Velsipity: Package Insert / Prescribing Info
Package insert / product label
Generic name: etrasimod
Dosage form: tablet, film coated
Drug class: Selective immunosuppressants
Medically reviewed by Drugs.com. Last updated on Aug 25, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
VELSIPITY™ (etrasimod) tablets, for oral use
Initial U.S. Approval: 2023
Indications and Usage for Velsipity
VELSIPITY is a sphingosine 1-phosphate receptor modulator indicated for the treatment of moderately to severely active ulcerative colitis in adults. (1)
Velsipity Dosage and Administration
Dosage Forms and Strengths
Tablets: 2 mg of etrasimod (3)
Contraindications
- •
- In the last 6 months, experienced myocardial infarction, unstable angina pectoris, stroke, transient ischemic attack, decompensated heart failure requiring hospitalization, or Class III or IV heart failure. (4, 5.2)
- •
- History or presence of Mobitz type II second-degree or third-degree atrioventricular (AV) block, sick sinus syndrome, or sino-atrial block, unless the patient has a functioning pacemaker. (4, 5.2)
Warnings and Precautions
- •
- Infections: May increase the risk of infections. Obtain a complete blood count (CBC) before initiation of treatment. Monitor for infection during treatment and for 5 weeks after discontinuation. Consider interruption of treatment if a serious infection develops. Avoid use of live attenuated vaccines during and for up to 5 weeks after treatment. (5.1)
- •
- Bradyarrhythmia and Atrioventricular Conduction Delays: May result in a transient decrease in heart rate and AV conduction delays. Obtain an electrocardiogram (ECG) to assess for preexisting cardiac conduction abnormalities before starting treatment. Consider cardiology consultation for conduction abnormalities or concomitant use with other drugs that decrease heart rate. (2.1, 5.2, 7)
- •
- Liver Injury: Elevations of aminotransferases may occur. Obtain transaminase and bilirubin levels before initiating VELSIPITY. Discontinue if significant liver injury is confirmed. (2.1, 5.3)
- •
- Macular Edema: May increase the risk of macular edema. Obtain a baseline evaluation of the fundus, including the macula, near the start of treatment with VELSIPITY. Periodically conduct an evaluation of the fundus, including the macula, while on therapy and any time there is a change in vision. Consider discontinuing VELSIPITY if macular edema develops. (2.1, 5.4)
- •
- Increased Blood Pressure: Monitor blood pressure during treatment. (5.5)
- •
- Fetal Risk: May cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for one week after stopping VELSIPITY. (5.6, 8.3)
- •
- Cutaneous Malignancies: Obtain a skin examination prior to or shortly after the start of treatment and periodically during treatment, especially if risk factors. Promptly evaluate suspicious skin lesions. (2.1, 5.7)
- •
- Posterior Reversible Encephalopathy Syndrome (PRES): If symptoms develop, obtain a physical and neurological exam, and consider MRI. (5.8)
- •
- Respiratory Effects: May cause a decline in pulmonary function. Assess pulmonary function (e.g., spirometry) if clinically indicated. (5.9)
- •
- Unintended Additive Immune System Effects from Prior Treatment with Immunosuppressive or Immune-Modulating Drugs: Consider the half-life and mode of action of prior therapies. (5.10)
- •
- Immune System Effects After Stopping VELSIPITY: If using concomitant immunosuppressants, monitor patients for infectious complications for up to 5 weeks after the last dose of VELSIPITY. (5.11)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥5%) are: headache, elevated liver tests, and dizziness. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See full prescribing information for a list of clinically important drug interactions. (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2025
Full Prescribing Information
1. Indications and Usage for Velsipity
VELSIPITY is indicated for the treatment of moderately to severely active ulcerative colitis (UC) in adults.
2. Velsipity Dosage and Administration
2.1 Assessments, Medications, and Vaccinations Prior to First Dose of VELSIPITY
Before initiation of treatment with VELSIPITY, assess the following:
Complete Blood Count
Obtain a recent (i.e., within the last 6 months or after discontinuation of prior UC therapy) complete blood count (CBC), including lymphocyte count [see Warnings and Precautions (5.1)].
Cardiac Evaluation
Obtain an electrocardiogram (ECG) to determine whether preexisting conduction abnormalities are present. In patients with certain preexisting conditions, advice from a cardiologist should be sought [see Warnings and Precautions (5.2)].
Liver Function Tests
Obtain recent (i.e., within the last 6 months) transaminase and bilirubin levels [see Warnings and Precautions (5.3)].
Ophthalmic Assessment
Obtain a baseline evaluation of the fundus, including the macula, near the start of treatment with VELSIPITY [see Warnings and Precautions (5.4)].
Skin Examination
Obtain a skin examination prior to or shortly after initiation of VELSIPITY. If a suspicious skin lesion is observed, it should be promptly evaluated [see Warnings and Precautions (5.7)].
Current or Prior Medications
- •
- Determine if patients are taking drugs that could slow heart rate or atrioventricular (AV) conduction [see Warnings and Precautions (5.2) and Drug Interactions (7)].
- •
- If patients are taking anti-neoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies, or if there is a history of prior use of these drugs, consider possible unintended additive immunosuppressive effects before initiating treatment with VELSIPITY [see Warnings and Precautions (5.10) and Drug Interactions (7)].
Vaccinations
Patients without a healthcare professional-confirmed history of varicella (chickenpox) or without documentation of a full course of vaccination against varicella zoster virus (VZV) should be tested for antibodies to VZV before initiating VELSIPITY; VZV vaccination of antibody-negative patients is recommended prior to commencing treatment with VELSIPITY.
If live attenuated vaccine immunizations are required, administer at least 4 weeks prior to initiation of VELSIPITY.
Update immunizations in agreement with current immunization guidelines prior to initiating VELSIPITY therapy [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
- •
- The recommended dosage of VELSIPITY is 2 mg orally once daily.
- •
- Swallow the tablet whole, with or without food [see Clinical Pharmacology (12.3)].
- •
- If a dose is missed, take the missed dose at the next scheduled time; do not double the next dose.
3. Dosage Forms and Strengths
Tablets: 2 mg of etrasimod as a round, green, film-coated tablet, debossed with “ETR” on one side and “2” on the other side.
4. Contraindications
VELSIPITY is contraindicated in patients who:
- •
- In the last 6 months, have experienced a myocardial infarction, unstable angina pectoris, stroke, transient ischemic attack (TIA), decompensated heart failure requiring hospitalization, or Class III or IV heart failure [see Warnings and Precautions (5.2)].
- •
- Have a history or presence of Mobitz type II second-degree or third-degree AV block, sick sinus syndrome, or sino-atrial block, unless the patient has a functioning pacemaker [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Infections
Risk of Infections
VELSIPITY causes a mean reduction in peripheral blood lymphocyte count to approximately 45% of baseline values at Week 52 because of reversible sequestration of lymphocytes in lymphoid tissues [see Clinical Pharmacology (12.2)]. VELSIPITY may, therefore, increase the susceptibility to infections. Life-threatening and rare fatal infections have been reported in association with other sphingosine 1-phosphate (S1P) receptor modulators.
Before initiating treatment, obtain a recent (i.e., within 6 months or after discontinuation of prior UC therapy) CBC, including lymphocyte count.
Delay initiation of VELSIPITY in patients with an active infection until the infection is resolved.
In UC-1, the overall rate of infections in subjects treated with VELSIPITY was 24.9% compared to 22.2% in subjects who received placebo. In pooled data from UC-2 and UC-3, the overall rate of infections in subjects treated with VELSIPITY was 14.0% compared to 11.8% in subjects who received placebo. The most common infections were urinary tract infections and herpes viral infections in UC-1, and urinary tract infections in UC-2 and UC-3 [see Adverse Reactions (6.1)].
The proportion of subjects treated with VELSIPITY who experienced lymphocyte counts less than 0.2 x 109/L was 5.5% in UC-1 and 0.6% in UC-2 and UC-3. These events did not lead to treatment discontinuation. Peripheral blood absolute lymphocyte counts returned to the normal range in 90% of subjects within 4 to 5 weeks of stopping therapy [see Clinical Pharmacology (12.2)].
Consider interruption of treatment with VELSIPITY if a patient develops a serious infection.
Because residual pharmacodynamic effects, such as lowering effects on peripheral lymphocyte count, may persist up to 5 weeks after discontinuation of VELSIPITY, vigilance for infection should be continued throughout this period.
Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML) is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically occurs in patients who are immunocompromised, and that usually leads to death or severe disability. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
PML has been reported in multiple sclerosis (MS) patients treated with S1P receptor modulators and has been associated with some risk factors (e.g., immunocompromised patients, polytherapy with immunosuppressants, and duration of use). Based on data from patients with MS, longer treatment duration increases the risk of PML in patients treated with S1P receptor modulators, and the majority of PML cases have occurred in patients treated with S1P receptor modulators for at least 18 months. VELSIPITY is not indicated for the treatment of MS. Physicians should be vigilant for clinical symptoms or unexplained neurologic findings that may be suggestive of PML. MRI findings may be apparent before clinical signs or symptoms. If PML is suspected, treatment with VELSIPITY should be suspended until PML has been excluded by an appropriate diagnostic evaluation.
If PML is confirmed, discontinue treatment with VELSIPITY.
Immune reconstitution inflammatory syndrome (IRIS) has been reported in MS patients treated with S1P receptor modulators who developed PML and subsequently discontinued treatment. IRIS presents as a clinical decline in the patient’s condition that may be rapid, can lead to serious neurological complications or death, and is often associated with characteristic changes on MRI. The time to onset of IRIS in patients with PML was generally within a few months after S1P receptor modulator discontinuation. Monitoring for development of IRIS and appropriate treatment of the associated inflammation should be undertaken.
Herpes Viral Infections
Herpes simplex encephalitis, varicella zoster meningitis, and localized herpes viral infections have been reported with S1P receptor modulators. In UC-1, herpes zoster was reported in 0.7% of subjects treated with VELSIPITY and in none of the subjects who received placebo. Patients without a healthcare professional-confirmed history of varicella (chickenpox), or without documentation of a full course of vaccination against varicella zoster virus (VZV), should be tested for antibodies to VZV before initiating VELSIPITY (see Vaccinations).
Cryptococcal Infection
Cases of fatal cryptococcal meningitis (CM) and disseminated cryptococcal infections have been reported with S1P receptor modulators. Physicians should be vigilant for clinical symptoms or signs of CM. Patients with symptoms or signs consistent with a cryptococcal infection should undergo prompt diagnostic evaluation and treatment. VELSIPITY treatment should be suspended until a cryptococcal infection has been excluded. If CM is diagnosed, appropriate treatment should be initiated.
Prior and Concomitant Treatment with Anti-neoplastic, Immune-modulating, or Non-corticosteroid Immunosuppressive Therapies
VELSIPITY has not been studied in combination with anti-neoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies. Avoid concomitant administration of these therapies with VELSIPITY and in the weeks following administration because of the risk of additive immunosuppressive effects [see Warnings and Precautions (5.10)].
Patients without a healthcare professional-confirmed history of varicella (chickenpox) or without documentation of a full course of vaccination against varicella zoster virus (VZV) should be tested for antibodies to VZV before initiating VELSIPITY. A full course of VZV vaccination of antibody-negative patients is recommended prior to commencing treatment with VELSIPITY, following which initiation of treatment with VELSIPITY should be postponed for 4 weeks to allow the full effect of vaccination to occur.
No clinical data are available on the safety and efficacy of vaccinations in patients taking VELSIPITY. Vaccinations may be less effective if administered during VELSIPITY treatment.
If live attenuated vaccine immunizations are required, administer at least 4 weeks prior to initiation of VELSIPITY. Avoid the use of live attenuated vaccines during and for 5 weeks after treatment with VELSIPITY.
Update immunizations in agreement with current immunization guidelines prior to initiating VELSIPITY therapy.
5.2 Bradyarrhythmia and Atrioventricular Conduction Delays
Initiation of VELSIPITY may result in a transient decrease in heart rate and AV conduction delays [see Clinical Pharmacology (12.2)].
Reduction in Heart Rate
Initiation of VELSIPITY may result in a transient decrease in heart rate. After the first dose of VELSIPITY 2 mg, subjects with UC experienced the greatest mean decrease from baseline in heart rate of 7.2 bpm at Hour 3 in UC-1 and Hour 2 in UC-2.
In UC-1, bradycardia was reported on the day of treatment initiation in 1% of subjects treated with VELSIPITY compared to none in subjects who received placebo. On Day 2, bradycardia was reported in 1 subject (0.3%) treated with VELSIPITY compared to none in subjects who received placebo. In UC-2 and UC-3, bradycardia was reported on the day of treatment initiation in 2.9% of subjects treated with VELSIPITY compared to none in subjects who received placebo. On Day 2, bradycardia was reported in 1 subject (0.3%) treated with VELSIPITY compared to none in subjects who received placebo. Subjects who experienced bradycardia were generally asymptomatic. Few subjects experienced symptoms, such as dizziness, and these symptoms resolved without intervention.
Atrioventricular Conduction Delays
Initiation of VELSIPITY may result in transient AV conduction delays.
On the day of treatment initiation of VELSIPITY 2 mg, first- or second-degree Mobitz type I AV blocks were observed in 0.7% of VELSIPITY-treated subjects compared to none in placebo in UC-1, and in 0.8% of VELSIPITY-treated subjects compared to none in placebo in UC-2 and UC-3. In UC-1, UC-2, and UC-3, Mobitz type II second- or third-degree AV blocks were not reported in VELSIPITY-treated subjects.
If treatment with VELSIPITY is considered, advice from a cardiologist should be sought for those individuals:
- •
- With significant QT prolongation (QTcF ≥450 msec in males, ≥470 msec in females)
- •
- With arrhythmias requiring treatment with Class Ia or Class III anti-arrhythmic drugs or QT prolonging drugs [see Drug Interactions (7)]
- •
- With unstable ischemic heart disease, Class I or II heart failure, history of cardiac arrest, cerebrovascular disease, or uncontrolled hypertension [see Contraindications (4)]
- •
- With resting heart rate of less than 50 bpm
- •
- With history of symptomatic bradycardia, recurrent cardiogenic syncope, or severe untreated sleep apnea
- •
- With history of Mobitz type I second-degree AV block, unless the patient has a functioning pacemaker [see Contraindications (4)]
5.3 Liver Injury
Elevations of aminotransferases may occur in patients receiving VELSIPITY. In UC-1, elevations of alanine transaminase (ALT) greater than 3-fold the upper limit of normal (ULN) occurred in 4.5% of subjects who received VELSIPITY and 2.1% of subjects who received placebo. In UC-2 and UC-3, elevations of ALT greater than 3-fold the ULN occurred in 2.5% of subjects who received VELSIPITY and 0.5% of subjects who received placebo.
Obtain transaminase and bilirubin levels, if not recently available (i.e., within last 6 months), before initiation of VELSIPITY.
Obtain transaminases and bilirubin in patients who develop symptoms suggestive of hepatic dysfunction, such as unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine. Discontinue VELSIPITY if significant liver injury is confirmed.
5.4 Macular Edema
S1P receptor modulators, including VELSIPITY, have been associated with an increased risk of macular edema. Obtain a baseline evaluation of the fundus, including the macula, near the start of treatment with VELSIPITY. Periodically conduct an evaluation of the fundus, including the macula, while on therapy and any time there is a change in vision.
Macular edema over an extended period of time (i.e., 6 months) can lead to permanent visual loss. Consider discontinuing VELSIPITY if macular edema develops.
5.5 Increased Blood Pressure
In UC-1 and UC-2 and UC-3, subjects treated with VELSIPITY had an average increase of approximately 1 to 4 mm Hg in systolic blood pressure and approximately 1 to 2 mm Hg in diastolic blood pressure compared to <1.5 mm Hg and <1 mm Hg in subjects receiving placebo, respectively. The increase was first detected after 2 weeks of treatment and remained within the specified average range of BP increases throughout treatment. Hypertension was reported as an adverse reaction in UC-1 [see Adverse Reactions (6.1)].
Monitor blood pressure during treatment with VELSIPITY and manage appropriately.
5.6 Fetal Risk
Based on animal studies, VELSIPITY may cause fetal harm when administered to a pregnant woman. In animal reproduction studies conducted in rats and rabbits, embryofetal toxicity was observed with administration of etrasimod at clinically relevant doses. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception to avoid pregnancy during and for one week after stopping VELSIPITY [see Use in Specific Populations (8.1, 8.3)].
5.7 Cutaneous Malignancies
The risk of cutaneous malignancies (including basal cell carcinoma, squamous cell carcinoma, and melanoma) is increased in patients treated with S1P receptor modulators in controlled trials. Kaposi’s sarcoma and Merkel cell carcinoma have also been reported in patients treated with S1P receptor modulators in the postmarketing setting.
Skin examinations are recommended prior to or shortly after the start of treatment and periodically thereafter for all patients, particularly those with risk factors for skin cancer. Providers and patients are advised to monitor for suspicious skin lesions. If a suspicious skin lesion is observed, it should be promptly evaluated. As usual for patients with increased risk for skin cancer, exposure to sunlight and ultraviolet (UV) light should be limited by wearing protective clothing and using a sunscreen with a high protection factor. Concomitant phototherapy with UV-B radiation or PUVA-photochemotherapy is not recommended in patients taking VELSIPITY.
5.8 Posterior Reversible Encephalopathy Syndrome
Rare cases of posterior reversible encephalopathy syndrome (PRES) have been reported in patients receiving S1P receptor modulators. If a patient develops any neurological or psychiatric symptoms/signs (e.g., cognitive deficits, behavioral changes, cortical visual disturbances, or any other neurological cortical symptoms/signs), any symptom/sign suggestive of an increase of intracranial pressure, or accelerated neurological deterioration, the physician should promptly schedule a complete physical and neurological examination and should consider an MRI. Symptoms of PRES are usually reversible but may evolve into ischemic stroke or cerebral hemorrhage. Delay in diagnosis and treatment may lead to permanent neurological sequelae. If PRES is suspected, discontinue treatment with VELSIPITY.
5.9 Respiratory Effects
Reductions in absolute forced expiratory volume over 1 second (FEV1) were observed in subjects treated with VELSIPITY as early as 3 months after treatment initiation. In UC-1, the decline in absolute FEV1 from baseline in subjects treated with VELSIPITY compared to placebo was 79 mL (95% CI: -152, -5) at 3 months. In UC-2, reductions in absolute FEV1 were not observed. There is insufficient information to determine the reversibility of the decrease in FEV1 after drug discontinuation. In UC-1 and UC-2, subjects with UC and asthma and/or chronic obstructive pulmonary disease were treated with VELSIPITY; however, interpretation of changes in pulmonary function test measures in this population are limited due to small sample sizes. Spirometric evaluation of respiratory function should be performed during therapy with VELSIPITY if clinically indicated.
5.10 Unintended Additive Immune System Effects from Prior Treatment with Immunosuppressive or Immune-Modulating Drugs
When switching to VELSIPITY from drugs with prolonged immune effects, consider the half-life and mode of action of these drugs to avoid unintended additive immunosuppressive effects [see Drug Interactions (7)].
5.11 Immune System Effects After Stopping VELSIPITY
After stopping VELSIPITY, lymphocyte counts returned to the normal range in 90% of subjects within 4 to 5 weeks of stopping VELSIPITY [see Clinical Pharmacology (12.2)]. Use of immunosuppressants within this period may lead to an additive effect on the immune system, and therefore monitor patients receiving concomitant immunosuppressants for infectious complications up to 5 weeks after the last dose of VELSIPITY [see Drug Interactions (7)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- •
- Infections [see Warnings and Precautions (5.1)]
- •
- Bradyarrhythmia and Atrioventricular Conduction Delays [see Warnings and Precautions (5.2)]
- •
- Liver Injury [see Warnings and Precautions (5.3)]
- •
- Macular Edema [see Warnings and Precautions (5.4)]
- •
- Increased Blood Pressure [see Warnings and Precautions (5.5)]
- •
- Fetal Risk [see Warnings and Precautions (5.6)]
- •
- Cutaneous Malignancies [see Warnings and Precautions (5.7)]
- •
- Posterior Reversible Encephalopathy Syndrome [see Warnings and Precautions (5.8)]
- •
- Respiratory Effects [see Warnings and Precautions (5.9)]
- •
- Unintended Additive Immune System Effects from Prior Treatment with Immunosuppressive or Immune-Modulating Drugs [see Warnings and Precautions (5.10)]
- •
- Immune System Effects After Stopping VELSIPITY [see Warnings and Precautions (5.11)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of VELSIPITY 2 mg once daily in subjects with moderately to severely active ulcerative colitis was evaluated in two randomized, placebo-controlled studies of 52 weeks (UC-1) and 12 weeks (UC-2) duration [see Clinical Studies (14)]. Additional safety data were obtained from a randomized, double-blind, placebo-controlled dose-finding study of 12 weeks duration (UC-3).
In the 52-week study (UC-1), 433 subjects were enrolled of whom 289 received VELSIPITY 2 mg once daily. In the 12-week studies (UC-2 and UC-3), 458 subjects were enrolled of whom 288 received VELSIPITY 2 mg once daily.
Table 1 summarizes the adverse reactions reported in at least 2% of subjects and at a higher rate than placebo during UC-1.
|
||
|
Adverse Reaction |
VELSIPITY 2 mg Once Daily N = 289 % |
Placebo N = 144 % |
|
Headache† |
9 |
5 |
|
Elevated liver tests‡ |
6 |
5 |
|
Dizziness§ |
5 |
2 |
|
Arthralgia |
4 |
2 |
|
Hypertension¶ |
3 |
1 |
|
Urinary tract infection# |
3 |
2 |
|
Nausea |
3 |
1 |
|
HypercholesterolemiaÞ |
3 |
0 |
|
Herpes viral infectionß |
2 |
1 |
Table 2 summarizes the adverse reactions reported in at least 2% of subjects and at a higher rate than placebo during UC-2 and UC-3.
|
||
|
Adverse Reaction |
VELSIPITY 2 mg Once Daily N = 288 %† |
Placebo N = 170 %† |
|
Headache‡ |
6 |
4 |
|
Elevated liver tests§ |
5 |
<1 |
|
Nausea |
4 |
2 |
|
Bradycardia¶ |
3 |
0 |
|
Urinary tract infection# |
3 |
0 |
Ophthalmologic Findings
In UC-1, for subjects with a baseline and follow-up examination, a decrease in visual acuity was reported in 2.6% (4/156) of subjects who received VELSIPITY and no subjects who received placebo [see Warnings and Precautions (5.4)].
Related/similar drugs
7. Drug Interactions
Etrasimod is primarily metabolized by CYP2C8, CYP2C9, and CYP3A4. Table 3 includes drugs with clinically important drug interactions when administered concomitantly with VELSIPITY and instructions for preventing or managing them. Consult the labeling of concomitantly used drugs to obtain further information.
The effect of concomitant use of VELSIPITY with a combination of separate drugs that are moderate to strong inhibitors or inducers of either CYP2C8, CYP2C9, or CYP3A4 is unknown. However, based on the information below, a similar clinically significant change in exposure cannot be ruled out when two or more metabolic pathways are affected.
|
Anti-Arrhythmic Drugs and QT Prolonging Drugs |
|
|
Clinical Impact |
A transient decrease in heart rate and AV conduction delays may occur when initiating VELSIPITY [see Warnings and Precautions (5.2)]. Because of the potential additive effect on heart rate, VELSIPITY may increase the risk of QT prolongation and Torsades de Pointes with concomitant use of Class Ia and Class III anti-arrhythmic drugs and QT prolonging drugs. |
|
Prevention or Management |
Seek the advice of a cardiologist before initiating VELSIPITY treatment with Class Ia (e.g., quinidine, procainamide), Class III anti-arrhythmic drugs (e.g., amiodarone, sotalol), or other drugs that prolong the QT interval. |
|
Beta-Blockers or Calcium Channel Blockers |
|
|
Clinical Impact |
A transient decrease in heart rate and AV conduction delays may occur when initiating VELSIPITY [see Warnings and Precautions (5.2)]. Concomitant use of VELSIPITY in patients receiving stable beta blocker treatment did not result in additive effects on heart rate reduction [see Clinical Pharmacology (12.2)]. However, the risk of additive heart rate reduction following initiation of beta blocker therapy with stable VELSIPITY treatment or concomitant use with other drugs that may decrease heart rate is unknown. |
|
Prevention or Management |
VELSIPITY can be initiated in patients receiving stable doses of beta blocker treatment. Seek the advice of a cardiologist before initiating a beta blocker in a patient receiving stable VELSIPITY treatment or concomitant use with other drugs that may decrease heart rate (e.g., calcium channel blockers). |
|
Anti-Neoplastic, Immune-Modulating, or Non-Corticosteroid Immunosuppressive Therapies |
|
|
Clinical Impact |
Risk of additive immune system effects with VELSIPITY VELSIPITY has not been studied in combination with anti-neoplastic, immune-modulating, or non-corticosteroid immunosuppressive therapies. |
|
Prevention or Management |
Avoid concomitant administration during and in the weeks following administration of VELSIPITY. When switching from drugs with prolonged immune effects, consider the half-life and mode of action of these drugs to avoid unintended additive immunosuppressive effects [see Warnings and Precautions (5.10)]. |
|
Moderate to Strong Inhibitors of CYP2C9 and CYP3A4 |
|
|
Clinical Impact |
Increased exposure of etrasimod was observed with concomitant use with a drug that is a moderate inhibitor of CYP2C9 and a moderate inhibitor of CYP3A4 (i.e., fluconazole) [see Clinical Pharmacology (12.3)]. |
|
Prevention or Management |
Concomitant use with a drug that is a moderate to strong inhibitor of CYP2C9 and a moderate to strong inhibitor of CYP3A4 is not recommended. |
|
CYP2C9 Poor Metabolizers Using Moderate to Strong Inhibitors of CYP2C8 or CYP3A4 |
|
|
Clinical Impact |
Increased exposure of etrasimod in patients who are CYP2C9 poor metabolizers is expected with concomitant use of moderate to strong inhibitors of CYP2C8 or CYP3A4 [see Clinical Pharmacology (12.3, 12.5)]. |
|
Prevention or Management |
Concomitant use not recommended. |
|
Rifampin |
|
|
Clinical Impact |
Concomitant use with a drug that is a combined CYP3A4 (strong), CYP2C8 (moderate) and CYP2C9 (moderate) inducer (i.e., rifampin) decreases exposure to etrasimod [see Clinical Pharmacology (12.3)]. |
|
Prevention or Management |
Concomitant use not recommended. |
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in females exposed to VELSIPITY during pregnancy. Pregnant females exposed to VELSIPITY and healthcare providers are encouraged to contact the pregnancy registry by calling 1-800-616-3791.
Risk Summary
Based on findings from animal studies, VELSIPITY may cause fetal harm when administered to a pregnant woman. Available data from reports of pregnancies from the clinical development program with VELSIPITY are insufficient to identify a drug-associated risk of major birth defects, miscarriage or other adverse maternal or fetal outcomes. There are risks to the mother and the fetus associated with increased disease activity in women with inflammatory bowel disease during pregnancy (see Clinical Considerations).
In animal reproduction studies, administration of etrasimod during organogenesis produced adverse effects on development, including embryolethality and fetal malformations, in both rats and rabbits at maternal exposures 5 and 6 times, respectively, the exposure at the maximum recommended human dose (MRHD). Administration of VELSIPITY to pregnant rats during organogenesis through lactation resulted in decreased pup growth and viability at maternal exposures 5 times the exposure at the MRHD, as well as impaired reproductive performance in first generation offspring, including decreased implantations and increased pre-implantation loss at maternal exposures 24 times the exposure at the MRHD (see Data).
The background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data suggest that the risk of adverse pregnancy outcomes in women with inflammatory bowel disease is associated with increased disease activity. Adverse pregnancy outcomes include preterm delivery (before 37 weeks of gestation), low birth weight (less than 2500 g) infants, and small for gestational age at birth.
Animal Data
In an embryo-fetal development study in pregnant rats, etrasimod was orally administered at 1, 2, or 4 mg/kg/day (5, 11, and 21 times the exposure at the MRHD of 2 mg, based on AUC comparison) during the period of organogenesis, from gestation day 6 to 17. No maternal toxicity was observed up to 21 times the exposure at the MRHD. Increased post-implantation loss with a corresponding decrease in the number of viable fetuses was observed at 4 mg/kg/day (21 times the exposure at the MRHD). Etrasimod-related fetal external and/or visceral malformations were noted at all dose levels (≥5 times the exposure at the MRHD).
In an embryo-fetal development study in pregnant rabbits, etrasimod was orally administered at 2, 10, or 20 mg/kg/day (0.8, 6, and 11 times the exposure at the MRHD of 2 mg, based on AUC comparison) during the period of organogenesis, from gestation day 7 to 20. Increased post-implantation loss with a corresponding decrease in the number of viable fetuses was observed at 10 and 20 mg/kg/day (6 and 11 times the exposure at the MRHD). Etrasimod-related fetal malformations including aortic arch defects and fused sternebrae were noted at 10 and/or 20 mg/kg/day (6 and 11 times the exposure at the MRHD). There were no adverse effects on embryofetal development at 2 mg/kg/day (less than the exposure at the MRHD).
In a pre- and post-natal development study in rats, etrasimod was orally administered at 0.4, 2, or 4 mg/kg/day (2, 10, and 24 times the exposure at the MRHD of 2 mg, based on AUC comparison) throughout pregnancy and lactation, from gestation day 6 through lactation day 20. Mortality during delivery and impaired maternal performance including increased post-implantation loss, increased number of females with stillborn pups, increased number of stillborn pups per litter, decreased viability index, and/or decreased lactation index was observed at 2 and 4 mg/kg/day (10 and 24 times the exposure at the MRHD). Etrasimod was detected in the plasma of F1 offspring, indicating exposure from the milk of the lactating dam. Decreased pup body weights were observed during the preweaning period at all dose levels (maternal exposures ≥2 times the exposure at the MRHD), and decreased pup viability was observed at 2 and 4 mg/kg/day (maternal exposures 10 and 24 times the exposure at the MRHD). Reduced fertility and reproductive performance including reduction in implantations and increased preimplantation loss in F1 offspring occurred at the highest dose tested (maternal exposures 24 times the exposure at the MRHD).
8.2 Lactation
Risk Summary
There are no data on the presence of etrasimod in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. When etrasimod was orally administered to female rats during pregnancy and lactation, etrasimod was detected in the plasma of the offspring, suggesting excretion of etrasimod in milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VELSIPITY and any potential adverse effects on the breastfed infant from VELSIPITY or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Based on animal data, VELSIPITY may cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Contraception
Females
Before initiation of VELSIPITY treatment, females of reproductive potential should be counseled on the potential for a serious risk to the fetus and the need for effective contraception during treatment with VELSIPITY and for one week following the last dose [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of VELSIPITY in pediatric patients have not been established.
8.5 Geriatric Use
Of the 577 VELSIPITY-treated subjects in the three clinical trials (UC-1, UC-2, and UC-3), 30 subjects (5%) were 65 years of age and older, while 3 subjects (<1%) were 75 years of age and older. Clinical studies of VELSIPITY did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger adult subjects. The pharmacokinetics of etrasimod are similar in subjects 65 years of age and older compared to younger adult subjects [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
Etrasimod undergoes extensive hepatic metabolism. Exposure to etrasimod was similar in subjects with mild and moderate hepatic impairment (Child-Pugh A and B) compared to subjects with normal hepatic function; however, etrasimod exposure was increased in subjects with severe hepatic impairment (Child‑Pugh C) compared to subjects with normal hepatic function [see Clinical Pharmacology (12.3)].
Use of VELSIPITY in patients with severe hepatic impairment is not recommended. No dosage adjustment is needed in patients with mild to moderate hepatic impairment.
8.7 CYP2C9 Poor Metabolizers
Increased exposure of etrasimod in patients who are CYP2C9 poor metabolizers is expected with concomitant use of moderate to strong inhibitors of CYP2C8 or CYP3A4. Concomitant use of VELSIPITY is not recommended in these patients [see Clinical Pharmacology (12.3, 12.5)].
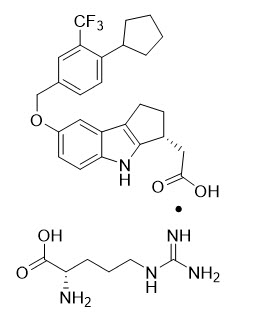
11. Velsipity Description
VELSIPITY contains etrasimod, a sphingosine 1-phosphate (S1P) receptor modulator, supplied as etrasimod arginine.
Etrasimod arginine is a white, off-white to light brown solid that is slightly soluble in water. The chemical name of etrasimod arginine is L-Arginine, (3R)-7-[[4-cyclopentyl-3-(trifluoromethyl)phenyl]methoxy]-1,2,3,4-tetrahydrocyclopent[b]indole-3-acetate (1:1) having a molecular formula of C32H40F3N5O5 and a molecular weight of 631.69 g/mol.
The chemical structure of etrasimod arginine is:
VELSIPITY is supplied for oral administration as 2 mg tablets. Each tablet contains 2 mg etrasimod (equivalent to 2.76 mg etrasimod arginine) and the following inactive ingredients: magnesium stearate, mannitol, microcrystalline cellulose, and sodium starch glycolate, with a film coating containing FD&C blue #1/brilliant blue FCF aluminum lake, FD&C blue #2/indigo carmine aluminum lake, FD&C yellow #5/tartrazine aluminum lake, macrogol 4000 JP/PEG 3350, polyvinyl alcohol (partially hydrolyzed), talc, and titanium dioxide.
12. Velsipity - Clinical Pharmacology
12.1 Mechanism of Action
Etrasimod is a sphingosine 1-phosphate (S1P) receptor modulator that binds with high affinity to S1P receptors 1, 4, and 5 (S1P1,4,5). Etrasimod has minimal activity on S1P3 (25-fold lower than Cmax at the recommended dose) and no activity on S1P2. Etrasimod partially and reversibly blocks the capacity of lymphocytes to egress from lymphoid organs, reducing the number of lymphocytes in peripheral blood. The mechanism by which etrasimod exerts therapeutic effects in UC is unknown but may involve the reduction of lymphocyte migration into the intestines.
12.2 Pharmacodynamics
Reduction in Blood Lymphocyte Counts
VELSIPITY causes a reduction in peripheral blood lymphocyte count [see Warnings and Precautions (5.1)]. In UC-1 and UC-2, mean lymphocyte counts decreased to approximately 50% of baseline at 2 weeks (approximate mean blood lymphocyte counts 0.9 x 109/L) and the lower lymphocyte counts were maintained during treatment with VELSIPITY.
Dose-response relationship analysis indicates there is a dose-dependent reduction in blood lymphocyte counts. After discontinuing VELSIPITY 2 mg once daily, the median time for peripheral blood lymphocytes to return to the normal range was 2.6 weeks, with approximately 90% of subjects in the normal range within 4.7 weeks.
Reduction in Heart Rate and AV Conduction Delays
VELSIPITY may result in a transient decrease in heart rate and AV conduction upon treatment initiation [see Warnings and Precautions (5.2)]. In UC-1 and UC-2, the mean (SD) decrease in heart rate was 7.2 (8.98) bpm at 2 to 3 hours after the first dose of VELSIPITY on Day 1.
Cardiac Electrophysiology
At 2 times the maximum recommended dose, etrasimod does not cause clinically significant QTc interval prolongation.
Pulmonary Function
Reductions in absolute FEV1 were observed in subjects treated with VELSIPITY [see Warnings and Precautions (5.9)].
Drug Interaction Studies
No clinically significant differences in heart rate reduction were observed when etrasimod was used concomitantly with stable beta blocker treatment. The effect of concomitant use of etrasimod with a calcium channel blocker on heart rate reduction is unknown [see Drug Interactions (7)].
12.3 Pharmacokinetics
Etrasimod mean (SD) steady-state maximum plasma concentration (Cmax) was 113 (27.5) ng/mL and area under the time concentration curve at the dosing interval (AUCtau) was 2162 (488) ng*h/mL at the recommended dosage. Etrasimod Cmax and AUC are approximately dose proportional from 0.7 mg to 2 mg (0.35 times up to the recommended dosage). Etrasimod steady state is reached within 7 days with an accumulation of approximately 2- to 3-fold compared to the first dose.
Absorption
The median (range) time to reach etrasimod Cmax (Tmax) is approximately 4 hours (range 2 to 8 hours) after oral administration.
Effect of Food
No clinically significant differences in the pharmacokinetics of etrasimod were observed following administration of VELSIPITY with a high-fat meal (800 to 1000 calories) [see Dosage and Administration (2.2)].
Distribution
The mean apparent volume of distribution of etrasimod is 66 (24) L. Etrasimod plasma protein binding is 97.9%.
Elimination
The mean plasma elimination half-life (t1/2) of etrasimod is approximately 30 hours with an apparent steady-state oral clearance of approximately 1 L/h after oral administration.
Metabolism
Etrasimod is metabolized by oxidation and dehydrogenation mediated primarily by CYP2C8, CYP2C9, and CYP3A4, with a minor contribution by CYP2C19 and CYP2J2. Etrasimod also undergoes conjugation primarily mediated by UGTs, with a minor contribution by sulfotransferases. Unchanged etrasimod is the main circulating component in plasma.
Excretion
Approximately 82% of total radioactive etrasimod dose was recovered in the feces and 5% in the urine within 336 hours. Approximately 11% of administered radioactive dose was excreted as unchanged etrasimod in feces and none was excreted unchanged in urine.
Specific Populations
No clinically significant differences in the pharmacokinetics of etrasimod were observed based on age (>65 years), sex, body weight, race, ethnicity, disease (healthy subjects vs. subjects with ulcerative colitis), and severe renal impairment (eGFR ≤29 mL/min).
Patients with Hepatic Impairment
Etrasimod AUC increased by 13% in subjects with mild (Child-Pugh A), 29% in moderate (Child-Pugh B), and 57% in severe (Child-Pugh C) hepatic impairment, respectively, compared with subjects with normal liver function. No clinically significant difference in the unbound etrasimod AUC was observed [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Combined moderate CYP2C9 and CYP3A4 inhibitors: Concomitant use of etrasimod with steady-state fluconazole (moderate CYP2C9 and CYP3A4 inhibitor) increased etrasimod AUC by 84% [see Drug Interactions (7)].
Combined CYP3A4 (strong), CYP2C8 (moderate) and CYP2C9 (moderate) inducers: Concomitant use of etrasimod with rifampin (strong CYP3A4, moderate CYP2C8, and CYP2C9 inducer) decreased etrasimod AUC by 49% [see Drug Interactions (7)].
Oral Contraceptives: No clinically significant differences in the pharmacokinetics and pharmacodynamics of oral contraceptive containing 30 mcg ethinyl estradiol and 150 mcg levonorgestrel were observed when used concomitantly with etrasimod.
Other Drugs: Itraconazole (a P-gp and strong CYP3A inhibitor) increased etrasimod AUC by 32%. Gemfibrozil (an inhibitor of OATP1B1 and OAT3 and a strong inhibitor of CYP2C8) increased etrasimod AUC by 36%. These effects are unlikely to be clinically significant.
In Vitro Studies
Based on in vitro studies, etrasimod is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, nor an inducer of CYP1A2, CYP2B6, and CYP3A4 at clinically relevant concentrations.
Etrasimod is not an inhibitor of UGT1A3, UGT1A4, UGT1A9, UGT2B7, or UGT2B17 in vitro.
Etrasimod is not a substrate or an inhibitor of P-gp, BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2-K transporters.
12.5 Pharmacogenomics
CYP2C9 activity is decreased in individuals with genetic variants such as CYP2C9*2 and CYP2C9*3 alleles. The impact of CYP2C9 genetic variants on the pharmacokinetics of etrasimod has not been directly evaluated. CYP2C9 poor metabolizers (e.g., *2/*3, *3/*3) may have decreased clearance of etrasimod when VELSIPITY is used concomitantly with moderate to strong inhibitors of CYP2C8 or CYP3A4 [see Drug Interactions (7) and Use in Specific Populations (8.7)].
CYP2C9 intermediate metabolizers (e.g., *1/*2, *1/*3, *2/*2) may have decreased clearance of etrasimod when VELSIPITY is used concomitantly with moderate to strong inhibitors of CYP2C8 or CYP3A4; however, the effect on VELSIPITY exposure in CYP2C9 intermediate metabolizers with concomitant CYP2C8 or CYP3A4 inhibitors is not known.
The prevalence of the CYP2C9 poor metabolizer phenotype is approximately 2 to 3% in White populations, 0.5 to 4% in Asian populations, and <1% in African-American populations. Other decreased or nonfunctional CYP2C9 alleles (e.g., *5, *6, *8, *11) are more prevalent in African-American populations.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral carcinogenicity studies with etrasimod were conducted in mice and rats. In mice administered etrasimod (2, 6, or 20 mg/kg/day) for up to 104 weeks, there was an increase in hemangiosarcoma and hemangioma in males and females at 6 and 20 mg/kg/day. No increase in tumors was observed at the lowest tested dose of 2 mg/kg/day (approximately 19 times the exposure at the MRHD of 2 mg, based on AUC comparison). In rats, oral administration of etrasimod (2, 6, or 20 mg/kg/day) for up to 91 weeks did not result in an increase in tumors (male and female exposures 80 and 179 times, respectively, the exposure at the MRHD).
Mutagenesis
Etrasimod was negative in a battery of in vitro (Ames, chromosomal aberration in human peripheral blood lymphocytes) and in vivo (rat micronucleus) assays.
Impairment of Fertility
Etrasimod administered orally to male rats at 25, 100, or 200 mg/kg/day from pre-mating through mating had no adverse effects on male fertility at exposures up to 467 times the exposure at the MRHD of 2 mg, based on AUC comparison. Etrasimod administered orally to female rats at 1, 2, or 4 mg/kg/day from pre-mating to implantation had no adverse effects on female fertility at exposures up to 21 times the exposure at the MRHD.
14. Clinical Studies
The efficacy of VELSIPITY was evaluated in 2 randomized, double-blind, placebo-controlled clinical studies [UC-1 (NCT03945188) and UC-2 (NCT03996369)] in adult subjects with moderately to severely active ulcerative colitis who had an inadequate response, loss of response, or intolerance to one or more of the following treatment options: oral aminosalicylates, corticosteroids, thiopurines, Janus kinase (JAK) inhibitors, or biologic therapies (e.g., TNF blocker, anti-integrin, anti-IL12/23). UC-1 was a 52-week study and UC-2 was a 12-week study. In both studies, subjects were randomized to VELSIPITY or placebo and continued on treatment for the entire duration of the study.
Disease severity was assessed based on the modified Mayo score (mMS), a 3-component Mayo score (0 to 9) which consists of the following subscores (0 to 3 for each subscore): stool frequency (SF), rectal bleeding (RB), and findings on centrally read endoscopy score (ES). An ES of 2 was defined by marked erythema, lack of vascular pattern, any friability, and/or erosions, and a score of 3 was defined by spontaneous bleeding and ulceration.
Subjects in these studies may have received other concomitant UC therapies including stable daily doses of oral aminosalicylates and/or oral corticosteroids (≤20 mg/day prednisone, ≤9 mg/day budesonide, or equivalent steroid). Concomitant treatment with immunomodulators (e.g., thiopurines, methotrexate), biologic therapies, JAK inhibitors, rectal 5-ASA, or rectal corticosteroids was not permitted.
Endpoints and Results Study UC-1
In UC-1, efficacy was evaluated in 408 adult subjects with a baseline mMS of 5 to 9 (median of 7), including a centrally read endoscopy subscore of 2 or 3. Subjects were randomized 2:1 to receive VELSIPITY 2 mg or placebo administered orally once daily. Subjects had a mean age of 41 years (range 18 to 78 years); 45% were female; and 89% identified as White, 7% as Asian, 2% as Black or African American, 1% as American Indian or Alaska Native, and 1% did not report their racial group. A total of 30% of subjects had prior exposure to biologic/JAK inhibitors and a total of 14% of subjects had exposure to >1 biologic/JAK inhibitor. At baseline, 68% of subjects were receiving oral aminosalicylates and 31% of subjects were receiving oral corticosteroids.
The coprimary endpoints were the proportion of subjects achieving clinical remission at Week 12 and at Week 52. The secondary endpoints included the proportion of subjects achieving endoscopic improvement, histologic-endoscopic mucosal improvement, corticosteroid-free clinical remission, and maintenance of clinical remission (see Table 4).
| CI = confidence interval | |||
|
|||
|
|
|
|
Treatment Difference* (95% CI) |
|
Coprimary Endpoints |
|||
|
Clinical Remission† at Week 12 |
|||
|
Total population |
N = 134 7% |
N = 274 27% |
20%‡ (13%, 27%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 10% |
N = 194 31% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 2% |
N = 80 18% |
|
|
Clinical Remission† at Week 52 |
|||
|
Total population |
N = 134 7% |
N = 274 32% |
26%‡ (19%, 33%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 8% |
N = 194 37% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 5% |
N = 80 21% |
|
|
Week 12 Endpoints |
|||
|
Endoscopic Improvement§ |
|||
|
Total population |
N = 134 14% |
N = 274 35% |
21%‡ (13%, 29%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 18% |
N = 194 39% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 5% |
N = 80 25% |
|
|
Histologic-Endoscopic Mucosal Improvement¶ |
|||
|
Total population |
N = 134 4% |
N = 274 21% |
17%‡ (11%, 23%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 6% |
N = 194 24% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 0% |
N = 80 14% |
|
|
Week 52 Endpoints |
|||
|
Endoscopic Improvement§ |
|||
|
Total population |
N = 134 10% |
N = 274 37% |
27%‡ (19%, 34%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 13% |
N = 194 40% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 5% |
N = 80 30% |
|
|
Histologic-Endoscopic Mucosal Improvement¶ |
|||
|
Total population |
N = 134 8% |
N = 274 27% |
18%‡ (11%, 25%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 11% |
N = 194 28% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 2% |
N = 80 23% |
|
|
Corticosteroid-free Clinical Remission# |
|||
|
Total population |
N = 134 7% |
N = 274 32% |
26%‡ (19%, 33%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 8% |
N = 194 37% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 5% |
N = 80 21% |
|
|
Maintenance of Clinical RemissionÞ |
|||
|
Total population |
N = 134 2% |
N = 274 18% |
16%‡ (11%, 21%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 93 2% |
N = 194 22% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 41 2% |
N = 80 10% | |
The relationship of histologic-endoscopic mucosal improvement at Week 12 or Week 52 to disease progression and longer-term outcomes after Week 52 was not evaluated in Study UC-1.
Clinical Response
A greater proportion of subjects treated with VELSIPITY compared to placebo achieved clinical response, defined as a ≥2 point and ≥30% decrease from baseline in mMS, and a ≥1 point decrease from baseline in RB subscore or an absolute RB subscore ≤1 at Week 12 (62% vs 34%).
Stool Frequency and Rectal Bleeding Subscores
Decreases in SF subscores were observed as early as Week 2 and decreases in RB subscores were observed as early as Week 4 in subjects treated with VELSIPITY compared to placebo.
Endoscopic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. A greater proportion of subjects treated with VELSIPITY compared to placebo achieved endoscopic remission by Week 12 (15% vs 4%), Week 52 (26% vs 6%), and both Week 12 and Week 52 (11% vs 1%).
Endpoints and Results Study UC-2
In UC-2, efficacy was evaluated in 333 adult subjects with a baseline mMS of 5 to 9 (median of 7), including a centrally read endoscopy subscore of 2 or 3. Subjects were randomized 2:1 to receive VELSIPITY 2 mg or placebo administered orally once daily. Subjects had a mean age of 41 years (range 18 to 73 years); 41% were female; and 76% identified as White, 19% as Asian, 2% as American Indian or Alaska Native, 1% as Black or African American, and 2% as multiple racial groups or did not report their racial group. A total of 34% of subjects had prior exposure to biologic/JAK inhibitors and a total of 17% of subjects had exposure to >1 biologic/JAK inhibitor. At baseline, 66% of subjects were receiving oral aminosalicylates and 28% of subjects were receiving oral corticosteroids.
The primary endpoint was the proportion of subjects achieving clinical remission at Week 12. The secondary endpoints included the proportion of subjects achieving endoscopic improvement and histologic-endoscopic mucosal improvement at Week 12 (see Table 5).
| CI = confidence interval | |||
|
|||
|
|
|
|
Treatment Difference* (95% CI) |
|
Clinical Remission† |
|||
|
Total population |
N = 112 15% |
N = 221 26% |
11%‡ (3%, 20%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 74 16% |
N = 147 30% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 38 13% |
N = 74 19% |
|
|
Endoscopic Improvement§ |
|||
|
Total population |
N = 112 19% |
N = 221 30% |
12%‡ (3%, 21%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 74 19% |
N = 147 34% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 38 18% |
N = 74 23% |
|
|
Histologic-Endoscopic Mucosal Improvement¶ |
|||
|
Total population |
N = 112 9% |
N = 221 16% |
8%‡ (1%, 15%) |
|
No prior biologic/ JAK inhibitor exposure |
N = 74 11% |
N = 147 19% | |
|
Prior biologic/ JAK inhibitor exposure |
N = 38 5% |
N = 74 11% | |
The relationship of histologic-endoscopic mucosal improvement at Week 12 to disease progression and longer-term outcomes after Week 12 was not evaluated in Study UC-2.
Clinical Response
A greater proportion of subjects treated with VELSIPITY compared to placebo achieved clinical response, defined as a ≥2 point and ≥30% decrease from baseline in mMS, and a ≥1 point decrease from baseline in RB subscore or an absolute RB subscore ≤1 at Week 12 (62% vs 41%).
Stool Frequency and Rectal Bleeding Subscores
Decreases in SF subscores were observed as early as Week 2 and decreases in RB subscores were observed as early as Week 4 in subjects treated with VELSIPITY compared to placebo.
Endoscopic Assessment
Normalization of the endoscopic appearance of the mucosa (endoscopic remission) was defined as ES of 0. A greater proportion of subjects treated with VELSIPITY compared to placebo achieved endoscopic remission by Week 12 (17% vs 8%).
16. How is Velsipity supplied
How Supplied
VELSIPITY tablet is available as a round, green film-coated 2 mg etrasimod tablet, debossed with “ETR” on one side and “2” on the other side.
VELSIPITY tablets are packed in a child-resistant 100 mL white, round, high-density polyethylene (HDPE) bottle containing 4 g of silica gel desiccant integrated directly into the 45 mm polypropylene (PP) cap.
|
Dosage Form |
Strength |
Description |
NDC Number |
|
Tablets |
2 mg of etrasimod |
30 count bottle |
0069-0274-30 |
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Risk of Infections
Inform patients that they may be more likely to get infections, some of which could be life-threatening, when taking VELSIPITY and for 5 weeks after stopping it, and that they should contact their healthcare provider if they develop symptoms of infection [see Warnings and Precautions (5.1)]. Inform patients that prior or concomitant use of drugs that suppress the immune system may increase the risk of infection.
Advise patients that some vaccines containing live virus (live attenuated vaccines) should be avoided during treatment with VELSIPITY. Advise patients that if immunizations are planned, they should be administered at least 4 weeks prior to initiation of VELSIPITY. Inform patients that the use of live attenuated vaccines should be avoided during and for 5 weeks after treatment with VELSIPITY.
Cardiac Effects
Advise patients that initiation of VELSIPITY treatment may result in transient decrease in heart rate [see Warnings and Precautions (5.2)].
Liver Injury
Inform patients that VELSIPITY may increase liver enzymes. Advise patients that they should contact their healthcare provider if they have any unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine [see Warnings and Precautions (5.3)].
Macular Edema
Advise patients that VELSIPITY may cause macular edema, and that they should obtain an eye exam near the start of treatment with VELSIPITY, have their eyes monitored periodically by an eye care professional while receiving therapy, and contact their healthcare provider if they experience any changes in their vision while taking VELSIPITY [see Warnings and Precautions (5.4)].
Fetal Risk
VELSIPITY may cause fetal harm. Advise females to immediately inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with VELSIPITY and for one week after stopping VELSIPITY [see Use in Specific Populations (8.3)].
Pregnancy and Pregnancy Registry
Advise patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to VELSIPITY during pregnancy [see Use in Specific Populations (8.1)].
Cutaneous Malignancies
Advise patients to limit exposure to sunlight and ultraviolet (UV) light, wear protective clothing, and use a sunscreen with a high protection factor. Concomitant phototherapy with UV-B radiation or PUVA‑photochemotherapy is not recommended in patients taking VELSIPITY. If a suspicious skin lesion is observed, patients should immediately report it to their healthcare provider [see Warnings and Precautions (5.7)].
Posterior Reversible Encephalopathy Syndrome
Advise patients to immediately report to their healthcare provider any symptoms involving sudden onset of severe headache, altered mental status, visual disturbances, or seizure. Inform patients that delayed treatment could lead to permanent neurological consequences [see Warnings and Precautions (5.8)].
Respiratory Effects
Advise patients that they should contact their healthcare provider if they experience new onset or worsening dyspnea [see Warnings and Precautions (5.9)].
Immune System Effects after Stopping VELSIPITY
Advise patients that VELSIPITY continues to have effects, such as lowering effects on peripheral lymphocyte count, for up to 5 weeks after the last dose, and to monitor for signs and symptoms of infection during that time [see Warnings and Precautions (5.11)].
This product’s labeling may have been updated. For the most recent prescribing information, please visit www.pfizer.com.
For Medical Information about VELSIPITY, please visit www.pfizermedinfo.com or call 1-800-438-1985.

| This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 06/2024 | |||||
|
MEDICATION GUIDE
|
|||||
|
Read this Medication Guide before you start taking VELSIPITY and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or treatment. |
|||||
|
What is the most important information I should know about VELSIPITY?
|
|||||
|
|||||
|
|
||||
|
Your healthcare provider may delay or stop your VELSIPITY treatment if you have an infection.
|
|||||
|
|
||||
|
See "What are the possible side effects of VELSIPITY?" for more information about side effects. |
|||||
|
What is VELSIPITY?
It is not known if VELSIPITY is safe and effective in children. |
|||||
|
Do not take VELSIPITY if you:
Talk to your healthcare provider before taking VELSIPITY if you have any of these conditions or do not know if you have any of these conditions. |
|||||
|
Before taking VELSIPITY, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
You should not receive live vaccines at least 4 weeks before starting VELSIPITY, during treatment with VELSIPITY and for 5 weeks after you stop taking VELSIPITY. Talk to your healthcare provider before you receive a vaccine during treatment and for 5 weeks after treatment with VELSIPITY. If you receive a live vaccine, you may get the infection the vaccine was meant to prevent. Vaccines may not work as well when given during treatment with VELSIPITY. Know the medicines you take. Keep a list of your medicines to show the list to your healthcare provider and pharmacist when you get a new medicine. |
|||||
|
How should I take VELSIPITY?
|
|||||
|
What are the possible side effects of VELSIPITY?
|
|||||
|
|
||||
|
|||||
|
|
||||
The most common side effects of VELSIPITY include headache, elevated liver tests, and dizziness. |
|||||
|
How should I store VELSIPITY?
Keep VELSIPITY and all medicines out of the reach of children. |
|||||
|
General information about the safe and effective use of VELSIPITY.
|
|||||
|
What are the ingredients in VELSIPITY?
|
|||||

| VELSIPITY
etrasimod tablet, film coated |
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
|
||||||||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Pfizer Ireland Pharmaceuticals Unlimited Company | 985052076 | API MANUFACTURE(0069-0274) , ANALYSIS(0069-0274) | |
More about Velsipity (etrasimod)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: selective immunosuppressants
- Breastfeeding
- En español
