Exemestane Tablets: Package Insert / Prescribing Info
Package insert / product label
Dosage form: tablet
Drug classes: Aromatase inhibitors, Hormones / antineoplastics
Medically reviewed by Drugs.com. Last updated on Aug 10, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
EXEMESTANE tablets, for oral use
Initial U.S. Approval: 1999
Indications and Usage for Exemestane Tablets
Exemestane is an aromatase inhibitor indicated for:
- adjuvant treatment of postmenopausal women with estrogen-receptor positive early breast cancer who have received two to three years of tamoxifen and are switched to exemestane tablets for completion of a total of five consecutive years of adjuvant hormonal therapy (14.1).
- treatment of advanced breast cancer in postmenopausal women whose disease has progressed following tamoxifen therapy (14.2).
Exemestane Tablets Dosage and Administration
Recommended Dose: One 25 mg tablet once daily after a meal (2.1).
Dosage Forms and Strengths
Tablets: 25 mg (3)
Contraindications
Patients with a known hypersensitivity to the drug or to any of the excipients (4).
Warnings and Precautions
- Reductions in bone mineral density (BMD) over time are seen with exemestane use (5.1).
- Routine assessment of 25-hydroxy vitamin D levels prior to the start of aromatase inhibitor treatment should be performed (5.2).
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception (5.6, 8.1, 8.3).
Adverse Reactions/Side Effects
- Early breast cancer: Adverse reactions occurring in ≥10% of patients in any treatment group (exemestane vs. tamoxifen) were hot flushes (21% vs. 20%), fatigue (16% vs. 15%), arthralgia (15% vs. 9%), headache (13% vs. 11%), insomnia (12% vs. 9%), and increased sweating (12% vs. 10%). Discontinuation rates due to AEs were similar between exemestane and tamoxifen (6% vs. 5%). Incidences of cardiac ischemic events (myocardial infarction, angina, and myocardial ischemia) were exemestane 1.6%, tamoxifen 0.6%. Incidence of cardiac failure: exemestane 0.4%, tamoxifen 0.3% (6, 6.1).
- Advanced breast cancer: Most common adverse reactions were mild to moderate and included hot flushes (13% vs. 5%), nausea (9% vs. 5%), fatigue (8% vs. 10%), increased sweating (4% vs. 8%), and increased appetite (3% vs. 6%) for exemestane and megestrol acetate, respectively (6, 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Northstar Rx LLC at 1-800-206-7821 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
Lactation: Advise not to breastfeed (8.2).
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2025
Full Prescribing Information
1. Indications and Usage for Exemestane Tablets
1.1 Adjuvant Treatment of Postmenopausal Women
Exemestane tablets are indicated for adjuvant treatment of postmenopausal women with estrogen-receptor positive early breast cancer who have received two to three years of tamoxifen and are switched to exemestane tablets for completion of a total of five consecutive years of adjuvant hormonal therapy [see Clinical Studies (14.1)].
1.2 Advanced Breast Cancer in Postmenopausal Women
Exemestane tablets are indicated for the treatment of advanced breast cancer in postmenopausal women whose disease has progressed following tamoxifen therapy [see Clinical Studies (14.2)].
2. Exemestane Tablets Dosage and Administration
2.1 Recommended Dose
The recommended dose of exemestane tablets in early and advanced breast cancer is one 25 mg tablet once daily after a meal.
- adjuvant treatment of postmenopausal women with estrogen-receptor positive early breast cancer who have received two to three years of tamoxifen and are switched to exemestane tablets for completion of a total of five consecutive years of adjuvant hormonal therapy.
- the treatment of advanced breast cancer in postmenopausal women whose disease has progressed following tamoxifen therapy.
2.2 Dose Modifications
Concomitant use of strong CYP 3A4 inducers decreases exemestane exposure, For patients receiving exemestane tablets with a strong CYP 3A4 inducer such as rifampicin or phenytoin, the recommended dose of exemestane tablets is 50 mg once daily after a meal [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
Exemestane tablets, USP are white to off-white, round, biconvex, bevel-edged tablets debossed with “25” on one side and plain on the other side. Each tablet contains 25 mg of exemestane USP.
4. Contraindications
Exemestane is contraindicated in patients with a known hypersensitivity to the drug or to any of the excipients.
5. Warnings and Precautions
5.1 Reductions in Bone Mineral Density (BMD)
Reductions in bone mineral density (BMD) over time are seen with exemestane use. Table 1 describes changes in BMD from baseline to 24 months in patients receiving exemestane compared to patients receiving tamoxifen (IES) or placebo (027). Concomitant use of bisphosphonates, vitamin D supplementation, and calcium was not allowed.
Table 1. Percent Change in BMD from Baseline to 24 months, Exemestane vs. Control1
| | IES
| 027
|
||
| BMD
| Exemestane
N=29 | Tamoxifen1
N=38 | Exemestane
N=59 | Placebo1
N=65 |
| Lumbar spine (%) | -3.1 | -0.2 | -3.5 | -2.4 |
| Femoral neck (%) | -4.2 | -0.3 | -4.6 | -2.6 |
During adjuvant treatment with exemestane, women with osteoporosis or at risk of osteoporosis should have their bone mineral density formally assessed by bone densitometry at the commencement of treatment. Monitor patients for bone mineral density loss and treat as appropriate.
5.2 Vitamin D Assessment
Routine assessment of 25-hydroxy vitamin D levels prior to the start of aromatase inhibitor treatment should be performed, due to the high prevalence of vitamin D deficiency in women with early breast cancer (EBC). Women with vitamin D deficiency should receive supplementation with vitamin D.
5.3 Administration with Estrogen-Containing Agents
Exemestane should not be coadministered with systemic estrogen-containing agents as these could interfere with its pharmacologic action.
5.4 Laboratory Abnormalities
In patients with early breast cancer, the incidence of hematological abnormalities of Common Toxicity Criteria (CTC) grade ≥1 was lower in the exemestane treatment group, compared with tamoxifen. Incidence of CTC grade 3 or 4 abnormalities was low (approximately 0.1%) in both treatment groups. Approximately 20% of patients receiving exemestane in clinical studies in advanced breast cancer experienced CTC grade 3 or 4 lymphocytopenia. Of these patients, 89% had a pre-existing lower grade lymphopenia. Forty percent of patients either recovered or improved to a lesser severity while on treatment. Patients did not have a significant increase in viral infections, and no opportunistic infections were observed. Elevations of serum levels of AST, ALT, alkaline phosphatase, and gamma glutamyl transferase >5 times the upper value of the normal range (i.e., ≥ CTC grade 3) have been rarely reported in patients treated for advanced breast cancer but appear mostly attributable to the underlying presence of liver and/or bone metastases. In the comparative study in advanced breast cancer patients, CTC grade 3 or 4 elevation of gamma glutamyl transferase without documented evidence of liver metastasis was reported in 2.7% of patients treated with exemestane and in 1.8% of patients treated with megestrol acetate.
In patients with early breast cancer, elevations in bilirubin, alkaline phosphatase, and creatinine were more common in those receiving exemestane than either tamoxifen or placebo. Treatment-emergent bilirubin elevations (any CTC grade) occurred in 5% of exemestane patients and 0.8% of tamoxifen patients on the Intergroup Exemestane Study (IES), and in 7% of exemestane treated patients vs. 0% of placebo treated patients in the 027 study. CTC grade 3 to 4 increases in bilirubin occurred in 0.9% of exemestane treated patients compared to 0.1% of tamoxifen treated patients. Alkaline phosphatase elevations of any CTC grade occurred in 15% of exemestane treated patients on the IES compared to 2.6% of tamoxifen treated patients, and in 14% of exemestane treated patients compared to 7% of placebo treated patients in study 027. Creatinine elevations occurred in 6% of exemestane treated patients and 4.3% of tamoxifen treated patients on the IES and in 6% of exemestane treated patients and 0% of placebo treated patients in study 027.
5.5 Use in Premenopausal Women
Exemestane is not indicated for the treatment of breast cancer in premenopausal women.
5.6 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, exemestane can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of exemestane to pregnant rats and rabbits caused increased incidence of abortions and embryo-fetal toxicity. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with exemestane and for 1 month after the last dose [see Use in Specific Populations (8.1), (8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Reductions in Bone Mineral Density (BMD) [see Warnings and Precautions (5.1)]
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adjuvant Therapy
The data described below reflect exposure to exemestane in 2325 postmenopausal women with early breast cancer. Exemestane tolerability in postmenopausal women with early breast cancer was evaluated in two well-controlled trials: the IES study [See Clinical Studies (14.1)] and the 027 study (a randomized, placebo-controlled, double-blind, parallel group study specifically designed to assess the effects of exemestane on bone metabolism, hormones, lipids, and coagulation factors over 2 years of treatment).
The median duration of adjuvant treatment was 27.4 months and 27.3 months for patients receiving exemestane or tamoxifen, respectively, within the IES study and 23.9 months for patients receiving exemestane or placebo within the 027 study. Median duration of observation after randomization for exemestane was 34.5 months and for tamoxifen was 34.6 months. Median duration of observation was 30 months for both groups in the 027 study.
Certain adverse reactions, which were expected based on the known pharmacological properties and side effect profiles of test drugs, were actively sought through a positive checklist. Signs and symptoms were graded for severity using CTC in both studies. Within the IES study, the presence of some illnesses/conditions was monitored through a positive checklist without assessment of severity. These included myocardial infarction, other cardiovascular disorders, gynecological disorders, osteoporosis, osteoporotic fractures, other primary cancer, and hospitalizations.
Within the IES study, discontinuations due to adverse reactions occurred in 6% and 5% of patients receiving exemestane and tamoxifen, respectively, and in 12% and 4.1% of patients receiving exemestane or placebo respectively within study 027.
Deaths due to any cause were reported for 1.3% of the exemestane treated patients and 1.4% of the tamoxifen treated patients within the IES study. There were 6 deaths due to stroke on the exemestane arm compared to 2 on tamoxifen. There were 5 deaths due to cardiac failure on the exemestane arm compared to 2 on tamoxifen.
The incidence of cardiac ischemic events (myocardial infarction, angina, and myocardial ischemia) was 1.6% in exemestane treated patients and 0.6% in tamoxifen treated patients in the IES study. Cardiac failure was observed in 0.4% of exemestane treated patients and 0.3% of tamoxifen treated patients.
In the adjuvant treatment of early breast cancer, the most common adverse reactions occurring in ≥10% of patients in any treatment group (exemestane vs. tamoxifen) were hot flushes (21% vs. 20%), fatigue (16% vs. 15%), arthralgia (15% vs. 9%), headache (13% vs. 11%), insomnia (12% vs. 9%), and increased sweating (12% vs. 10%). Discontinuation rates due to AEs were similar between exemestane and tamoxifen (6% vs. 5%). Incidences of cardiac ischemic events (myocardial infarction, angina, and myocardial ischemia) were exemestane 1.6%, tamoxifen 0.6%. Incidence of cardiac failure: exemestane 0.4%, tamoxifen 0.3%.
Treatment-emergent adverse reactions and illnesses including all causalities and occurring with an incidence of ≥5% in either treatment group of the IES study during or within one month of the end of treatment are shown in Table 2.
Table 2. Incidence (%) of Adverse Reactions of all Grades1 and Illnesses Occurring in (≥5%) of Patients in Any Treatment Group in Study IES in Postmenopausal Women with Early Breast Cancer
|
| % of patients
|
|
| Body system and Adverse Reaction by MedDRA dictionary
| Exemestane
25 mg daily (N=2252) | Tamoxifen
20 mg daily2 (N=2280) |
| Eye
Visual disturbances3 | 5 | 3.8 |
| Gastrointestinal
Nausea3 | 9 | 9 |
| General Disorders
Fatigue3 | 16 | 15 |
| Musculoskeletal
Arthralgia Pain in limb Back pain Osteoarthritis | 15 9 9 6 | 9 6 7 4.5 |
| Nervous System
Headache3 Dizziness3 | 13 10 | 11 8 |
| Psychiatric
Insomnia3 Depression | 12 6 | 9 6 |
| Skin & Subcutaneous Tissue
Increased sweating3 | 12 | 10 |
| Vascular
Hot flushes3 Hypertension | 21 10 | 20 8 |
1 Graded according to Common Toxicity Criteria;
2 75 patients received tamoxifen 30 mg daily;
3 Event actively sought.
In the IES study, as compared to tamoxifen, exemestane was associated with a higher incidence of events in musculoskeletal disorders and in nervous system disorders, including the following events occurring with frequency lower than 5% (osteoporosis [4.6% vs. 2.8%], osteochondrosis [0.3% vs. 0%] and stenosing tenosynovitis (trigger finger) [0.3% vs. 0%], paresthesia [2.6% vs. 0.9%], carpal tunnel syndrome [2.4% vs. 0.2%], and neuropathy [0.6% vs. 0.1%]). Diarrhea was also more frequent in the exemestane group (4.2% vs. 2.2%). Clinical fractures were reported in 94 patients receiving exemestane (4.2%) and 71 patients receiving tamoxifen (3.1%). After a median duration of therapy of about 30 months and a median follow-up of about 52 months, gastric ulcer was observed at a slightly higher frequency in the exemestane group compared to tamoxifen (0.7% vs. <0.1%). The majority of patients on exemestane with gastric ulcer received concomitant treatment with non-steroidal anti-inflammatory agents and/or had a prior history.
Tamoxifen was associated with a higher incidence of muscle cramps [3.1% vs. 1.5%], thromboembolism [2.0% vs. 0.9%], endometrial hyperplasia [1.7% vs. 0.6%], and uterine polyps [2.4% vs. 0.4%].
Common adverse reactions occurring in study 027 are described in Table 3.
Table 3. Incidence of Selected Treatment-Emergent Adverse Reactions of all CTC Grades* Occurring in ≥ 5% of Patients in Either Arm in Study 027
| Adverse Reaction
| Exemestane
N=73 (% incidence) | Placebo
N=73 (% incidence) |
| Hot flushes | 33 | 25 |
| Arthralgia | 29 | 29 |
| Increased sweating | 18 | 21 |
| Alopecia | 15 | 4.1 |
| Hypertension | 15 | 7 |
| Insomnia | 14 | 15 |
| Nausea | 12 | 16 |
| Fatigue | 11 | 19 |
| Abdominal pain | 11 | 14 |
| Depression | 10 | 7 |
| Diarrhea | 10 | 1.4 |
| Dizziness | 10 | 10 |
| Dermatitis | 8 | 1.4 |
| Headache | 7 | 4.1 |
| Myalgia | 6 | 4.1 |
| Edema | 6 | 7 |
* Most events were CTC grade 1 to 2
Treatment of Advanced Breast Cancer
A total of 1058 patients were treated with exemestane 25 mg once daily in the clinical trials program. One death was considered possibly related to treatment with exemestane; an 80-year-old woman with known coronary artery disease had a myocardial infarction with multiple organ failure after 9 weeks on study treatment. In the clinical trials program, 3% of the patients discontinued treatment with exemestane because of adverse reactions, 2.7% of patients discontinued exemestane within the first 10 weeks of treatment.
In the comparative study, adverse reactions were assessed for 358 patients treated with exemestane and 400 patients treated with megestrol acetate. Fewer patients receiving exemestane discontinued treatment because of adverse reactions than those treated with megestrol acetate (2% vs. 5%). Adverse reactions that were considered drug related or of indeterminate cause included hot flashes (13% vs. 5%), nausea (9% vs. 5%), fatigue (8% vs. 10%), increased sweating (4% vs. 8%), and increased appetite (3% vs. 6%) for exemestane and megestrol acetate, respectively. The proportion of patients experiencing an excessive weight gain (>10% of their baseline weight) was significantly higher with megestrol acetate than with exemestane (17% vs. 8%).
In the treatment of advanced breast cancer, the most common adverse reactions included hot flushes (13% vs. 5%), nausea (9% vs. 5%), fatigue (8% vs. 10%), increased sweating (4% vs. 8%), and increased appetite (3% vs. 6%) for exemestane and megestrol acetate, respectively.
Table 4 shows the adverse reactions of all CTC grades, regardless of causality, reported in 5% or greater of patients in the study treated either with exemestane or megestrol acetate.
Table 4. Incidence (%) of Adverse Reactions of all Grades* and Causes Occurring in ≥5% of Advanced Breast Cancer Patients In Each Treatment Arm in the Comparative Study
| Body system and Adverse Reaction by WHO ART dictionary
| Exemestane
25 mg once daily (N=358) | Megestrol Acetate
40 mg QID (N=400) |
| Autonomic Nervous
Increased sweating | 6 | 9 |
| Body as a Whole
Fatigue Hot flashes Pain Influenza-like symptoms Edema (includes edema, peripheral edema, leg edema) | 22 13 13 6 7 | 29 6 13 5 6 |
| Cardiovascular
Hypertension | 5 | 6 |
| Nervous
Depression Insomnia Anxiety Dizziness Headache | 13 11 10 8 8 | 9 9 11 6 7 |
| Gastrointestinal
Nausea Vomiting Abdominal pain Anorexia Constipation Diarrhea Increased appetite | 18 7 6 6 5 4 3 | 12 4 11 5 8 5 6 |
| Respiratory
Dyspnea Coughing | 10 6 | 15 7 |
* Graded according to Common Toxicity Criteria
Adverse reactions of any cause (from 2% to 5%) reported in the comparative study for patients receiving exemestane 25 mg once daily were fever, generalized weakness, paresthesia, pathological fracture, bronchitis, sinusitis, rash, itching, urinary tract infection, and lymphedema.
Additional adverse reactions of any cause observed in the overall clinical trials program (N = 1058) in 5% or greater of patients treated with exemestane 25 mg once daily but not in the comparative study included pain at tumor sites (8%), asthenia (6%), and fever (5%). Adverse reactions of any cause reported in 2% to 5% of all patients treated with exemestane 25 mg in the overall clinical trials program but not in the comparative study included chest pain, hypoesthesia, confusion, dyspepsia, arthralgia, back pain, skeletal pain, infection, upper respiratory tract infection, pharyngitis, rhinitis, and alopecia.
6.2 Post-Marketing Experience
The following adverse reactions have been identified during post approval use of exemestane. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune system disorders- hypersensitivity
Hepatobiliary disorders- hepatitis including cholestatic hepatitis
Nervous system disorders- paresthesia
Musculoskeletal and connective tissue disorder- tendon disorders including tendon rupture, tendonitis, and tenosynovitis
Skin and subcutaneous tissue disorders- acute generalized exanthematous pustulosis, urticaria, pruritus
Related/similar drugs
7. Drug Interactions
Drugs That Induce CYP 3A4
Co-medications that induce CYP 3A4 (e.g., rifampicin, phenytoin, carbamazepine, phenobarbital, or St. John’s wort) may significantly decrease exposure to exemestane. Dose modification is recommended for patients who are also receiving a strong CYP 3A4 inducer [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and its mechanism of action, exemestane can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited human data from case reports are insufficient to inform a drug-associated risk. In animal reproduction studies, administration of exemestane to pregnant rats and rabbits caused increased incidence of abortions, embryo-fetal toxicity, and prolonged gestation with abnormal or difficult labor [see Data]. Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In animal reproduction studies in rats and rabbits, exemestane caused embryo-fetal toxicity, and was abortifacient. Radioactivity related to 14C-exemestane crossed the placenta of rats following oral administration of 1 mg/kg exemestane. The concentration of exemestane and its metabolites was approximately equivalent in maternal and fetal blood. When rats were administered exemestane from 14 days prior to mating until either days 15 or 20 of gestation, and resuming for the 21 days of lactation, an increase in placental weight was seen at 4 mg/kg/day (approximately 1.5 times the recommended human daily dose on a mg/m2 basis). Increased resorptions, reduced number of live fetuses, decreased fetal weight, retarded ossification, prolonged gestation and abnormal or difficult labor was observed at doses equal to or greater than 20 mg/kg/day (approximately 7.5 times the recommended human daily dose on a mg/m2 basis). Daily doses of exemestane, given to rabbits during organogenesis, caused a decrease in placental weight at 90 mg/kg/day (approximately 70 times the recommended human daily dose on a mg/m2 basis) and in the presence of maternal toxicity, abortions, an increase in resorptions, and a reduction in fetal body weight were seen at 270 mg/kg/day. No malformations were noted when exemestane was administered to pregnant rats or rabbits during the organogenesis period at doses up to 810 and 270 mg/kg/day, respectively (approximately 320 and 210 times the recommended human dose on a mg/m2 basis, respectively).
8.2 Lactation
Risk Summary
There is no information on the presence of exemestane in human milk, or on its effects on the breastfed infant or milk production. Exemestane is present in rat milk at concentrations similar to maternal plasma [see Data]. Because of the potential for serious adverse reactions in breast-fed infants from exemestane, advise a woman not to breastfeed during treatment with exemestane and for 1 month after the final dose.
Data
Radioactivity related to exemestane appeared in rat milk within 15 minutes of oral administration of radiolabeled exemestane. Concentrations of exemestane and its metabolites were approximately equivalent in the milk and plasma of rats for 24 hours after a single oral dose of 1 mg/kg 14C-exemestane.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy testing is recommended for females of reproductive potential within seven days prior to initiating exemestane.
Contraception
Females
Exemestane can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with exemestane and for 1 month after the final dose.
Infertility
Based on findings in animals, male and female fertility may be impaired by treatment with exemestane [see Nonclinical Toxicology (13.1)].
8.6 Hepatic Impairment
The AUC of exemestane was increased in subjects with moderate or severe hepatic impairment (Childs-Pugh B or C) [see Clinical Pharmacology (12.3)]. However, based on experience with exemestane at repeated doses up to 200 mg daily that demonstrated a moderate increase in non life-threatening adverse reactions, dosage adjustment does not appear to be necessary.
8.7 Renal Impairment
The AUC of exemestane was increased in subjects with moderate or severe renal impairment (creatinine clearance <35 mL/min/1.73 m2) [see Clinical Pharmacology (12.3)]. However, based on experience with exemestane at repeated doses up to 200 mg daily that demonstrated a moderate increase in non life-threatening adverse reactions, dosage adjustment does not appear to be necessary.
10. Overdosage
Clinical trials have been conducted with exemestane given as a single dose to healthy female volunteers at doses as high as 800 mg and daily for 12 weeks to postmenopausal women with advanced breast cancer at doses as high as 600 mg. These dosages were well tolerated. There is no specific antidote to overdosage and treatment must be symptomatic. General supportive care, including frequent monitoring of vital signs and close observation of the patient, is indicated.
A male child (age unknown) accidentally ingested a 25 mg tablet of exemestane. The initial physical examination was normal, but blood tests performed 1 hour after ingestion indicated leucocytosis (WBC 25000/mm3 with 90% neutrophils). Blood tests were repeated 4 days after the incident and were normal. No treatment was given.
In mice, mortality was observed after a single oral dose of exemestane of 3,200 mg/kg, the lowest dose tested (about 640 times the recommended human dose on a mg/m2 basis). In rats and dogs, mortality was observed after single oral doses of exemestane of 5,000 mg/kg (about 2000 times the recommended human dose on a mg/m2 basis) and of 3,000 mg/kg (about 4000 times the recommended human dose on a mg/m2 basis), respectively.
Convulsions were observed after single doses of exemestane of 400 mg/kg and 3,000 mg/kg in mice and dogs (approximately 80 and 4000 times the recommended human dose on a mg/m2 basis), respectively.
11. Exemestane Tablets Description
Exemestane tablets, USP for oral administration contain 25 mg of exemestane USP, an irreversible, steroidal aromatase inactivator. Exemestane is chemically described as 6-methylenandrosta-1,4-diene-3,17-dione. Its molecular formula is C20H24O2 and its structural formula is as follows:
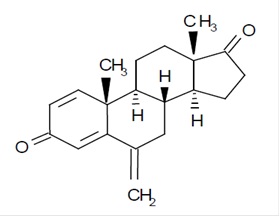
The active ingredient is a white or almost white crystalline powder with a molecular weight of 296.41. Exemestane is freely soluble in N, N-dimethyl formamide, soluble in methanol and practically insoluble in water.
Each exemestane tablet, USP contains the following inactive ingredients: colloidal silicon dioxide, corn starch, crospovidone, hydroxypropyl cellulose, lecithin (soya), magnesium stearate, microcrystalline cellulose, polysorbate 80, polyvinyl alcohol-part. hydrolyzed, povidone, pregelatinized starch (corn), sodium starch glycolate, talc, titanium dioxide and xanthan gum.
12. Exemestane Tablets - Clinical Pharmacology
12.1 Mechanism of Action
Breast cancer cell growth may be estrogen-dependent. Aromatase is the principal enzyme that converts androgens to estrogens both in pre- and postmenopausal women. While the main source of estrogen (primarily estradiol) is the ovary in premenopausal women, the principal source of circulating estrogens in postmenopausal women is from conversion of adrenal and ovarian androgens (androstenedione and testosterone) to estrogens (estrone and estradiol) by the aromatase enzyme in peripheral tissues.
Exemestane is an irreversible, steroidal aromatase inactivator, structurally related to the natural substrate androstenedione. It acts as a false substrate for the aromatase enzyme, and is processed to an intermediate that binds irreversibly to the active site of the enzyme, causing its inactivation, an effect also known as “suicide inhibition.” Exemestane significantly lowers circulating estrogen concentrations in postmenopausal women, but has no detectable effect on adrenal biosynthesis of corticosteroids or aldosterone. Exemestane has no effect on other enzymes involved in the steroidogenic pathway up to a concentration at least 600 times higher than that inhibiting the aromatase enzyme.
12.2 Pharmacodynamics
Effect on Estrogens
Multiple doses of exemestane ranging from 0.5 to 600 mg/day were administered to postmenopausal women with advanced breast cancer. Plasma estrogen (estradiol, estrone, and estrone sulfate) suppression was seen starting at a 5 mg daily dose of exemestane, with a maximum suppression of at least 85% to 95% achieved at a 25 mg dose. Exemestane 25 mg daily reduced whole body aromatization (as measured by injecting radiolabeled androstenedione) by 98% in postmenopausal women with breast cancer. After a single dose of exemestane 25 mg, the maximal suppression of circulating estrogens occurred 2 to 3 days after dosing and persisted for 4 to 5 days.
Effect on Corticosteroids
In multiple-dose trials of doses up to 200 mg daily, exemestane selectivity was assessed by examining its effect on adrenal steroids. Exemestane did not affect cortisol or aldosterone secretion at baseline or in response to ACTH at any dose. Thus, no glucocorticoid or mineralocorticoid replacement therapy is necessary with exemestane treatment.
Other Endocrine Effects
Exemestane does not bind significantly to steroidal receptors, except for a slight affinity for the androgen receptor (0.28% relative to dihydrotestosterone). The binding affinity of its 17-dihydrometabolite for the androgen receptor, however, is 100 times that of the parent compound. Daily doses of exemestane up to 25 mg had no significant effect on circulating levels of androstenedione, dehydroepiandrosterone sulfate, or 17-hydroxyprogesterone, and were associated with small decreases in circulating levels of testosterone. Increases in testosterone and androstenedione levels have been observed at daily doses of 200 mg or more. A dose-dependent decrease in sex hormone binding globulin (SHBG) has been observed with daily exemestane doses of 2.5 mg or higher. Slight, nondose-dependent increases in serum luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels have been observed even at low doses as a consequence of feedback at the pituitary level. Exemestane 25 mg daily had no significant effect on thyroid function [free triiodothyronine (FT3), free thyroxine (FT4), and thyroid stimulating hormone (TSH)].
Coagulation and Lipid Effects
In study 027 of postmenopausal women with early breast cancer treated with exemestane (N=73) or placebo (N=73), there was no change in the coagulation parameters activated partial thromboplastin time [APTT], prothrombin time [PT], and fibrinogen. Plasma HDL cholesterol was decreased 6 to 9% in exemestane treated patients; total cholesterol, LDL cholesterol, triglycerides, apolipoprotein-A1, apolipoprotein-B, and lipoprotein-a were unchanged. An 18% increase in homocysteine levels was also observed in exemestane treated patients compared with a 12% increase seen with placebo.
12.3 Pharmacokinetics
Following oral administration to healthy postmenopausal women, plasma concentrations of exemestane decline polyexponentially with a mean terminal half-life of about 24 hours. The pharmacokinetics of exemestane are dose proportional after single (10 to 200 mg) or repeated oral doses (0.5 to 50 mg). Following repeated daily doses of exemestane 25 mg, plasma concentrations of unchanged drug are similar to levels measured after a single dose. Pharmacokinetic parameters in postmenopausal women with advanced breast cancer following single or repeated doses have been compared with those in healthy, postmenopausal women. After repeated dosing, the average oral clearance in women with advanced breast cancer was 45% lower than the oral clearance in healthy postmenopausal women, with corresponding higher systemic exposure. Mean AUC values following repeated doses in women with breast cancer (75.4 ng∙h/mL) were about twice those in healthy women (41.4 ng∙h/mL).
Absorption
Following oral administration, exemestane appeared to be absorbed more rapidly in women with breast cancer than in the healthy women, with a mean tmax of 1.2 hours in the women with breast cancer and 2.9 hours in healthy women. Approximately 42% of radiolabeled exemestane was absorbed from the gastrointestinal tract. A high-fat breakfast increased AUC and Cmax of exemestane by 59% and 39%, respectively, compared to fasted state.
Distribution
Exemestane is distributed extensively into tissues. Exemestane is 90% bound to plasma proteins and the fraction bound is independent of the total concentration. Albumin and α11-acid glycoprotein both contribute to the binding. The distribution of exemestane and its metabolites into blood cells is negligible.
Metabolism
Exemestane is extensively metabolized, with levels of the unchanged drug in plasma accounting for less than 10% of the total radioactivity. The initial steps in the metabolism of exemestane are oxidation of the methylene group in position 6 and reduction of the 17-keto group with subsequent formation of many secondary metabolites. Each metabolite accounts only for a limited amount of drug-related material. The metabolites are inactive or inhibit aromatase with decreased potency compared with the parent drug. One metabolite may have androgenic activity [see Clinical Pharmacology (12.2)]. Studies using human liver preparations indicate that cytochrome P 450 3A4 (CYP 3A4) is the principal isoenzyme involved in the oxidation of exemestane. Exemestane is metabolized also by aldoketoreductases.
Elimination
Following administration of radiolabeled exemestane to healthy postmenopausal women, the cumulative amounts of radioactivity excreted in urine and feces were similar (42 ± 3% in urine and 42 ± 6% in feces over a 1-week collection period). The amount of drug excreted unchanged in urine was less than 1% of the dose.
Specific Populations
Geriatric: Healthy postmenopausal women aged 43 to 68 years were studied in the pharmacokinetic trials. Age-related alterations in exemestane pharmacokinetics were not seen over this age range.
Gender: The pharmacokinetics of exemestane following administration of a single, 25 mg tablet to fasted healthy males (mean age 32 years) were similar to the pharmacokinetics of exemestane in fasted healthy postmenopausal women (mean age 55 years).
Race: The influence of race on exemestane pharmacokinetics has not been evaluated.
Hepatic Impairment: The pharmacokinetics of exemestane have been investigated in subjects with moderate or severe hepatic impairment (Childs-Pugh B or C). Following a single 25 mg oral dose, the AUC of exemestane was approximately 3 times higher than that observed in healthy volunteers.
Renal Impairment: The AUC of exemestane after a single 25 mg dose was approximately 3 times higher in subjects with moderate or severe renal insufficiency (creatinine clearance <35 mL/min/1.73 m2) compared with the AUC in healthy volunteers.
Pediatric: The pharmacokinetics of exemestane have not been studied in pediatric patients.
Drug Interaction Studies
Exemestane does not inhibit any of the major CYP isoenzymes, including CYP 1A2, 2C9, 2D6, 2E1, and 3A4.
In a pharmacokinetic interaction study of 10 healthy postmenopausal volunteers pretreated with potent CYP 3A4 inducer rifampicin 600 mg daily for 14 days followed by a single dose of exemestane 25 mg, the mean plasma Cmax and AUC 0–∞ of exemestane were decreased by 41% and 54%, respectively [see Dosage and Administration (2.2) and Drug Interactions (7)].
In a clinical pharmacokinetic study, coadministration of ketoconazole, a potent inhibitor of CYP 3A4, has no significant effect on exemestane pharmacokinetics. Although no other formal drug-drug interaction studies with inhibitors have been conducted, significant effects on exemestane clearance by CYP isoenzyme inhibitors appear unlikely.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
A 2-year carcinogenicity study in mice at doses of 50, 150, and 450 mg/kg/day exemestane (gavage), resulted in an increased incidence of hepatocellular adenomas and/or carcinomas in both genders at the high dose level. Plasma AUC(0–24hr) at the high dose were 2,575 ± 386 and 5,667 ± 1,833 ng.hr/mL in males and females (approx. 34 and 75 fold the AUC in postmenopausal patients at the recommended clinical dose). An increased incidence of renal tubular adenomas was observed in male mice at the high dose of 450 mg/kg/day. Since the doses tested in mice did not achieve an MTD, neoplastic findings in organs other than liver and kidneys remain unknown.
A separate carcinogenicity study was conducted in rats at the doses of 30, 100, and 315 mg/kg/day exemestane (gavage) for 92 weeks in males and 2 years in females. No evidence of carcinogenic activity up to the highest dose tested of 315 mg/kg/day was observed in females. The male rat study was inconclusive since it was terminated prematurely at Week 92. At the highest dose, plasma AUC(0–24hr) levels in male (1,418 ± 287 ng.hr/mL) and female (2,318 ± 1,067 ng.hr/mL) rats were 19 and 31 fold higher than those measured in postmenopausal cancer patients receiving the recommended clinical dose.
Exemestane was not mutagenic in vitro in bacteria (Ames test) or mammalian cells (V79 Chinese hamster lung cells). Exemestane was clastogenic in human lymphocytes in vitro without metabolic activation but was not clastogenic in vivo (micronucleus assay in mouse bone marrow). Exemestane did not increase unscheduled DNA synthesis in rat hepatocytes when tested in vitro.
In a pilot reproductive study in rats, male rats were treated with doses of 125 to 1,000 mg/kg/day exemestane, beginning 63 days prior to and during cohabitation. Untreated female rats showed reduced fertility when mated to males treated with ≥500 mg/kg/day exemestane (≥200 times the recommended human dose on a mg/m2 basis). In a separate study, exemestane was given to female rats at 4 to 100 mg/kg/day beginning 14 days prior to mating and through day 15 or 20 of gestation. Exemestane increased the placental weights at ≥4 mg/kg/day (≥1.5 times the human dose on a mg/m2 basis). Exemestane showed no effects on ovarian function, mating behavior, and conception rate in rats given doses up to 20 mg/kg/day (approximately 8 times the recommended human dose on a mg/m2 basis); however, decreases in mean litter size and fetal body weight, along with delayed ossification were evidenced at ≥20 mg/kg/day. In general toxicology studies, changes in the ovary, including hyperplasia, an increase in the incidence of ovarian cysts, and a decrease in corpora lutea were observed with variable frequency in mice, rats, and dogs at doses that ranged from 3 to 20 times the human dose on a mg/m2 basis.
14. Clinical Studies
14.1 Adjuvant Treatment in Early Breast Cancer
The Intergroup Exemestane Study 031 (IES) was a randomized, double-blind, multicenter, multinational study comparing exemestane (25 mg/day) vs. tamoxifen (20 or 30 mg/day) in postmenopausal women with early breast cancer. Patients who remained disease-free after receiving adjuvant tamoxifen therapy for 2 to 3 years were randomized to receive an additional 3 or 2 years of exemestane or tamoxifen to complete a total of 5 years of hormonal therapy.
The primary objective of the study was to determine whether, in terms of disease-free survival, it was more effective to switch to exemestane rather than continuing tamoxifen therapy for the remainder of five years. Disease-free survival was defined as the time from randomization to time of local or distant recurrence of breast cancer, contralateral invasive breast cancer, or death from any cause.
The secondary objectives were to compare the two regimens in terms of overall survival and long-term tolerability. Time to contralateral invasive breast cancer and distant recurrence-free survival were also evaluated.
A total of 4724 patients in the intent-to-treat (ITT) analysis were randomized to exemestane (exemestane tablets) 25 mg once daily (N = 2352) or to continue to receive tamoxifen once daily at the same dose received before randomization (N = 2372). Demographics and baseline tumor characteristics are presented in Table 5. Prior breast cancer therapy is summarized in Table 6.
Table 5. Demographic and Baseline Tumor Characteristics from the IES Study of Postmenopausal Women with Early Breast Cancer (ITT Population)
|
Parameter | Exemestane
(N = 2352) | Tamoxifen
(N = 2372) |
| Age (years):
Median age (range) | 63.0 (38.0 to 96.0) | 63.0 (31.0 to 90.0) |
| Race, n (%): Caucasian Hispanic Asian Black Other/not reported | 2315 (98.4) 13 (0.6) 10 (0.4) 7 (0.3) 7 (0.3) | 2333 (98.4) 13 (0.5) 9 (0.4) 10 (0.4) 7 (0.3) |
| Nodal status, n (%):
Negative Positive 1 to 3 Positive nodes 4 to 9 Positive nodes >9 Positive nodes Not reported Unknown or missing | 1217 (51.7) 1051 (44.7) 721 (30.7) 239 (10.2) 88 (3.7) 3 (0.1) 84 (3.6) | 1228 (51.8) 1044 (44.0) 708 (29.8) 244 (10.3) 86 (3.6) 6 (0.3) 100 (4.2) |
| Histologic type, n (%):
Infiltrating ductal Infiltrating lobular Other Unknown or missing | 1777 (75.6) 341 (14.5) 231 (9.8) 3 (0.1) | 1830 (77.2) 321 (13.5) 213 (9.0) 8 (0.3) |
| Receptor status*, n (%):
ER and PgR Positive ER Positive and PgR Negative/Unknown ER Unknown and PgR Positive**/Unknown ER Negative and PgR Positive ER Negative and PgR Negative/Unknown (none positive) Missing | 1331 (56.6) 677 (28.8) 288 (12.2) 6 (0.3) 48 (2.0) 2 (0.1) | 1319 (55.6) 692 (29.2) 291 (12.3) 7 (0.3) 58 (2.4) 5 (0.2) |
| Tumor Size, n (%):
≤ 0.5 cm > 0.5 to 1.0 cm > 1.0 to 2 cm > 2.0 to 5.0 cm > 5.0 cm Not reported | 58 (2.5) 315 (13.4) 1031 (43.8) 833 (35.4) 62 (2.6) 53 (2.3) | 46 (1.9) 302 (12.7) 1033 (43.5) 883 (37.2) 59 (2.5) 49 (2.1) |
| Tumor Grade, n (%):
G1 G2 G3 G4 Unknown/Not Assessed/Not reported | 397 (16.9) 977 (41.5) 454 (19.3) 23 (1.0) 501 (21.3) | 393 (16.6) 1007 (42.5) 428 (18.0) 19 (0.8) 525 (22.1) |
* Results for receptor status include the results of the post-randomization testing of specimens from subjects for whom receptor status was unknown at randomization.
** Only one subject in the exemestane group had unknown ER status and positive PgR status.
Table 6. Prior Breast Cancer Therapy of Patients in the IES Study of Postmenopausal Women with Early Breast Cancer (ITT Population)
| Parameter
| Exemestane (N = 2352)
| Tamoxifen (N = 2372)
|
| Type of surgery, n (%):
Mastectomy Breast-conserving Unknown or missing | 1232 (52.4) 1116 (47.4) 4 (0.2) | 1242 (52.4) 1123 (47.3) 7 (0.3) |
| Radiotherapy to the breast, n (%):
Yes No Not reported | 1524 (64.8) 824 (35.5) 4 (0.2) | 1523 (64.2) 843 (35.5) 6 (0.3) |
| Prior therapy, n (%):
Chemotherapy Hormone replacement therapy Bisphosphonates | 774 (32.9) 567 (24.1) 43 (1.8) | 769 (32.4) 561 (23.7) 34 (1.4) |
| Duration of tamoxifen therapy at randomization (months):
Median (range) | 28.5 (15.8 to 52.2) | 28.4 (15.6 to 63.0) |
| Tamoxifen dose, n (%):
20 mg 30 mg* Not reported | 2270 (96.5) 78 (3.3) 4 (0.2) | 2287 (96.4) 75 (3.2) 10 (0.4) |
*The 30 mg dose was used only in Denmark, where this dose was the standard of care.
After a median duration of therapy of 27 months and with a median follow-up of 34.5 months, 520 events were reported, 213 in the exemestane group and 307 in the tamoxifen group (Table 7).
Table 7. Primary Endpoint Events (ITT Population)
| Event
| First Events
N (%) |
|
| | Exemestane
(N = 2352) | Tamoxifen
(N = 2372) |
| Loco-regional recurrence | 34 (1.45) | 45 (1.90) |
| Distant recurrence | 126 (5.36) | 183 (7.72) |
| Second primary – contralateral breast cancer | 7 (0.30) | 25 (1.05) |
| Death – breast cancer | 1 (0.04) | 6 (0.25) |
| Death – other reason | 41 (1.74) | 43 (1.81) |
| Death – missing/unknown | 3 (0.13) | 5 (0.21) |
| Ipsilateral breast cancer | 1 (0.04) | 0 |
| Total number of events
| 213 (9.06)
| 307 (12.94)
|
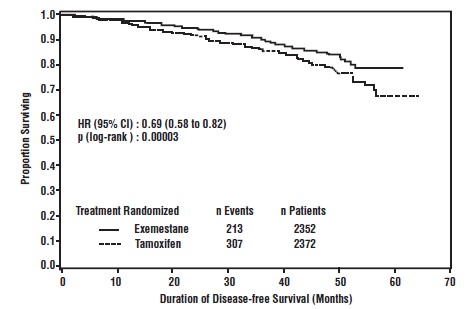
Disease-free survival in the intent-to-treat population was statistically significantly improved [Hazard Ratio (HR) = 0.69, 95% CI: 0.58, 0.82, P = 0.00003, Table 8, Figure 1] in the exemestane arm compared to the tamoxifen arm. In the hormone receptor-positive subpopulation representing about 85% of the trial patients, disease-free survival was also statistically significantly improved (HR = 0.65, 95% CI: 0.53, 0.79, P = 0.00001) in the exemestane arm compared to the tamoxifen arm. Consistent results were observed in the subgroups of patients with node negative or positive disease, and patients who had or had not received prior chemotherapy.
An overall survival update at 119 months median follow-up showed no significant difference between the two groups, with 467 deaths (19.9%) occurring in the exemestane group and 510 deaths (21.5%) in the tamoxifen group.
Table 8. Efficacy Results from the IES Study in Postmenopausal Women with Early Breast Cancer
| ITT Population
| Hazard Ratio
(95% CI) | p-value
(log-rank test) |
| Disease-free survival Time to contralateral breast cancer Distant recurrence-free survival Overall survival | 0.69 (0.58 to 0.82) 0.32 (0.15 to 0.72) 0.74 (0.62 to 0.90) 0.91 (0.81 to 1.04) | 0.00003 0.00340 0.00207 0.16* |
| ER and/or PgR positive
| | |
| Disease-free survival Time to contralateral breast cancer Distant recurrence-free survival Overall survival | 0.65 (0.53 to 0.79) 0.22 (0.08 to 0.57) 0.73 (0.59 to 0.90) 0.89 (0.78 to 1.02) | 0.00001 0.00069 0.00367 0.09065* |
*Not adjusted for multiple testing.
Figure 1. Disease-Free Survival in the IES Study of Postmenopausal Women with Early Breast Cancer (ITT Population)
14.2 Treatment of Advanced Breast Cancer
Exemestane 25 mg administered once daily was evaluated in a randomized double-blind, multicenter, multinational comparative study and in two multicenter single-arm studies of postmenopausal women with advanced breast cancer who had disease progression after treatment with tamoxifen for metastatic disease or as adjuvant therapy. Some patients also have received prior cytotoxic therapy, either as adjuvant treatment or for metastatic disease.
The primary purpose of the three studies was evaluation of objective response rate (complete response [CR] and partial response [PR]). Time to tumor progression and overall survival were also assessed in the comparative trial. Response rates were assessed based on World Health Organization (WHO) criteria, and in the comparative study, were submitted to an external review committee that was blinded to patient treatment. In the comparative study, 769 patients were randomized to receive exemestane tablets 25 mg once daily (N = 366) or megestrol acetate 40 mg four times daily (N = 403). Demographics and baseline characteristics are presented in Table 9.
Table 9. Demographics and Baseline Characteristics from the Comparative Study of Postmenopausal Women with Advanced Breast Cancer Whose Disease Had Progressed after Tamoxifen Therapy
| Parameter
| Exemestane
(N = 366) | Megestrol Acetate
(N = 403) |
| Median Age (range) | 65 (35 to 89) | 65 (30 to 91) |
| ECOG Performance Status 0 1 2 |
167 (46%) 162 (44%) 34 (9%) |
187 (46%) 172 (43%) 42 (10%) |
| Receptor Status ER and/or PgR + ER and PgR unknown Responders to prior tamoxifen NE for response to prior tamoxifen | 246 (67%) 116 (32%) 68 (19%) 46 (13%) | 274 (68%) 128 (32%) 85 (21%) 41 (10%) |
| Site of Metastasis Visceral ± other sites Bone only Soft tissue only Bone & soft tissue | 207 (57%) 61 (17%) 54 (15%) 43 (12%) | 239 (59%) 73 (18%) 51 (13%) 38 (9%) |
| Measurable Disease | 287 (78%) | 314 (78%) |
| Prior Tamoxifen Therapy Adjuvant or Neoadjuvant Advanced Disease, Outcome CR, PR, or SD ≥ 6 months SD < 6 months, PD or NE |
145 (40%) 179 (49%) 42 (12%) |
152 (38%) 210 (52%) 41 (10%) |
| Prior Chemotherapy For advanced disease ± adjuvant Adjuvant only No chemotherapy | 58 (16%) 104 (28%) 203 (56%) | 67 (17%) 108 (27%) 226 (56%) |
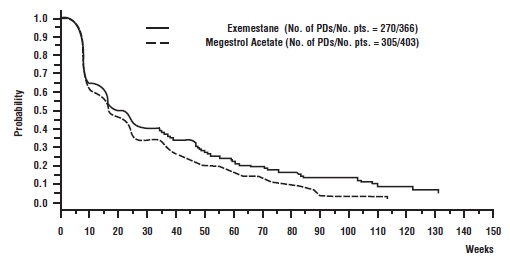
The efficacy results from the comparative study are shown in Table 10. The objective response rates observed in the two treatment arms showed that exemestane was not different from megestrol acetate. Response rates for exemestane from the two single-arm trials were 23.4% and 28.1%.
Table 10. Efficacy Results from the Comparative Study of Postmenopausal Women with Advanced Breast Cancer Whose Disease Had Progressed after Tamoxifen Therapy
| Response Characteristics
| Exemestane
(N=366) | Megestrol Acetate
(N=403) |
| Objective Response Rate = CR + PR (%) Difference in Response Rate (AR-MA) 95% C.I. | 15.0 | 12.4 |
| 2.6 7.5, -2.3 |
||
| CR (%) PR (%) SD ≥ 24 Weeks (%) Median Duration of Response (weeks) Median TTP (weeks) | 2.2 12.8 21.3 76.1 20.3 | 1.2 11.2 21.1 71.0 16.6 |
| Hazard Ratio (AR-MA) | 0.84 |
|
| Abbreviations: CR = complete response, PR = partial response, SD = stable disease (no change), TTP = time to tumor progression, C.I. = confidence interval, MA = megestrol acetate, AR = exemestane |
||
There were too few deaths occurring across treatment groups to draw conclusions on overall survival differences. The Kaplan-Meier curve for time to tumor progression in the comparative study is shown in Figure 2.
Figure 2. Time to Tumor Progression in the Comparative Study of Postmenopausal Women With Advanced Breast Cancer Whose Disease Had Progressed After Tamoxifen Therapy
16. How is Exemestane Tablets supplied
Exemestane tablets, USP are white to off-white, round, biconvex, bevel-edged tablets debossed with “25” on one side and plain on the other side. Each tablet contains 25 mg of exemestane USP.
Exemestane tablets, USP are packaged in HDPE bottles with a child-resistant screw cap, supplied in packs of 30 tablets.
Bottles of 30 NDC 72603-329-01
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Bone Effects
Advise patients that exemestane lowers the level of estrogen in the body. This may lead to reduction in bone mineral density (BMD) over time. The lower the BMD, the greater the risk of osteoporosis and fracture [see Warnings and Precautions (5.1)].
Other Estrogen-Containing Agents
Advise patients that they should not take estrogen-containing agents while they are taking exemestane as these could interfere with its pharmacologic action [see Warnings and Precautions (5.3)].
Use in Premenopausal Women
Advise patients that exemestane is not for use for the treatment of breast cancer in premenopausal women [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that exposure during pregnancy or within 1 month prior to conception can result in fetal harm. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception while taking exemestane and for 1 month after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with exemestane and for 1 month after the last dose [see Use in Specific Populations (8.2)].
Manufactured for:
Northstar Rx LLC
Memphis, TN 38141.
Manufactured by:
Eugia Pharma Specialities Limited
Hyderabad - 500032
India
| Patient Information
Exemestane Tablets, USP (ex-e-MES-tane) |
| What are exemestane tablets? Exemestane tablets are used in women who are past menopause for the treatment of:
o have had other treatments for breast cancer, and o have taken tamoxifen for 2 to 3 years, and o are switching to exemestane tablets to complete 5 years in a row of hormonal therapy.
|
| Do not take exemestane tablets if you are allergic to exemestane or any of the ingredients in exemestane tablets. See the end of this leaflet for a complete list of ingredients in exemestane tablets. |
Before you take exemestane tablets, tell your doctor about all your medical conditions, including if you:
|
How should I take exemestane tablets?
|
| What are the possible side effects of exemestane tablets? Exemestane tablets may cause serious side effects, including:
• hot flashes • feeling tired • joint pain • headache • trouble sleeping • increased sweating The most common side effects of exemestane tablets in women with advanced breast cancer include: • hot flashes • nausea • feeling tired • increased sweating • increased appetite Your doctor will do blood tests to check your vitamin D level before starting treatment with exemestane tablets. Exemestane tablets may cause decreased fertility in males and females. Talk to your doctor if you have concerns about fertility. These are not all the possible side effects of exemestane tablets. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
How should I store exemestane tablets?
|
| General information about the safe and effective use of exemestane tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use exemestane tablets for a condition for which it was not prescribed. Do not give exemestane tablets to other people, even if they have the same symptoms you have. They may harm them. You can ask your pharmacist or doctor for information about exemestane tablets that is written for health professionals. |
| What is in exemestane tablets? Active ingredient: exemestane Inactive ingredients: colloidal silicon dioxide, corn starch, crospovidone, hydroxypropyl cellulose, lecithin (soya), magnesium stearate, microcrystalline cellulose, polysorbate 80, polyvinyl alcohol-part. hydrolyzed, povidone, pregelatinized starch (corn), sodium starch glycolate, talc, titanium dioxide and xanthan gum. Manufactured for: Northstar Rx LLC Memphis, TN 38141. Manufactured by: Eugia Pharma Specialities Limited Hyderabad - 500032 India For more information, please contact Northstar Rx LLC at 1-800-206-7821. |
This Patient Information has been approved by the U.S. Food and Drug Administration
Revised: May 2025

| EXEMESTANE
exemestane tablet, film coated |
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||
| Labeler - NorthStar Rx LLC (830546433) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| EUGIA Pharma Specialities Limited | 872201704 | ANALYSIS(72603-329) , MANUFACTURE(72603-329) , PACK(72603-329) | |
Frequently asked questions
- Which is better - Aromasin or Femara?
- Aromasin vs Femara - how do they compare?
- How long do you need to take Aromasin?
- Should you take Aromasin with food?
More about exemestane
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (120)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: aromatase inhibitors
- Breastfeeding
- En español
