Lithostat: Package Insert / Prescribing Info
Package insert / product label
Generic name: acetohydroxamic acid
Dosage form: tablet
Drug class: Miscellaneous genitourinary tract agents
Medically reviewed by Drugs.com. Last updated on Jan 14, 2024.
On This Page
Lithostat Description
Acetohydroxamic acid (AHA) is a stable, synthetic compound derived from hydroxylamine and ethyl acetate. Its molecular structure is similar to urea:
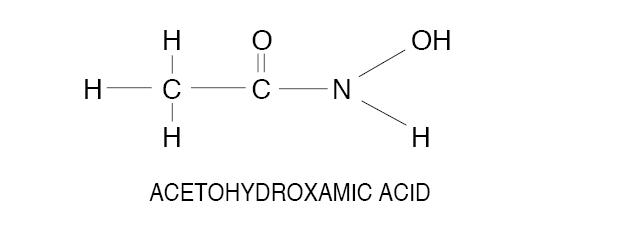
AHA is weakly acidic, highly soluble in water, and chelates metals - notably iron. The molecular weight is 75.068. AHA has a pKa of 9.32 and a melting point of 89-91° C. AHA is a urease inhibitor. Available as 250 mg tablets.
Lithostat - Clinical Pharmacology
AHA reversibly inhibits the bacterial enzyme urease, thereby inhibiting the hydrolysis of urea and production of ammonia in urine infected with urea-splitting organisms. The reduced ammonia levels and decreased pH enhance the effectiveness of antimicrobial agents and allow an increased cure rate of these infections.
AHA is well absorbed from the gastrointestinal tract after oral administration; peak blood levels occur from 0.25 to 1 hour after dosing. The compound is distributed throughout body water, and there is no known binding to any tissue. AHA chelates with dietary iron within the gut. This reaction may interfere with absorption of AHA and with iron. Concomitant hypochromic anemia should be treated with intramuscular iron.
In rodents, the metabolic fate of AHA is well known; 55% is excreted unchanged in urine, 25% is excreted as acetamide or acetate and 7% is excreted by the lungs as carbon dioxide. Less than 1% is excreted in the feces. Approximately 5% of the administered dose is unaccounted for. In rodents, AHA shows a dose-related change in pharmacokinetics; with increasing dose, there is an increase in the half-life and an increase in the percent of the administered dose recovered in urine as unchanged AHA.
Pharmacokinetics in man are generally similar to rodents including the dose-related increase in half-life, but they are not as well characterized as in the rodent. Thirty-six to sixty-five percent (36-65%) of the oral dosage is excreted unchanged in the urine. It is unaltered AHA in the urine that provides the therapeutic effect, but the precise concentration of AHA in urine that is necessary to inhibit urease is incompletely delineated. Therapeutic benefit may be obtained from concentrations as low as 8 mcg/ml; higher concentrations (i.e., 30 mcg/ml) are expected to provide more complete inhibition of urease. The plasma half-life of AHA is approximately 5-10 hours in subjects with normal renal function and is prolonged in patients with reduced renal function.
Acetohydroxamic acid has been evaluated clinically in patients with urea-splitting urinary infections, often accompanied by struvite stone disease, that were recalcitrant to other forms of medical and surgical management. In these clinical trials, AHA reduced the pathologically elevated urinary ammonia and pH levels that result from the hydrolysis of urea by the enzyme, urease.
AHA does not acidify urine directly nor does it have a direct antibacterial effect. The usefulness of reducing ammonia levels and decreasing urinary pH is suggested by single (not yet replicated) clinical trials in which urease inhibition 1) allowed successful antibiotic treatment of urea-splitting Proteus infections after surgical removal of struvite stones in patients not cured by 3 months of antibacterial treatment alone, and 2) reduced the rate of stone growth in patients who were not candidates for surgical removal of stones.
Indications and Usage for Lithostat
Acetohydroxamic acid is indicated as adjunctive therapy in patients with chronic urea-splitting urinary infection. AHA is intended to decrease urinary ammonia and alkalinity, but it should not be used in lieu of curative surgical treatment (for patients with stones) or antimicrobial treatment. Long-term treatment with AHA may be warranted to maintain urease inhibition as long as urea-splitting infection is present. Experience with AHA does not go beyond 7 years. A patient package insert should be distributed to each patient who receives AHA.
Acetohydroxamic acid
should not be used in:
a. patients whose physical state and disease are amenable to definitive surgery and appropriate antimicrobial agents
b. patients whose urine is infected by non-urease producing organisms
c. patients whose urinary infections can be controlled by culture-specific oral antimicrobial agents
d. patients whose renal function is poor (i.e., serum creatinine more than 2.5 mg/dl and/or creatinine clearance less than 20 ml/min)
e. female patients who do not evidence a satisfactory method of contraception
f. patients who are pregnant
Acetohydroxamic acid may cause fetal harm when administered to a pregnant woman. AHA was teratogenic (retarded and/or clubbed rear leg at 750 mg/kg and above and exencephaly and encephalocele at 1,500 mg/kg) when given intraperitoneally to rats.
AHA is contraindicated in women who are or may become pregnant. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be informed of the potential hazard to the fetus.
Warnings
A Coombs negative hemolytic anemia has occurred in patients receiving AHA. Gastrointestinal upset characterized by nausea, vomiting, anorexia and generalized malaise have accompanied the most severe forms of hemolytic anemia. Approximately 15% of patients receiving AHA have had only laboratory findings of an anemia. However, most patients developed a mild reticulocytosis. The untoward reactions have reverted to normal following cessation of treatment. A complete blood count, including a reticulocyte count, is recommended after two weeks of treatment. If the reticulocyte count exceeds 6%, a reduced dosage should be entertained. A CBC and reticulocyte count are recommended at 3-month intervals for the duration of treatment.
Precautions
General
Hematologic Effects: Bone marrow depression (leukopenia, anemia, and thrombocytopenia) has occurred in experimental animals receiving large doses of AHA, but has not been seen in man to date. AHA is a known inhibitor of DNA synthesis and also chelates metals - notably iron. Its bone marrow suppressant effect is probably related to its ability to inhibit DNA synthesis, but anemia could also be related to depletion of iron stores. To date, the only clinical effect noted has been hemolysis, with a decrease in the circulating red blood cells, hemoglobin and hematocrit. Abnormalities in platelet or white blood cell count have not been noted. However, clinical monitoring of the platelet and white cell count is recommended.
Monitoring Liver Function: Abnormalities of liver function have not been reported to date. However, a chloro-benzene derivative of acetohydroxamic acid caused significant liver dysfunction in an unrelated study. Therefore, close monitoring of liver function is recommended. (See Carcinogenesis for discussion of possible hepatic carcinogenesis.)
Use In Patients With Renal Impairment: Since AHA is eliminated primarily by the kidneys, patients with significantly impaired renal function should be closely monitored, and a reduction of daily dose may be needed to avoid excessive drug accumulation. (See Dosage and Administration.)
Drug Interactions
AHA has been used concomitantly with insulin, oral and parenteral antibiotics, and progestational agents. No clinically significant interactions have been noted, but until wider clinical experience is obtained, AHA should be used with caution in patients receiving other therapeutic agents.
AHA taken in association with alcoholic beverages has resulted in a rash. (See Adverse Reactions.)
AHA chelates heavy metals-notably iron. The absorption of iron and AHA from the intestinal lumen may be reduced when both drugs are taken concomitantly. When iron administration is indicated, intramuscular iron is probably the product of choice.
CARCINOGENESIS, MUTAGENESIS, IMPAIRMENT OF FERTILITY
Well controlled, long-term animal studies that identify the carcinogenic potential of AHA treatment have not been conducted. Acetamide, a metabolite of AHA, has been shown to cause hepatocellular carcinoma in rats at oral doses 1,500 times the human dose. AHA is cytotoxic and was positive for mutagenicity in the Ames test.
NURSING MOTHERS
It is not known whether AHA is secreted in human milk. Because many drugs are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from AHA, a decision should be made to discontinue nursing or the drug, taking into account the significance of the drug to the mother’s well being.
PEDIATRIC USE
Children with chronic, recalcitrant, urea-splitting urinary infection may benefit from treatment with AHA. However, detailed studies involving dosage and dose intervals in children have not been established. Children have tolerated a dose of 10 mg/kg/day, taken in two or three divided doses, satisfactorily for periods up to one year. Close monitoring of such patients is mandatory.
Adverse Reactions/Side Effects
Experience with AHA is limited. About 150 patients have been treated, most for periods of more than a year.
Adverse reactions have occurred in up to thirty percent (30%) of the patients receiving AHA. In some instances the reactions were symptomatic; in others only changes in laboratory parameters were noted. Adverse reactions seem to be more prevalent in patients with preexisting thrombophlebitis or phlebothrombosis and/or in patients with advanced degrees of renal insufficiency. The risk of adverse reactions is highest during the first year of treatment. Chronic treatment does not seem to increase the risk nor the severity of adverse reactions.
The following reactions have been reported:
NEUROLOGICAL: Mild headaches are commonly reported (about 30%) during the first 48 hours of treatment. These headaches are mild, responsive to oral salicylate-type analgesics, and usually disappear spontaneously. The headaches have not been associated with vertigo, tinnitus, or visual or auditory abnormalities. Tremulousness and nervousness have also been reported.
GASTROINTESTINAL: Gastrointestinal symptoms, nausea, vomiting, anorexia, and malaise have occurred in 20-25% of patients. In most patients the symptoms were mild, transitory, and did not result in interruption of treatment. Approximately 3% of patients developed a hemolytic anemia of sufficient magnitude to warrant interruption in treatment; several of these patients also had symptoms of gastrointestinal upset.
HEMATOLOGICAL: Approximately 15% of patients have had laboratory findings characteristic of a hemolytic anemia. A mild reticulocytosis (5- 6%) without anemia, is even more prevalent. The laboratory findings are occasionally accompanied by systemic symptoms such as malaise, lethargy and fatigue, and gastrointestinal symptoms. Symptoms and laboratory findings have invariably improved following cessation of treatment with AHA. The hematological abnormalities are more prevalent in patients with advanced renal failure.
DERMATOLOGICAL: A nonpruritic, macular skin rash has occurred in the upper extremities and on the face of several patients taking AHA on a long-term basis, usually when AHA has been taken concomitantly with alcoholic beverages, but in a few patients in the absence of alcohol consumption. The rash commonly appears 30-45 minutes after ingestion of alcoholic beverages; it characteristically disappears spontaneously in 30-60 minutes. The rash may be associated with a general sensation of warmth. In some patients the rash is sufficiently severe to warrant discontinuation of treatment, but most patients have continued treatment, avoiding alcohol or using smaller quantities of it. Alopecia has also been reported in patients taking AHA.
CARDIOVASCULAR: Superficial phlebitis involving the lower extremities has occurred in several patients on AHA during the early (Phase II) clinical trials. Several of the affected patients had had phlebitic episodes prior to treatment. One patient developed deep vein thrombosis of the lower extremities. The patient with phlebothrombosis had an associated traumatic injury to the groin. It is unclear whether the phlebitis was related to or exacerbated by treatment with AHA. No patient in the three (3) year controlled (Phase III) clinical trial developed phlebitis. In all instances these vascular abnormalities returned to normal following appropriate medical therapy. Embolic phenomena have been reported in three patients taking AHA in the Phase II trial. The phlebitis and emboli resolved following discontinuation of AHA and implementation of appropriate medical therapy. Several patients have resumed treatment with AHA without ill effect. Palpitations have also been reported in patients taking AHA.
RESPIRATORY: No symptoms have been reported. Radiographic evidence of small pulmonary emboli has been seen in three patients with phlebitis in their lower legs.
PSYCHIATRIC: Depression, anxiety, nervousness, and tremulousness have been observed in approximately 20% of patients taking AHA. In most patients the symptoms were mild and transitory, but in about 6% of patients the symptoms were sufficiently distressing to warrant interruption or discontinuation of treatment.
Related/similar drugs
Overdosage
Acute deliberate overdosage in man has not occurred, but would be expected to induce the following symptoms: anorexia, malaise, lethargy, diminished sense of well being, tremulousness, anxiety, nausea and vomiting. Laboratory findings are likely to include an elevated reticulocyte count and a severe hemolytic reaction requiring hospitalization, symptomatic treatment, and possibly blood transfusions. Concomitant reduction in platelets and/or white blood cells should be anticipated.
Milder overdosages resulting in hemolysis have occurred in an occasional patient with reduced renal function after several weeks or months of continuous treatment.
The acute LD 50 of AHA in animals (rats) is 4.8 gm/kg.
Recommended treatment for an overdosage reaction consists of (1) cessation of treatment, (2) close monitoring of hematologic status, (3) symptomatic treatment, and (4) blood transfusions as required by the clinical circumstances. The drug is probably dialyzable, but this property has not been tested clinically.
Lithostat Dosage and Administration
AHA should be administered orally, one tablet 3-4 times a day in a total daily dose of 10-15 mg/kg/day. The recommended starting dose is 12 mg/kg/day, administered at 6-8 hour intervals at a time when the stomach is empty. The maximum daily dose should be no more than 1.5 grams, regardless of body weight.
The dosage should be reduced in patients with reduced renal function. Patients whose serum creatinine is greater than 1.8 mg/dl should take no more than 1.0 gm/day; such patients should be dosed at q-12-h intervals. Further reductions in dosage to prevent the accumulation of toxic concentrations in the blood may also be desirable. Insufficient data exists to accurately characterize the optimum dose and/or dose interval in patients with moderate degrees of renal insufficiency.
Patients with advanced renal insufficiency (i.e., serum creatinine more than 2.5 mg/dl) should not be treated with AHA. The risk of accumulation of toxic blood levels of AHA seems to be greater than the chances for a beneficial effect in such patients.
In children an initial dose of 10 mg/kg/day is recommended. Close monitoring of the patient’s clinical condition and hematologic status is recommended. Titration of the dose to higher or lower levels may be required to obtain an optimum therapeutic effect and/or to reduce the risk of side effects.
How is Lithostat supplied
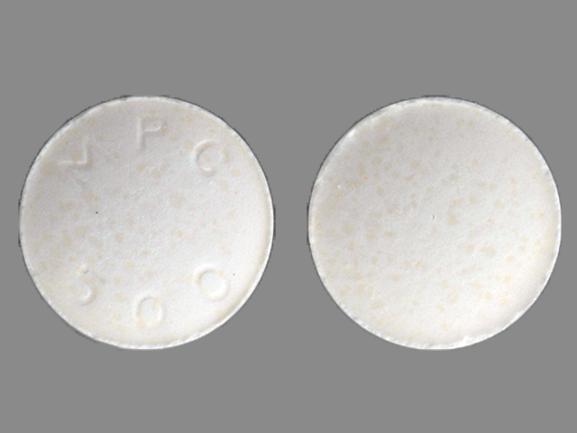
LITHOSTAT ®, NDC 0178-0500-01, is available for oral administration as 250 mg white, round tablets, in unit of use packages of 100 tablets. Each LITHOSTAT ®tablet is debossed MPC 500 on one side and blank on the other side. LITHOSTAT ®should be stored in a dry place at room temperature, 15° - 30°C (59° - 86°F). Container should be closed tightly.
L050001R0620
PATIENT INFORMATION
PLEASE READ THIS INFORMATION BEFORE USING THIS DRUG.
GENERAL INFORMATION:It has been known for many years that urinary infection may cause the formation of urinary stones. As these stones form, bacteria are trapped within the stones. The trapped bacteria cause the stones to grow, and the stones protect the bacteria from antibiotics. Surgical removal of the stone attempts to break this vicious cycle - many times successfully. However, if infection persists or if a small stone fragment persists, then there is an increased risk of stone recurrence. Multiple operations to remove kidney stones may result in damage and scarring of the kidney. In some situations removal of the kidney may be necessary.
In some instances stones may form initially as a result of non-infectious (i.e., metabolic) causes. If a metabolic stone becomes infected, then an “infection stone” may grow onto the “metabolic stone.” Stone analysis and/or biochemical tests will usually determine which factors are present.
Experimental investigations have identified an enzyme called urease which is made by some (but not all) bacteria. Urease reacts with urine to make ammonia. Ammonia changes the acidity of the urine and the change in acidity encourages stone formation. LITHOSTAT
®(acetohydroxamic acid) inhibits urease and thereby reduces urinary ammonia. In some instances, LITHOSTAT
®enhances the effectiveness of antibiotics and thereby makes urinary infection easier to control.
WHAT IS LITHOSTAT
®?
LITHOSTAT
®is a drug which prevents the excessive buildup of ammonia in your urine, which controls the acidity and alkalinity (pH) of your urine. The cause of excessive ammonia and alkalinity in your urine is a bacterial infection.
WHAT CAN LITHOSTAT ®DO? Treatment with LITHOSTAT ®is prescribed to decrease urinary ammonia. This may increase the chance of controlling your infection with antibiotics and may help the treatment of your kidney stones. Dissolution of existing stones is unlikely.
LITHOSTAT ®should not be used in place of surgical treatment. Surgical removal of all stones and elimination of all infection with antibiotics offers the possibility of curative treatment. LITHOSTAT ®is likely to be more effective after large stones or obstructing stones have been removed.
WHAT ARE THE PROBLEMS OR SIDE EFFECTS WITH LITHOSTAT ®? The complete spectrum of side effects induced by LITHOSTAT ®(acetohydroxamic acid) is unknown. However, some side effects which have been reported to date have been headaches, abdominal discomfort, nausea, loss of hair, shakiness, and anemia. Lifethreatening problems (blood clot in the legs) occurred in several patients with advanced disease in early investigation. In more extensive later investigations, this problem has not occurred. No patient has died as a consequence of taking LITHOSTAT ®. The most serious side effects seem to occur in patients with poor kidney function and/or in patients with a previous history of these conditions.
Problems related to LITHOSTAT ®have disappeared following cessation of the drug and initiation of appropriate medical treatment. Most patients have resumed treatment without ill effect.
A flushing skin reaction (i.e., redness, warmth, and tingling) has occurred in several patients who consumed alcohol during treatment with LITHOSTAT ®. The reaction persisted approximately 30 minutes and disappeared without treatment. The cause and significance of this reaction are unknown. Consequently, patients are encouraged to abstain from consumption of alcoholic beverages while being treated with LITHOSTAT ®.
In animal studies doses of LITHOSTAT ®about 20 times the maximum human dose have caused fetal abnormalities (birth defects) indicating a potential for such an adverse effect in an exposed human fetus. Therefore, LITHOSTAT ®should not be given to pregnant women or to any sexually active woman of child-baring age, not using a highly effective method of contraception (oral contraceptive or IUD).
An acceptable long-term study of the cancer causing potential of LITHOSTAT ®has not been conducted, but a known metabolite of LITHOSTAT ®, acetamide, is carcinogenic (cancer-causing) to the liver in rats at doses about 80 times the maximum human dose of LITHOSTAT ®. LITHOSTAT ®thus must be considered a potential human carcinogen. LITHOSTAT ®kills tissue cells grown in tissue culture and alters genetic material in cells grown in culture.
LITHOSTAT ®may induce other adverse reactions which have not yet been recognized.
Unusual symptoms should be reported to your physician. Mild symptoms usually do not warrant discontinuation of treatment. Severe symptoms may necessitate temporary cessation of treatment and/or alteration of dosage.
WHAT ABOUT TAKING OTHER DRUGS WITH LITHOSTAT ®? Only take those drugs prescribed by your physician. Do not take prescription drugs or over the counter preparations without your physician‘s specific prescription or recommendation. Drugs that contain iron should not be taken at the same time as LITHOSTAT ®, (acetohydroxamic acid).
LITHOSTAT ®reacts with iron, and may not be absorbed into the bloodstream. Both the iron you take and the LITHOSTAT ®you take may be ineffective if both drugs are taken together.
HOW IMPORTANT IS MY DAILY DOSAGE OF LITHOSTAT®?If you fail to follow your daily dosage schedule with LITHOSTAT ®you will probably suffer a setback in treatment effectiveness and new kidney stone formation is likely. LITHOSTAT ®plus antibiotic therapy must be taken exactly as your physician prescribes it for optimum effectiveness.
IN CONCLUSION:Your daily dosage of LITHOSTAT ®(acetohydroxamic acid) is important to the proper treatment of your condition. Any unusual side effects should be reported to your physician at once.

| LITHOSTAT
acetohydroxamic acid tablet |
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
|
||||||||||||||||||
| Labeler - Mission Pharmacal Company (008117095) |
| Registrant - Mission Pharmacal Company (927726893) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Mission Pharmacal Company | 927726893 | manufacture(0178-0500) | |
More about Lithostat (acetohydroxamic acid)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: miscellaneous genitourinary tract agents
- En español