Ampyra: Package Insert / Prescribing Info
Package insert / product label
Generic name: dalfampridine
Dosage form: tablet, film coated, extended release
Drug class: Miscellaneous central nervous system agents
Medically reviewed by Drugs.com. Last updated on May 26, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
AMPYRA ® (dalfampridine) extended-release tablets, for oral use
Initial U.S. Approval: 2010
Indications and Usage for Ampyra
Ampyra Dosage and Administration
- The maximum recommended dosage is 10 mg twice daily (approximately 12 hours apart). There is no evidence of additional benefit with doses greater than 10 mg twice daily. Adverse reactions, including seizures, were more frequent at higher doses. (2.1)
- Take with or without food. Administer tablets whole; do not divide, crush, chew, or dissolve (2.2)
- Patients should not take double or extra doses if they miss a dose. (2.2)
- Estimated creatinine clearance (CrCl) should be known before initiating treatment with AMPYRA. In patients with mild renal impairment (CrCl 51–80 mL/min), AMPYRA may reach plasma levels associated with a greater risk of seizures, and the potential benefits of AMPYRA should be carefully considered against the risk of seizures in these patients (2.3, 5.2, 8.6)
Dosage Forms and Strengths
10 mg tablets (3)
Contraindications
Warnings and Precautions
- AMPYRA can cause seizures; the risk of seizures increases with increasing AMPYRA doses; discontinue AMPYRA and do not restart if a seizure occurs (5.1)
- Avoid concomitant use with other forms of 4-aminopyridine (4-AP, fampridine), since the active ingredient is the same (5.3)
- AMPYRA can cause anaphylaxis. Discontinue and do not restart AMPYRA if this occurs (5.4)
Adverse Reactions/Side Effects
The most common adverse events (incidence ≥2% and at a rate greater than the placebo rate) for AMPYRA were urinary tract infection, insomnia, dizziness, headache, nausea, asthenia, back pain, balance disorder, multiple sclerosis relapse, paresthesia, nasopharyngitis, constipation, dyspepsia, and pharyngolaryngeal pain (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Merz Pharmaceuticals, LLC at 1-800-367-5109 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
OCT2 Inhibitors: Concomitant use may cause an increased exposure and potential risk of seizures (7.1)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2025
Full Prescribing Information
1. Indications and Usage for Ampyra
AMPYRA is indicated as a treatment to improve walking in adult patients with multiple sclerosis (MS). This was demonstrated by an increase in walking speed [see Clinical Studies (14)].
2. Ampyra Dosage and Administration
2.1 Dosage Information
The maximum recommended dosage of AMPYRA is one 10 mg tablet twice daily and should not be exceeded. Take doses approximately 12 hours apart.
There is no evidence of additional benefit at doses greater than 10 mg twice daily. Adverse reactions, including seizures, and discontinuations because of adverse reactions were more frequent at higher doses.
2.2 Administration Instructions
AMPYRA can be taken with or without food. Administer tablets whole; do not divide, crush, chew, or dissolve AMPYRA tablets.
If a dose is missed, patients should not take double or extra doses.
2.3 Renal Monitoring Prior to and During Treatment
Estimated creatinine clearance (CrCl) should be known before initiating treatment with AMPYRA, and monitored at least annually during treatment with AMPYRA. CrCl can be estimated using the following equation (multiply by 0.85 for women):

2.4 Dosage in Patients with Renal Impairment
In patients with mild renal impairment (CrCl 51–80 mL/min), AMPYRA plasma levels may approach those seen at a dose of 15 mg twice daily, a dose that is 1.5 times the maximum recommended dose and may be associated with an increased risk of seizures. As mild renal impairment is common after age 50, estimating CrCl is particularly important in these patients. The potential benefits of AMPYRA should be carefully considered against the risk of seizures in these patients [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. AMPYRA is contraindicated in patients with moderate or severe renal impairment (CrCl≤50 mL/min).
3. Dosage Forms and Strengths
AMPYRA is available in a 10 mg strength and is a film-coated, white to off-white, biconvex, oval shaped, non-scored tablet with flat edge, debossed with "A10" on one side.
4. Contraindications
The use of AMPYRA is contraindicated in the following conditions:
- History of seizure [see Warnings and Precautions (5.1)]
- Moderate or severe renal impairment (CrCl≤50 mL/min) [see Warnings and Precautions (5.2)]
- History of hypersensitivity to AMPYRA or 4-aminopyridine; reactions have included anaphylaxis [see Warnings and Precautions (5.4)]
5. Warnings and Precautions
5.1 Seizures
AMPYRA can cause seizures. Increased incidence of seizures has been observed at 20 mg twice daily (2 times the maximum recommended dosage) in controlled clinical studies of 9–14 weeks duration with dalfampridine in patients with MS. In open-label extension trials in MS patients, the incidence of seizures during treatment with dalfampridine 15 mg twice daily (1.7/100PY) was over 4 times higher than the incidence during treatment with 10 mg twice daily (0.4/100PY). In the post-marketing period seizures have been reported. The majority of seizures occurred at the recommended dose and in patients without a history of seizures, and generally within days to weeks of starting therapy.
AMPYRA has not been evaluated in patients with a history of seizures or with evidence of epileptiform activity on an EEG, as these patients were excluded from clinical trials. The risk of seizures in patients with epileptiform activity on an EEG is unknown, and could be substantially higher than that observed in AMPYRA clinical studies. Permanently discontinue AMPYRA in patients who have a seizure while on treatment. AMPYRA is contraindicated in patients with a history of seizures [see Contraindications (4)] .
5.2 Renal Impairment
AMPYRA is eliminated through the kidneys primarily as unchanged drug [see Clinical Pharmacology (12.3)].
Because patients with moderate to severe renal impairment (CrCl ≤50mL/min) would require a dose lower than 10 mg twice daily and no strength smaller than 10 mg is available, AMPYRA is contraindicated in these patients [see Contraindications (4)] .
In patients with mild renal impairment (CrCl 51–80 mL/min), AMPYRA plasma levels may approach those seen at a dose of 15 mg twice daily, a dose that may be associated with an increased risk of seizures [see Warnings and Precautions (5.1)] .
5.3 Concurrent Treatment with Other Forms of 4-Aminopyridine
Avoid concomitant use with other forms of 4-aminopyridine (4-AP, fampridine) since the active ingredient is the same. Instruct patients to discontinue use of any product containing 4-aminopyridine prior to initiating treatment with AMPYRA in order to reduce the potential for dose-related adverse reactions.
5.4 Anaphylaxis
AMPYRA can cause anaphylaxis and severe allergic reactions. Signs and symptoms have included respiratory compromise, urticaria, and angioedema of the throat and or tongue. AMPYRA is contraindicated in patients with a history of hypersensitivity to AMPYRA or 4-aminopyridine. Inform patients of the signs and symptoms of anaphylaxis and instruct them to discontinue AMPYRA and seek immediate medical care should these signs and symptoms occur.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described in more detail elsewhere in the labeling:
- Seizures [see Warnings and Precautions (5.1)]
- Anaphylaxis [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
In three placebo-controlled clinical trials of up to 14 weeks duration, 4% (15/400) of patients treated with AMPYRA 10 mg twice daily experienced one or more adverse reactions leading to discontinuation, compared to 2% (5/238) of placebo-treated patients. The adverse reactions leading to discontinuation of at least 2 patients treated with AMPYRA and that led to discontinuation more frequently compared to placebo were headache (AMPYRA 0.5%, placebo 0%), balance disorder (AMPYRA 0.5%, placebo 0%), dizziness (AMPYRA 0.5%, placebo 0%), and confusional state (AMPYRA 0.3%, placebo 0%).
Table 1 lists adverse reactions that occurred in ≥2% of patients treated with AMPYRA 10 mg twice daily, and more frequently than in placebo-treated patients, in controlled clinical trials.
| Adverse Reaction |
Placebo % |
AMPYRA % |
| Urinary tract infection | 8 | 12 |
| Insomnia | 4 | 9 |
| Dizziness | 4 | 7 |
| Headache | 4 | 7 |
| Nausea | 3 | 7 |
| Asthenia | 4 | 7 |
| Back pain | 2 | 5 |
| Balance disorder | 1 | 5 |
| Multiple sclerosis relapse | 3 | 4 |
| Paresthesia | 3 | 4 |
| Nasopharyngitis | 2 | 4 |
| Constipation | 2 | 3 |
| Dyspepsia | 1 | 2 |
| Pharyngolaryngeal pain | 1 | 2 |
Other Adverse Reactions
AMPYRA has been evaluated in a total of 1,952 subjects, including 917 MS patients. A total of 741 patients have been treated with AMPYRA for over six months, 501 for over one year and 352 for over two years. The experience in open-label clinical trials is consistent with the safety profile observed in the placebo-controlled clinical trials. As in controlled clinical trials, a dose-dependent increase in the incidence of seizures has been observed in open-label clinical trials with AMPYRA in patients with MS as follows: AMPYRA 10 mg twice daily 0.41 per 100 person-years (95% confidence interval 0.13–0.96); dalfampridine 15 mg twice daily 1.7 per 100 person-years (95% confidence interval 0.21–6.28).
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use with dalfampridine. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure: vomiting, vertigo.
Related/similar drugs
7. Drug Interactions
7.1 OCT2 Inhibitors
Concurrent treatment with OCT2 inhibitors, such as cimetidine, may cause increased exposure to dalfampridine [see Clinical Pharmacology (12.3)]. Elevated levels of dalfampridine increase the risk of seizures [see Warnings and Precautions (5.1, 5.2)]. The potential benefits of taking OCT2 inhibitors concurrently with AMPYRA should be considered against the risk of seizures in these patients.
7.2 Baclofen
No interaction was identified between dalfampridine and baclofen [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with use of AMPYRA in pregnant women. Administration of dalfampridine to animals during pregnancy and lactation resulted in decreased offspring viability and growth at clinically relevant doses [see Data]. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of dalfampridine to pregnant rats and rabbits throughout organogenesis resulted in no evidence of developmental toxicity in either species. The highest doses tested (10 mg/kg/day in rats, 5 mg/kg/day in rabbits), which were associated with maternal toxicity, are approximately 5 times the MRHD on a body surface area (mg/m2) basis.
Oral administration of dalfampridine (0, 1, 3, and 9 to 6 mg/kg/day; high dose reduced during the second week of dosing) to female rats throughout pregnancy and lactation resulted in decreased offspring viability at the highest dose tested and decreased body weight in offspring at the mid and high doses. The no-effect dose for pre- and postnatal developmental toxicity in rats (1 mg/kg/day) is less than the MRHD on a mg/m2 basis.
8.2 Lactation
Risk Summary
There are no data on the presence of dalfampridine in human milk, the effects of dalfampridine on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for AMPYRA and any potential adverse effects on the breastfed infant from AMPYRA or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness in patients younger than 18 years of age have not been established.
8.5 Geriatric Use
Clinical studies of AMPYRA did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently than younger subjects. A population PK analysis showed that dalfampridine clearance modestly decreased with increasing age, but not sufficiently to necessitate a modification of dose with age. Other reported clinical experience has identified no differences in responses between the elderly and younger patients.
AMPYRA is known to be substantially excreted by the kidneys and the risk of adverse reactions, including seizures, is greater with increasing exposure of dalfampridine. Because elderly patients are more likely to have decreased renal function, it is particularly important to know the estimated creatinine clearance (CrCl) in these patients [see Warnings and Precautions (5.2)].
8.6 Impaired Renal Function
Clearance of dalfampridine is decreased in patients with renal impairment and is significantly correlated with creatinine clearance (CrCl) [see Clinical Pharmacology (12.3)]. AMPYRA is contraindicated in patients with moderate or severe renal impairment (CrCl ≤50 mL/min) [see Contraindications (4)]. The risk of seizures in patients with mild renal impairment (CrCl 51–80 mL/min) is unknown, but dalfampridine plasma levels in these patients may approach those seen at a dose of 15 mg twice daily, a dose that may be associated with an increased risk of seizures. If unknown, estimated creatinine clearance should be calculated prior to initiating treatment with AMPYRA [see Dosage and Administration (2.3) and Warnings and Precautions (5.2)] .
10. Overdosage
Three cases of overdose were reported in controlled clinical trials with AMPYRA, involving two MS patients. The first patient took six times the currently recommended dose (60 mg) and was taken to the emergency room with altered mental state. The second patient took 40 mg doses on two separate occasions. In the first instance, she experienced a complex partial seizure and, in the second instance, a period of confusion. Both patients recovered by the following day without sequelae.
Several cases of overdose are found in the scientific literature in which various formulations of dalfampridine were used, resulting in numerous adverse events including seizure, confusion, tremulousness, diaphoresis, and amnesia. In some instances, patients developed status epilepticus, requiring intensive supportive care and were responsive to standard therapy for seizures. In one published case report, an MS patient who ingested 300 mg of 4-aminopyridine (dalfampridine) developed a condition that resembled limbic encephalitis. This patient developed weakness, reduced awareness, memory loss, hypophonic speech, and temporal lobe hyperintensities on MRI. The patient's speech and language and ambulation improved over time, and an MRI at 4 months after the overdose no longer showed signal abnormalities. At one year, the patient continued to have difficulty with short term memory and learning new tasks.
11. Ampyra Description
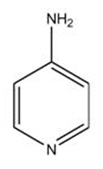
AMPYRA (dalfampridine) is a potassium channel blocker, available in a 10 mg tablet strength. Each tablet contains 10 mg dalfampridine, formulated as an extended-release tablet for twice-daily oral administration. Dalfampridine is also known by its chemical name, 4-aminopyridine, with the following structure:
AMPYRA (dalfampridine) extended-release tablets are available in a 10 mg strength and are white to off-white, biconvex, oval shaped, film-coated, non-scored tablets with flat edge, debossed with "A10" on one side, containing 10 mg of dalfampridine. Inactive ingredients consist of colloidal silicon dioxide, hydroxypropyl methylcellulose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, and titanium dioxide.
Dalfampridine is a fine white powder with a molecular weight of 94.1, CAS 504-24-5, and a molecular formula of C 5H 6N 2. At ambient conditions, dalfampridine is soluble in water, methanol, acetone, tetrahydrofuran, isopropanol, acetonitrile, N,N-dimethylformamide, dimethylsulfoxide, and ethanol.
12. Ampyra - Clinical Pharmacology
12.1 Mechanism of action
The mechanism by which dalfampridine exerts its therapeutic effect has not been fully elucidated. Dalfampridine is a broad spectrum potassium channel blocker. In animal studies, dalfampridine has been shown to increase conduction of action potentials in demyelinated axons through inhibition of potassium channels.
12.2 Pharmacodynamics
AMPYRA does not prolong the QTc interval and does not have a clinically important effect on QRS duration.
12.3 Pharmacokinetics
Absorption and Distribution
Orally administered dalfampridine is rapidly and completely absorbed from the gastrointestinal tract. Absolute bioavailability of extended release AMPYRA tablets has not been assessed, but relative bioavailability is 96% when compared to an aqueous oral solution. The extended release tablet delays absorption of dalfampridine relative to the solution formulation, giving a slower rise to a lower peak concentration (Cmax), with no effect on the extent of absorption (AUC). Single AMPYRA tablet 10 mg doses administered to healthy volunteers in a fasted state gave peak concentrations ranging from 17.3 ng/mL to 21.6 ng/mL occurring 3 to 4 hours post-administration (Tmax). In comparison, Cmax with the same 10 mg dose of dalfampridine in an oral solution was 42.7 ng/mL and occurred approximately 1.3 hours after dosing. Exposure increased proportionally with dose.
Dalfampridine is largely unbound to plasma proteins (97–99%). The apparent volume of distribution is 2.6 L/kg.
There is no apparent difference in pharmacokinetic parameter values following administration of AMPYRA tablets to either healthy volunteers or patients with MS.
When dalfampridine is taken with food, there is a slight increase in Cmax (12–17%) and a slight decrease in AUC (4–7%). These changes in exposure are not clinically significant, and therefore the drug may be taken with or without food [see Dosage and Administration (2.2)].
Metabolism and Elimination
Dalfampridine and metabolites elimination is nearly complete after 24 hours, with 95.9% of the dose recovered in urine and 0.5% recovered in feces. Most of the excreted radioactivity in urine was parent drug (90.3%). Two metabolites were identified: 3-hydroxy-4-aminopyridine (4.3%) and 3-hydroxy-4-aminopyridine sulfate (2.6%). These metabolites have been shown to have no pharmacologic activity on potassium channels.
The apparent elimination half-life of dalfampridine following administration of the extended release tablet formulation of AMPYRA is 5.2 to 6.5 hours. The plasma half-life of the sulfate conjugate is approximately 7.6 hours and the half-life of 3-hydroxy-4-aminopyridine could not be calculated because concentrations for most subjects were close to or below the limit of quantitation.
In vitro studies with human liver microsomes indicate that CYP2E1 was the major enzyme responsible for the 3-hydroxylation of dalfampridine. The identity of the CYP enzymes suspected of playing a minor role in the 3-hydroxylation of dalfampridine could not be established unequivocally.
Specific Populations
Pediatric
The safety and effectiveness in patients younger than 18 years of age have not been established.
Geriatric
A population pharmacokinetic analysis showed that dalfampridine clearance modestly decreased with increasing age, but not sufficiently to necessitate a modification of dose.
Gender
A population pharmacokinetic analysis suggested that female patients would be expected to have higher maximum dalfampridine plasma concentration than male patients. The magnitude of these differences is small and does not necessitate any dose modification.
Renal Impairment [see Contraindications (4) and Warnings and Precautions (5.2)].
The pharmacokinetics of dalfampridine was studied in 9 male and 11 female subjects with varying degrees of renal function. Elimination of the drug is significantly correlated with the creatinine clearance. Total body clearance of dalfampridine was reduced by about 45 % in patients with mild renal impairment (CrCl 51–80 mL/min), by about 50% in patients with moderate renal impairment (CrCl = 30–50 mL/min), and by about 75% in patients with severe renal impairment (CrCl <30 mL/min). The terminal half-life of dalfampridine is about 3.3 times longer in patients with severe renal impairment but is not prolonged in patients with mild or moderate renal impairment.
Hepatic Impairment
The pharmacokinetics of dalfampridine in hepatically impaired subjects has not been studied. Since dalfampridine is primarily excreted unchanged in the urine, hepatic impairment is not expected to significantly affect dalfampridine pharmacokinetics or recommended dosing.
Race
There were too few non-Caucasians in the patient population to evaluate the effect of race.
Drug Interactions
Effects of Co-administered Drugs on Dalfampridine
Interferon
Dalfampridine kinetics were not affected by co-administration of subcutaneous injections of 8 million units interferon beta-1b.
Baclofen
Based on a population analysis, dalfampridine kinetics were not affected by baclofen.
Cimetidine
In a single-dose clinical study, 23 healthy volunteers took the OCT2 inhibitor cimetidine 400 mg every 6 hours concurrently with dalfampridine 10 mg single dose. The test-reference ratio for AUC0–∞ was 125% (90% confidence interval: 121% to 130%) due to a reduction in the clearance of dalfampridine [see Drug Interactions (7.1)].
Effects of Dalfampridine on Co-administered Drugs
In vitro data with human liver microsomes showed that dalfampridine was not a direct or time-dependent inhibitor of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5. Dalfampridine is not likely to affect the pharmacokinetics of drugs that are substrates of these enzymes.
Other in vitro studies with cultured human hepatocytes with 0.025 μM, 0.25 μM, 2.5 μM, and 25 μM dalfampridine had little or no effect on CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2E1, or CYP3A4/5 enzyme activities. Consequently, the potential for dalfampridine to induce human hepatocytes at therapeutic concentrations is remote.
In vitro, dalfampridine is not a substrate or an inhibitor for the p-glycoprotein transporter. The pharmacokinetics of AMPYRA are unlikely to be affected by drugs that inhibit the p-glycoprotein transporter, and dalfampridine is not likely to affect the pharmacokinetics of drugs that are substrates of the p-glycoprotein transporter.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year dietary carcinogenicity studies of dalfampridine were conducted in mice and rats. In mice, the doses tested (approximately 2, 12.5, and 80 mg/kg/day) were associated with plasma exposures (AUC) up to 11 times the plasma AUC in humans at the maximum recommended human dose (MRHD) of 20 mg/day. There was no evidence of drug-related carcinogenicity.
In rats, the doses tested (approximately 2, 6, and 18 mg/kg/day) were approximately 1, 3, and 9 times the MRHD on a body surface area (mg/m2) basis. There was a significant increase in uterine polyps at the highest dose tested.
Mutagenesis
Dalfampridine was negative in in vitro (bacterial reverse mutation, mouse lymphoma tk, chromosomal aberration) and in vivo (mouse bone marrow, rat erythrocyte micronucleus) genetic toxicology assays.
Impairment of Fertility
Oral administration of dalfampridine (0, 1, 3, and 9 mg/kg/day) to male and female rats prior to and throughout mating, and continuing in females through early pregnancy (to gestation day 13) or throughout pregnancy and lactation resulted in no adverse effects on fertility. Reduced offspring viability and body weight were observed at 9 mg/kg/day. The no-effect dose for adverse effects on fertility (9 mg/kg/day) and reproductive performance (3 mg/kg/day) are 4 times and similar to, respectively, the MRHD on a mg/m2 basis.
14. Clinical Studies
The effectiveness of AMPYRA in improving walking in patients with multiple sclerosis was evaluated in two adequate and well controlled trials involving 540 patients. Patients in these two clinical trials had a mean disease duration of 13 years and a mean Kurtzke Expanded Disability Status Scale (EDSS) score of 6.
Trial 1 was a randomized, placebo-controlled, parallel group, 21-week study (one week post screening, two-week, single-blind placebo run-in, 14-week double-blind treatment, and 4-week no treatment follow-up) in 301 patients with multiple sclerosis at 33 centers in the U.S. and Canada: 229 patients assigned to AMPYRA 10 mg twice daily and 72 patients assigned to placebo. A total of 283 patients (212 AMPYRA and 71 placebo) completed all study visits. Patient inclusion criteria included the ability to walk 25 feet in 8–45 seconds. Patient exclusion criteria included a history of seizures or evidence of epileptiform activity on a screening EEG, and onset of an MS exacerbation within 60 days.
Trial 2 was a randomized, placebo-controlled, parallel group, 14-week study (one week post-screening, two weeks of single-blind, placebo run-in, nine weeks of double-blind treatment, and two weeks of no-treatment follow-up) in 239 patients with multiple sclerosis at 39 centers in the U.S. and Canada: 120 patients assigned to 10 mg twice daily and 119 assigned to placebo. A total of 227 patients (113 AMPYRA and 114 placebo) completed all study visits. The patient inclusion and exclusion criteria used in Trial 1 were employed in Trial 2, and in addition patients with severe renal impairment were also excluded.
The primary measure of efficacy in both trials was walking speed (in feet per second) as measured by the Timed 25-foot Walk (T25FW), using a responder analysis. A responder was defined as a patient who showed faster walking speed for at least three visits out of a possible four during the double-blind period than the maximum value achieved in the five non-double-blind no treatment visits (four before the double-blind period and one after).
A significantly greater proportion of patients taking AMPYRA 10 mg twice daily were responders, compared to patients taking placebo, as measured by the T25FW (Trial 1: 34.8% vs. 8.3%; Trial 2: 42.9% vs. 9.3%). The increased response rate in the AMPYRA group was observed across all four major types of MS disease course.
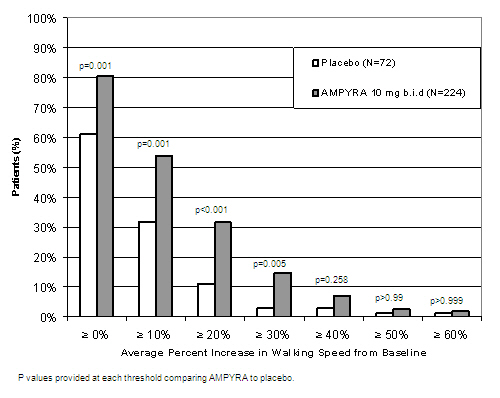
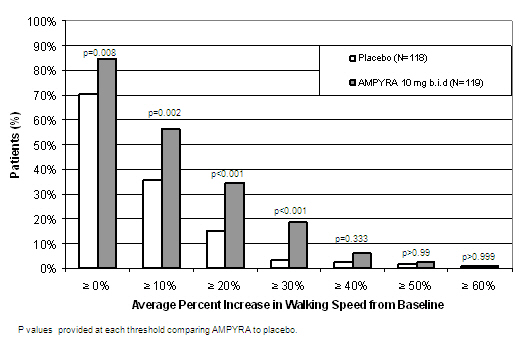
During the double-blind treatment period, a significantly greater proportion of patients taking AMPYRA 10 mg twice daily had increases in walking speed of at least 10%, 20%, or 30% from baseline, compared to placebo (Figure 1 and Figure 2).
| Figure 1: Average walking speed change (%) from baseline during the double-blind phase of Trial 1 |
|
|
| Figure 2: Average walking speed change (%) from baseline during the double-blind phase of Trial 2 |
|
|
In Trial 1 and Trial 2, consistent improvements in walking speed were shown to be associated with improvements on a patient self-assessment of ambulatory disability, the 12-item Multiple Sclerosis Walking Scale (MSWS-12), for both drug and placebo treated patients. However, a drug-placebo difference was not established for that outcome measure.
The majority of patients in these trials (63%) were using immunomodulatory drugs (interferons, glatiramer acetate, or natalizumab), but the magnitude of improvement in walking ability was independent of concomitant treatment with these drugs. No differences in effectiveness based on degree of impairment, age, gender, or body mass index were detected. There were too few non-Caucasians in the patient population to evaluate the effect of race.
16. How is Ampyra supplied
AMPYRA (dalfampridine) extended release tablets, 10 mg are film-coated, white to off-white, biconvex, oval shaped, non-scored tablets with flat edge. The tablets are identified by a debossed code "A10" on one side and are available in bottles of 60.
- NDC 0259-5010-60 bottles of 60 tablets
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Risk of Seizures
Inform patients that AMPYRA can cause seizures, and that they must discontinue use of AMPYRA if they experience a seizure [see Warnings and Precautions (5.1)].
AMPYRA dosing
Instruct patients to take AMPYRA exactly as prescribed. Instruct patients not to take a double dose after they miss a dose, as this would increase their risk of seizure. Instruct patients not to take more than 2 tablets in a 24-hour period and to make sure that there is an approximate 12-hour interval between doses [see Dosage and Administration (2.1, 2.2)].
Anaphylaxis
Advise patients to discontinue AMPYRA and seek medical care if they develop signs and symptoms of anaphylaxis [see Warnings and Precautions (5.4)].
Effects on Driving or Using Machinery
Counsel patients that central nervous system-related adverse reactions, such as vertigo and dizziness, associated with the use of AMPYRA might impair their ability to drive or use machinery should they develop these symptoms.
Drug Interactions
Instruct patients to notify their healthcare provider prior to starting any new medication, including over-the-counter drugs.
Storage
Advise patients to store AMPYRA at 25°C (77°F), with excursions permitted to 15ºC to 30ºC (59ºF to 86ºF). Advise patients to safely throw away AMPYRA that is out of date or no longer needed.
Marketed by:
Merz Pharmaceuticals, LLC
Raleigh, NC 27615
AMPYRA® is a registered trademark of Merz Pharmaceuticals, LLC
©2025 Merz Pharmaceuticals, LLC. All rights reserved.
FPI-0052 v2.0
MEDICATION GUIDE
AMPYRA® (am-PEER-ah)
(dalfampridine)
Extended Release Tablets
Read this Medication Guide before you start taking AMPYRA and each time you get a refill. There may be new information. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about AMPYRA?
AMPYRA can cause seizures.
- You could have a seizure even if you never had a seizure before.
- Your chance of having a seizure is higher if you take too much AMPYRA or if your kidneys have a mild decrease of function, which is common after age 50.
- Your doctor may do a blood test to check how well your kidneys are working, if that is not known before you start taking AMPYRA.
- Do not take AMPYRA if you have ever had a seizure.
- Before taking AMPYRA tell your doctor if you have kidney problems.
- Take AMPYRA exactly as prescribed by your doctor. See "How should I take AMPYRA?"
Stop taking AMPYRA and call your doctor right away if you have a seizure while taking AMPYRA.
What is AMPYRA?
AMPYRA is a prescription medicine used to help improve walking in adults with multiple sclerosis (MS). This was shown by an increase in walking speed.
It is not known if AMPYRA is safe or effective in children less than 18 years of age.
Who should not take AMPYRA?
Do not take AMPYRA if you:
- have ever had a seizure
- have certain types of kidney problems
- are allergic to dalfampridine (4-aminopyridine), the active ingredient in AMPYRA
What should I tell my doctor before taking AMPYRA?
Before you take AMPYRA, tell your doctor if you:
- have any other medical conditions
- are taking compounded 4-aminopyridine (fampridine, 4-AP)
- are taking any other medicines, including over-the-counter medicines such as cimetidine
- are pregnant or plan to become pregnant. It is not known if AMPYRA will harm your unborn baby.
- are breast-feeding or plan to breast-feed. It is not known if AMPYRA passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you take AMPYRA.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take AMPYRA?
- Take AMPYRA exactly as your doctor tells you to take it. Do not change your dose of AMPYRA.
- Take one tablet of AMPYRA 2 times each day about 12 hours apart. Do not take more than 2 tablets of AMPYRA in a 24-hour period.
- Take AMPYRA tablets whole. Do not break, crush, chew or dissolve AMPYRA tablets before swallowing. If you cannot swallow AMPYRA tablets whole, tell your doctor.
- AMPYRA is released slowly over time. If the tablet is broken, the medicine may be released too fast. This can raise your chance of having a seizure.
- AMPYRA can be taken with or without food.
- If you miss a dose of AMPYRA, do not make up the missed dose. Do not take 2 doses at the same time. Take your next dose at your regular scheduled time.
- If you take too much AMPYRA, call your doctor or go to the nearest hospital emergency room right away.
- Do not take AMPYRA together with other aminopyridine medications, including compounded 4-AP (sometimes called 4-aminopyridine, fampridine).
What should I avoid while taking AMPYRA?
AMPYRA may cause dizziness or vertigo. If you have these symptoms, do not drive, operate machinery, or do other dangerous activities.
What are the possible side effects of AMPYRA?
AMPYRA may cause serious side effects, including:
- serious allergic reactions. Stop taking AMPYRA and call your doctor right away or get emergency medical help if you have:
- shortness of breath or trouble breathing
- swelling of your throat or tongue
- hives
See "What is the most important information I should know about AMPYRA?"
The most common side effects of AMPYRA include:
- urinary tract infection
- trouble sleeping (insomnia)
- dizziness
- headache
- nausea
- weakness
- back pain
- problems with balance
- multiple sclerosis relapse
- burning, tingling or itching of your skin
- irritation in your nose and throat
- constipation
- indigestion
- pain in your throat
Tell your doctor if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of AMPYRA. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to the FDA at 1-800-FDA-1088.
How should I store AMPYRA?
- Store AMPYRA at 59ºF to 86ºF (15ºC to 30ºC).
- Safely throw away AMPYRA that is out of date or no longer needed.
Keep AMPYRA and all medicines out of the reach of children.
General Information about the safe and effective use of AMPYRA
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use AMPYRA for a condition for which it was not prescribed. Do not give AMPYRA to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about AMPYRA. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about AMPYRA that is written for health professionals.
For more information, go to www.AMPYRA.com or call 1-800-367-5109.
What are the ingredients in AMPYRA?
Active ingredient: dalfampridine (previously called fampridine)
Inactive ingredients: colloidal silicon dioxide, hydroxypropyl methylcellulose, magnesium stearate, microcrystalline cellulose, polyethylene glycol, and titanium dioxide.

| AMPYRA
dalfampridine tablet, film coated, extended release |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Merz Pharmaceuticals, LLC (126209282) |
| Registrant - Patheon Inc., Toronto Regional Operations (240769596) |
More about Ampyra (dalfampridine)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (77)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: miscellaneous central nervous system agents
- Breastfeeding
- En español
