Axert: Package Insert / Prescribing Info
Package insert / product label
Generic name: almotriptan malate
Dosage form: tablet, coated
Drug class: Antimigraine agents
Medically reviewed by Drugs.com. Last updated on May 19, 2025.
The Axert brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
AXERT® (almotriptan malate) Tablets for Oral Use
Initial U.S. Approval: 2001
Indications and Usage for Axert
AXERT® is a 5HT1B/1D receptor agonist (triptan) indicated for:
- Acute treatment of migraine attacks in adults with a history of migraine with or without aura (1.1)
- Acute treatment of migraine headache pain in adolescents age 12 to 17 years with a history of migraine with or without aura, and who have migraine attacks usually lasting 4 hours or more (1.1)
Important limitations:
- Use only after a clear diagnosis of migraine has been established (1.2)
- In adolescents age 12 to 17 years, efficacy of AXERT® on migraine-associated symptoms was not established (1.2)
- Not intended for the prophylactic therapy of migraine (1.2)
- Not indicated for the treatment of cluster headache (1.2)
Axert Dosage and Administration
- Adults and adolescents age 12 to 17 years: 6.25 mg or 12.5 mg single dose; may repeat after 2 hours if headache returns; benefit of second dose in patients who have failed to respond to first dose has not been established; maximum daily dose 25 mg (2.1)
- Patients with hepatic or severe renal impairment: 6.25 mg starting dose; maximum daily dose 12.5 mg (2.2, 2.3)
Dosage Forms and Strengths
Tablets: 6.25 mg and 12.5 mg (3)
Contraindications
- Ischemic heart disease, coronary artery vasospasm, or other significant underlying cardiovascular disease (4.1)
- Cerebrovascular syndromes (e.g., history of stroke or TIA) (4.2)
- Peripheral vascular disease (including ischemic bowel disease) (4.3)
- Uncontrolled hypertension (4.4)
- Do not use AXERT® within 24 hours of an ergotamine-containing, or ergot-type medication, or of another 5-HT1 agonist, e.g., another triptan (4.5, 4.6)
- Hemiplegic or basilar migraine (4.7)
- Known hypersensitivity to AXERT® (4.8)
Warnings and Precautions
- Serious adverse cardiac events, including acute myocardial infarction and life-threatening disturbances of cardiac rhythm (5.1)
- It is strongly recommended that AXERT® not be given to patients in whom unrecognized coronary artery disease (CAD) is predicted by the presence of risk factors. In very rare cases, serious cardiovascular events have been reported in association with AXERT® use in the absence of known cardiovascular disease. If AXERT® is considered, patients should first have a cardiovascular evaluation. If the evaluation is satisfactory, first dose should take place in a physician's office setting (5.1)
- Sensations of pain, tightness, pressure, and heaviness in the chest, throat, neck, and jaw: generally not associated with myocardial ischemia, but patients with signs or symptoms suggestive of angina should be evaluated for the presence of CAD (5.2)
- Cerebrovascular events, some fatal (5.3)
- Gastrointestinal ischemic events and peripheral vasospastic reactions (e.g., Raynaud's syndrome) (5.4)
- Potentially life-threatening serotonin syndrome, particularly in combination with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). Monitor patients for neurologic changes and gastrointestinal symptoms if concomitant treatment is clinically warranted (5.5, 7.3)
- Medication overuse headache: Detoxification may be necessary (5.6)
- Increase in blood pressure, very rarely associated with significant clinical events (4.4, 5.7)
- Use with caution in patients with a known hypersensitivity to sulfonamides (5.8)
Adverse Reactions/Side Effects
The most common adverse reactions (≥1% and greater than placebo) are:
- In adults: nausea, dry mouth and paresthesia (6.1)
- In adolescents: dizziness, somnolence, headache, paresthesia, nausea and vomiting (6.2)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Pharmaceuticals, Inc at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Do not use AXERT® and ergotamine-containing or ergot-type medications within 24 hours of each other (4.5, 7.1)
- Do not use AXERT® and other 5-HT1 agonists (e.g., triptans) within 24 hours of each other (4.6, 7.2)
- SSRI or SNRI: life-threatening serotonin syndrome reported during combined use with triptans (5.5, 7.3)
- Ketoconazole: use single dose of AXERT® 6.25 mg; maximum AXERT® daily dose 12.5 mg (7.4)
Use In Specific Populations
- Pregnancy: based on animal data, may cause fetal harm (8.1)
- Nursing mothers: use AXERT® with caution (8.3)
- Pediatric use: AXERT® has not been studied in children under 12 years (8.4)
- Geriatric use: insufficient safety and efficacy data; use with caution, usually starting with the 6.25 mg dose (8.5)
- Hepatic impairment: use single 6.25 mg tablet as a starting dose; maximum daily dose 12.5 mg (2.2, 8.6)
- Severe renal impairment: use single 6.25 mg tablet as a starting dose; maximum daily dose 12.5 mg (2.3, 8.7)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 5/2017
Full Prescribing Information
1. Indications and Usage for Axert
1.1 Acute Treatment of Migraine Attacks
1.2 Important Limitations
AXERT® should only be used where a clear diagnosis of migraine has been established. If a patient has no response for the first migraine attack treated with AXERT®, the diagnosis of migraine should be reconsidered before AXERT® is administered to treat any subsequent attacks.
In adolescents age 12 to 17 years, efficacy of AXERT® on migraine-associated symptoms (nausea, photophobia, and phonophobia) was not established. AXERT® is not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine [see Contraindications (4.7)].
Safety and effectiveness of AXERT® have not been established for cluster headache which is present in an older, predominantly male population.
2. Axert Dosage and Administration
2.1 Acute Treatment of Migraine Attacks
The recommended dose of AXERT® (almotriptan malate) in adults and adolescents age 12 to 17 years is 6.25 mg to 12.5 mg, with the 12.5 mg dose tending to be a more effective dose in adults. As individuals may vary in their response to different doses of AXERT®, the choice of dose should be made on an individual basis.
If the headache is relieved after the initial AXERT® dose but returns, the dose may be repeated after 2 hours. The effectiveness of a second dose has not been established in placebo-controlled trials. The maximum daily dose should not exceed 25 mg. The safety of treating an average of more than four migraines in a 30-day period has not been established.
2.2 Hepatic Impairment
The recommended starting dose of AXERT® in patients with hepatic impairment is 6.25 mg. The maximum daily dose should not exceed 12.5 mg over a 24-hour period [see Warnings and Precautions (5.9) and Clinical Pharmacology (12.3)].
2.3 Renal Impairment
The recommended starting dose of AXERT® in patients with severe renal impairment is 6.25 mg. The maximum daily dose should not exceed 12.5 mg over a 24-hour period [see Warnings and Precautions (5.9) and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
AXERT® (almotriptan malate) Tablets are available as white, coated, circular, biconvex tablets in the following dosage strengths:
6.25 mg tablet with red code imprint "2080"
12.5 mg tablet with blue stylized imprint "A."
4. Contraindications
4.1 Ischemic or Vasospastic Coronary Artery Disease, or Other Significant Underlying Cardiovascular Disease
Do not use AXERT® (almotriptan malate) in patients with ischemic heart disease (angina pectoris, history of myocardial infarction, or documented silent ischemia), or in patients who have symptoms or findings consistent with ischemic heart disease, coronary artery vasospasm, including Prinzmetal's variant angina, or other significant underlying cardiovascular disease [see Warnings and Precautions (5.1)].
4.2 Cerebrovascular Syndromes
Do not use AXERT® in patients with cerebrovascular syndromes including (but not limited to) stroke of any type as well as transient ischemic attacks [see Warnings and Precautions (5.3)].
4.3 Peripheral Vascular Disease
Do not use AXERT® in patients with peripheral vascular disease including (but not limited to) ischemic bowel disease [see Warnings and Precautions (5.4)].
4.4 Uncontrolled Hypertension
Because AXERT® may increase blood pressure, do not use AXERT® in patients with uncontrolled hypertension [see Warnings and Precautions (5.7)].
4.5 Ergotamine-Containing and Ergot-Type Medications
Do not use AXERT® and ergotamine-containing or ergot-derived medications like dihydroergotamine, ergotamine tartrate, or methysergide within 24 hours of each other [see Drug Interactions (7.1)].
4.6 Concomitant Use With 5-HT1 Agonists (e.g., Triptans)
AXERT® and other 5-HT1 agonists (e.g., triptans) should not be administered within 24 hours of each other [see Warnings and Precautions (5.1) and (5.2)].
5. Warnings and Precautions
5.1 Risk of Myocardial Ischemia and Infarction and Other Adverse Cardiac Events
Cardiac Events and Fatalities with 5-HT1 Agonists
Serious adverse cardiac events, including acute myocardial infarction, have been reported within a few hours following administration of AXERT® (almotriptan malate). Life-threatening disturbances of cardiac rhythm and death have been reported within a few hours following the administration of other triptans. Considering the extent of use of triptans in patients with migraine, the incidence of these events is extremely low.
AXERT® can cause coronary vasospasm; at least one of these events occurred in a patient with no cardiac history and with documented absence of coronary artery disease. Because of the close proximity of the events to use of AXERT®, a causal relationship cannot be excluded. Patients who experience signs or symptoms suggestive of angina following dosing should be evaluated for the presence of coronary artery disease (CAD) or a predisposition to Prinzmetal's variant angina before receiving additional doses of medication, and should be monitored electrocardiographically if dosing is resumed and similar symptoms recur.
Premarketing Experience with AXERT® in Adults
Among the 3865 subjects/patients who received AXERT® in premarketing clinical trials, one patient was hospitalized for observation after a scheduled electrocardiogram (ECG) was found to be abnormal (negative T-waves on the left leads) 48 hours after taking a single 6.25 mg dose of almotriptan. The patient, a 48-year-old female, had previously taken 3 other doses for earlier migraine attacks. Myocardial enzymes at the time of the abnormal ECG were normal. The patient was diagnosed as having had myocardial ischemia and that she had a family history of coronary disease. An ECG performed 2 days later was normal, as was a follow-up coronary angiography. The patient recovered without incident.
Postmarketing Experience with AXERT® in Adults
Serious cardiovascular events have been reported in association with the use of AXERT®. The uncontrolled nature of postmarketing surveillance, however, makes it impossible to definitively determine the proportion of the reported cases that were actually caused by almotriptan or to reliably assess causation in individual cases [see Adverse Reactions (6.3)].
Patients with Documented Coronary Artery Disease
Because of the potential of this class of compound (5-HT1 agonists) to cause coronary vasospasm, AXERT® should not be given to patients with documented ischemic or vasospastic coronary artery disease [see Contraindications (4.1)].
Patients with Risk Factors for CAD
It is strongly recommended that AXERT® not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, or male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of coronary artery and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient's medical history, electrocardiographic or other investigations reveal findings indicative of, or consistent with, coronary artery vasospasm or myocardial ischemia, AXERT® should not be administered [see Contraindications (4.1)].
For patients with risk factors predictive of CAD, who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of AXERT® take place in the setting of a physician's office or similar medically staffed and equipped facility unless the patient has previously received AXERT®. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining on the first occasion of use an ECG during the interval immediately following AXERT®, in these patients with risk factors. It is recommended that patients who are intermittent long-term users of AXERT® and who have or acquire risk factors predictive of CAD, as described above, undergo periodic interval cardiovascular evaluation as they continue to use AXERT®.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease will be inadvertently exposed to AXERT®. The ability of cardiac diagnostic procedures to detect all cardiovascular diseases or predisposition to coronary artery vasospasm is modest at best. Cardiovascular events associated with triptan treatment have occurred in patients with no cardiac history and with documented absence of coronary artery disease.
5.2 Sensations of Pain, Tightness, Pressure in the Chest and/or Throat, Neck, and Jaw
As with other 5-HT1 agonists, sensations of tightness, pain, pressure, and heaviness in the precordium, throat, neck, and jaw have been reported after treatment with AXERT®. Because 5-HT1 agonists may cause coronary vasospasm, patients who experience signs or symptoms suggestive of angina following dosing should be evaluated for the presence of CAD or a predisposition to Prinzmetal's variant angina before receiving additional doses of medication, and should be monitored electrocardiographically if dosing is resumed and similar symptoms occur. Patients shown to have CAD and those with Prinzmetal's variant angina should not receive 5-HT1 agonists [see Contraindications (4.1) and Warnings and Precautions (5.1)].
5.3 Cerebrovascular Events and Fatalities
Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with other triptans and some events have resulted in fatalities. In a number of cases, it appeared possible that the cerebrovascular events were primary, the triptan having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine, when they were not. As with other acute migraine therapies, before treating headaches in patients not previously diagnosed as migraineurs and in migraineurs who present with atypical symptoms, care should be taken to exclude other potentially serious neurological conditions. It should be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g., stroke, hemorrhage, and transient ischemic attack) [see Contraindications (4.2)].
5.4 Other Vasospasm-Related Events, Including Peripheral Vascular Ischemia and Colonic Ischemia
Triptans, including AXERT®, may cause vasospastic reactions other than coronary artery vasospasm, such as peripheral and gastrointestinal vascular ischemia with abdominal pain and bloody diarrhea. Very rare reports of transient and permanent blindness and significant partial vision loss have been reported with the use of triptans. Visual disorders may also be part of a migraine attack. Patients who experience symptoms or signs suggestive of decreased arterial flow following the use of any triptan, such as ischemic bowel syndrome or Raynaud's syndrome, are candidates for further evaluation [see Contraindications (4.3)].
5.5 Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome may occur with triptans, including AXERT®, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). If concomitant treatment with AXERT® and an SSRI (e.g., fluoxetine, paroxetine, sertraline, fluvoxamine, citalopram, escitalopram) or SNRI (e.g., venlafaxine, duloxetine) is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea) [See Drug Interactions (7.3)].
5.6 Medication Overuse Headache
Overuse of acute migraine drugs (e.g., ergotamine, triptans, opioids, or combination of these drugs for 10 or more days per month) may lead to exacerbation of headache (medication overuse headache). Medication overuse headache may present as migraine-like daily headaches or as a marked increase in frequency of migraine attacks. Detoxification of patients, including withdrawal of the overused drugs, and treatment of withdrawal symptoms (which often includes a transient worsening of headache) may be necessary.
5.7 Increases in Blood Pressure
As with other triptans, significant elevations in systemic blood pressure have been reported on rare occasions with AXERT® use in patients with and without a history of hypertension; very rarely these increases in blood pressure have been associated with significant clinical events. AXERT® is contraindicated in patients with uncontrolled hypertension [see Contraindications (4.4)]. In normotensive healthy subjects and patients with hypertension controlled by medication, small, but clinically insignificant, increases in mean systolic (0.21 and 4.87 mm Hg, respectively) and diastolic (1.35 and 0.26 mm Hg, respectively) blood pressure relative to placebo were seen over the first 4 hours after oral administration of 12.5 mg of almotriptan.
An 18% increase in mean pulmonary artery pressure was seen following dosing with another triptan in a study evaluating subjects undergoing cardiac catheterization.
5.8 Hypersensitivity to Sulfonamides
Caution should be exercised when prescribing AXERT® to patients with known hypersensitivity to sulfonamides. The chemical structure of almotriptan contains a sulfonyl group, which is structurally different from a sulfonamide. Cross-sensitivity to almotriptan in patients allergic to sulfonamides has not been systematically evaluated.
5.9 Impaired Hepatic or Renal Function
AXERT® should be administered with caution to patients with diseases that may alter the absorption, metabolism, or excretion of drugs, such as those with impaired hepatic or renal function [see Dosage and Administration (2.2), (2.3) and Clinical Pharmacology (12.3)].
5.10 Binding to Melanin-Containing Tissues
When pigmented rats were given a single oral dose of 5 mg/kg of radiolabeled almotriptan, the elimination half-life of radioactivity from the eye was 22 days. This finding suggests that almotriptan and/or its metabolites may bind to melanin in the eye. Because almotriptan could accumulate in melanin-rich tissues over time, there is the possibility that it could cause toxicity in these tissues with extended use. However, no adverse retinal effects related to treatment with almotriptan were noted in a 52-week toxicity study in dogs given up to 12.5 mg/kg/day (resulting in exposure [AUC] to parent drug approximately 20 times that in humans receiving the maximum recommended human dose of 25 mg/day). Although no systematic monitoring of ophthalmologic function was undertaken in clinical trials, and no specific recommendations for ophthalmologic monitoring are offered, prescribers should be aware of the possibility of long-term ophthalmologic effects.
5.11 Corneal Opacities
Three male dogs (out of a total of 14 treated) in a 52-week toxicity study of oral almotriptan developed slight corneal opacities that were noted after 51 weeks, but not after 25 weeks of treatment. The doses at which this occurred were 2, 5, and 12.5 mg/kg/day. The opacity reversed after a 4-week drug-free period in the affected dog treated with the highest dose. Systemic exposure (plasma AUC) to parent drug at 2 mg/kg/day was approximately 2.5 times the exposure in humans receiving the maximum recommended human daily dose of 25 mg. A no-effect dose was not established.
6. Adverse Reactions/Side Effects
Serious cardiac reactions, including myocardial infarction, have occurred following the use of AXERT® (almotriptan malate) Tablets. These reactions are extremely rare and most have been reported in patients with risk factors predictive of CAD. Reactions reported in association with triptans have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation [see Contraindications (4.1) and Warnings and Precautions (5.1)].
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Risk of Myocardial Ischemia and Infarction and Other Adverse Cardiac Events [see Warnings and Precautions (5.1)]
- Sensations of Pain, Tightness, Pressure in the Chest and/or Throat, Neck, and Jaw [see Warnings and Precautions (5.2)]
- Cerebrovascular Events and Fatalities [see Warnings and Precautions (5.3)]
- Other Vasospasm-Related Events, Including Peripheral Vascular Ischemia and Colonic Ischemia [see Warnings and Precautions (5.4)]
- Serotonin Syndrome [see Warnings and Precautions (5.5)]
- Increases in Blood Pressure [see Warnings and Precautions (5.7)]
Adverse events were assessed in controlled clinical trials that included 1840 adult patients who received one or two doses of AXERT® and 386 adult patients who received placebo. The most common adverse reactions during treatment with AXERT® were nausea, somnolence, headache, paresthesia, and dry mouth. In long-term open-label studies where patients were allowed to treat multiple attacks for up to 1 year, 5% (63 out of 1347 patients) withdrew due to adverse experiences.
Adverse events were assessed in controlled clinical trials that included 362 adolescent patients who received AXERT® and 172 adolescent patients who received placebo. The most common adverse reactions during treatment with AXERT® were dizziness, somnolence, headache, paresthesia, nausea, and vomiting. In a long-term, open-label study where patients were allowed to treat multiple attacks for up to 1 year, 2% (10 out of 420 adolescent patients) withdrew due to adverse events.
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
6.1 Commonly-Observed Adverse Reactions in Double-Blind, Placebo-Controlled AXERT® Clinical Trials
Adults
Table 1 lists the adverse events that occurred in at least 1% of the adult patients treated with AXERT®, and at an incidence greater than in patients treated with placebo, regardless of drug relationship.
| System/Organ Class Adverse Event | AXERT® 6.25 mg (n=527) % | AXERT® 12.5 mg (n=1313) % | Placebo (n=386) % |
|---|---|---|---|
| Digestive Disorders | |||
| Nausea | 1 | 2 | 1 |
| Dry mouth | 1 | 1 | 0.5 |
| Nervous System Disorders | |||
| Paresthesia | 1 | 1 | 0.5 |
The incidence of adverse events in controlled clinical trials was not affected by gender, weight, age, presence of aura, or use of prophylactic medications or oral contraceptives. There were insufficient data to assess the effect of race on the incidence of adverse events.
Adolescents
Table 2 lists the adverse reactions reported by 1% or more of AXERT®-treated adolescents age 12 to 17 years in 1 placebo-controlled, double-blind clinical trial.
| System/Organ Class Adverse Reaction | AXERT® 6.25 mg (n=180) % | AXERT® 12.5 mg (n=182) % | Placebo (n=172) % |
|---|---|---|---|
| Nervous System Disorders | |||
| Dizziness | 4 | 3 | 2 |
| Somnolence | <1 | 5 | 2 |
| Headache | 1 | 2 | 1 |
| Paresthesia | <1 | 1 | <1 |
| Gastrointestinal Disorders | |||
| Nausea | 1 | 3 | 0 |
| Vomiting | 2 | 0 | <1 |
6.2 Other Adverse Reactions Observed in AXERT® Clinical Trials
In the paragraphs that follow, the frequencies of less commonly reported adverse clinical reactions are presented. The reports include adverse reactions in 5 adult controlled studies and 1 adolescent controlled study. Variability associated with adverse reaction reporting, the terminology used to describe adverse reactions, etc., limit the value of the quantitative frequency estimates provided. Reaction frequencies are calculated as the number of patients who used AXERT® and reported a reaction divided by the total number of patients exposed to AXERT® (n=3047, all doses). All reported reactions are included except those already listed in the previous table, those too general to be informative, and those not reasonably associated with the use of the drug. Reactions are further classified within system organ class and enumerated in order of decreasing frequency using the following definitions: frequent adverse reactions are those occurring in 1/100 or more patients, infrequent adverse reactions are those occurring in fewer than 1/100 to 1/1000 patients, and rare adverse reactions are those occurring in fewer than 1/1000 patients.
Body: Frequent: Headache. Infrequent: Abdominal cramp or pain, Asthenia, Chills, Back pain, Chest pain, Neck pain, Fatigue, and Rigid neck. Rare: Fever and Photosensitivity reaction.
Cardiovascular: Infrequent: Vasodilation, Palpitations, and Tachycardia. Rare: Hypertension and Syncope.
Digestive: Infrequent: Diarrhea, Vomiting, Dyspepsia, Gastroenteritis, and Increased thirst. Rare: Colitis, Gastritis, Esophageal reflux, and Increased salivation.
Metabolic: Infrequent: Hyperglycemia and Increased serum creatine phosphokinase. Rare: Increased gamma glutamyl transpeptidase and Hypercholesteremia.
Musculo-Skeletal: Infrequent: Myalgia. Rare: Arthralgia, Arthritis, Myopathy, and Muscle weakness.
Nervous: Frequent: Dizziness and Somnolence. Infrequent: Tremor, Vertigo, Anxiety, Hypoesthesia, Restlessness, CNS stimulation, and Shakiness. Rare: Change in dreams, Impaired concentration, Abnormal coordination, Depressive symptoms, Euphoria, Hyperreflexia, Hypertonia, Nervousness, Neuropathy, Nightmares, Nystagmus, and Insomnia.
Respiratory: Infrequent: Pharyngitis, Rhinitis, Dyspnea, Laryngismus, Sinusitis, and Bronchitis. Rare: Hyperventilation, Laryngitis, Sneezing, and Epistaxis.
Skin: Infrequent: Diaphoresis, Pruritus, and Rash. Rare: Dermatitis and Erythema.
Special Senses: Infrequent: Ear pain and Tinnitus. Rare: Diplopia, Dry eyes, Eye pain, Otitis media, Parosmia, Scotoma, Conjunctivitis, Eye irritation, Hyperacusis, and Taste alteration.
Urogenital: Infrequent: Dysmenorrhea.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of AXERT®. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity reactions (including angioedema, anaphylactic reactions and anaphylactic shock)
Psychiatric Disorders: Confusional state, Restlessness
Nervous System Disorders: Hemiplegia, Hypoesthesia, Seizures
Eye Disorders: Blepharospasm, Visual impairment, Vision blurred
Ear and Labyrinth Disorders: Vertigo
Cardiac Disorders: Acute myocardial infarction, Coronary artery vasospasm, Angina pectoris, Tachycardia
Gastrointestinal Disorders: Abdominal discomfort, Abdominal pain, Abdominal pain upper, Colitis, Hypoesthesia oral, Swollen tongue
Skin and Subcutaneous Tissue Disorders: Cold sweat, Erythema, Hyperhidrosis
Musculoskeletal, Connective Tissue, and Bone Disorders: Arthralgia, Myalgia, Pain in extremity
Reproductive System and Breast Disorders: Breast pain
General Disorders: Malaise, Peripheral coldness.
Related/similar drugs
7. Drug Interactions
7.1 Ergot-Containing Drugs
These drugs have been reported to cause prolonged vasospastic reactions. Because, in theory, vasospastic effects may be additive, ergotamine-containing or ergot-type medications (like dihydroergotamine, ergotamine tartrate, or methysergide) and AXERT® (almotriptan malate) should not be used within 24 hours of each other [see Contraindications (4.5)].
7.2 5-HT1 Agonists (e.g., Triptans)
Concomitant use of other 5-HT1 agonists (e.g., triptans) within 24 hours of treatment with AXERT® is contraindicated [see Contraindications (4.6)].
7.3 Selective Serotonin Reuptake Inhibitors/Serotonin Norepinephrine Reuptake Inhibitors
Cases of life-threatening serotonin syndrome have been reported during combined use of triptans and selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs) [see Warnings and Precautions (5.5), Clinical Pharmacology (12.3)].
7.4 Ketoconazole and Other Potent CYP3A4 Inhibitors
Co-administration of almotriptan and oral ketoconazole, a potent CYP3A4 inhibitor, resulted in an approximately 60% increase in exposure of almotriptan. Increased exposures to almotriptan may be expected when almotriptan is used concomitantly with other potent CYP3A4 inhibitors [see Clinical Pharmacology (12.3)].
In patients concomitantly using potent CYP3A4 inhibitors, the recommended starting dose of AXERT® is 6.25 mg. The maximum daily dose should not exceed 12.5 mg within a 24-hour period. Concomitant use of AXERT® and potent CYP3A4 inhibitors should be avoided in patients with renal or hepatic impairment [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Category C
In animal studies, almotriptan produced developmental toxicity (increased embryolethality and fetal skeletal variations, and decreased offspring body weight) at doses greater than those used clinically. There are no adequate and well-controlled studies in pregnant women; therefore, AXERT® (almotriptan malate) should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
When almotriptan (125, 250, 500, or 1000 mg/kg/day) was administered orally to pregnant rats throughout the period of organogenesis, increased incidences of fetal skeletal variations (decreased ossification) were noted at a dose of 250 mg/kg/day or greater and an increase in embryolethality was seen at the highest dose. The no-effect dose for embryo-fetal developmental toxicity in rats (125 mg/kg/day) is approximately 100 times the maximum recommended human dose (MRHD) of 25 mg/day on a body surface area (mg/m2) basis. Similar studies in pregnant rabbits conducted with almotriptan (oral doses of 5, 20, or 60 mg/kg/day) demonstrated increases in embryolethality at the highest dose. The no-effect dose for embryo-fetal developmental toxicity in rabbits (20 mg/kg/day) is approximately 15 times the MRHD on a mg/m2 basis. When almotriptan (25, 100, or 400 mg/kg/day) was administered orally to rats throughout the periods of gestation and lactation, gestation length was increased and litter size and offspring body weight were decreased at the highest dose. The decrease in pup weight persisted throughout lactation. The no-effect dose in this study (100 mg/kg/day) is 40 times the MRHD on a mg/m2 basis.
8.3 Nursing Mothers
It is not known whether almotriptan is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when AXERT® is administered to a nursing woman. Levels of almotriptan in rat milk were up to 7 times higher than in rat plasma.
8.4 Pediatric Use
Safety and efficacy of AXERT® in pediatric patients under the age of 12 years have not been established. The pharmacokinetics, efficacy, and safety of AXERT® have been evaluated in adolescent patients, age 12 to 17 years [see Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
In a clinical study, AXERT® 6.25 mg and 12.5 mg were found to be effective for the relief of migraine headache pain in adolescent patients age 12 to 17 years. Efficacy on migraine-associated symptoms (nausea, photophobia, and phonophobia) was not established. The most common adverse reactions (incidence of ≥1%) associated with AXERT® treatment were dizziness, somnolence, headache, paresthesia, nausea, and vomiting [see Adverse Reactions (6.1)]. The safety and tolerability profile of AXERT® treatment in adolescents is similar to the profile observed in adults.
Postmarketing experience with other triptans include a limited number of reports that describe pediatric patients who have experienced clinically serious adverse events that are similar in nature to those reported rarely in adults.
8.5 Geriatric Use
Clinical studies of AXERT® did not include sufficient numbers of subjects age 65 and over to determine whether they respond differently from younger subjects. Clearance of almotriptan was lower in elderly volunteers than in younger individuals, but there were no observed differences in the safety and tolerability between the two populations [see Clinical Pharmacology (12.3)]. In general, dose selection for an elderly patient should be cautious, usually starting at the low dose, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy. The recommended dose of AXERT® for elderly patients with normal renal function for their age is the same as that recommended for younger adults.
8.6 Hepatic Impairment
The recommended starting dose of AXERT® in patients with hepatic impairment is 6.25 mg. The maximum daily dose should not exceed 12.5 mg over a 24-hour period [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
The recommended starting dose of AXERT® in patients with severe renal impairment is 6.25 mg. The maximum daily dose should not exceed 12.5 mg over a 24-hour period [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
10. Overdosage
10.1 Signs and Symptoms
Patients and volunteers receiving single oral doses of 100 to 150 mg of almotriptan did not experience significant adverse events. Six additional normal volunteers received single oral doses of 200 mg without serious adverse events. During clinical trials with AXERT® (almotriptan malate), one patient ingested 62.5 mg in a 5-hour period and another patient ingested 100 mg in a 38-hour period. Neither patient experienced adverse reactions.
Based on the pharmacology of triptans, hypertension or other more serious cardiovascular symptoms could occur after overdosage.
10.2 Recommended Treatment
There is no specific antidote to AXERT®. In cases of severe intoxication, intensive care procedures are recommended, including establishing and maintaining a patent airway, ensuring adequate oxygenation and ventilation, and monitoring and support of the cardiovascular system.
Clinical and electrocardiographic monitoring should be continued for at least 20 hours even if clinical symptoms are not observed.
It is unknown what effect hemodialysis or peritoneal dialysis has on plasma concentrations of almotriptan.
11. Axert Description
AXERT® (almotriptan malate) Tablets contain almotriptan malate, a selective 5-hydroxytryptamine1B/1D (5-HT1B/1D) receptor agonist. Almotriptan malate is chemically designated as 1-[[[3-[2-(Dimethylamino)ethyl]-1H-indol-5-yl]methyl]sulfonyl]pyrrolidine (±)-hydroxybutanedioate (1:1) and its structural formula is:
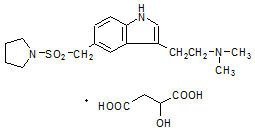
Its empirical formula is C17H25N3O2S-C4H6O5, representing a molecular weight of 469.56. Almotriptan is a white to slightly yellow crystalline powder that is soluble in water. AXERT® for oral administration contains almotriptan malate equivalent to 6.25 or 12.5 mg of almotriptan. Each compressed tablet contains the following inactive ingredients: carnauba wax, cellulose, FD&C Blue No. 2 (12.5 mg only), hypromellose, iron oxide (6.25 mg only), mannitol, polyethylene glycol, povidone, propylene glycol, sodium starch glycolate, sodium stearyl fumarate and titanium dioxide.
12. Axert - Clinical Pharmacology
12.1 Mechanism of Action
Almotriptan binds with high affinity to 5-HT1D, 5-HT1B, and 5-HT1F receptors. Almotriptan has weak affinity for 5-HT1A and 5-HT7 receptors, but has no significant affinity or pharmacological activity at 5-HT2, 5-HT3, 5-HT4, 5-HT6; alpha or beta adrenergic; adenosine (A1, A2); angiotensin (AT1, AT2); dopamine (D1, D2); endothelin (ETA, ETB); or tachykinin (NK1, NK2, NK3) binding sites.
12.2 Pharmacodynamics
Current theories on the etiology of migraine headache suggest that symptoms are due to local cranial vasodilatation and/or to the release of vasoactive and pro-inflammatory peptides from sensory nerve endings in an activated trigeminal system. The therapeutic activity of almotriptan in migraine can most likely be attributed to agonist effects at 5-HT1B/1D receptors on the extracerebral, intracranial blood vessels that become dilated during a migraine attack and on nerve terminals in the trigeminal system. Activation of these receptors results in cranial vessel constriction, inhibition of neuropeptide release, and reduced transmission in trigeminal pain pathways.
12.3 Pharmacokinetics
Absorption
The absolute bioavailability of almotriptan is about 70%, with peak plasma levels occurring 1 to 3 hours after administration; food does not affect pharmacokinetics.
Distribution
Almotriptan is minimally protein bound (approximately 35%) and the mean apparent volume of distribution is approximately 180 to 200 liters.
Metabolism
Almotriptan is metabolized by two major and one minor pathways. Monoamine oxidase (MAO)-mediated oxidative deamination (approximately 27% of the dose), and cytochrome P450-mediated oxidation (approximately 12% of the dose) are the major routes of metabolism, while flavin monooxygenase is the minor route. MAO-A is responsible for the formation of the indoleacetic acid metabolite, whereas cytochrome P450 (3A4 and 2D6) catalyzes the hydroxylation of the pyrrolidine ring to an intermediate that is further oxidized by aldehyde dehydrogenase to the gamma-aminobutyric acid derivative. Both metabolites are inactive.
Excretion
Almotriptan has a mean half-life of 3 to 4 hours. Almotriptan is eliminated primarily by renal excretion (about 75% of the oral dose), with approximately 40% of an administered dose excreted unchanged in urine. Renal clearance exceeds the glomerular filtration rate by approximately 3-fold, indicating an active mechanism. Approximately 13% of the administered dose is excreted via feces, both unchanged and metabolized.
Drug-Drug Interactions
All drug interaction studies were performed in healthy volunteers using a single 12.5 mg dose of almotriptan and multiple doses of the other drug.
Monoamine Oxidase Inhibitors
Co-administration of almotriptan and moclobemide (150 mg twice daily for 8 days) resulted in a 27% decrease in almotriptan clearance and an increase in Cmax of approximately 6%. No dose adjustment is necessary.
Propranolol
Co-administration of almotriptan and propranolol (80 mg twice daily for 7 days) resulted in no significant changes in the pharmacokinetics of almotriptan.
Fluoxetine
Co-administration of almotriptan and fluoxetine (60 mg daily for 8 days), a potent inhibitor of CYP2D6, had no effect on almotriptan clearance, but maximal concentrations of almotriptan were increased 18%. This difference is not clinically significant.
Verapamil
Co-administration of almotriptan and verapamil (120 mg sustained-release tablets twice daily for 7 days), an inhibitor of CYP3A4, resulted in a 20% increase in the area under the plasma concentration-time curve, and in a 24% increase in maximal plasma concentrations of almotriptan. Neither of these changes is clinically significant. No dose adjustment is necessary.
Ketoconazole and other Potent CYP3A4 Inhibitors
Co-administration of almotriptan and ketoconazole, a potent CYP3A4 inhibitor, resulted in an approximately 60% increase in exposure of almotriptan. Increased exposures to almotriptan may be expected when almotriptan is used with other potent CYP3A4 inhibitors.
Special Populations
Geriatric
Renal and total clearance, and amount of drug excreted in the urine, were lower in elderly healthy volunteers (age 65 to 76 years) than in younger healthy volunteers (age 19 to 34 years), resulting in longer terminal half-life (3.7 hours vs. 3.2 hours) and a 25% higher area under the plasma concentration-time curve in the elderly subjects. The differences, however, do not appear to be clinically significant.
Pediatric
A pharmacokinetics study of almotriptan was conducted in adolescents (12 to 17 years) and adults (18 to 55 years) with or without a history of migraine. No differences were observed in the rate or extent of absorption of almotriptan in adolescents compared with adults.
Race
No significant differences were observed in pharmacokinetic parameters between Caucasian and African-American volunteers.
Hepatic Impairment
The pharmacokinetics of almotriptan have not been assessed in patients with hepatic impairment. Based on the known mechanisms of clearance of almotriptan, the maximum decrease expected in almotriptan clearance due to hepatic impairment would be 60% [see Dosage and Administration (2.2)].
Renal Impairment
The clearance of almotriptan was approximately 65% lower in patients with severe renal impairment (Cl/F=19.8 L/hour; creatinine clearance between 10 and 30 mL/min) and approximately 40% lower in patients with moderate renal impairment (Cl/F=34.2 L/hour; creatinine clearance between 31 and 71 mL/min) than in healthy volunteers (Cl/F=57 L/hour). Maximal plasma concentrations of almotriptan increased by approximately 80% in these patients [see Dosage and Administration (2.3)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Almotriptan was administered to mice and rats for up to 103–104 weeks at oral doses up to 250 mg/kg/day and 75 mg/kg/day, respectively. These doses were associated with plasma exposures (AUC) to parent drug that were approximately 40 and 80 times, in mice and rats respectively, the plasma AUC in humans at the maximum recommended human dose (MRHD) of 25 mg/day. Because of high mortality rates in both studies, which reached statistical significance in high-dose female mice, all female rats, all male mice, and high-dose female mice were terminated between weeks 96 and 98. There was no increase in tumors related to almotriptan administration.
Mutagenesis
Almotriptan was not mutagenic in two in vitro gene mutation assays, the Ames test, and the mouse lymphoma tk assay. Almotriptan was not clastogenic in an in vivo mouse micronucleus assay.
Impairment of Fertility
When male and female rats received almotriptan (25, 100, or 400 mg/kg/day) orally prior to and during mating and gestation, prolongation of the estrous cycle was observed at the mid-dose and greater, and fertility was impaired at the highest dose. Subsequent mating of treated with untreated animals indicated that the decrease in fertility was due to an effect on females. The no-effect dose for reproductive toxicity in rats (25 mg/kg/day) is approximately 10 times the MRHD on a mg/m2 basis.
14. Clinical Studies
14.1 Adults
The efficacy of AXERT® (almotriptan malate) was established in three multi-center, randomized, double-blind, placebo-controlled European trials. Patients enrolled in these studies were primarily female (86%) and Caucasian (more than 98%), with a mean age of 41 years (range of 18 to 72). Patients were instructed to treat a moderate to severe migraine headache. Two hours after taking one dose of study medication, patients evaluated their headache pain. If the pain had not decreased in severity to mild or no pain, the patient was allowed to take an escape medication. If the pain had decreased to mild or no pain at 2 hours but subsequently increased in severity between 2 and 24 hours, it was considered a relapse and the patient was instructed to take a second dose of study medication. Associated symptoms of nausea, vomiting, photophobia, and phonophobia were also evaluated.
In these studies, the percentage of patients achieving a response (mild or no pain) 2 hours after treatment was significantly greater in patients who received either AXERT® 6.25 mg or 12.5 mg, compared with those who received placebo. A higher percentage of patients reported pain relief after treatment with the 12.5 mg dose than with the 6.25 mg dose. Doses greater than 12.5 mg did not lead to a significantly better response. These results are summarized in Table 3.
| Placebo | AXERT®
6.25 mg | AXERT®
12.5 mg |
|
|---|---|---|---|
| Study 1 | 33.8% (n = 80) | 55.4%*
(n = 166) | 58.5%†
(n = 164) |
| Study 2 | 40.0% (n = 95) | --- | 57.1%‡
(n =175) |
| Study 3 | 33.0% (n = 176) | 55.6%†
(n = 360) | 64.9%†
(n = 370) |
The estimated probability of achieving pain relief within 2 hours following initial treatment with AXERT® in adults is shown in Figure 1.
Figure 1. Estimated Probability of Achieving an Initial Headache Response (Mild or no Pain) in 2 Hours in Adults
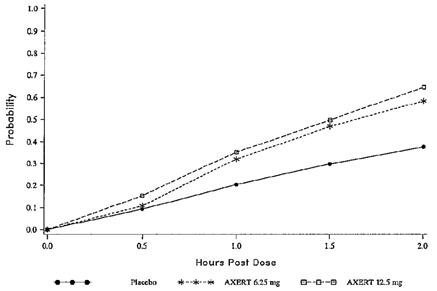
This Kaplan-Meier plot is based on data obtained in the three placebo-controlled clinical trials that provided evidence of efficacy (Studies 1, 2, and 3). Patients not achieving pain relief by 2 hours were censored at 2 hours.
For patients with migraine-associated photophobia, phonophobia, nausea, and vomiting at baseline, there was a decreased incidence of these symptoms following administration of AXERT® compared with placebo.
Two to 24 hours following the initial dose of study medication, patients were allowed to take an escape medication or a second dose of study medication for pain response. The estimated probability of patients taking escape medication or a second dose of study medication over the 24 hours following the initial dose of study medication is shown in Figure 2.
Figure 2. Estimated Probability of Adult Patients Taking Escape Medication or a Second Dose of Study Medication Over the 24 Hours Following the Initial Dose of Study Treatment
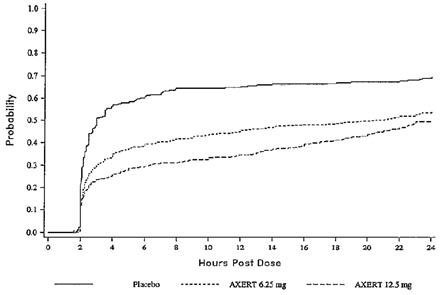
This Kaplan-Meier plot is based on data obtained in the three placebo-controlled trials that provided evidence of efficacy (Studies 1, 2, and 3). Patients not using additional treatment were censored at 24 hours. Remedication was not allowed within 2 hours after the initial dose of AXERT®.
The efficacy of AXERT® was unaffected by the presence of aura; by gender, weight, or age of the patient; or by concomitant use of common migraine prophylactic drugs (e.g., beta-blockers, calcium channel blockers, and tricyclic antidepressants); or oral contraceptives. There were insufficient data to assess the effect of race on efficacy.
14.2 Adolescents Age 12 to 17 Years
The efficacy of AXERT® in adolescent patients age 12 to 17 years was evaluated in a double-blind, randomized, placebo-controlled study. Patients enrolled in that study had at least a 1-year history of migraine attacks with or without aura usually lasting 4 hours or more (when untreated). Patients enrolled in the study were primarily females (60%) and Caucasian (75%), while 15% of patients were black, and 10% were of other races. Patients were instructed to treat a moderate to severe migraine headache. Two hours after taking one dose of study medication, patients evaluated their headache pain. Associated symptoms of nausea, photophobia, and phonophobia were also evaluated.
In this study, the percentage of patients achieving a pain relief response (mild or no pain) 2 hours after treatment was statistically significantly greater in patients who received AXERT® 6.25 mg or 12.5 mg compared with those who received placebo. There was no additional benefit on pain relief provided by the 12.5 mg dose. The 2-hour pain relief results are summarized in Table 4.
| Placebo | AXERT® 6.25 mg | AXERT® 12.5 mg | |
|---|---|---|---|
| Study 1 | 55.3% (n/N = 94/170) | 71.8%*
(n/N = 127/177) | 72.9%†
(n/N = 132/181) |
The estimated probability of achieving pain relief within 2 hours following initial treatment with AXERT® in adolescents age 12 to 17 years is shown in Figure 3.
Figure 3. Estimated Probability of Achieving an Initial Headache Response (Mild or no Pain) in 2 Hours in the Adolescent Study
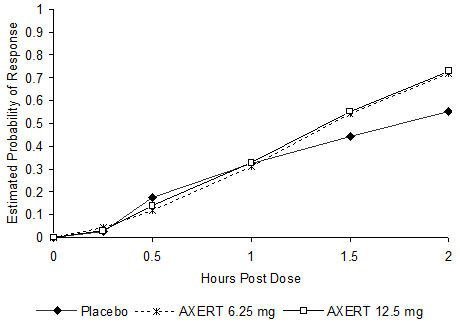
The prevalence of the migraine-associated symptoms (nausea, photophobia, and phonophobia) at 2 hours after taking the dose was not significantly different between patients who received AXERT® 6.25 mg or 12.5 mg and those who received placebo.
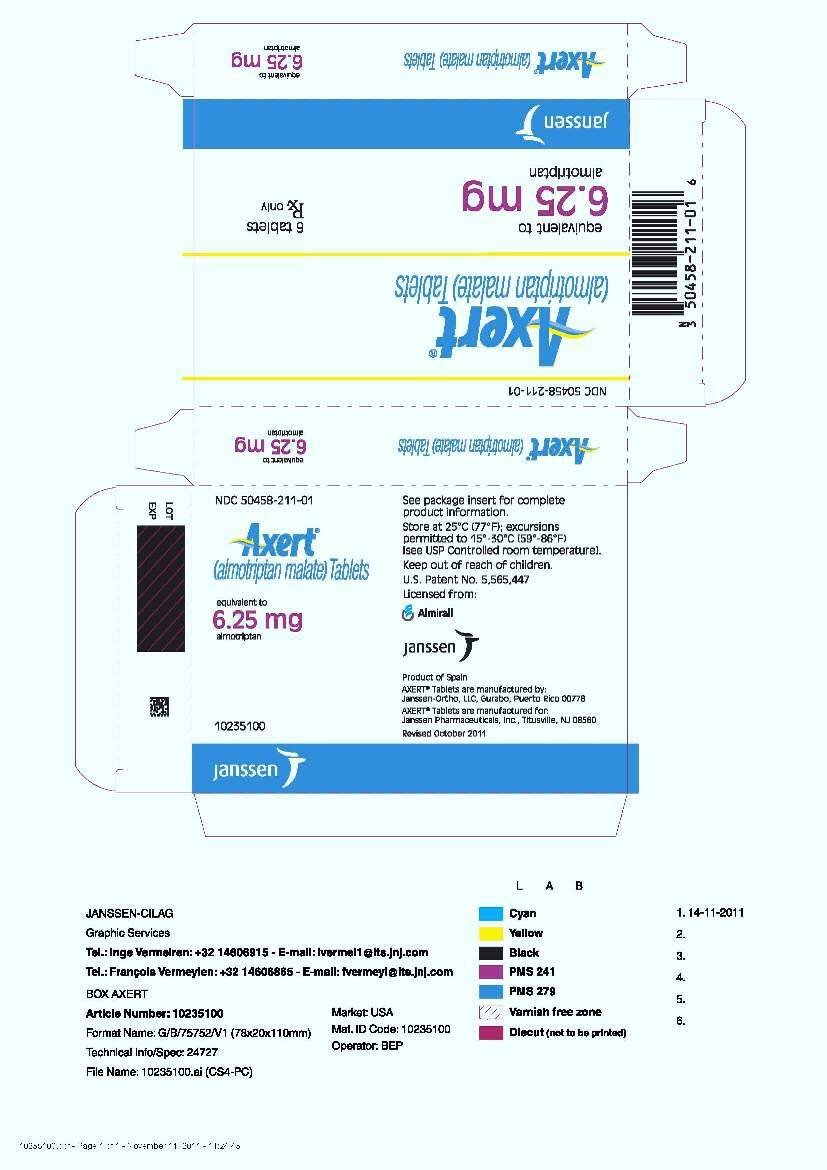
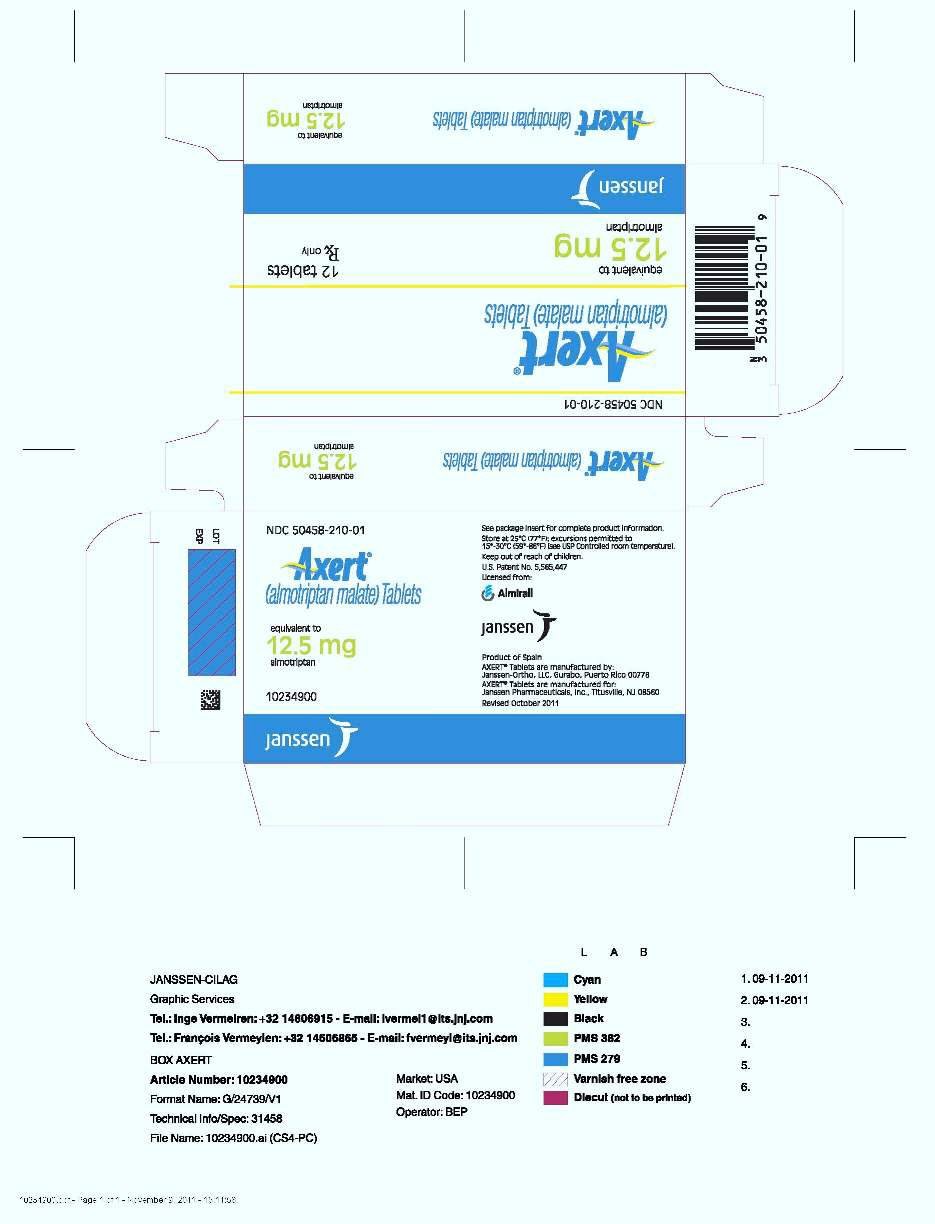
16. How is Axert supplied
AXERT® (almotriptan malate) Tablets are available as follows:
6.25 mg: White, coated, circular, biconvex tablets with red code imprint "2080."
| Unit Dose (aluminum blister pack) | |
| 6 tablets | NDC 50458-211-01 |
12.5 mg: White, coated, circular, biconvex tablets with blue stylized imprint "A."
| Unit Dose (aluminum blister pack) | |
| 12 tablets | NDC 50458-210-01 |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Drug Interactions
Advise patients to talk with their physician or pharmacist before taking any new medicines, including prescription and non-prescription drugs and supplements [see Contraindications (4.5) and (4.6) and Drug Interactions (7)].
Hypersensitivity
Inform patients to tell their physician if they develop a rash, itching, or breathing difficulties after taking AXERT® [see Warnings and Precautions (5.8)].
Risk of Myocardial Ischemia and/or Infarction, Other Adverse Cardiac Events, Other Vasospasm-Related Events, and Cerebrovascular Events
Inform patients that AXERT® (almotriptan malate) may cause serious cardiovascular side effects such as myocardial infarction or stroke, which may result in hospitalization and even death. Although serious cardiovascular events can occur without warning symptoms, patients should be alert for the signs and symptoms of chest pain, shortness of breath, weakness, or slurring of speech, and should ask for medical advice when observing any indicative signs or symptoms. Apprise the patient of the importance of this follow-up [see Warnings and Precautions (5.1), (5.2), (5.3), and (5.4)].
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome with the use of AXERT® or other triptans, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs) [see Warnings and Precautions (5.5)].
Medication Overuse Headache
Inform patients that use of acute migraine drugs for 10 or more days per month may lead to an exacerbation of headache and encourage patients to record headache frequency and drug use (e.g., by keeping a headache diary) [see Warnings and Precautions (5.6)].
Pregnancy
Advise patients to notify their physician if they become pregnant during treatment or intend to become pregnant [see Use in Specific Populations (8.1)].
Nursing Mothers
Advise patients to notify their physician if they are breastfeeding or plan to breastfeed [see Use in Specific Populations (8.3)].
Ability to Operate Machinery or Vehicles
Counsel patients that AXERT® may cause dizziness, somnolence, visual disturbances, and other CNS symptoms that can interfere with driving or operating machinery. Accordingly, advise the patient not to drive, operate complex machinery, or engage in other hazardous activities until they have gained sufficient experience with AXERT® to gauge whether it affects their mental or visual performance adversely.
Product of Spain
AXERT® Tablets are manufactured by:
Janssen-Ortho, LLC
Gurabo, Puerto Rico 00778
AXERT® Tablets are manufactured for:
Janssen Pharmaceuticals, Inc.
Titusville, NJ 08560
Licensed from: Almirall, S.A.
U.S. Patent No. 5,565,447
© Janssen Pharmaceuticals, Inc. 2009
7560705
| This Patient Information has been approved by the U.S. Food and Drug Administration | Revised : May 2017 |
| PATIENT INFORMATION AXERT® (AX-ERT) (almotriptan malate) tablets |
|
|
What is the most important information I should know about AXERT? AXERT can cause serious side effects, including: Heart attack and other heart problems. Heart problems may lead to death. Stop taking AXERT and get emergency medical help right away if you have any of the following symptoms of a heart attack:
AXERT is not for people with risk factors for heart disease unless a heart exam is done and shows no problem. You have a higher risk for heart disease if you:
|
|
|
What is AXERT?
|
|
|
Do not take AXERT if you:
Ask your doctor if you are not sure if your medicine is listed above. |
|
|
Before you take AXERT, tell your doctor about all of your medical conditions, including if you:
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your doctor if you take medicines called:
Also tell your doctor if you take certain other types of medicines used to treat fungal infection (such as ketoconazole or itraconazole) or to treat HIV/AIDS (such as ritonavir). Ask your doctor or pharmacist for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine. |
|
|
How should I take AXERT?
|
|
|
What should I avoid while taking AXERT? AXERT may cause dizziness, sleepiness, and problems seeing. Do not drive, operate machinery, or do other dangerous activities until you know how AXERT affects you. |
|
|
What are the possible side effects of AXERT? AXERT may cause serious side effects, including:
The most common side effects of AXERT in adults are:
The most common side effects of AXERT in adolescents are:
These are not all the possible side effects of AXERT. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
How should I store AXERT?
Keep AXERT and all medicines out of the reach of children. |
|
|
General information about the safe and effective use of AXERT. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use AXERT for a condition for which it was not prescribed. Do not give AXERT to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about AXERT that is written for health professionals. |
|
|
What are the ingredients in AXERT? Active ingredient: almotriptan malate Inactive ingredients: carnauba wax, cellulose, FD&C Blue No. 2 (12.5 mg only), hypromellose, iron oxide (6.25 mg only), mannitol, polyethylene glycol, povidone, propylene glycol, sodium starch glycolate, sodium stearyl fumarate and titanium dioxide. Manufactured by: Janssen-Ortho, LLC Gurabo, Puerto Rico 00778 For more information, go to www.AXERT.com or call 1-800-526-7736. |
|
| AXERT
almotriptan malate tablet, coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| AXERT
almotriptan malate tablet, coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Janssen Pharmaceuticals, Inc. (063137772) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Janssen-Ortho LLC | 805887986 | ANALYSIS(50458-211, 50458-210) , MANUFACTURE(50458-211, 50458-210) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Ranke Quimica, S.L. | 563076157 | API MANUFACTURE(50458-211, 50458-210) | |
More about Axert (almotriptan)
- Check interactions
- Compare alternatives
- Reviews (29)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: antimigraine agents
- Breastfeeding
