
|
|
Rashb | 54 | 0 | 0 | 44 | 3 | 0 | 5 | 0 | 0 |
|
|
Palmar-plantar erythrodysesthesia | 10 | 0 | 0 | 8 | <1 | 0 | <1 | 0 | 0 |
|
|
Alopecia | 10 | 0 | 0 | 7 | 0 | 0 | 2 | 0 | 0 |
|
|
General disorders and administration site conditions | | | | | | | | | |
|
|
Fatigue | 12 | <1 | 0 | 14 | 2 | 0 | 10 | 0 | 0 |
|
|
Musculoskeletal and connective tissue disorders | | | | | | | | | |
|
|
Arthralgia | 13 | <1 | 0 | 14 | 0 | 0 | 12 | 0 | 0 |
|
|
Pain in extremity | 7 | <1 | 0 | 10 | 0 | 0 | 3 | 0 | 0 |
|
|
Respiratory, thoracic, and mediastinal disorders | | | | | | | | | |
|
|
Cough | 8 | 0 | 0 | 8 | 0 | 0 | 15 | 0 | 0 |
|
|
Metabolism and nutrition dis orders | | | | | | | | | |
|
|
Decreased appetite | 18 | 0 | 0 | 13 | 0 | 0 | 3 | 0 | 0 |
|
|
Infections and infestations | | | | | | | | | |
|
|
Paronychia | 30 | 0 | 0 | 15 | 2 | 0 | 0 | 0 | 0 |
|
|
Investigations | | | | | | | | | |
|
|
Alanine aminotransferase increased | 7 | 0 | 0 | 15 | 3 | <1 | 6 | 4 | 0 |
|
|
Aspartate aminotransferase increased | 6 | 0 | 0 | 17 | 5 | 0 | 9 | 4 | 0 |
|
|
Nervous system disorders | | | | | | | | | |
|
|
Headache | 5 | 0 | 0 | 16 | 2 | 0 | 10 | <1 | 0 |
|
Decreases in Left Ventricular Ejection Fraction: Due to potential cardiac toxicity with HER2 (ErbB2) inhibitors, LVEF was monitored in clinical trials at approximately 8-week intervals. LVEF decreases were defined as signs or symptoms of deterioration in left ventricular cardiac function that are greater than or equal to Grade 3 (NCI CTCAE v3.0), or a greater than or equal to 20% decrease in left ventricular cardiac ejection fraction relative to baseline which is below the institution's lower limit of normal. Among 198 patients who received combination treatment with lapatinib tablets/capecitabine, 3 experienced Grade 2 and one had Grade 3 LVEF adverse reactions (NCI CTCAE v3.0). Among 654 patients who received combination treatment with lapatinib tablets/letrozole, 26 patients experienced Grade 1 or 2 and 6 patients had Grade 3 or 4 LVEF adverse reactions [see Warnings and Precautions (5.1)].
Hepatotoxicity: Lapatinib tablets have been associated with hepatotoxicity [see Boxed Warning and Warnings and Precautions (5.2)].
Interstitial Lung Disease/Pneumonitis: Lapatinib tablets has been associated with interstitial lung disease and pneumonitis in monotherapy or in combination with other chemotherapies [see Warnings and Precautions (5.5)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of lapatinib tablets. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity reaction, including anaphylaxis [see Contraindications (4)].
Skin and Subcutaneous Tissue Disorders: Nail disorders, including paronychia. Severe cutaneous adverse reactions, including Stevens-Johnson Syndrome (SJS), toxic epidermal necrolysis (TEN), skin fissures.
Cardiac Disorders: Ventricular arrhythmias/Torsades de Pointes (TdP) Electrocardiogram (ECG) QT prolongation.
Related/similar drugs
7. Drug Interactions
7.1 Effects of Lapatinib on Drug-Metabolizing Enzymes and Drug Transport Systems
Lapatinib inhibits CYP3A4, CYP2C8, and P-glycoprotein (P-gp, ABCB1) in vitro at clinically relevant concentrations and is a weak inhibitor of CYP3A4 in vivo. Caution should be exercised and dose reduction of the concomitant substrate drug should be considered when dosing lapatinib tablets concurrently with medications with narrow therapeutic windows that are substrates of CYP3A4, CYP2C8, or P-gp. Lapatinib did not significantly inhibit the following enzymes in human liver microsomes: CYP1A2, CYP2C9, CYP2C19, and CYP2D6 or UGT enzymes in vitro, however, the clinical significance is unknown.
Midazolam: Following coadministration of lapatinib tablets and midazolam (CYP3A4 substrate), 24-hour systemic exposure (AUC) of orally administered midazolam increased 45%, while 24-hour AUC of intravenously administered midazolam increased 22%.
Paclitaxel: In cancer patients receiving lapatinib tablets and paclitaxel (CYP2C8 and P-gp substrate), 24-hour systemic exposure (AUC) of paclitaxel was increased 23%. This increase in paclitaxel exposure may have been underestimated from the in vivo evaluation due to study design limitations.
Digoxin: Following coadministration of lapatinib tablets and digoxin (P-gp substrate), systemic AUC of an oral digoxin dose increased approximately 2.8-fold. Serum digoxin concentrations should be monitored prior to initiation of lapatinib tablets and throughout coadministration. If digoxin serum concentration is greater than 1.2 ng/mL, the digoxin dose should be reduced by half.
7.2 Drugs That Inhibit or Induce Cytochrome P450 3A4 Enzymes
Lapatinib undergoes extensive metabolism by CYP3A4, and concomitant administration of strong inhibitors or inducers of CYP3A4 alter lapatinib concentrations significantly (see Ketoconazole and Carbamazepine sections, below). Dose adjustment of lapatinib should be considered for patients who must receive concomitant strong inhibitors or concomitant strong inducers of CYP3A4 enzymes [see Dosage and Administration (2.2)].
Ketoconazole: In healthy subjects receiving ketoconazole, a CYP3A4 inhibitor, at 200 mg twice daily for 7 days, systemic exposure (AUC) to lapatinib was increased to approximately 3.6-fold of control and half-life increased to 1.7-fold of control.
Carbamazepine: In healthy subjects receiving the CYP3A4 inducer, carbamazepine, at 100 mg twice daily for 3 days and 200 mg twice daily for 17 days, systemic exposure (AUC) to lapatinib was decreased approximately 72%.
7.3 Drugs That Inhibit Drug Transport Systems
Lapatinib is a substrate of the efflux transporter P-glycoprotein (P-gp, ABCB1). If lapatinib tablets are administered with drugs that inhibit P-gp, increased concentrations of lapatinib are likely, and caution should be exercised.
7.4 Acid-Reducing Agents
The aqueous solubility of lapatinib is pH dependent, with higher pH resulting in lower solubility. However, esomeprazole, a proton pump inhibitor, administered at a dose of 40 mg once daily for 7 days, did not result in a clinically meaningful reduction in lapatinib steady-state exposure.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and its mechanism of action, lapatinib tablets can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].There are no available human data to inform of the drug-associated risks. In an animal reproduction study, administration of lapatinib to pregnant rats during organogenesis and through lactation led to death of offspring within the first 4 days after birth at maternal exposures that were ≥ 3.3 times the human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine. When administered to pregnant animals during the period of organogenesis, lapatinib caused fetal anomalies (rats) or abortions (rabbits) at maternally toxic doses. (see Data).
Advise pregnant women and females of reproductive potential of the potential risk to the fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown; however, in the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In embryo-fetal development studies in rats and rabbits, pregnant animals received oral doses of lapatinib at 30, 60, and 120 mg/kg/day during the period of organogenesis. Minor anomalies (left-sided umbilical artery, cervical rib, and precocious ossification) occurred in rats at the maternally toxic dose of 120 mg/kg/day (approximately 6.4 times the human clinical exposure based on AUC following 1,250 mg dose of lapatinib plus capecitabine). In rabbits, lapatinib was associated with maternal toxicity at 60 and 120 mg/kg/day (approximately 0.07 and 0.2 times the human clinical exposure, respectively, based on AUC following 1,250 mg dose of lapatinib plus capecitabine) and abortions at 120 mg/kg/day. Maternal toxicity was associated with decreased fetal body weights and minor skeletal variations.
In a pre- and post-natal development study, rats were given oral doses of 20, 60, and 120 mg/kg/day during gestation through lactation up to weaning. In rats, doses of 60 and 120 mg/kg/day (approximately 3.3 and 6.4 times the human clinical exposure, respectively, based on AUC following 1,250 mg dose of lapatinib plus capecitabine) led to decrease in F1 postnatal survival (91% and 34% of the pups died by the fourth day after birth, at 60 and 120 mg/kg/day, respectively).
8.2 Lactation
Risk Summary
There are no data on the presence of lapatinib in human milk, or its effects on the breastfed child, or milk production. Because of the potential for serious adverse reactions in a breastfed child from lapatinib tablets, advise lactating women not to breastfeed during treatment with lapatinib tablets and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of lapatinib tablets.
Contraception
Females
Based on findings in animal studies, lapatinib tablets can cause fetal harm when administered to a pregnant woman[see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with lapatinib tablets and for 1 week after the last dose.
Males
Based on findings in animal reproduction studies, advise male patients with female partners of reproductive potential to use effective contraception during treatment with lapatinib tablets and for 1 week after the last dose[see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of lapatinib tablets in pediatric patients have not been established.
8.5 Geriatric Use
Of the total number of metastatic breast cancer patients in clinical studies of lapatinib tablets in combination with capecitabine (N = 198), 17% were 65 years of age and older, and 1% were 75 years of age and older. Of the total number of hormone receptor-positive, HER2-positive metastatic breast cancer patients in clinical studies of lapatinib tablets in combination with letrozole (N = 642), 44% were 65 years of age and older, and 12% were 75 years of age and older. No overall differences in safety or effectiveness were observed between elderly subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Lapatinib pharmacokinetics have not been specifically studied in patients with renal impairment or in patients undergoing hemodialysis. There is no experience with lapatinib tablets in patients with severe renal impairment. However, renal impairment is unlikely to affect the pharmacokinetics of lapatinib given that less than 2% (lapatinib and metabolites) of an administered dose is eliminated by the kidneys.
8.7 Hepatic Impairment
The pharmacokinetics of lapatinib were examined in subjects with pre-existing moderate (n = 8) or severe (n = 4) hepatic impairment (Child-Pugh Class B/C, respectively) and in 8 healthy control subjects. Systemic exposure (AUC) to lapatinib after a single oral 100-mg dose increased approximately 14% and 63% in subjects with moderate and severe preexisting hepatic impairment, respectively. Administration of lapatinib tablets in patients with severe hepatic impairment should be undertaken with caution due to increased exposure to the drug. A dose reduction should be considered for patients with severe preexisting hepatic impairment [see Dosage and Administration (2.2)] .In patients who develop severe hepatotoxicity while on therapy, lapatinib tablets should be discontinued and patients should not be retreated with lapatinib tablets [see Warnings and Precautions (5.2)] .
10. Overdosage
There is no known antidote for overdoses of lapatinib tablets. The maximum oral doses of lapatinib that have been administered in clinical trials are 1,800 mg once daily. More frequent ingestion of lapatinib tablets could result in serum concentrations exceeding those observed in clinical trials and could result in increased toxicity. Therefore, missed doses should not be replaced and dosing should resume with the next scheduled daily dose.
Asymptomatic and symptomatic cases of overdose have been reported. The doses ranged from 2,500 to 9,000 mg daily and where reported, the duration varied between 1 and 17 days. Symptoms observed include lapatinib-associated events [see Adverse Reactions (6.1)] and in some cases sore scalp, sinus tachycardia (with otherwise normal ECG), and/or mucosal inflammation.
Because lapatinib is not significantly renally excreted and is highly bound to plasma proteins, hemodialysis would not be expected to be an effective method to enhance the elimination of lapatinib.
Treatment of overdose with lapatinib tablets should consist of general supportive measures.
11. Lapatinib Description
Lapatinib is a small molecule and a member of the 4-anilinoquinazoline class of kinase inhibitors. It is present as the ditosylate salt, with chemical name N-(3 chloro-4-[(3-fluorophenyl)methyl]oxy}phenyl)-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furanyl]-4-quinazolinamine bis(4-methylbenzenesulfonate). It has the molecular formula C29H26ClFN4O4S (C7H8O3S)2 and a molecular weight of 925.48 g/mol. Lapatinib ditosylate has the following chemical structure:
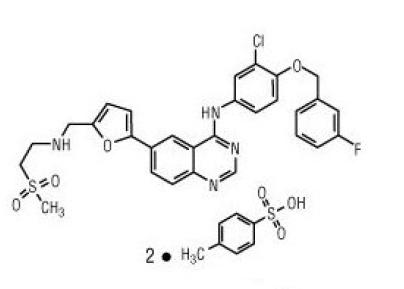
Lapatinib is a yellow solid, and its solubility in water is 0.007 mg/mL and in 0.1N HCl is 0.001 mg/mL at 25°C.
Each 250 mg lapatinib film coated tablet contains 398 mg lapatinib ditosylate equivalent to 250 mg lapatinib free base.
The inactive ingredients of lapatinib tablets are: FD & C Yellow # 6/Sunset yellow FCF Aluminum lake, hypromellose, lecithin (soy), magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, polyvinyl alcohol, povidone, sodium starch glycolate, talc, titanium dioxide, xanthan gum.
12. Lapatinib - Clinical Pharmacology
12.1 Mechanism of Action
Lapatinib is a 4-anilinoquinazoline kinase inhibitor of the intracellular tyrosine kinase domains of both Epidermal Growth Factor Receptor (EGFR [ErbB1]) and of Human Epidermal Receptor Type 2 (HER2 [ErbB2]) receptors (estimated Kiapp values of 3nM and 13nM, respectively) with a dissociation half-life of greater than or equal to 300 minutes. Lapatinib inhibits ErbB-driven tumor cell growth in vitro and in various animal models.
An additive effect was demonstrated in an in vitro study when lapatinib and 5-FU (the active metabolite of capecitabine) were used in combination in the 4-tumor cell lines tested. The growth inhibitory effects of lapatinib were evaluated in trastuzumab-conditioned cell lines. Lapatinib retained significant activity against breast cancer cell lines selected for long-term growth in trastuzumab-containing medium in vitro. These in vitro findings suggest non-cross-resistance between these two agents.
Hormone receptor-positive breast cancer cells (with ER [Estrogen Receptor] and/or PgR [Progesterone Receptor]) that coexpress the HER2 tend to be resistant to established endocrine therapies. Similarly, hormone receptor-positive breast cancer cells that initially lack EGFR or HER2 upregulate these receptor proteins as the tumor becomes resistant to endocrine therapy.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of lapatinib on the QT-interval was evaluated in a single-blind, placebo-controlled, single sequence (placebo and active treatment) crossover study in patients with advanced solid tumors (N = 58). During the 4-day treatment period, three doses of matching placebo were administered 12 hours apart in the morning and evening on Day 1 and in the morning on Day 2. This was followed by three doses of lapatinib 2,000 mg (1.3 – 1.6 times the recommended dosage) administered in the same way. Measurements, including ECGs and pharmacokinetic samples were done at baseline and at the same time points on Day 2 and Day 4. In the evaluable population of subjects who had complete dosing and ECG assessments (N = 37), the maximum mean ΔΔQTcF (90% CI) of 8.75 ms (4.08, 13.42) was observed 10 hours after ingestion of the third dose of lapatinib 2000 mg. The ΔΔQTcF exceeded the 5 ms threshold and the upper bound 90% CIs exceeded the 10 ms threshold at multiple time points.
There was a concentration-dependent increase in QTcF effects [see Warnings and Precautions (5.6)].
12.3 Pharmacokinetics
Absorption: Absorption following oral administration of lapatinib tablets is incomplete and variable. Serum concentrations appear after a median lag time of 0.25 hours (range 0 to 1.5 hour). Peak plasma concentrations (Cmax) of lapatinib are achieved approximately 4 hours after administration. Daily dosing of lapatinib tablets results in achievement of steady-state within 6 to 7 days, indicating an effective half-life of 24 hours.
At the dose of 1,250 mg daily, steady state geometric mean [95% confidence interval (CI)] values of Cmax were 2.43 mcg/mL (1.57 to 3.77 mcg/mL) and AUC were 36.2 mcg.h/mL (23.4 to 56 mcg.h/mL).
Divided daily doses of lapatinib tablets resulted in approximately 2-fold higher exposure at steady-state (steady-state AUC) compared to the same total dose administered once daily.
Systemic exposure to lapatinib is increased when administered with food. Lapatinib AUC values were approximately 3- and 4-fold higher (Cmax approximately 2.5- and 3-fold higher) when administered with a low-fat (5% fat-500 calories) or with a high-fat (50% fat-1000 calories) meal, respectively.
Distribution: Lapatinib is highly bound (greater than 99%) to albumin and alpha-1 acid glycoprotein. In vitro studies indicate that lapatinib is a substrate for the transporters breast cancer-resistance protein (BCRP, ABCG2) and P-glycoprotein (P-gp, ABCB1). Lapatinib has also been shown to inhibit P-gp, BCRP, and the hepatic uptake transporter OATP 1B1, in vitro at clinically relevant concentrations.
Metabolism: Lapatinib undergoes extensive metabolism, primarily by CYP3A4 and CYP3A5, with minor contributions from CYP2C19 and CYP2C8 to a variety of oxidated metabolites, none of which accounts for more than 14% of the dose recovered in the feces or 10% of lapatinib concentration in plasma.
Elimination: At clinical doses, the terminal phase half-life following a single dose was 14.2 hours; accumulation with repeated dosing indicates an effective half-life of 24 hours.
Elimination of lapatinib is predominantly through metabolism by CYP3A4/5 with negligible ( less than 2%) renal excretion. Recovery of parent lapatinib in feces accounts for a median of 27% (range 3 % to 67%) of an oral dose.
Effects of Age, Gender, or Race: Studies of the effects of age, gender, or race on the pharmacokinetics of lapatinib have not been performed.
12.5 Pharmacogenomics
The HLA alleles DQA1*02:01 and DRB1*07:01 were associated with hepatotoxicity reactions in a genetic substudy of a monotherapy trial with lapatinib tablets (n = 1194). Severe liver injury (ALT greater than 5 times the upper limit of the normal, NCI CTCAE Grade 3) occurred in 2% of patients overall; the incidence of severe liver injury among DQA1*02:01 or DRB1*07:01 allele carriers was 8% verses 0.5% in non-carriers. These HLA alleles are present in approximately 15% to 25% of Caucasian, Asian, African, and Hispanic populations and 1% in Japanese populations. Liver function should be monitored in all patients receiving therapy with lapatinib tablets regardless of genotype.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In carcinogenicity studies, lapatinib was administered orally for up to 104 weeks at doses of 75 and 150 mg/kg/day in male mice and 75, 150, and 300 mg/kg/day in female mice (approximately 0.7 to 2 times the expected human clinical exposure based on AUC for a clinical dose of 1,250 mg/day plus capecitabine) and 60, 120, 240 and 500 mg/kg/day (approximately 0.6 to 2.3 times the expected human clinical exposure based on AUC) in male rats, and 20, 60, and 180 mg/kg/day (approximately 1.4 to 10 times the expected human clinical exposure based on AUC for a clinical dose of 1,250 mg/day plus capecitabine) in female rats. There was no evidence of carcinogenicity in mice. In male rats, there was an increased incidence of whole body combined hemangiomas and hemangiosarcomas.
Lapatinib was not clastogenic or mutagenic in the Chinese hamster ovary chromosome aberration assay, microbial mutagenesis (Ames) assay, human peripheral lymphocyte chromosome aberration assay or the in vivo rat bone marrow chromosome aberration assay at single doses up to 2,000 mg/kg.
There were no effects on male or female rat mating or fertility at doses up to 120 mg/kg/day in females and 180 mg/kg/day in males (approximately 6.4 times and 2.6 times the expected human clinical exposure based on AUC following 1,250-mg dose of lapatinib plus capecitabine, respectively). The effect of lapatinib on human fertility is unknown. However, when female rats were given oral doses of lapatinib during breeding and through the first 6 days of gestation, a significant decrease in the number of live fetuses was seen at 120 mg/kg/day and in the fetal body weights at greater than or equal to 60 mg/kg/day (approximately 6.4 times and 3.3 times the expected human clinical exposure based on AUC following 1,250- mg dose of lapatinib plus capecitabine, respectively).
13.2 Animal Toxicology and/or Pharmacology
In 104-week repeat-dose studies in rodents, severe skin lesions that led to lethality were seen at the highest doses tested (300 mg/kg/day) in male mice and female rats. There was also an increase in renal infarcts and papillary necrosis in female rats at greater than or equal to 60 mg/kg/day and greater than or equal to180 mg/kg/day, respectively (approximately 7 and 10 times the expected human clinical exposure based on AUC, respectively). The relevance of these findings for humans is uncertain .
14. Clinical Studies
14.1 HER2-Positive Metastatic Breast Cancer
The efficacy and safety of lapatinib tablets in combination with capecitabine in breast cancer were evaluated in a randomized, Phase 3 trial. Patients eligible for enrollment had HER2 (ErbB2) overexpressing (IHC 3+ or IHC 2+ confirmed by FISH), locally advanced or metastatic breast cancer, progressing after prior treatment that included anthracyclines, taxanes, and trastuzumab.
Patients were randomized to receive either lapatinib tablets 1,250 mg once daily (continuously) plus capecitabine 2,000 mg/m2/day on Days 1 - 14 every 21 days, or to receive capecitabine alone at a dose of 2,500 mg/m2/day on Days 1 - 14 every 21 days. The endpoint was time to progression (TTP). TTP was defined as time from randomization to tumor progression or death related to breast cancer. Based on the results of a pre-specified interim analysis, further enrollment was discontinued. Three hundred and ninety-nine (399) patients were enrolled in this study. The median age was 53 years and 14% were older than 65 years. Ninety-one percent (91%) were Caucasian. Ninety-seven percent (97%) had stage IV breast cancer, 48% were estrogen receptor+ (ER+) or progesterone receptor+ (PR+), and 95% were ErbB2 IHC 3+ or IHC 2+ with FISH confirmation. Approximately 95% of patients had prior treatment with anthracyclines, taxanes, and trastuzumab.
Efficacy analyses 4 months after the interim analysis are presented in Table 6, Figure 1, and Figure 2.
Table 6: Efficacy Results
| TTP=Time to progression. CI = Confidence Interval a The time from last tumor assessment to the data cut-off date was greater than 100 days in approximately 30% of patients in the independent assessment. The pre-specified assessment interval was 42 or 84 days. |
||||||
|
|
Independent Assessmenta |
Investigator Assessment |
||||
|
Lapatinib Tablets 1,250 mg/day+ Capecitabine 2,000 mg/m2/day |
Capecitabine 2,500 mg/m2/day |
Lapatinib Tablets 1,250 mg/day+ Capecitabine 2,000 mg/m2/day |
Capecitabine 2,500 mg/m2/day |
|||
|
(N=198) |
(N=201) |
(N=198) |
(N=201) |
|||
|
Number of TTP events |
82 |
102 |
121 |
126 |
||
|
Median TTP, weeks(25th, 75th, percentile), weeks |
27.1 (17.4, 49.4) |
18.6 (9.1, 36.9) |
23.9 (12.0, 44.0) |
18.3 (6.9, 35.7) |
||
|
Hazard Ratio (HR) (95% CI) P value |
0.57 (0.43, 0.77) 0.00013 |
0.72 (0.56, 0.92) 0.00762 |
||||
|
Response Rate (%) (95% CI) |
23.7 (18.0, 30.3) |
13.9 (9.5, 19.5) |
31.8 (25.4, 38.8) |
17.4 (12.4, 23.4) |
||
Figure 1: Kaplan-Meier Estimates for Independent Review Panel-evaluated Time to Progression
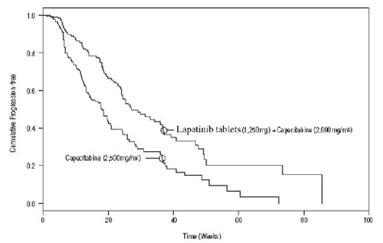
Figure 2: Kaplan-Meier Estimates for Investigator Assessment Time to Progression
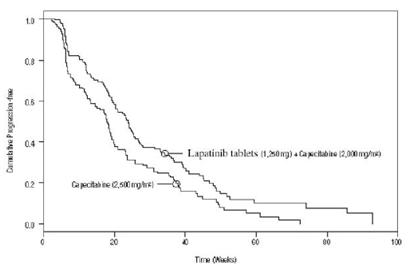
At the time of above efficacy analysis, the (OS) data were not mature (32% events). However, based on the TTP results, the study was unblinded and patients receiving capecitabine alone were allowed to cross over to treatment with lapatinib tablets plus capecitabine. The survival data were followed for an additional 2 years to be mature and the analysis is summarized in Table 7.
Table 7: Overall Survival Data
| Abbreviation CI, confidence interval. |
||||||||||||||
|
|
Lapatinib Tablets 1,250 mg/day+ Capecitabine 2,000 mg/m2/ day (N=207) |
Capecitabine 2,500 mg/m2/day (N=201) |
||||||||||||
|
Overall Survival |
|
|
||||||||||||
|
Died |
76% |
82% |
||||||||||||
|
Median Overall Survival (weeks) |
75.0 |
65.9 |
||||||||||||
| Hazard ratio, 95% CI (P value) |
0.89 (0.71, 1.10) 0.276 |
|||||||||||||
Clinical Studies Describing Limitations of Use: In two randomized trials, lapatinib tablets- based chemotherapy regimens have been shown to be less effective than trastuzumab-based chemotherapy regimens. The first randomized, open-label study compared the safety and efficacy of lapatinib tablets in combination with capecitabine relative to trastuzumab in combination with capecitabine in women with HER2-positive metastatic breast cancer (N=540). The study was stopped early based on the findings of a pre-planned interim analysis showing a low incidence of CNS events (primary endpoint) and superior efficacy of the trastuzumab plus capecitabine. The median progression-free survival was 6.6 months in the group receiving lapatinib tablets in combination with capecitabine compared with 8.0 months in the group receiving the trastuzumab combination [HR = 1.30 (95% CI: 1.04, 1.64)]. Overall survival was analyzed when 26% of deaths occurred in the group receiving lapatinib tablets in combination with capecitabine and 22% in the group receiving the trastuzumab combination [HR = 1.34 (95% CI: 0.95, 1.92)].
The second randomized, open-label study compared the safety and efficacy of taxane-based chemotherapy plus lapatinib tablets to taxane-based chemotherapy plus trastuzumab as first-line therapy in women with HER2-positive, metastatic breast cancer (N=652). The study was stopped early based on the findings of a pre-planned interim analysis. The median progression-free survival was 11.3 months in the trastuzumab combination treatment arm compared to 9.0 months in patients treated with lapatinib tablets in the combination arm for the intent-to-treat population [HR= 1.37 (95% CI: 1.13, 1.65)].
14.2 Hormone Receptor-Positive, HER2-Positive Metastatic Breast Cancer
The efficacy and safety of lapatinib tablets in combination with letrozole were evaluated in a double-blind, placebo-controlled, multi-center study. A total of 1286 postmenopausal women with hormone receptor positive (ER positive and/or PgR-positive) metastatic breast cancer, who had not received prior therapy for metastatic disease, were randomly assigned to receive either lapatinib tablets (1,500 mg once daily) plus letrozole (2.5 mg once daily) (n = 642) or letrozole (2.5 mg once daily) alone (n = 644). Of all patients randomized to treatment, 219 (17%) patients had tumors overexpressing the HER2 receptor, defined as fluorescence in situ hybridization (FISH) greater than or equal to 2 or 3+ immunohistochemistry (IHC). There were 952 (74%) patients who were HER2-negative and 115 (9%) patients did not have their HER2 receptor status confirmed. The primary objective was to evaluate and compare progression-free survival (PFS) in the HER2-positive population. Progression-free survival was defined as the interval of time between date of randomization and the earlier date of first documented sign of disease progression or death due to any cause.
The baseline demographic and disease characteristics were balanced between the two treatment arms. The median age was 63 years and 45% were 65 years of age or older. Eighty-four percent (84%) of the patients were white. Approximately 50% of the HER2-positive population had prior adjuvant/neo-adjuvant chemotherapy and 56% had prior hormonal therapy. Only 2 patients had prior trastuzumab.
In the HER2-positive subgroup (n = 219), the addition of lapatinib tablets to letrozole resulted in an improvement in PFS. In the HER2-negative subgroup, there was no improvement in PFS of the combination of lapatinib tablets plus letrozole combination compared to the letrozole plus placebo. Overall response rate (ORR) was also improved with combination of lapatinib tablets plus letrozole. The OS data were not mature. Efficacy analyses for the hormone receptor-positive, HER2-positive and HER2-negative subgroups are presented in Table 8 and Figure 3.
Table 8. Efficacy Results
| Abbreviations: CI, confidence interval, PFS progression-free survival a Kaplan-Meier estimate. |
||||
|
|
HER2-Positive Population |
HER2-Negative Population |
||
|
Lapatinib Tablets 1,500 mg/day + Letrozole 2.5 mg/day |
Letrozole 2.5 mg/day |
Lapatinib Tablets 1500 mg/day + Letrozole 2.5 mg/day |
Letrozole 2.5 mg /day |
|
|
(N = 111) |
(N = 108) |
(N = 478) |
(N = 474) |
|
|
Median PFSa, weeks (95% CI) |
35.4 (24.1, 39.4) |
13.0 (12.0, 23.7) |
59.7 (48.6, 69.7) |
58.3 (47.9, 62.0) |
|
Hazard Ratio (95% CI) P value |
0.71 (0.53, 0.96) 0.019 |
0.90 (0.77, 1.05) 0.188 |
||
|
Response Rate (%) (95% CI) |
27.9 (19.8, 37.2) |
14.8 (8.7, 22.9) |
32.6 (28.4, 37.0) |
31.6 (27.5, 36.0) |
Figure 3: Kaplan-Meier Estimates for Progression-Free Survival for the HER2-Positive Population
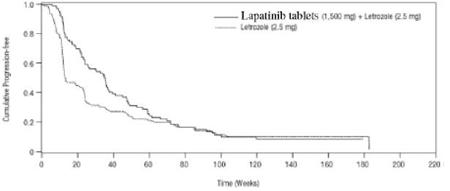
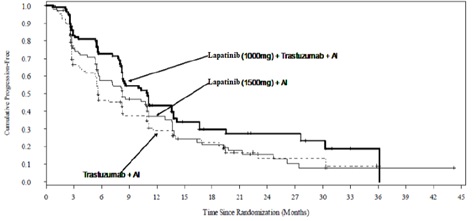
The efficacy and safety of lapatinib tablets, in combination with an aromatase inhibitor (AI), were confirmed in another randomized Phase 3 trial. Patients enrolled were post-menopausal women who had hormone receptor positive (HR+)/HER2-positive, metastatic breast cancer, which had progressed after prior trastuzumab containing chemotherapy and endocrine therapies. A total of 355 patients were randomized in a 1:1:1 ratio to lapatinib tablets 1000 mg + trastuzumab + AI (N = 120), or trastuzumab + AI arm (N = 117), or lapatinib tablets 1500 mg + AI (N = 118). In the trastuzumab-containing arms, trastuzumab was administered with a loading dose of 8 mg/kg IV followed by the maintenance dose of 6 mg/kg every 3 weeks. In the AI-containing arms, the AIs were administered at doses of letrozole 2.5 mg once daily, or exemestane 25 mg once daily, or anastrozole 1 mg once daily.
The study was designed to evaluate a potential benefit in Progression Free Survival (PFS) when double versus single HER2 targeted therapy was administered in combination with an AI (letrozole, exemestane, or anastrozole). The major efficacy outcome measure was PFS based on local radiology/investigator’s assessment comparing lapatinib tablets + trastuzumab + AI versus trastuzumab + AI. The median age was 56 years (range 30-84). The majority of the patients treated on this trial (70%) were Caucasian.
Efficacy results are presented in Table 9 and Figure 4. The OS data were not mature (23% of patients had died).
Table 9. Efficacy Results
| Lapatinib Tablets
(1,000 mg) + Trastuzumab + AI |
Trastuzumab + AI |
Lapatinib Tablets (1,500 mg) + AI |
|
| (N = 120) | (N = 117) | (N = 118) | |
| Median PFSa, months (95% CI) |
11.0 (8.3, 13.8) |
5.6 (5.4, 8.3) |
8.3 (5.8,11.1) |
| Hazard Ratio (95% CI) P-valueb |
0.62 (0.45, 0.88) 0.0040 | ----------- | |
| ----------- |
0.85 (0.62, 1.17) 0.2921c |
||
| Response Rate (%)d
(95% CI) | 22.5 (15.4, 31.0) | 8.5 (4.2, 15.2) | 12.7 (7.3, 20.1) |
| Abbreviations: CI, confidence interval; PFS, progression-free survival. a Kaplan-Meier estimate. b Stratified log-rank test. c Nominal p-value. No multiplicity adjustment. d Confirmed CR + PR; subjects with unknown or missing response were treated as non-responders. |
|||
Figure 4. Kaplan-Meier Estimates for Progression-Free Survival
16. How is Lapatinib supplied
Lapatinib tablets 250 mg are orange colored, oval shaped, film-coated tablets, debossed “NTL” on one side and plain on another side and are available in:
Bottles of 150 tablets: NDC 68180-801-36
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients of the following:
Decreased Left Ventricular Ejection Fraction (LVEF)
- Lapatinib tablets have been reported to decrease left ventricular ejection fraction which may result in shortness of breath, palpitations, and/or fatigue [see Warnings and Precautions (5.1)]. Advise patients to inform their healthcare provider if they develop these symptoms while taking lapatinib tablets.
Hepatotoxicity and Hepatic Impairment
- Periodic laboratory testing will be performed while taking lapatinib tablets. Advise patients to report signs and symptoms of liver dysfunction to their healthcare provider right away [see Warnings and Precautions (5.2)].
Diarrhea
- Lapatinib tablets often causes diarrhea which may be severe in some cases [see Warnings and Precautions (5.4)]. Instruct patients on how to manage and/or prevent diarrhea and to inform their healthcare provider immediately if there is any change in bowel patterns or severe diarrhea occurs during treatment with lapatinib tablets.
Interstitial Lung Disease/Pneumonitis
- Advise patients to report pulmonary signs or symptoms indicative of ILD or pneumonitis [see Warnings and Precautions (5.5)]
Severe Cutaneous Reactions
- Advise patients to report severe cutaneous reactions to their healthcare provider if they develop these symptoms while taking lapatinib tablets [see Warnings and Precautions (5.7)].
Drug and Food Interactions
- Lapatinib tablets may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products [see Drug Interactions (7)].
- Lapatinib tablets may interact with grapefruit. Advise patients not to take lapatinib tablets with grapefruit products [see Dosage and Administration (2.2), Drug Interactions (7.2)].
Dosing Administration
- Lapatinib tablets should be taken at least one hour before or one hour after a meal, in contrast to capecitabine which should be taken with food or within 30 minutes after food. The dose of Lapatinib tablets should be taken once daily. Dividing the daily dose is not recommended [see Dosage and Administration (2.1)].
Embryo-Fetal Toxicity
- Inform female patients of the risk to a fetus and potential loss of the pregnancy. Advise females to inform their healthcare provider if they are pregnant or become pregnant [see Use in Specific Populations (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with Lapatinib tablets and for 1 week after the last dose.
- Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 1 week following the last dose [see Warning and Precautions (5.8) and Use in Specific Populations (8.1, 8.3)].
Lactation
- Advise patients not to breastfeed during treatment and for 1 week after the last dose of lapatinib tablets [see Use in Specific Populations (8.2)]
Manufactured by:
Natco Pharma Limited
India
Distributed by:
Lupin Pharmaceuticals, Inc.
Naples, FL 34108
United States.
Rev.: 06/2025
Medication Guide
PATIENT INFORMATION
LAPATINIB (la-PA-ti-nib) tablets
What are lapatinib tablets?
Lapatinib tablets are prescription medicine used:
- with the medicine capecitabine to treat people with breast cancer that is advanced or that has spread to other parts of the body (metastatic), and:
- that is HER2-positive (tumors that produce large amounts of a protein called human epidermal growth factor receptor-2), and
- who have already had certain other breast cancer treatments Before taking lapatinib tablets with capecitabine, your breast cancer should have gotten worse (progressed) with the medicine trastuzumab.
- with the medicine letrozole to treat women who:
- have gone through the change of life (postmenopausal), and
- who have metastatic breast cancer that is hormone receptor-positive, HER2-positive, and hormonal therapy is a treatment option for them.
It is not known if lapatinib tablets are safe and effective in children.
Do not take lapatinib tablets if you are allergic to any of the ingredients in lapatinib tablets. A complete list of ingredients in lapatinib tablets can be found at the end of this Patient Information.
Before you take lapatinib tablets, tell your healthcare provider about all of your medical conditions, including if you:
- have heart problems
- have liver problems. You may need a lower dose of lapatinib tablets.
For females, tell your healthcare provider if you:
- are pregnant or plan to become pregnant. Lapatinib tablets can harm your unborn baby. Your healthcare provider should check to see if you are pregnant before you start taking lapatinib tablets. You should use effective birth control (contraception) during treatment with lapatinib tablets and for 1 week after the last dose of lapatinib tablets. Tell your healthcare provider right away if you become pregnant during treatment with lapatinib tablets.
- are breastfeeding or plan to breastfeed. It is not known if lapatinib passes into your breast milk. You should not breastfeed during treatment with lapatinib tablets and for 1 week after the last dose of lapatinib tablets. Talk to your healthcare provider about the best way to feed your baby if you breastfeed.
For males with female partners who are able to become pregnant:
- use effective contraception during treatment with lapatinib tablets and for 1 week after the last dose.
- if your female partner becomes pregnant during treatment with lapatinib tablets, tell your healthcare provider right away.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Lapatinib tablets may affect the way other medicines work, and other medicines may affect the way lapatinib tablets works. Know the medicines you take. Keep a list of your medicines with you to show your healthcare provider and pharmacist when you get a new medicine. Do not take other medicines during treatment with lapatinib tablets without first talking with your healthcare provider.
How should I take lapatinib tablets?
- Take lapatinib tablets exactly as your healthcare provider tells you to take it. Your healthcare provider may change your dose of lapatinib tablets if needed.
- For people with advanced or metastatic breast cancer, lapatinib tablets and capecitabine are taken in 21-day cycles. The usual dose of lapatinib tablets is 1,250 mg (5 tablets) taken by mouth all at the same time,1 time a day on days 1 to 21.
- Your healthcare provider will tell you the dose of capecitabine you should take and when you should take it.
- Take capecitabine with food or within 30 minutes after food.
- For people with hormone receptor-positive, HER2-positive breast cancer, lapatinib tablets and letrozole are taken every day. The usual dose of lapatinib tablets is 1,500 mg (6 tablets) taken by mouth all at the same time, 1 time a day. Your healthcare provider will tell you the dose of letrozole you should take and when you should take it.
- Take lapatinib tablets at least 1 hour before, or at least 1 hour after a meal.
- Avoid eating or drinking grapefruit products during treatment with lapatinib tablets.
- If you miss a dose of lapatinib tablets, take your next dose at your regular time the next day.
If you take too much lapatinib tablets, call your healthcare provider or go to the nearest hospital emergency room right away
What are the possible side effects of lapatinib tablets?
Lapatinib tablets may cause serious side effects, including:
-
heart problems, including decreased pumping of blood from the heart and an abnormal heartbeat. Signs and symptoms of an abnormal heartbeat include:
- feeling like your heart is pounding or racing
- dizziness
- tiredness
- feeling lightheaded
- shortness of breath
Your healthcare provider should check your heart function before you start taking lapatinib tablets and during treatment.
-
liver problems. Liver problems can be severe and deaths have happened. Signs and symptoms of liver problems include:
- itching
- yellowing of your skin or the white part of your eyes
- dark urine
- pain or discomfort in the right upper stomach area
Your healthcare provider should do blood tests to check your liver before you start taking lapatinib tablets and during treatment.
- diarrhea. Diarrhea is common with lapatinib tablets and may sometimes be severe. Severe diarrhea can cause loss of body fluid (dehydration) and some deaths have happened. Call your healthcare provider right away if you have a change in bowel pattern or if you have severe diarrhea. Follow your healthcare provider’s instructions for what to do to help prevent or treat diarrhea.
- lung problems. Symptoms of a lung problem with lapatinib tablets include a cough that will not go away or shortness of breath.
- severe skin reactions. Lapatinib tablets may cause severe skin reactions. Tell your healthcare provider right away if you develop a rash, red skin, blistering of the lips, eyes, or mouth, peeling of the skin, fever or any combination of these. As severe skin reactions can be life-threatening, your healthcare provider may tell you to stop taking lapatinib tablets.
Call your healthcare provider right away if you have any of the signs or symptoms of the serious side effects listed above.
Common side effects of lapatinib tablets in combination with capecitabine or letrozole include:
- diarrhea
- red, painful hands and feet
- nausea
- rash
- vomiting
- inflamed mouth, digestive tract and airways
- mouth sores
- headache
- unusual hair loss or thinning
- shortness of breath
- dry skin
- itching
- tiredness
- painful arms, legs and back
- loss of appetite
- indigestion
- nose bleeds
- nail disorder such as nail bed changes, nail pain, infection and swelling of the cuticles
- difficulty sleeping
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of lapatinib tablets. For more information, ask your healthcare provider or pharmacist.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also get side effects from the other medicines taken with lapatinib tablets. Talk to your healthcare provider about possible side effects you may get during treatment.
How should I store lapatinib tablets?
- Store lapatinib tablets at room temperature between 68° F to 77°F (20° C to 25°C).
- Do not keep medicine that is out of date or that you no longer need.
Keep lapatinib tablets and all medicines out of the reach of children.
General information about the safe and effective use of lapatinib tablets.
Medicines are sometimes prescribed for purposes other than those listed in Patient Information leaflet. Do not use lapatinib tablets for a condition for which it was not prescribed. Do not give lapatinib tablets to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about lapatinib tablets that is written for health professionals.
For more information, you may also call Lupin Pharmaceuticals, Inc. at toll-free number, 1-800-399-2561.
What are the ingredients in lapatinib tablets?
Active Ingredient: Lapatinib ditosylate.
Inactive Ingredients: FD & C Yellow # 6/Sunset yellow FCF Aluminum lake, hypromellose, lecithin (soy), magnesium stearate, microcrystalline cellulose, polyethylene glycol, polysorbate 80, polyvinyl alcohol, povidone, sodium starch glycolate, talc, titanium dioxide, xanthan gum.
Manufactured by:
Natco Pharma Limited
India
Distributed by:
Lupin Pharmaceuticals, Inc.
Naples, FL 34108
United States.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: June/2025
PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
150 Tablets
NDC 68180-801-36
250 mg
RX Only
Each film coated tablet contains 398 mg lapatinib ditosylate equivalent to 250 mg lapatinib.
Usual Dosage: See accompanying prescribing information.
Important: Use safety closures when dispensing this product unless otherwise directed by physicians or requested by purchaser.
Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F).

| LAPATINIB
lapatinib tablet |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Lupin Pharmaceuticals, Inc. (089153071) |
| Registrant - NATCO PHARMA LIMITED (650224736) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Natco Pharma Limited-Pharma Division | 918588174 | MANUFACTURE(68180-801) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| NATCO PHARMA LIMITED | 675484209 | MANUFACTURE(68180-801) | |
Frequently asked questions
More about lapatinib
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: EGFR inhibitors
- Breastfeeding
- En español
