Optison: Package Insert / Prescribing Info
Package insert / product label
Generic name: human albumin microspheres
Dosage form: injection, solution
Drug class: Ultrasound contrast media
Medically reviewed by Drugs.com. Last updated on Aug 31, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
OPTISON (perflutren protein-type A microspheres) injectable suspension, for intravenous use
Initial U.S. Approval: 1997
WARNING: SERIOUS CARDIOPULMONARY REACTIONS
See full prescribing information for complete boxed warning.
Serious cardiopulmonary reactions, including fatalities, have occurred uncommonly during or following perflutren-containing microsphere administration. Most serious reactions occur within 30 minutes of administration (5.1).
Recent Major Changes
| Indications and Usage (1) | 5/2025 |
| Dosage and Administration, Recommended Dosage (2.1) | 5/2025 |
| Dosage and Administration, Preparation Instructions (2.2) | 5/2025 |
| Dosage and Administration, Administration Instructions (2.3) | 5/2025 |
| Warnings and Precautions, Transmissible Infectious Agents (5.5) - Removed | 5/2025 |
Indications and Usage for Optison
OPTISON is an ultrasound contrast agent indicated for use in adult and pediatric patients with suboptimal echocardiograms to opacify the left ventricle and to improve the delineation of the left ventricular endocardial borders (1).
Optison Dosage and Administration
- Adults
- 0.5 mL intravenously at a rate not exceeding 1 mL/s.
- If contrast enhancement is inadequate after the dose of 0.5 mL, additional doses of 0.5 mL may be repeated up to a total of 5 mL in a 10-minute period with a maximum total dose of 8.7 mL in any one patient study (2.1).
- Pediatric Patients
- 28 kg or less: 0.2 mL diluted with 0.2 mL of 0.9% Sodium Chloride Injection.
- 29 kg to 40 kg: 0.3 mL diluted with 0.3 mL of 0.9% Sodium Chloride Injection.
- 41 kg or more: 0.4 mL diluted with 0.4 mL of 0.9% Sodium Chloride Injection.
- Administer intravenously at a rate not exceeding 0.05 mL/s.
- If contrast enhancement is inadequate after the initial dose, up to four additional doses of the same diluted volume may be repeated for further contrast enhancement as needed (2.1).
- Follow the OPTISON injection with a flush of 0.9% Sodium Chloride Injection or 5% Dextrose Injection (2.3).
- See full prescribing information for preparation instructions (2.2).
Dosage Forms and Strengths
Injectable suspension: 5-8 ×108/mL protein-type A microspheres, 10 mg/mL albumin human, and 0.22 ± 0.11 mg/mL perflutren in 3 mL single-patient use vials (3)
Contraindications
Known or suspected hypersensitivity to perflutren or albumin (4)
Warnings and Precautions
Hypersensitivity Reactions: Serious anaphylactic reactions have been observed. Always have cardiopulmonary resuscitation personnel and equipment readily available prior to OPTISON administration and monitor all patients for hypersensitivity reactions (5.2).
Adverse Reactions/Side Effects
Common adverse reactions (incidence ≥ 0.5%) were: headache, nausea and/or vomiting, warm sensation or flushing, dizziness, dysgeusia, chills or fever, flu-like symptoms, malaise/weakness/fatigue, chest pain, dyspnea, injection site discomfort, and erythema (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact GE HealthCare at 1-800-654-0118 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2025
Full Prescribing Information
WARNING: SERIOUS CARDIOPULMONARY REACTIONS
Serious cardiopulmonary reactions, including fatalities, have occurred uncommonly during or following perflutren-containing microsphere administration. Most serious reactions occur within 30 minutes of administration [see Warnings and Precautions (5.1)].
- Assess all patients for the presence of any condition that precludes OPTISON administration [see Contraindications (4)].
- Always have resuscitation equipment and trained personnel readily available [see Warnings and Precautions (5.1)].
1. Indications and Usage for Optison
OPTISON is indicated for use in adult and pediatric patients with suboptimal echocardiograms to opacify the left ventricle and to improve the delineation of the left ventricular endocardial borders.
2. Optison Dosage and Administration
2.1 Recommended Dosage
Adults
- The recommended dose in adults is 0.5 mL administered intravenously at a rate not exceeding 1 mL/s [see Dosage and Administration (2.3)].
- If the contrast enhancement is inadequate after the dose of 0.5 mL, additional doses of 0.5 mL may be repeated for further contrast enhancement as needed.
- The maximum total dose is 5 mL in any 10-minute period.
- The maximum total dose is 8.7 mL in any one patient study.
Pediatric Patients
- The recommended dose by body weight in pediatric patients is shown in Table 1.
- Administer by intravenous injection at a rate not exceeding 0.05 mL/s [see Dosage and Administration (2.3)].
- If the contrast enhancement is inadequate after the initial dose, up to four additional doses of the same diluted volume may be repeated for further contrast enhancement as needed.
-
OPTISON must be diluted with 0.9% Sodium Chloride Injection to form a 1:1 dilution for pediatric administration [see Dosage and Administration (2.2)].
Table 1. Recommended Dose of OPTISON by Body Weight in Pediatric Patients Body Weight Dose 28 kg or less 0.2 mL of OPTISON diluted with 0.2 mL of 0.9% Sodium Chloride Injection 29 kg to 40 kg 0.3 mL of OPTISON diluted with 0.3 mL of 0.9% Sodium Chloride Injection 41 kg or more 0.4 mL of OPTISON diluted with 0.4 mL of 0.9% Sodium Chloride Injection
2.2 Preparation Instructions
General
- Visually inspect the OPTISON vial. Do not use if the container has been damaged, the protective seal and/or rubber cap have been entered, or the upper white layer is absent (may indicate the microspheres have been damaged and may result in poor or no echo contrast).
- Invert the vial and gently rotate to resuspend the microspheres. This process will allow the product to come to room temperature (20°C to 25°C or 68°F to 77°F) before use.
- Inspect the vial for complete resuspension. Do not use if the suspension appears to be clear rather than opaque and milky-white.
- Vent the OPTISON vial with a sterile vent spike or with a sterile 18-gauge needle before withdrawing the OPTISON suspension into the injection syringe.
- Do not inject air into the vial.
- Use the product within one minute of suspension. If one minute is exceeded, resuspend by inverting and gently rotating the syringe for no less than 10 seconds. Failure to adequately resuspend OPTISON may cause inadequate delivery of the microspheres and may result in inadequate contrast.
Dilution for Pediatric Administration
- Invert the OPTISON vial and gently rotate to resuspend the microspheres.
- Draw equal volumes of OPTISON and 0.9% Sodium Chloride Injection into the same syringe to form a 1:1 dilution.
- Mix gently by holding the syringe horizontally between the palms and rolling it gently back and forth for at least 10 seconds.
- If not used within 10 minutes after dilution, discard the diluted product.
2.3 Administration Instructions
- OPTISON is for intravenous use only and must not be administered by intra-arterial injection [see Warnings and Precautions (5.3)].
- Inspect visually for foreign particulate matter and discoloration prior to administration, whenever suspension and container permit. Do not inject if the suspension is not opaque and milky-white, or foreign particulate matter is present.
- For adults, inject through a 20-gauge or larger angiocatheter into a peripheral vein at a rate not exceeding 1 mL/s as a faster rate may reduce performance of the OPTISON microspheres.
- For pediatric patients, inject through a 24-gauge or larger angiocatheter into a peripheral vein at a rate not exceeding 0.05 mL/s as a faster rate may reduce performance of the OPTISON microspheres.
- Suggested methods of administration include: a short extension tubing, heparin lock, or intravenous line, all with a 3-way stopcock.
- Do not aspirate blood back into the OPTISON-containing syringe before administration; this may promote the formation of a blood clot within the syringe.
- For short extension tubing or heparin lock, fill one syringe with 10 mL of 0.9% Sodium Chloride Injection for adults or 5 mL of 0.9% Sodium Chloride Injection for pediatric patients and flush the line for patency before and after the injection of OPTISON.
- For a continuous intravenous line, open an intravenous line with 0.9% Sodium Chloride Injection or 5% Dextrose Injection to maintain vascular patency. Flush the line in its entirety immediately after injection of OPTISON.
- Each vial is for single-patient use. Discard unused portion.
3. Dosage Forms and Strengths
Injectable suspension: 5-8×108/mL protein-type A microspheres, 10 mg/mL albumin human, and 0.22 ± 0.11 mg/mL perflutren as a clear liquid lower layer, a white liquid upper layer, and a headspace filled with perflutren gas in 3 mL single-patient use vial; after resuspension, OPTISON is a sterile, homogeneous, opaque, and milky-white injectable suspension.
4. Contraindications
OPTISON is contraindicated in patients with known or suspected hypersensitivity to perflutren or albumin [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Serious Cardiopulmonary Reactions
Serious cardiopulmonary reactions including fatalities have occurred uncommonly during or shortly following perflutren-containing microsphere administration, typically within 30 minutes of administration. The risk for these reactions may be increased among patients with unstable cardiopulmonary conditions (acute myocardial infarction, acute coronary artery syndromes, worsening or unstable congestive heart failure, or serious ventricular arrhythmias).
The reported reactions to perflutren-containing microspheres include: fatal cardiac or respiratory arrest, shock, syncope, symptomatic arrhythmias (atrial fibrillation, tachycardia, bradycardia, supraventricular tachycardia, ventricular fibrillation, ventricular tachycardia), hypertension, hypotension, dyspnea, hypoxia, chest pain, respiratory distress, stridor, wheezing, loss of consciousness and convulsions [see Adverse Reactions (6.2)].
Always have cardiopulmonary resuscitation personnel and equipment readily available prior to OPTISON administration and monitor all patients for acute reactions.
5.2 Hypersensitivity Reactions
Serious anaphylactic reactions have been observed during or shortly following perflutren-containing microsphere administration including: shock, hypersensitivity, bronchospasm, throat tightness, angioedema, edema (pharyngeal, palatal, mouth, peripheral, localized), swelling (face, eye, lip, tongue upper airway), facial hypoesthesia, rash, urticaria, pruritus, flushing, and erythema have occurred in patients with no prior exposure to perflutren-containing microsphere products. Always have cardiopulmonary resuscitation personnel and equipment readily available prior to OPTISON administration and monitor all patients for hypersensitivity reactions [see Adverse Reactions (6.2)].
5.3 Systemic Embolization
When administering OPTISON to patients with a cardiac shunt, microspheres can bypass filtering of the lung and enter the arterial circulation. Assess patients with shunts for embolic phenomena following OPTISON administration. OPTISON is only for intravenous administration; do not administer OPTISON by intra-arterial injection.
5.4 Ventricular Arrhythmia Related to High Mechanical Index
High ultrasound mechanical index values may cause microsphere rupture and lead to ventricular arrhythmias. Additionally, end-systolic triggering with high mechanical indices has been reported to cause ventricular arrhythmias. OPTISON is not recommended for use at mechanical indices greater than 0.8.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in the labeling:
- Serious Cardiopulmonary Reactions [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults
The safety of OPTISON was evaluated in 279 adult patients in clinical studies. These patients were 69% male, 31% female, 71% White, 19% Black or African American, 9% Hispanic or Latino, and 1% other racial or ethnic groups. Adverse reactions reported in ≥ 0.5% of patients who received OPTISON by intravenous injection are given in Table 2.
| Adverse Reaction | OPTISON n=279 % |
|---|---|
| Body as a Whole | |
| Headache | 5.4 |
| Warm Sensation/Flushing | 3.6 |
| Chills/fever | 1.4 |
| Flu-like Symptoms | 1.1 |
| Malaise/Weakness/Fatigue | 1.1 |
| Cardiovascular System | |
| Dizziness | 2.5 |
| Chest Pain | 1.1 |
| Digestive System | |
| Nausea and/or Vomiting | 4.3 |
| Respiratory System | |
| Dyspnea | 1.1 |
| Skin & Appendages | |
| Injection Site Discomfort | 1.1 |
| Erythema | 0.7 |
| Special Senses | |
| Dysgeusia | 1.8 |
Adverse reactions reported in < 0.5% of patients who received OPTISON included:
Body as a Whole: induration, discoloration at the injection site
Cardiovascular system: premature ventricular contractions, palpitations
Digestive system: dry mouth
Immune system disorders: hypersensitivity
Musculoskeletal and connective tissue disorders: back pain, arthralgia, body or muscle aches
Nervous system: tremor, paresthesia, irritableness
Respiratory system: oxygen saturation decline due to coughing, wheezing
Skin and appendages: urticaria, rash, pruritus
Special Senses: tinnitus, visual blurring, photophobia, burning sensation in the eyes
Adverse Reactions in Pediatric Patients
Overall, the safety profile observed in pediatric patients from the clinical study was consistent with the safety profile in adult patients [see Clinical Studies (14.2)].
6.2 Postmarketing Experience
Adverse Reactions from Observational Studies
In a prospective, post-marketing safety surveillance study of OPTISON used in routine clinical practice, a total of 1,039 patients received OPTISON. These patients had an average age of 59.9 years (min, max: 20, 97) and were 62% male, 38% female, 83% White, 14% Black or African American, 2% Asian, and 1.5% other racial or ethnic groups. Overall, 17% of patients reported at least one adverse event. No deaths or serious adverse reactions were reported in this study.
Adverse Reactions from Postmarking Spontaneous Reports
The following adverse reactions have been identified during the postmarketing use of OPTISON or other perflutren-containing microspheres. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Cardiopulmonary
Fatal cardiac or respiratory arrest, shock, syncope, symptomatic arrhythmias (atrial fibrillation, tachycardia, bradycardia, supraventricular tachycardia, ventricular fibrillation, ventricular tachycardia), hypertension, hypotension, dyspnea, hypoxia, chest pain, respiratory distress or decreased oxygenation, stridor, wheezing.
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no data with OPTISON use in pregnant women to inform any drug-associated risks. No adverse developmental outcomes were observed in animal reproduction studies with intravenous administration of OPTISON to pregnant rats and rabbits during organogenesis at doses up to at least 5 and 10 times the recommended human dose based on body surface area (see Data).
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
OPTISON was administered intravenously to rats at doses of 0.25, 5 and 10 mL/kg/day (approximately 0.2, 5, and 10 times the recommended maximum human dose of 8.7 mL, respectively, based on body surface area) and to rabbits at 0.25, 2.5, and 5 mL/kg/day (approximately 0.5, 5, and 10 times the recommended maximum human dose, respectively, based on body surface area) during organogenesis. No significant findings attributable solely to a direct effect on the fetus were detected in the studies.
8.2 Lactation
There are no data on the presence of perflutren protein-type A microspheres in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OPTISON and any potential adverse effects on the breastfed infant from OPTISON or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of OPTISON to opacify the left ventricle and improve the delineation of the left ventricular endocardial borders have been established in pediatric patients with suboptimal echocardiograms. Use of OPTISON in pediatric patients is supported by evidence from adequate and well-controlled studies in adults and additional efficacy and safety data from a clinical study in 37 pediatric patients aged 9 to 17 years old [see Adverse Reactions (6.1) and Clinical Studies (14.1, 14.2)].
8.5 Geriatric Use
Of the total number of subjects in a clinical study of OPTISON, 35% were 65 and over, while 14% were 75 and over. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
11. Optison Description
OPTISON (perflutren protein-type A microspheres) injectable suspension is an ultrasound contrast agent for intravenous use.
Perflutren is chemically characterized as 1,1,1,2,2,3,3,3-perflutren with a molecular weight of 188, an empirical formula of C3F8, and the following structural formula:
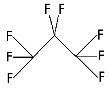
Each mL contains 5-8×108 protein-type A microspheres, 10 mg albumin human, 0.22 ± 0.11 mg perflutren, and the following inactive ingredients: 0.2 mg N-acetyltryptophan and 0.12 mg caprylic acid in 0.9% aqueous sodium chloride. The headspace of the vial is filled with perflutren gas. The pH is adjusted to 6.4 to 7.4. The protein in the microsphere shell makes up approximately 5% to 7% (w/w) of the total protein in the suspension. The microsphere particle size parameters are listed in Table 3.
OPTISON is supplied as a clear liquid lower layer, a white liquid upper layer, and a headspace filled with perflutren gas, and after resuspension, OPTISION is a sterile, homogeneous, opaque, and milky-white suspension.
| Parameter | |
|---|---|
| Mean diameter (range) | 3 to 4.5 µm |
| Percent less than 10 µm | 95% |
| Maximum diameter | 32 µm |
12. Optison - Clinical Pharmacology
12.1 Mechanism of Action
The OPTISON microspheres create an echogenic contrast effect in the blood. The acoustic impedance of the OPTISON microspheres is much lower than that of the blood. Therefore, impinging ultrasound waves are scattered and reflected at the microsphere-blood interface and ultimately may be visualized in the ultrasound image. At the frequencies used in adult echocardiography (2 MHz to 5 MHz), the microspheres resonate which further increases the extent of ultrasound scattering and reflection.
12.2 Pharmacodynamics
Contrast Enhancement Duration in Adults
The median duration of OPTISON contrast enhancement for each of the four doses of OPTISON, 0.2 (40% of recommended dose), 0.5, 3, and 5 mL, were approximately one, two, four, and five minutes, respectively [see Clinical Studies (14.1)].
Pulmonary Hemodynamic Effects
The effect of OPTISON on pulmonary hemodynamics was studied in a prospective, open-label study of 30 patients scheduled for pulmonary artery catheterization, including 19 with an elevated baseline pulmonary arterial systolic pressure (PASP) (>35 mmHg) and 11 with a normal PASP (≤35 mmHg). Systemic hemodynamic parameters and ECGs were also evaluated. No clinically important pulmonary hemodynamic, systemic hemodynamic, or ECG changes were observed.
12.3 Pharmacokinetics
After injection of OPTISON, diffusion of the perflutren gas out of the microspheres is limited by the low partition coefficient of the gas in blood that contributes to the persistence of the microspheres.
The pharmacokinetics of the intact microspheres of OPTISON in humans are unknown.
Distribution
The binding of perflutren to plasma proteins and its partitioning into blood cells are unknown. However, perflutren protein binding is expected to be minimal due to the low partition coefficient of the gas in blood.
Elimination
Following intravenous injection, perflutren is cleared with a pulmonary elimination half-life of 1.3 ± 0.69 minutes (mean ± SD).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Animal studies were not carried out to determine the carcinogenic potential of OPTISON.
Mutagenesis
The result of the following genotoxicity studies with OPTISON were negative: 1) Salmonella/Escherichia coli reverse mutation assay, 2) in vitro mammalian chromosome aberration assay using Chinese hamster ovary cells (CHO) with and without metabolic activation, 3) CHO/HGPRT forward mutation assay, and 4) in vivo mammalian micronucleus assay.
14. Clinical Studies
14.1 Echocardiography in Adults
The effectiveness of OPTISON was evaluated in two identical multicenter, controlled, dose escalation studies in 203 adult patients (Study A: n=101, Study B: n=102) with sub-optimal non-contrast echocardiography defined as having at least two out of six segments of the left ventricular endocardial border inadequately delineated in the apical 4-chamber view. These patients were 79% male, 21% female, 64% White, 25% Black or African American, 10% Hispanic or Latino, and 1% other race or ethnic group. The patients had a mean age of 61 years (range: 21 years to 83 years), a mean weight of 196 lbs. (range: 117 lbs. to 342 lbs.), a mean height of 68 inches (range: 47 inches to 78 inches), and a mean body surface area of 2 m2 (range: 1.4 m2 to 2.6 m2). Approximately 23% of the patients had chronic pulmonary disease, and 17% had congestive and dilated cardiomyopathy with left ventricular ejection fractions (LVEFs) between 20% and 40% (by previous echocardiography). Patients with a LVEF of less than 20% or with New York Heart Association Class IV heart failure were not included in the studies.
After non-contrast imaging, OPTISON was administered in increasing increments as four doses (0.2, 0.5, 3, and 5 mL) with at least 10 minutes between each dose. Ultrasound settings were optimized for the baseline (non-contrast) apical 4-chamber view and remained unchanged for the contrast imaging. Static echocardiographic images and video-tape segments were interpreted by a reader who was blinded to the patient's clinical history and to the dose of OPTISON. Left ventricular endocardial border delineation and left ventricular opacification were assessed before and after OPTISON administration by the measurement of visualized endocardial border length and ventricular opacification.
In comparison to non-contrast ultrasound, OPTISON significantly increased the length of endocardial border that could be visualized both at end-systole and end-diastole (see Table 4). In these patients there was a trend towards less visualization in women. OPTISON increased left ventricular opacification (peak intensity) in the mid-chamber and apical views (see Table 5). The imaging effects of OPTISON on endocardial border delineation and left ventricular opacification were similar at doses between 0.5 mL and 5 mL and were also similar among patients with or without pulmonary disease and dilated cardiomyopathy.
| Study | OPTISON dose | Length at End-Systole (cm) | Length at End-Diastole (cm) | ||
|---|---|---|---|---|---|
| n | mean ± S.D. | n | mean ± S.D. | ||
|
|||||
| Study A (n=101) | 0 mL (baseline) | 87 | 7.7 ± 3.0 | 86 | 9.3 ± 3.4 |
| 0.5 mL | 86 | 12.0 ± 4.9 | 91 | 15.8 ± 5.1 | |
| Study B (n=102) | 0 mL (baseline) | 89 | 8.1 ± 3.4 | 89 | 9.6 ± 3.7 |
| 0.5 mL | 95 | 12.4 ± 4.9 | 97 | 16.4 ± 4.6 | |
| Study | OPTISON dose | Mid-Chamber | Apex | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Intensity at End-Diastole | Intensity at End-Systole | Intensity at End-Diastole | Intensity at End-Systole | ||||||
| n | mean ± S.D. | n | mean ± S.D. | n | mean ± S.D. | n | mean ± S.D. | ||
|
|||||||||
| Study A (n=101) | 0 mL (baseline) | 91 | 39.5 ± 16.9 | 91 | 40.0 ± 18.1 | 91 | 46.7 ± 19.7 | 91 | 46.9 ± 20.1 |
| 0.5 mL | 91 | 57.3 ± 26.8 | 90 | 57.4 ± 26.7 | 91 | 67.0 ± 30.1 | 90 | 64.1 ± 30.2 | |
| Study B (n=102) | 0 mL (baseline) | 95 | 40.4 ± 17.4 | 95 | 40.9 ± 17.5 | 95 | 43.7 ± 19.9 | 95 | 45.0 ± 19.6 |
| 0.5 mL | 97 | 53.3 ± 20.7 | 96 | 53.6 ± 21.0 | 97 | 64.4 ± 25.3 | 96 | 61.6 ± 26.7 | |
14.2 Echocardiography in Pediatric Patients
The effectiveness of OPTISON was evaluated in a multicenter open-label clinical trial (NCT03740997) of 37 pediatric patients aged 9 to 17 years who were clinically indicated for transthoracic echocardiography and had suboptimal non-contrast echocardiography, defined as at least two contiguous segments in any view that could not be visualized.
These patients were 62% male, 38% female, 70% White, 8% Black or African American, 3% Asian, 3% American Indian or Alaska Native, 5% multiple races, 8% unknown race, and 3% race not reported. Ethnicity distribution was 49% Hispanic or Latino, 47% not Hispanic or Latino, and 3% not reported.
After non-contrast echocardiography, two increasing doses of OPTISON (dose level 1 and 2) based on body weight were administered with at least 10 minutes between doses. Contrast and non-contrast images for each patient were evaluated by three independent readers who were blinded to clinical information. Readers assessed visualization of 12 segments of the left ventricular wall in standard apical 4- and 2-chamber views on a 4-point Endocardial Border Delineation scale (0 to 3, indicating no, poor, fair, and good/optimal visualization). Segments scored 2 or 3 were considered visualized. The mean number of visualized segments of left ventricular wall under non-contrast echocardiography, echocardiography following OPTISON at dose level 2 (0.2 mL, 0.3 mL, or 0.4 mL, depending on body weight), and the difference (OPTISON at dose level 2 minus non-contrast) for the 37 patients in the full analysis set are shown in Table 6.
| Reader | Non-Contrast Mean (SD) | OPTISON Dose Level 2† Mean (SD) | Difference Between OPTISON Dose Level 2 & Non-Contrast Mean (95% CI)‡ |
|---|---|---|---|
|
|||
| Reader 1 | 3.3 (3.4) | 9.2 (4.3) | 5.9 (4.4, 7.5) |
| Reader 2 | 2.8 (2.8) | 9.0 (4.7) | 6.2 (4.4, 8.1) |
| Reader 3 | 4.6 (5.0) | 9.3 (4.1) | 4.6 (2.6, 6.7) |
16. How is Optison supplied
How Supplied
OPTISON (perflutren protein-type A microspheres) injectable suspension is supplied at concentrations of 5-8×108/mL protein-type A microspheres, 10 mg/mL albumin human, and 0.22 ± 0.11 mg/mL perflutren as a clear liquid lower layer, a white liquid upper layer, and a headspace filled with perflutren gas in 3 mL single-patient use vials. After resuspension, OPTISON is a sterile, homogeneous, opaque, and milky-white injectable suspension.
| NDC 0407-2707-03 NDC 0407-2707-18 |
17. Patient Counseling Information
Hypersensitivity Reactions
Advise patients to inform their healthcare provider if they develop any symptoms of hypersensitivity after OPTISON administration including rash, wheezing, or shortness of breath [see Warnings and Precautions (5.2)].

| OPTISON
PERFLUTREN PROTEIN-TYPE A MICROSPHERES
human albumin microspheres and perflutren injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - GE Healthcare Inc. (053046579) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| GE Healthcare AS | 515048908 | manufacture(0407-2707) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Octapharma Pharmazeutika Produktionsgesellschaft, m.b.H | 301119178 | manufacture(0407-2707) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Octapharma AB | 558833179 | manufacture(0407-2707) | |
More about Optison (perflutren)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Side effects
- Dosage information
- During pregnancy
- Drug class: ultrasound contrast media
- Breastfeeding
- En español