Cystic Fibrosis
Medically reviewed by Drugs.com. Last updated on Aug 4, 2025.
What is cystic fibrosis (CF)?
CF is a lifelong condition that affects your lungs, digestive system, and other organs. Your mucus, tears, sweat, and saliva become so thick and sticky that they clog your lungs and digestive system. CF usually causes problems with breathing and with breaking down and absorbing food.
What are the signs and symptoms of CF?
You may have any of the following, depending on which organs are affected:
- Respiratory infections such as sinusitis, bronchitis, or pneumonia that happen often
- Cough, wheezing, and shortness of breath
- Clubbing of fingers or toes (large, blunt, and rounded)
- Weakness and fatigue
- Abdominal pain
How is CF diagnosed?
Your healthcare provider will ask about your symptoms. Your provider will also ask if a parent, brother, or sister has CF. CF is a genetic disorder. Your provider may recommend genetic counseling to learn why you have CF. If you plan to have a baby, genetic counseling may show if your child is at risk for CF. You may need any of the following tests:
- A sweat chloride test shows the amount of chloride in your sweat. The amount of chloride will be high if you have CF.
- Blood tests may be used to find signs of infection and to test your kidney function. They may also show the gene that caused your CF.
- An x-ray will show inflammation and enlargement of your lungs. It will also show plugged airways and any fluid buildup.
- A bronchoscopy is a procedure to look inside your lungs to check for damage. A bronchoscope (thin tube with a light) is inserted into your mouth and moved down your throat to your lungs. Tissue and fluid may be collected from your airway or lungs to be tested.
How is CF treated?
CF cannot be cured. Treatment may help you breathe more easily, prevent infections, or absorb nutrients.
- Medicines:
- Antibiotics help fight or prevent an infection caused by bacteria.
- Mucus-thinning medicine is breathed in to help thin lung mucus so you can cough it up more easily.
- NSAIDs help decrease swelling and pain or fever. This medicine is available with or without a doctor's order. NSAIDs can cause stomach bleeding or kidney problems in certain people. If you take blood thinner medicine, always ask your healthcare provider if NSAIDs are safe for you. Always read the medicine label and follow directions.
- Steroid medicine helps decrease inflammation.
- Bronchodilators help open the air passages in your lungs, and help you breathe more easily.
- Pancreatic enzymes help your digestive system break down food and absorb nutrients.
- Cystic fibrosis transmembrane regulators (CFTR) may help improve your symptoms if your CF is caused by a certain protein mutation. Your healthcare provider or CF specialist can give you more information about CFTRs.
- Extra oxygen may be needed if your blood oxygen level is lower than it should be. You may get oxygen through a mask placed over your nose and mouth or through small tubes placed in your nostrils.
- Surgery may be needed if you have severe damage to organs, such as your liver or lungs. Ask your provider for more information about surgery.
Treatment options
The following list of medications are related to or used in the treatment of this condition.
Related medications
What can I do to breathe more easily?
- Use airway clearance exercises to help remove mucus so you can breathe more easily. Your provider will show you how to do the exercises. These exercises may be used with machines or devices to help decrease your symptoms and risk for infection. Your healthcare provider may also recommend pulmonary rehabilitation (rehab) to help you improve lung function. Pulmonary rehab is usually 3 times each week for a few months.
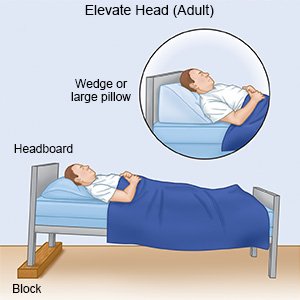
- Rest or sleep with your head elevated to help keep your airway open. Use pillows or foam wedges to elevate your head.
- Be physically active, as directed. Ask your provider about the best activity plan for you. Physical activity can help loosen secretions in your airways and lungs, and help you breathe more easily.

- Do not smoke. Nicotine and other chemicals in cigarettes and cigars can cause lung damage. Ask your healthcare provider for information if you currently smoke and need help to quit. E-cigarettes or smokeless tobacco still contain nicotine. Talk to your healthcare provider before you use these products.
What can I do to stay healthy?
- Ask about vaccines you may need. Get the flu vaccine as soon as recommended each year, usually in September or October. Get all recommended COVID-19 vaccine doses and booster shots. The pneumonia vaccine may also be recommended. Ask about other vaccines you may need, and when to get them.
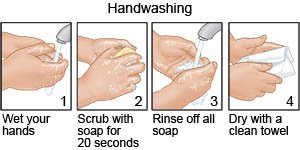
- Prevent the spread of germs. Avoid people who have a cold or the flu. Cover your mouth when you cough. Cough into a tissue or the bend of your elbow so you do not spread germs from your hands. Wash your hands often. Use soap and water. Wash your hands after you use the bathroom, change a child's diapers, or sneeze. Wash before you prepare or eat food.
- Eat a variety of healthy foods. Healthy foods include fruits, vegetables, whole-grain breads, low-fat dairy products, beans, lean meats, and fish. You may need to eat foods that have extra calories, fat, vitamins, or calcium. Ask your provider if you need to be on a special diet.

- Get screening tests as directed. Screening means you are checked for complications of CF before you show symptoms. You may be screened for CF-related diabetes at least 1 time each year. Talk to your provider about other screening tests you may need and how often.
Call your local emergency number (911 in the US) if:
- You cough up blood.
- You have chest pain or sudden trouble breathing.
When should I seek immediate care?
- You are coughing or wheezing more than usual.
- You have chest congestion and the mucus you cough up is a different color than before.
- Your lips or fingernails turn blue or white.
- You have severe abdominal pain.
When should I call my doctor?
- You have a fever.
- You have less appetite than usual, or you lose weight without trying.
- You have chills or feel weak or achy.
- You have trouble sleeping.
- You urinate less, have a dry mouth or cracked lips, or feel dizzy.
- You have questions or concerns about your condition or care.
Care Agreement
You have the right to help plan your care. Learn about your health condition and how it may be treated. Discuss treatment options with your healthcare providers to decide what care you want to receive. You always have the right to refuse treatment. The above information is an educational aid only. It is not intended as medical advice for individual conditions or treatments. Talk to your doctor, nurse or pharmacist before following any medical regimen to see if it is safe and effective for you.© Copyright Merative 2025 Information is for End User's use only and may not be sold, redistributed or otherwise used for commercial purposes.
Learn more about Cystic Fibrosis
Treatment options
Care guides
Symptoms and treatments
Further information
Always consult your healthcare provider to ensure the information displayed on this page applies to your personal circumstances.