Prosom: Package Insert / Prescribing Info
Package insert / product label
Generic name: estazolam
Dosage form: Tablets CS-IV
Drug class: Benzodiazepines
Medically reviewed by Drugs.com. Last updated on Mar 25, 2025.
The Prosom brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
Prosom Description
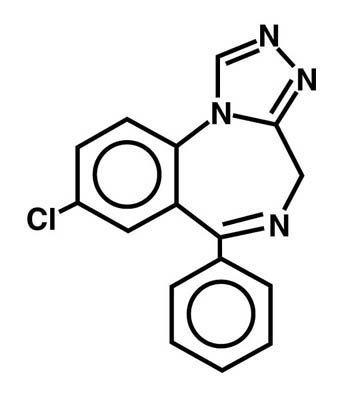
ProSom (estazolam), a triazolobenzodiazepine derivative, is an oral hypnotic agent. Estazolam occurs as a fine, white, odorless powder that is soluble in alcohol and practically insoluble in water. The chemical name for estazolam is 8−chloro−6−phenyl−4H−s−triazolo[4,3-α] [1,4]benzodiazepine. The empirical formula is C16H11ClN4. The structural formula is represented as follows:
ProSom tablets are scored and contain either 1 mg or 2 mg of estazolam.
Prosom - Clinical Pharmacology
Pharmacokinetics
Absorption
ProSom tablets have been found to be equivalent in absorption to an orally administered solution of estazolam. In healthy subjects who received up to three times the recommended dose of ProSom, peak estazolam plasma concentrations occurred within two hours after dosing (range 0.5 to 6.0 hours) and were proportional to the administered dose, suggesting linear pharmacokinetics over the dosage range tested.
Metabolism
Estazolam is extensively metabolized. Only two metabolites (1-oxo-estazolam & 4 − hydroxy − estazolam) were detected in human plasma up to 18 hrs.
The pharmacologic activity of estazolam is primarily from the parent drug. The elimination of the parent drug takes place via hepatic metabolism of estazolam to hydroxylated and other metabolites that are eliminated largely in the urine both free and conjugated. In humans, greater than 70% of a single dose of estazolam was recovered in the urine as metabolites. Less than 5% of a 2 mg dose of estazolam was excreted unchanged in the urine, with only 4% of the dose appearing in the feces. The principal urinary excretion product is an unidentified metabolite, presumed to be a metabolic product of 4-hydroxy-estazolam, accounting for at least 27% of the administered dose. 4 − hydroxy-estazolam is the major metabolite in plasma, with concentrations approaching 12% of those of the parent eight hours after administration. Urinary 4 − hydroxy − estazolam and 1-oxo-estazolam account for 11.9% and 4.4% of the dose respectively. In vitro studies with human liver microsomes indicate that the biotransformation of estazolam to the major circulating metabolite 4 − hydroxy − estazolam is mediated by cytochrome P450 3A (CYP3A). While 4-hydroxy-estazolam and the lesser metabolite, 1-oxo-estazolam, have some pharmacologic activity, their low potencies and low concentrations preclude any significant contribution to the hypnotic effect of ProSom.
Elimination
The range of estimates for the mean elimination half-life of estazolam varied from 10 to 24 hours. Radiolabel mass balance studies indicate that the main route of excretion is via the kidneys. After 5 days, 87% of the administered radioactivity was excreted in human urine. Less than 4% of the dose was excreted unchanged. Eleven metabolites were found in urine. Four metabolites were identified as 1-oxo-estazolam, 4' − hydroxy − estazolam, 4-hydroxy-estazolam, and benzophenone, as free metabolites and glucuronides. The predominant metabolite in urine (17% of the administered dose) has not been identified, but is likely to be a metabolite of 4-hydroxy-estazolam.
Special Populations
In a small study (N=8) using various doses in older subjects (59 to 68 years), peak estazolam concentrations were found to be similar to those observed in younger subjects with a mean elimination half-life of 18.4 hours (range 13.5 to 34.6 hours). The influence of hepatic or renal impairment on the pharmacokinetics of estazolam has not been studied.
Cigarette Smoking
The clearance of benzodiazepines is accelerated in smokers compared to nonsmokers, and there is evidence that this occurs with estazolam. This decrease in half-life, presumably due to enzyme induction by smoking, is consistent with other drugs with similar hepatic clearance characteristics. In all subjects and at all doses, the mean elimination half-life appeared to be independent of the dose.
Drug-Drug Interaction
The metabolism of estazolam to the major circulating metabolite 4-hydroxy-estazolam is catalyzed by CYP3A. While no in vivo drug-drug interaction studies were conducted between estazolam and inhibitors/inducers of CYP3A, compounds that are potent CYP3A inhibitors (such as ketoconazole, itraconazole, nefazodone, fluvoxamine, and erythromycin) would be expected to increase plasma estazolam concentrations and CYP3A inducers (such as carbamazepine, phenytoin, rifampin and barbiturates) would be expected to decrease estazolam concentrations.
Drug Interaction with Fluoxetine
A multiple-dose study was conducted to assess the effect of fluoxetine 20 mg BID on the pharmacokinetics of estazolam 2 mg QHS after seven days. The pharmacokinetics of estazolam (Cmax and AUC) were not affected during multiple-dose fluoxetine, suggesting no clinically significant pharmacokinetic interaction.
The Ability of Estazolam to Induce or Inhibit Human Enzyme Systems
The results from in vitro human liver microsomal studies suggest that at therapeutic concentrations, estazolam has no significant inhibitory effect on the major human cytochrome P450 enzyme activities (i.e., CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A). The ability of estazolam to induce human hepatic enzyme systems has not been determined.
Pharmacodynamics
Postulated relationship between elimination rate of benzodiazepine hypnotics and their profile of common untoward effects
The type and duration of hypnotic effects and the profile of unwanted effects during administration of benzodiazepine drugs may be influenced by the biologic half-life of administered drug and any active metabolites formed. If half-lives are long, drug or metabolites may accumulate during periods of nightly administration and may be associated with impairments of cognitive and/or motor performance during waking hours; the possibility of interaction with other psychoactive drugs or alcohol will be increased. In contrast, if half-lives are short, drug and metabolites will be cleared before the next dose is ingested, and carry-over effects related to excessive sedation or CNS depression should be minimal or absent. However, during nightly use for an extended period, pharmacodynamic tolerance or adaptation to some effects of benzodiazepine hypnotics may develop. If the drug has a short elimination half-life, it is possible that a relative deficiency of the drug or its active metabolites (i.e., in relationship to the receptor site) may occur at some point in the interval between each night's use. This sequence of events may account for two clinical findings reported to occur after several weeks of nightly use of rapidly eliminated benzodiazepine hypnotics, namely, increased wakefulness during the last third of the night and increased daytime anxiety in selected patients.
Clinical Studies
Controlled Trials Supporting Efficacy
In three 7-night, double-blind, parallel-group trials comparing estazolam 1 mg and/or 2 mg with placebo in adult outpatients with chronic insomnia, estazolam 2 mg was consistently superior to placebo in subjective measures of sleep induction (latency) and sleep maintenance (duration, number of awakenings, depth and quality of sleep); estazolam 1 mg was similarly superior to placebo on all measures of sleep maintenance, however, it significantly improved sleep induction in only one of two studies. In a similarly designed trial comparing estazolam 0.5 mg and 1 mg with placebo in geriatric outpatients with chronic insomnia, only the 1 mg estazolam dose was consistently superior to placebo in sleep induction (latency) and in only one measure of sleep maintenance (i.e., duration of sleep).
In a single-night, double-blind, parallel-group trial comparing estazolam 2 mg and placebo in patients admitted for elective surgery and requiring sleep medications, estazolam was superior to placebo in subjective measures of sleep induction and maintenance.
In a 12-week, double-blind, parallel-group trial including a comparison of estazolam 2 mg and placebo in adult outpatients with chronic insomnia, estazolam was superior to placebo in subjective measures of sleep induction (latency) and maintenance (duration, number of awakenings, total wake time during sleep) at Week 2, but produced consistent improvement over 12 weeks only for sleep duration and total wake time during sleep. Following withdrawal at Week 12, rebound insomnia was seen at the first withdrawal week, but there was no difference between drug and placebo by the second withdrawal week in all parameters except latency, for which normalization did not occur until the fourth withdrawal week.
Adult outpatients with chronic insomnia were evaluated in a sleep laboratory trial comparing four doses of estazolam (0.25, 0.50, 1.0 and 2.0 mg) and placebo, each administered for 2 nights in a crossover design. The higher estazolam doses were superior to placebo in most EEG measures of sleep induction and maintenance, especially at the 2 mg dose, but only for sleep duration in subjective measures of sleep.
Indications and Usage for Prosom
ProSom (estazolam) is indicated for the short-term management of insomnia characterized by difficulty in falling asleep, frequent nocturnal awakenings, and/or early morning awakenings. Both outpatient studies and a sleep laboratory study have shown that ProSom administered at bedtime improved sleep induction and sleep maintenance (see CLINICAL PHARMACOLOGY).
Because insomnia is often transient and intermittent, the prolonged administration of ProSom is generally neither necessary nor recommended. Since insomnia may be a symptom of several other disorders, the possibility that the complaint may be related to a condition for which there is a more specific treatment should be considered.
There is evidence to support the ability of ProSom to enhance the duration and quality of sleep for intervals up to 12 weeks (see CLINICAL PHARMACOLOGY).
Contraindications
Benzodiazepines may cause fetal damage when administered during pregnancy. An increased risk of congenital malformations associated with the use of diazepam and chlordiazepoxide during the first trimester of pregnancy has been suggested in several studies. Transplacental distribution has resulted in neonatal CNS depression and also withdrawal phenomena following the ingestion of therapeutic doses of a benzodiazepine hypnotic during the last weeks of pregnancy.
ProSom is contraindicated in pregnant women. If there is a likelihood of the patient becoming pregnant while receiving ProSom she should be warned of the potential risk to the fetus and instructed to discontinue the drug prior to becoming pregnant. The possibility that a woman of childbearing potential is pregnant at the time of institution of therapy should be considered.
Estazolam is contraindicated with ketoconazole and itraconazole, since these medications significantly impair oxidative metabolism mediated by CYP3A (see WARNINGS and PRECAUTIONS - Drug Interactions).
Warnings
ProSom, like other benzodiazepines, has CNS depressant effects. For this reason, patients should be cautioned against engaging in hazardous occupations requiring complete mental alertness, such as operating machinery or driving a motor vehicle, after ingesting the drug, including potential impairment of the performance of such activities that may occur the day following ingestion of ProSom. Patients should also be cautioned about possible combined effects with alcohol and other CNS depressant drugs.
As with all benzodiazepines, amnesia, paradoxical reactions (e.g,, excitement, agitation, etc.), and other adverse behavioral effects may occur unpredictably.
There have been reports of withdrawal signs and symptoms of the type associated with withdrawal from CNS depressant drugs following the rapid decrease or the abrupt discontinuation of benzodiazepines (see DRUG ABUSE AND DEPENDENCE ).
Estazolam Interaction with Drugs that Inhibit Metabolism via Cytochrome P450 3A (CYP3A)
The metabolism of estazolam to the major circulating metabolite 4-hydroxy-estazolam and the metabolism of other triazolobenzodiazepines is catalyzed by CYP3A. Consequently, estazolam should be avoided in patients receiving ketoconazole and itraconazole, which are very potent inhibitors of CYP3A (see CONTRAINDICATIONS). With drugs inhibiting CYP3A to a lesser, but still significant degree, estazolam should be used only with caution and consideration of appropriate dosage reduction. The following are examples of drugs known to inhibit the metabolism of other related benzodiazepines, presumably through inhibition of CYP3A: nefazodone, fluvoxamine, cimetidine, diltiazem, isoniazide, and some macrolide antibiotics.
While no in vivo drug-drug interaction studies were conducted between estazolam and inducers of CYP3A, compounds that are potent CYP3A inducers (such as carbamazepine, phenytoin, rifampin, and barbiturates) would be expected to decrease estazolam concentrations.
Precautions
General
Impaired motor and/or cognitive performance attributable to the accumulation of benzodiazepines and their active metabolites following several days of repeated use at their recommended doses is a concern in certain vulnerable patients (e.g., those especially sensitive to the effects of benzodiazepines or those with a reduced capacity to metabolize and eliminate them) (see DOSAGE AND ADMINISTRATION).
Elderly or debilitated patients and those with impaired renal or hepatic function should be cautioned about these risks and advised to monitor themselves for signs of excessive sedation or impaired conditions.
ProSom appears to cause dose-related respiratory depression that is ordinarily not clinically relevant at recommended doses in patients with normal respiratory function. However, patients with compromised respiratory function may be at risk and should be monitored appropriately. As a class, benzodiazepines have the capacity to depress respiratory drive; there are insufficient data available, however, to characterize their relative potency in depressing respiratory drive at clinically recommended doses.
As with other benzodiazepines, ProSom should be administered with caution to patients exhibiting signs or symptoms of depression. Suicidal tendencies may be present in such patients and protective measures may be required. Intentional overdosage is more common in this group of patients; therefore, the least amount of drug that is feasible should be prescribed for the patient at any one time.
Information for Patients
To assure the safe and effective use of ProSom, the following information and instructions should be given to patients:
- Inform your physician about any alcohol consumption and medicine you are taking now, including drugs you may buy without a prescription. Alcohol should not be used during treatment with hypnotics.
- Inform your physician if you are planning to become pregnant, if you are pregnant, or if you become pregnant while you are taking this medicine.
- You should not take this medicine if you are nursing, as the drug may be excreted in breast milk.
- Until you experience the way this medicine affects you, do not drive a car, operate potentially dangerous machinery, or engage in hazardous occupations requiring complete mental alertness after taking this medicine.
- Since benzodiazepines may produce psychological and physical dependence, you should not increase the dose before consulting your physician. In addition, since the abrupt discontinuation of ProSom may be associated with temporary sleep disturbances, you should consult your physician before abruptly discontinuing doses of 2 mg per night or more.
Laboratory Tests
Laboratory tests are not ordinarily required in otherwise healthy patients. When treatment with ProSom is protracted, periodic blood counts, urinalyses, and blood chemistry analyses are advisable.
Drug Interactions
If ProSom is given concomitantly with other drugs acting on the central nervous system, careful consideration should be given to the pharmacology of all agents. The action of the benzodiazepines may be potentiated by anticonvulsants, antihistamines, alcohol, barbiturates, monoamine oxidase inhibitors, narcotics, phenothiazines, psychotropic medications, or other drugs that produce CNS depression. Smokers have an increased clearance of benzodiazepines as compared to nonsmokers; this was seen in studies with estazolam (see CLINICAL PHARMACOLOGY).
While no in vivo drug-drug interaction studies were conducted between estazolam and inducers of CYP3A, compounds that are potent CYP3A inducers (such as carbamazepine, phenytoin, rifampin, and barbiturates) would be expected to decrease estazolam concentrations.
Estazolam Interaction with Drugs that Inhibit Metabolism via Cytochrome P450 3A (CYP3A)
The metabolism of estazolam to the major circulating metabolite 4-hydroxy-estazolam and the metabolism of other triazolobenzodiazepines is catalyzed by CYP3A. Consequently, estazolam should be avoided in patients receiving ketoconazole and itraconazole, which are very potent inhibitors of CYP3A (see CONTRAINDICATIONS). With drugs inhibiting CYP3A to a lesser, but still significant degree, estazolam should be used only with caution and consideration of appropriate dosage reduction. The following are examples of drugs known to inhibit the metabolism of other related benzodiazepines, presumably through inhibition of CYP3A: nefazodone, fluvoxamine, cimetidine, diltiazem, isoniazide, and some macrolide antibiotics.
Drug Interaction with Fluoxetine
A multiple-dose study was conducted to assess the effect of fluoxetine 20 mg BID on the pharmacokinetics of estazolam 2 mg QHS after seven days. The pharmacokinetics of estazolam (Cmax and AUC) were not affected during multiple-dose fluoxetine, suggesting no clinically significant pharmacokinetic interaction.
Estazolam Interaction with Other Drugs that are Metabolized by Cytochrome P450 (CYP)
At clinically relevant concentrations, in vitro studies indicate that estazolam (0.6 µM) was not inhibitory towards the major cytochrome P450 isoforms CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A. Therefore, based on these in vitro data, estazolam is very unlikely to inhibit the biotransformation of other drugs metabolized by these CYP isoforms.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted in mice and rats at dietary doses of 0.8, 3, and 10 mg/kg/day and 0.5, 2, and 10 mg/kg/day, respectively. Evidence of tumorigenicity was not observed in either study. Incidence of hyperplastic liver nodules increased in female mice given the mid- and high-dose levels. The significance of such nodules in mice is not known at this time.
In vitro and in vivo mutagenicity tests including the Ames test, DNA repair in B. subtilis, in vivo cytogenetics in mice and rats, and the dominant lethal test in mice did not show a mutagenic potential for estazolam.
Fertility in male and female rats was not affected by doses up to 30 times the usual recommended human dose.
Pregnancy
- Teratogenic Effects: Pregnancy Category X (see CONTRAINDICATIONS).
- Nonteratogenic Effects: The child born of a mother taking benzodiazepines may be at some risk for withdrawal symptoms during the postnatal period. Neonatal flaccidity has been reported in an infant born of a mother who received benzodiazepines during pregnancy.
Nursing Mothers
Human studies have not been conducted; however, studies in lactating rats indicate that estazolam and/or its metabolites are secreted in the milk. The use of ProSom in nursing mothers is not recommended.
Pediatric Use
Safety and effectiveness in pediatric patients below the age of 18 have not been established.
Geriatric Use
Approximately 18% of individuals participating in the premarketing clinical trials of ProSom were 60 years of age or older. Overall, the adverse event profile did not differ substantively from that observed in younger individuals. Care should be exercised when prescribing benzodiazepines to small or debilitated elderly patients (see DOSAGE AND ADMINISTRATION).
Adverse Reactions/Side Effects
Commonly Observed
The most commonly observed adverse events associated with the use of ProSom, not seen at an equivalent incidence among placebo-treated patients were somnolence, hypokinesia, dizziness, and abnormal coordination.
Associated with Discontinuation of Treatment
Approximately 3% of 1277 patients who received ProSom in US premarketing clinical trials discontinued treatment because of an adverse clinical event. The only event commonly associated with discontinuation, accounting for 1.3% of the total, was somnolence.
Incidence in Controlled Clinical Trials
The table below enumerates adverse events that occurred at an incidence of 1% or greater among patients with insomnia who received ProSom in 7-night, placebo-controlled trials. Events reported by investigators were classified into standard dictionary (COSTART) terms to establish event frequencies. Event frequencies reported were not corrected for the occurrence of these events at baseline. The frequencies were obtained from data pooled across six studies: ProSom, N=685; placebo, N=433. The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice in which patient characteristics and other factors differ from those that prevailed in these six clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigators involving related drug products and uses, since each group of drug trials was conducted under a different set of conditions. However, the cited figures provide the physician with a basis of estimating the relative contribution of drug and nondrug factors to the incidence of side effects in the population studied.
| Body System/ Adverse Event* | ProSom (N=685) | Placebo (N=433) |
|
* Events reported by at least 1% of ProSom patients. |
||
| Body as a Whole | ||
| Headache | 16 | 27 |
| Asthenia | 11 | 8 |
| Malaise | 5 | 5 |
| Lower extremity pain | 3 | 2 |
| Back pain | 2 | 2 |
| Body pain | 2 | 2 |
| Abdominal pain | 1 | 2 |
| Chest pain | 1 | 1 |
| Digestive System | ||
| Nausea | 4 | 5 |
| Dyspepsia | 2 | 2 |
| Musculoskeletal System | ||
| Stiffness | 1 | - |
| Nervous System | ||
| Somnolence | 42 | 27 |
| Hypokinesia | 8 | 4 |
| Nervousness | 8 | 11 |
| Dizziness | 7 | 3 |
| Coordination abnormal | 4 | 1 |
| Hangover | 3 | 2 |
| Confusion | 2 | - |
| Depression | 2 | 3 |
| Dream abnormal | 2 | 2 |
| Thinking abnormal | 2 | 1 |
| Respiratory System | ||
| Cold symptoms | 3 | 5 |
| Pharyngitis | 1 | 2 |
| Skin and Appendages | ||
| Pruritus | 1 | - |
Other Adverse Events
During clinical trials conducted by Abbott, some of which were not placebo − controlled, ProSom was administered to approximately 1300 patients. Untoward events associated with this exposure were recorded by clinical investigators using terminology of their own choosing. To provide a meaningful estimate of the proportion of individuals experiencing adverse events, similar types of untoward events must be grouped into a smaller number of standardized event categories. In the tabulations that follow, a standard COSTART dictionary terminology has been used to classify reported adverse events. The frequencies presented, therefore, represent the proportion of the 1277 individuals exposed to ProSom who experienced an event of the type cited on at least one occasion while receiving ProSom. All reported events are included except those already listed in the previous table, those COSTART terms too general to be informative, and those events where a drug cause was remote. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are defined as those occurring on one or more occasions in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in less than 1/1000 patients. It is important to emphasize that, although the events reported did occur during treatment with ProSom, they were not necessarily caused by it.
Nervous System
Special Senses
Urogenital System
Postintroduction Reports
Voluntary reports of non-US postmarketing experience with estazolam have included rare occurrences of photosensitivity, Stevens-Johnson syndrome, and agranulocytosis. Because of the uncontrolled nature of these spontaneous reports, a causal relationship to estazolam treatment has not been determined.
Related/similar drugs
Drug Abuse and Dependence
Abuse and Dependence
Withdrawal symptoms similar to those noted with sedatives/hypnotics and alcohol have occurred following the abrupt discontinuation of drugs in the benzodiazepine class. The symptoms can range from mild dysphoria and insomnia to a major syndrome that may include abdominal and muscle cramps, vomiting, sweating, tremors, and convulsions.
Although withdrawal symptoms are more commonly noted after the discontinuation of higher than therapeutic doses of benzodiazepines, a proportion of patients taking benzodiazepines chronically at therapeutic doses may become physically dependent on them. Available data, however, cannot provide a reliable estimate of the incidence of dependency or the relationship of the dependency to dose and duration of treatment. There is some evidence to suggest that gradual reduction of dosage will attenuate or eliminate some withdrawal phenomena. In most instances, withdrawal phenomena are relatively mild and transient; however, life-threatening events (e.g., seizures, delirium, etc.) have been reported.
Gradual withdrawal is the preferred course for any patient taking benzodiazepines for a prolonged period. Patients with a history of seizures, regardless of their concomitant antiseizure drug therapy, should not be withdrawn abruptly from benzodiazepines.
Individuals with a history of addiction to or abuse of drugs or alcohol should be under careful surveillance when receiving benzodiazepines because of the risk of habituation and dependence to such patients.
Overdosage
As with other benzodiazepines, experience with ProSom indicates that manifestations of overdosage include somnolence, respiratory depression, confusion, impaired coordination, slurred speech, and ultimately, coma. Patients have recovered from overdosage as high as 40 mg. As in the management of intentional overdose with any drug, the possibility should be considered that multiple agents may have been taken.
Gastric evacuation, either by the induction of emesis, lavage, or both, should be performed immediately. Maintenance of adequate ventilation is essential. General supportive care, including frequent monitoring of the vital signs and close observation of the patient, is indicated. Fluids should be administered intravenously to maintain blood pressure and encourage diuresis. The value of dialysis in treatment of benzodiazepine overdose has not been determined. The physician may wish to consider contacting a Poison Control Center for up-to-date information on the management of hypnotic drug product overdose.
Flumazenil, a specific benzodiazepine receptor antagonist, is indicated for the complete or partial reversal of the sedative effects of benzodiazepines and may be used in situations when an overdose with a benzodiazepine is known or suspected. Prior to the administration of flumazenil, necessary measures should be instituted to secure airway, ventilation, and intravenous access. Flumazenil is intended as an adjunct to, not as a substitute for, proper management of benzodiazepine overdose. Patients treated with flumazenil should be monitored for resedation, respiratory depression, and other residual benzodiazepine effects for an appropriate period after treatment. The prescriber should be aware of a risk of seizure in association with flumazenil treatment, particularly in long − term benzodiazepine users and in cyclic antidepressant overdose. The complete flumazenil package insert including CONTRAINDICATIONS, WARNINGS, and PRECAUTIONS should be consulted prior to use.
Prosom Dosage and Administration
The recommended initial dose for adults is 1 mg at bedtime; however, some patients may need a 2 mg dose. In healthy elderly patients, 1 mg is also the appropriate starting dose, but increases should be initiated with particular care. In small or debilitated older patients, a starting dose of 0.5 mg, while only marginally effective in the overall elderly population, should be considered.
| PROSOM
estazolam tablet |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| PROSOM
estazolam tablet |
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
|
||||||||||||||||||||||
| Labeler - Abbott Laboratories |
More about Prosom (estazolam)
- Check interactions
- Compare alternatives
- Reviews (2)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: benzodiazepines
- Breastfeeding
