Kevzara: Package Insert / Prescribing Info
Package insert / product label
Generic name: sarilumab
Dosage form: injection, solution
Drug classes: Antirheumatics, Interleukin inhibitors
Medically reviewed by Drugs.com. Last updated on May 26, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
KEVZARA (sarilumab) injection, for subcutaneous use
Initial U.S. Approval: 2017
WARNING: RISK OF SERIOUS INFECTIONS
See full prescribing information for complete boxed warning.
- Serious infections leading to hospitalization or death including bacterial, viral, invasive fungal, and other opportunistic infections have occurred in patients receiving KEVZARA. (5.1)
- If a serious infection develops, interrupt KEVZARA until the infection is controlled. (5.1)
- Cases of tuberculosis (TB) have been reported. Prior to starting KEVZARA, test for latent TB; if positive, start treatment for TB. (5.1)
- Closely monitor patients for signs and symptoms of infection during treatment with KEVZARA. (5.1)
Indications and Usage for Kevzara
KEVZARA® is an interleukin-6 (IL-6) receptor antagonist indicated for treatment of:
- adult patients with moderately to severely active rheumatoid arthritis (RA) who have had an inadequate response or intolerance to one or more disease-modifying antirheumatic drugs (DMARDs). (1.1)
- adult patients with polymyalgia rheumatica (PMR) who have had an inadequate response to corticosteroids or who cannot tolerate corticosteroid taper. (1.2)
- patients who weigh 63 kg or greater with active polyarticular juvenile idiopathic arthritis (pJIA). (1.3)
Kevzara Dosage and Administration
General Considerations for Administration
- KEVZARA initiation is not recommended in patients with ANC less than 2,000/mm3, platelets less than 150,000/mm3 or liver transaminases above 1.5 times ULN. See Full Prescribing Information (FPI) for complete information. (2.1)
Recommended Dosage in RA
- The recommended dosage is 200 mg subcutaneously, once every 2 weeks. (2.2)
- For RA, KEVZARA may be used as monotherapy or in combination with methotrexate (MTX) or other conventional DMARDs. (2.2)
Recommended Dosage in PMR
- The recommended dosage is 200 mg subcutaneously, once every two weeks in combination with a tapering course of corticosteroids. (2.3)
- For PMR, KEVZARA can be used as monotherapy following discontinuation of corticosteroids. (2.3)
Recommended Dosage in pJIA
- The recommended dosage is 200 mg given subcutaneously once every 2 weeks for pJIA patients who weigh 63 kg or greater using the 200 mg/1.14 mL pre-filled syringe. (2.4)
- For pJIA, KEVZARA can be used as monotherapy or in combination with conventional DMARDs. (2.4)
Dosage Modifications for Cytopenias, Abnormal Liver Enzymes, Infections
- See FPI for complete information. (2.6)
Dosage Forms and Strengths
Contraindications
KEVZARA is contraindicated in patients with known hypersensitivity to sarilumab or any of the inactive ingredients. (4)
Warnings and Precautions
- Serious Infections: Avoid KEVZARA use during an active infection. (5.1)
- Neutropenia, Thrombocytopenia, Elevated Liver Enzymes, Lipid Abnormalities: Monitor laboratory parameters. (5.2)
- Gastrointestinal (GI) Perforation: Risk may be increased with concurrent diverticulitis or concomitant use of NSAIDs or corticosteroids. Promptly evaluate acute abdominal signs or symptoms. (5.3)
- Hypersensitivity reactions. (5.5)
- Live vaccines: Avoid use with KEVZARA. (5.7, 7.3)
Adverse Reactions/Side Effects
Most common adverse reactions are:
- Rheumatoid Arthritis (incidence ≥3 %): neutropenia, increased ALT, injection site erythema, upper respiratory infections and urinary tract infections. (6.1)
- Polymyalgia Rheumatica (incidence ≥ 5%): neutropenia, leukopenia and injection site pruritus. (6.1)
- Polyarticular Juvenile Idiopathic Arthritis: nasopharyngitis, neutropenia, upper respiratory tract infection and injection site erythema. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
Use In Specific Populations
- Lactation: Discontinue drug or nursing taking into consideration importance of drug to mother. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2025
Full Prescribing Information
WARNING: RISK OF SERIOUS INFECTIONS
Patients treated with KEVZARA are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. Opportunistic infections have also been reported in patients receiving KEVZARA. Most patients who developed infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Avoid use of KEVZARA in patients with an active infection.
Reported infections include:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Patients should be tested for latent tuberculosis before KEVZARA use and during therapy. Treatment for latent infection should be initiated prior to KEVZARA use.
- Invasive fungal infections, such as candidiasis, and pneumocystis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral and other infections due to opportunistic pathogens.
Closely monitor patients for signs and symptoms of infection during treatment with KEVZARA. If a serious infection develops, interrupt KEVZARA until the infection is controlled.
Consider the risks and benefits of treatment with KEVZARA prior to initiating therapy in patients with chronic or recurrent infection.
1. Indications and Usage for Kevzara
1.1 Rheumatoid Arthritis (RA)
KEVZARA® is indicated for treatment of adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to one or more disease-modifying antirheumatic drugs (DMARDs).
2. Kevzara Dosage and Administration
2.1 General Considerations Prior to Administration
Not Recommended for Concomitant Use with Biological DMARDS
The concurrent use of KEVZARA with biological DMARDs such as tumor necrosis factor (TNF) antagonists, IL-1R antagonists, anti-CD20 monoclonal antibodies and selective co-stimulation modulators has not been studied. Avoid using KEVZARA with biological DMARDs because of the possibility of increased immunosuppression and increased risk of infection.
Recommended Evaluations Prior to Treatment
- Complete blood count (CBC): Treatment initiation with KEVZARA is not recommended in patients with an absolute neutrophil count (ANC) below 2000 per mm3, or platelet count below 150,000 per mm3. Monitor laboratory parameters [see Warnings and Precautions (5.2)].
- Liver function tests (LFT): Treatment initiation with KEVZARA is not recommended in patients with or who have alanine transaminase (ALT) or aspartate aminotransferase (AST) above 1.5 times the upper limit of normal (ULN). Monitor laboratory parameters [see Dosage and Administration (2.6) and Warnings and Precautions (5.2)].
- Lipid parameters (total cholesterol, LDL cholesterol, HDL cholesterol and/or triglycerides): Assess lipid parameters at baseline. Monitor laboratory parameters [see Warnings and Precautions (5.2)].
- Active and latent tuberculosis infection evaluation: Prior to initiating KEVZARA, test patients for active and latent tuberculosis (TB). KEVZARA should not be administered to patients with active TB. If positive for latent infection, consider treating for TB prior to KEVZARA use [see Warnings and Precautions (5.1)].
- Evaluate for infections: Avoid KEVZARA use in patients with active infections [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage for Rheumatoid Arthritis
The recommended dosage of KEVZARA is 200 mg once every two weeks given as a subcutaneous injection [see Dosage and Administration (2.1)].
KEVZARA may be used as monotherapy or in combination with methotrexate (MTX) or other conventional DMARDs.
Modify the dosage as recommended in Table 1 if the patient develops neutropenia, thrombocytopenia, or liver enzyme abnormalities [see Dosage and Administration (2.6), Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
2.3 Recommended Dosage for Polymyalgia Rheumatica
The recommended dosage of KEVZARA is 200 mg once every two weeks given as a subcutaneous injection, in combination with a tapering course of systemic corticosteroids [see Dosage and Administration (2.1)]. KEVZARA can be used as monotherapy following discontinuation of corticosteroids.
Discontinue KEVZARA if the patient develops neutropenia (using ANC results obtained at the end of the dosing interval), thrombocytopenia, or liver enzyme abnormalities [see Dosage and Administration (2.6), Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
2.4 Recommended Dosage for Polyarticular Juvenile Idiopathic Arthritis
The recommended dosage of KEVZARA for patients who weigh 63 kg and greater is 200 mg once every two weeks given as a subcutaneous injection (maximum dose 200 mg). Dosage in this patient population can be achieved by administering the 200 mg/1.14 mL pre-filled syringe. The pre-filled pen is not intended for use in pediatric patients [see Dosage and Administration (2.6), Warnings and Precautions (5.2) and Adverse Reactions (6.1)]. KEVZARA is not approved in pediatric patients weighing less than 63 kg because of the lack of an appropriate dosage form.
In patients with pJIA, KEVZARA can be used alone or in combination with conventional DMARDs.
2.5 Preparation and Administration Instructions
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. KEVZARA solution should be clear and colorless to pale yellow. Do not use if the solution is cloudy, discolored or contains particles, or if any part of the pre-filled syringe or pre-filled pen appears to be damaged.
Rotate injection sites with each injection. Do not inject into skin that is tender, damaged, or has bruises or scars.
Pre-filled Pen and Pre-filled Syringe
- KEVZARA is intended for use under the guidance of a healthcare professional. A patient may self-inject KEVZARA or the patient's caregiver may administer KEVZARA. Provide proper training to patients and/or caregivers on the preparation and administration of KEVZARA prior to use according to the Instructions for Use (IFU).
- Allow the pre-filled syringe to sit at room temperature for 30 minutes prior to subcutaneous injection. Do not warm KEVZARA in any other way.
- If using a pre-filled pen, allow the pre-filled pen to sit at room temperature for 60 minutes prior to subcutaneous injection. Do not warm KEVZARA in any other way.
- Instruct patients to inject the full amount in the syringe or pen (1.14 mL), which provides 200 mg or 150 mg of KEVZARA, according to the directions provided in the IFU.
- The ability of pediatric patients to self-inject with the pre-filled pen has not been tested.
2.6 Dosage Modifications for Cytopenias, Abnormal Liver Enzymes, or Infections
Dosage Modifications for Patients with Rheumatoid Arthritis
- Laboratory Abnormalities: Modify dosage in case of neutropenia, thrombocytopenia, or liver enzyme elevations as shown in Table 1 [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)]. For treatment initiation criteria, refer to the dosage recommendations for RA [see Dosage and Administration (2.1, 2.2)].
| Low Absolute Neutrophil Count (ANC) | |
| Lab Value (cells/mm3) | Recommendation |
| ANC greater than 1,000 | Maintain current dosage of KEVZARA. |
| ANC 500 to 1,000 | Hold treatment with KEVZARA until ANC greater than 1,000. KEVZARA can then be resumed at 150 mg every two weeks and increased to 200 mg every two weeks as clinically appropriate. |
| ANC less than 500 | Discontinue KEVZARA. |
| Low Platelet Count | |
| Lab Value (cells/mm3) | Recommendation |
| 50,000 to 100,000 | Hold treatment with KEVZARA until platelets greater than 100,000. KEVZARA can then be resumed at 150 mg every two weeks and increased to 200 mg every two weeks as clinically appropriate. |
| Less than 50,000 | If confirmed by repeat testing, discontinue KEVZARA. |
| Liver Enzyme Abnormalities | |
| Lab Value | Recommendation |
| ALT or AST greater than ULN to 3 times ULN | Consider dosage modification of concomitant DMARDs as clinically appropriate. |
| ALT or AST greater than 3 times ULN to 5 times ULN | Hold treatment with KEVZARA until ALT or AST less than 3 times ULN. KEVZARA can then be resumed at 150 mg every two weeks and increased to 200 mg every two weeks as clinically appropriate. |
| ALT or AST greater than 5 times ULN | Discontinue KEVZARA. |
- Infections: If a patient with RA develops a serious infection or an opportunistic infection, hold treatment with KEVZARA until the infection is controlled [see Warnings and Precautions (5.1)].
Dosage Modifications for Patients with Polymyalgia Rheumatica
-
Laboratory Abnormalities: Discontinue KEVZARA in patients with PMR who develop the following laboratory abnormalities [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)]:
- neutropenia (ANC below 1,000 per mm3 at the end of the dosing interval)
- thrombocytopenia (platelet count below 100,000 per mm3)
- AST or ALT elevations 3 times above the ULN
Dosage modifications have not been studied in patients with PMR with these conditions. For treatment initiation criteria, refer to the dosage recommendations for PMR [see Dosage and Administration (2.1, 2.3)]. - Infections: If a patient with PMR develops a serious infection or an opportunistic infection, hold treatment with KEVZARA until the infection is controlled [see Warnings and Precautions (5.1)].
Dosage Modification for Patients with Polyarticular Juvenile Idiopathic Arthritis
Dose reduction of KEVZARA has not been studied in the pJIA population. Discontinue KEVZARA if ALT >5 ULN, platelet count ≤50,000 cells/mm3, neutrophil count <500 cells/mm3 associated with infection. Hold KEVZARA dosing for ALT >3 to ≤5 ULN, platelet count >50,000 to ≤100,000 cells/mm3, and neutrophil count ≥500 to <1000 cells/mm3, and until the clinical condition has been evaluated. The decision to discontinue KEVZARA should be based upon the medical assessment of the individual patient. If appropriate, the dose of concomitant methotrexate and/or other medications should be modified or discontinued.
3. Dosage Forms and Strengths
Injection: 150 mg/1.14 mL and 200 mg/1.14 mL clear and colorless to pale-yellow solution in a single-dose pre-filled syringe.
Injection: 150 mg/1.14 mL and 200 mg/1.14 mL clear and colorless to pale-yellow solution in a single-dose pre-filled pen.
4. Contraindications
KEVZARA is contraindicated in patients with known hypersensitivity to sarilumab or any of the inactive ingredients [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
5. Warnings and Precautions
5.1 Serious Infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in patients receiving immunosuppressive agents including KEVZARA. Among opportunistic infections, tuberculosis, candidiasis, and pneumocystis were reported with KEVZARA. Some patients presented with disseminated rather than localized disease and were often taking concomitant immunosuppressants such as methotrexate or corticosteroids. The most frequently observed serious infections with KEVZARA in RA patients included pneumonia and cellulitis [see Adverse Reactions (6.1)]. While not reported in KEVZARA clinical studies, other serious infections (e.g., histoplasmosis, cryptococcus, aspergillosis) have been reported in patients receiving other immunosuppressive agents for the treatment of RA.
Avoid use of KEVZARA in patients with an active infection, including localized infections. Consider the risks and benefits of treatment prior to initiating KEVZARA in patients who have:
- chronic or recurrent infection;
- a history of serious or opportunistic infections;
- underlying conditions that may predispose them to infection;
- been exposed to tuberculosis; or
- lived in or traveled to areas of endemic tuberculosis or endemic mycoses.
Closely monitor patients for the development of signs and symptoms of infection during treatment with KEVZARA, as signs and symptoms of acute inflammation may be lessened due to suppression of the acute phase reactants [see Dosage and Administration (2.6), Adverse Reactions (6.1)].
Hold treatment with KEVZARA if a patient develops a serious infection or an opportunistic infection.
Perform prompt and complete diagnostic testing appropriate for an immunocompromised patient who develops a new infection during treatment with KEVZARA; initiate appropriate antimicrobial therapy, and closely monitor the patient.
Tuberculosis
Evaluate patients for tuberculosis (TB) risk factors and test for latent infection prior to initiating treatment with KEVZARA. Treat patients with latent TB with standard antimycobacterial therapy before initiating KEVZARA. Consider anti-TB therapy prior to initiation of KEVZARA in patients with a past history of latent or active TB in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent TB but having risk factors for TB infection. When considering anti-TB therapy, consultation with a physician with expertise in TB may be appropriate.
Closely monitor patients for the development of signs and symptoms of TB including patients who tested negative for latent TB infection prior to initiating therapy.
Viral Reactivation
Viral reactivation has been reported with immunosuppressive biologic therapies. Cases of herpes zoster were observed in clinical studies with KEVZARA [see Adverse Reactions (6.1)]. The risk of Hepatitis B reactivation with KEVZARA is unknown since patients who were at risk for reactivation were excluded.
5.2 Laboratory Abnormalities
Neutropenia
Treatment with KEVZARA was associated with a higher incidence of decrease in absolute neutrophil count (ANC), including neutropenia [see Adverse Reactions (6.1)].
- Assess neutrophil count prior to initiation of KEVZARA and monitor neutrophil count 4 to 8 weeks after start of therapy and every 3 months thereafter [see Clinical Pharmacology (12.2)]. For recommendations regarding initiating KEVZARA therapy and dosage modifications based on ANC results see Dosage and Administration (2.1 and 2.6).
- Based on the pharmacodynamics of the changes in ANC [see Clinical Pharmacology (12.2)], use results obtained at the end of the dosing interval when considering dosage modification.
Thrombocytopenia
Treatment with KEVZARA was associated with a reduction in platelet counts in clinical studies [see Adverse Reactions (6.1)].
- Assess platelet count prior to initiation of KEVZARA and monitor platelets 4 to 8 weeks after start of therapy and every 3 months thereafter. For recommendations regarding initiating KEVZARA therapy and dosage modifications based on platelet counts see Dosage and Administration (2.1 and 2.6).
Elevated Liver Enzymes
Treatment with KEVZARA was associated with a higher incidence of transaminase elevations. These elevations were transient and did not result in any clinically evident hepatic injury in clinical studies [see Adverse Reactions (6.1)]. Increased frequency and magnitude of these elevations were observed when potentially hepatotoxic drugs (e.g., MTX) were used in combination with KEVZARA.
- Assess ALT/AST levels prior to initiation of KEVZARA and monitor ALT and AST levels 4 to 8 weeks after start of therapy and every 3 months thereafter. When clinically indicated, consider other liver function tests such as bilirubin. For recommendations regarding initiating KEVZARA therapy and dosage modifications based on transaminase elevations see Dosage and Administration (2.1 and 2.6).
Lipid Abnormalities
Treatment with KEVZARA was associated with increases in lipid parameters such as LDL cholesterol, HDL cholesterol and/or triglycerides [see Adverse Reactions (6.1)].
- Assess lipid parameters approximately 4 to 8 weeks following initiation of treatment with KEVZARA, then at approximately 6-month intervals.
- Manage patients according to clinical guidelines for the management of hyperlipidemia.
5.3 Gastrointestinal Perforation
Gastrointestinal perforations have been reported in clinical studies, primarily as complications of diverticulitis. GI perforation risk may be increased with concurrent diverticulitis or concomitant use of NSAIDs or corticosteroids. Promptly evaluate patients presenting with new onset abdominal symptoms [see Adverse Reactions (6.1)].
5.4 Immunosuppression
Treatment with immunosuppressants may result in an increased risk of malignancies. The impact of treatment with KEVZARA on the development of malignancies is not known but malignancies were reported in clinical studies [see Adverse Reactions (6.1)].
5.5 Hypersensitivity Reactions
Hypersensitivity reactions have been reported in association with KEVZARA [see Adverse Reactions (6.1)]. Hypersensitivity reactions that required treatment discontinuation were reported in 0.3% of patients in controlled RA trials. Injection site rash, rash, and urticaria were the most frequent hypersensitivity reactions. Advise patients to seek immediate medical attention if they experience any symptoms of a hypersensitivity reaction. If anaphylaxis or other hypersensitivity reaction occurs, stop administration of KEVZARA immediately. Do not administer KEVZARA to patients with known hypersensitivity to sarilumab [see Contraindications (4) and Adverse Reactions (6.1)].
5.6 Active Hepatic Disease and Hepatic Impairment
Treatment with KEVZARA is not recommended in patients with active hepatic disease or hepatic impairment, as treatment with KEVZARA was associated with transaminase elevations [see Adverse Reactions (6.1), Use in Specific Populations (8.6)].
5.7 Live Vaccines
Avoid concurrent use of live vaccines during treatment with KEVZARA due to potentially increased risk of infections; clinical safety of live vaccines during KEVZARA treatment has not been established. No data are available on the secondary transmission of infection from persons receiving live vaccines to patients receiving KEVZARA. Prior to initiating treatment, it is recommended that all patients be brought up to date with all immunizations in agreement with current immunization guidelines. The interval between live vaccinations and initiation of KEVZARA therapy should be in accordance with current vaccination guidelines regarding immunosuppressive agents [see Drug Interactions (7.3)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in labeling:
- Serious infections [see Warnings and Precautions (5.1)]
- Neutropenia, thrombocytopenia, elevated liver enzymes, lipid abnormalities [see Warnings and Precautions (5.2)]
- Gastrointestinal perforation [see Warnings and Precautions (5.3)]
- Immunosuppression [see Warnings and Precautions (5.4)]
- Hypersensitivity reactions [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Rheumatoid Arthritis
All patients in the safety data described below had moderately to severely active rheumatoid arthritis.
The safety of KEVZARA in combination with conventional DMARDs was evaluated based on data from seven studies, of which two were placebo-controlled, consisting of 2887 patients (long-term safety population). Of these, 2170 patients received KEVZARA for at least 24 weeks, 1546 for at least 48 weeks, 1020 for at least 96 weeks, and 624 for at least 144 weeks.
The pre-rescue placebo-controlled population includes patients from the two Phase 3 efficacy studies (Studies 1 and 2) from weeks 0 to 16 for Study 1 and weeks 0 to 12 for Study 2, and was used to assess common adverse reactions and laboratory abnormalities prior to patients being permitted to switch from placebo to KEVZARA. In this population, 582 patients, 579 patients, and 579 patients received KEVZARA 200 mg, KEVZARA 150 mg, or placebo once every two weeks, respectively, in combination with conventional DMARDs.
The 52-week placebo-controlled population includes patients from one Phase 2 study of 12-week duration and two Phase 3 efficacy studies (one of 24-week duration and the other of 52-week duration). This placebo-controlled population includes all subjects from the double-blind, placebo-controlled periods from each study and was analyzed under their original randomization assignment. In this population, 661 patients, 660 patients, and 661 patients received KEVZARA 200 mg, KEVZARA 150 mg, or placebo once every two weeks, respectively, in combination with conventional DMARDs.
Most safety data are described for the pre-rescue population. For rarer events, the 52-week placebo-controlled population is used.
The most common serious adverse reactions were infections.
The most frequent adverse reactions (occurring in at least 3% of patients treated with KEVZARA in combination with DMARDs) observed with KEVZARA in the clinical studies were neutropenia, increased ALT, injection site erythema, upper respiratory infections, and urinary tract infections.
In the pre-rescue placebo-controlled population, premature discontinuation due to adverse reactions occurred in 8%, 6% and 3% of patients treated with KEVZARA 200 mg, KEVZARA 150 mg, and placebo, respectively.
The most common adverse reaction (greater than 1%) that resulted in discontinuation of therapy with KEVZARA was neutropenia.
The use of KEVZARA as monotherapy was assessed in 132 patients, of which 67 received KEVZARA 200 mg and 65 patients received KEVZARA 150 mg without concomitant DMARDs. The safety profile was generally consistent with that in the population receiving concomitant DMARDs.
Overall Infections
In the pre-rescue placebo-controlled population, the rate of infections in the 200 mg and 150 mg KEVZARA + DMARD group was 110 and 105 events per 100 patient-years, respectively, compared to 81 events per 100 patient-years in the placebo + DMARD group. The most commonly reported infections (2% to 4% of patients) were upper respiratory tract infections, urinary tract infections, and nasopharyngitis.
In the 52-week placebo-controlled population, 0.8% of patients (5 patients) treated with KEVZARA 200 mg + DMARD, 0.6% (4 patients) treated with KEVZARA 150 mg + DMARD and 0.5% (3 patients) treated with placebo + DMARD had an event of herpes zoster [see Warnings and Precautions (5.1)].
The overall rate of infections with KEVZARA + DMARD in the long-term safety population was consistent with rates in the controlled periods of the studies.
Serious Infections
In the pre-rescue population, the rate of serious infections in the 200 mg and 150 mg KEVZARA + DMARD group was 3.8 and 4.4 events per 100 patient-years, respectively, compared to 2.5 events per 100 patient-years in the placebo + DMARD group. In the 52-week placebo-controlled population, the rate of serious infections in the 200 mg and 150 mg KEVZARA + DMARD group was 4.3 and 3.0 events per 100 patient-years, respectively, compared to 3.1 events per 100 patient-years in the placebo + DMARD group.
In the long-term safety population, the overall rate of serious infections was consistent with rates in the controlled periods of the studies. The most frequently observed serious infections included pneumonia and cellulitis. Cases of opportunistic infection have been reported [see Warnings and Precautions (5.1)].
Gastrointestinal Perforation
In the 52-week placebo-controlled population, one patient on KEVZARA therapy experienced a gastrointestinal (GI) perforation (0.11 events per 100 patient-years).
In the long-term safety population, the overall rate of GI perforation was consistent with rates in the controlled periods of the studies. Reports of GI perforation were primarily reported as complications of diverticulitis including lower GI perforation and abscess. Most patients who developed GI perforations were taking concomitant nonsteroidal anti-inflammatory medications (NSAIDs) or corticosteroids. The contribution of these concomitant medications relative to KEVZARA in the development of GI perforations is not known [see Warnings and Precautions (5.3)].
Hypersensitivity Reactions
In the pre-rescue placebo-controlled population, the proportion of patients who discontinued treatment due to hypersensitivity reactions was higher among those treated with KEVZARA (0.3% in 200 mg, 0.2% in 150 mg) than placebo (0%). The rate of discontinuations due to hypersensitivity in the long-term safety population was consistent with the placebo-controlled period.
Injection Site Reactions
In the pre-rescue placebo-controlled population, injection site reactions were reported in 7% of patients receiving KEVZARA 200 mg, 6% receiving KEVZARA 150 mg, and 1% receiving placebo. These injection site reactions (including erythema and pruritus) were mild in severity for the majority of patients and necessitated drug discontinuation in 2 (0.2%) patients receiving KEVZARA.
Laboratory Abnormalities
Decreased neutrophil count
In the pre-rescue placebo-controlled population, decreases in neutrophil counts less than 1000 per mm3 occurred in 6% and 4% of patients in the 200 mg KEVZARA + DMARD and 150 mg KEVZARA + DMARD group, respectively, compared to no patients in the placebo + DMARD groups. Decreases in neutrophil counts less than 500 per mm3 occurred in 0.7% of patients in both the 200 mg KEVZARA + DMARD and 150 mg KEVZARA + DMARD groups. Decrease in ANC was not associated with the occurrence of infections, including serious infections.
In the long-term safety population, the observations on neutrophil counts were consistent with what was seen in the placebo-controlled clinical studies [see Warnings and Precautions (5.2)].
Decreased platelet count
In the pre-rescue placebo-controlled population, decreases in platelet counts less than 100,000 per mm3 occurred in 1% and 0.7% of patients on 200 mg and 150 mg KEVZARA + DMARD, respectively, compared to no patients on placebo + DMARD, without associated bleeding events.
In the long-term safety population, the observations on platelet counts were consistent with what was seen in the placebo-controlled clinical studies [see Warnings and Precautions (5.2)].
Elevated liver enzymes
Liver enzyme elevations in the pre-rescue placebo-controlled population (KEVZARA + DMARD or placebo + DMARD) are summarized in Table 2. In patients experiencing liver enzyme elevation, modification of treatment regimen, such as interruption of KEVZARA or reduction in dose, resulted in decrease or normalization of liver enzymes [see Dosage and Administration (2.6)]. These elevations were not associated with clinically relevant increases in direct bilirubin, nor were they associated with clinical evidence of hepatitis or hepatic impairment [see Warnings and Precautions (5.2)].
| Placebo + DMARD N=579 | KEVZARA 150 mg + DMARD N=579 | KEVZARA 200 mg + DMARD N=582 |
|
|---|---|---|---|
| ULN = Upper Limit of Normal | |||
|
|||
| AST | |||
| Greater than ULN to 3 times ULN or less | 15% | 27% | 30% |
| Greater than 3 times ULN to 5 times ULN | 0% | 1% | 1% |
| Greater than 5 times ULN | 0% | 0.7% | 0.2% |
| ALT | |||
| Greater than ULN to 3 times ULN or less | 25% | 38% | 43% |
| Greater than 3 times ULN to 5 times ULN | 1% | 4% | 3% |
| Greater than 5 times ULN | 0% | 1% | 0.7% |
Lipid Abnormalities
Lipid parameters (LDL, HDL, and triglycerides) were first assessed at 4 weeks following initiation of KEVZARA + DMARDs in the placebo-controlled population. Increases were observed at this time point with no additional increases observed thereafter. Changes in lipid parameters from baseline to Week 4 are summarized below:
- Mean LDL increased by 12 mg/dL in the KEVZARA 150 mg every two weeks + DMARD group and 16 mg/dL in the KEVZARA 200 mg every two weeks + DMARD group.
- Mean triglycerides increased by 20 mg/dL in the KEVZARA 150 mg every two weeks + DMARD group and 27 mg/dL in the KEVZARA 200 mg every two weeks + DMARD group.
- Mean HDL increased by 3 mg/dL in both the KEVZARA 150 mg every two weeks + DMARD and KEVZARA 200 mg every two weeks + DMARD groups.
In the long-term safety population, the observations in lipid parameters were consistent with what was observed in the placebo-controlled clinical studies.
Malignancies
In the 52-week placebo-controlled population, 9 malignancies (exposure-adjusted event rate of 1.0 event per 100 patient-years) were diagnosed in patients receiving KEVZARA + DMARD compared to 4 malignancies in patients in the control group (exposure-adjusted event rate of 1.0 event per 100 patient-years).
In the long-term safety population, the rate of malignancies was consistent with the rate observed in the placebo-controlled period [see Warnings and Precautions (5.4)].
Other Adverse Reactions
Adverse reactions occurring in 2% or more of patients on KEVZARA + DMARD and greater than those observed in patients on placebo + DMARD are summarized in Table 3.
| Adverse Reactions | Placebo + DMARD (N=579) | KEVZARA 150 mg + DMARD (N=579) | KEVZARA 200 mg + DMARD (N=582) |
|---|---|---|---|
| Neutropenia | 0.2% | 7% | 10% |
| Alanine aminotransferase increased | 2% | 5% | 5% |
| Injection site erythema | 0.9% | 5% | 4% |
| Injection site pruritus | 0.2% | 2% | 2% |
| Upper respiratory tract infection | 2% | 4% | 3% |
| Urinary tract infection | 2% | 3% | 3% |
| Hypertriglyceridemia | 0.5% | 3% | 1% |
| Leukopenia | 0% | 0.9% | 2% |
Medically relevant adverse reactions occurring at an incidence less than 2% in patients with rheumatoid arthritis treated with KEVZARA in controlled studies was oral herpes.
Polymyalgia Rheumatica
Safety has been studied in one Phase 3 study (Study 3) in 117 PMR patients of whom 59 received subcutaneous KEVZARA 200 mg [see Clinical Studies (14.2)]. Of these, 45 patients received KEVZARA for at least 24 weeks, 44 patients for at least 40 weeks, and 10 patients for at least 52 weeks. The total patient years duration in the KEVZARA PMR population was 47.37 patient years during the 12-month double blind, placebo-controlled study.
The common adverse reactions occurring in ≥5% of patients treated with KEVZARA were neutropenia (15.3%), leukopenia (6.8%), constipation (6.8%), rash pruritic (5.1%), myalgia (6.8%), fatigue (5.1%), and injection site pruritus (5.1%).
Serious adverse reactions of neutropenia occurred in 2 patients (3.4%) in the KEVZARA group compared to none in the placebo group. In both cases of neutropenia, the participants had a neutrophil count less than 500 per mm3 without any infections and resolved following permanent discontinuation of study drug.
The most common adverse reactions that resulted in permanent discontinuation of therapy with KEVZARA were neutropenia in 3 patients (5.1%) and infection in 3 separate patients (5.1%), including COVID-19 (n=1), intervertebral discitis (n=1), and pneumonia (n=1).
Overall Infections
In Study 3, the proportion of patients with infections was lower in the KEVZARA group (37.3%) compared to the placebo group (50.0%). Two patients (3.2%) in the KEVZARA group and 1 patient (1.7%) in the placebo group had an event of herpes zoster.
Injection Site Reactions
In Study 3, three patients (5.1%) in the KEVZARA group experienced injection site reactions of pruritus which were mild in severity. No patient in the placebo group experienced injection site reactions.
Laboratory Abnormalities
Decreased neutrophil count
In Study 3, decreases in neutrophil counts less than 1,000 per mm3 occurred in 12% of the KEVZARA treated group and no patient in the placebo treated group. Decreases in neutrophil counts less than 500 per mm3 occurred in 3.4% of patients in KEVZARA treated group compared to no patient in the placebo treated group.
Decreased platelet count
In Study 3, decreases in platelet counts between 75,000 to 100,000 per mm3 occurred in two patients (3.4%) in the KEVZARA group, compared to no patient in the placebo treated group. These platelet count decreases were transient and not associated with bleeding events.
Elevated liver enzymes
In Study 3, no KEVZARA treated patients had an ALT or AST greater than 3 times the upper limit of normal (ULN). In the placebo treated group, 2 patients had ALT elevations greater than 3 times the ULN.
Lipid Abnormalities
In Study 3, cholesterol levels ≥299.27 mg/dL were observed in 8/58 (13.8%) patients in the KEVZARA group compared to 4/58 (6.9%) patients in the placebo group. Triglycerides ≥407.4 mg/dL were observed in 3/58 (5.2%) patients in the KEVZARA group compared to 1/58 (1.7%) in the placebo group.
No significant differences in mean HDL between KEVZARA group and placebo group were observed. At Week 52, mean increase from baseline for LDL and triglycerides levels were observed in the KEVZARA group though both remained within the normal range.
Polyarticular Juvenile Idiopathic Arthritis (pJIA)
The safety of KEVZARA was studied in patients 2 to 17 years of age with pJIA who have had an inadequate response to current therapy (Study 4) [see Clinical Studies (14.3)]. A total of 93 patients received at least one administration of the recommended dose: 79 (84.9%) patients received the recommended dose for 52 weeks, 53 (57%) patients received it for 96 weeks and 30 (32.2%) patients received it for 156 weeks.
The overall median duration of study treatment for the recommended dose was 672 days. The cumulative exposure to treatment for patients who received the recommended dose at any time during the study was 184.1 patient-years.
The most common adverse drug reactions were nasopharyngitis, neutropenia, upper respiratory tract infection, and injection site erythema.
The most common adverse drug reaction that resulted in permanent discontinuation of therapy with KEVZARA was neutropenia (5.4%).
No new adverse reactions and safety concerns were identified in the pJIA population compared to the RA population.
Infections
In Study 4, the rate of infections was 146.6 events per 100 patient-years. The most common infections observed were nasopharyngitis (36.6%) and upper respiratory tract infections (URTI) (14.0%).
Injection Site Reactions
In Study 4, injection site reactions (ISRs) occurred in 13 (14.0%) patients and the most commonly reported ISR was injection site erythema (9.7%). The majority of these events were mild and none of the ISRs required patient withdrawal from treatment or dose interruption.
Laboratory Abnormalities
Decrease neutrophil count
In Study 4, decreases in neutrophil counts less than 1000 per mm3 occurred in 10/52 (19.2%) patients weighing in ≥30 kg and 20/41 (48.8%) patients weighing 10 to <30 kg. The frequency of decreased neutrophil count was higher until Week 12. Decrease in ANC was not associated with an occurrence of infections, including serious infections.
Decrease monocyte count
In Study 4, decrease in monocyte counts occurred in 4 (4.3%) patients and were mild in severity and non-serious.
Elevated liver enzymes
In Study 4, nine (9.7%) patients had ALT increases, including one (1.1%) patient who had ALT >3 times upper limit of normal (ULN) and up to ≤5 times ULN, and two (2.2%) patients who had ALT increases >5 times ULN and up to ≤10 times ULN that resulted in permanent discontinuation. All patients recovered.
Lipid Abnormalities
In Study 4, triglyceride levels of ≥150 mg/dL (1 × ULN) were observed in one (1.1%) patient. Three (3.2%) patients overall had elevation in triglycerides, and all were mild in severity and non-serious. No significant changes in mean LDL, HDL or total cholesterol were observed during the entire 156-week treatment period.
Related/similar drugs
7. Drug Interactions
7.1 Use with Other Drugs
Population pharmacokinetic analyses did not detect any effect of methotrexate (MTX) on sarilumab clearance. KEVZARA has not been investigated in combination with JAK inhibitors or biological DMARDs such as TNF antagonists [see Dosage and Administration (2.2)].
7.2 Interactions with CYP450 Substrates
Various in vitro and limited in vivo human studies have shown that cytokines and cytokine modulators can influence the expression and activity of specific cytochrome P450 (CYP) enzymes and therefore have the potential to alter the pharmacokinetics of concomitantly administered drugs that are substrates of these enzymes. Elevated interleukin-6 (IL-6) concentration may down-regulate CYP activity such as in patients with RA and hence increase drug levels compared to subjects without RA. Blockade of IL-6 signaling by IL-6Rα antagonists such as KEVZARA might reverse the inhibitory effect of IL-6 and restore CYP activity, leading to altered drug concentrations.
The modulation of IL-6 effect on CYP enzymes by KEVZARA may be clinically relevant for CYP substrates with a narrow therapeutic index, where the dose is individually adjusted. Upon initiation or discontinuation of KEVZARA, in patients being treated with CYP substrate medicinal products, perform therapeutic monitoring of effect (e.g., warfarin) or drug concentration (e.g., theophylline) and adjust the individual dose of the medicinal product as needed.
Exercise caution when co-administering KEVZARA with CYP3A4 substrate drugs where decrease in effectiveness is undesirable, e.g., oral contraceptives, lovastatin, atorvastatin, etc. The effect of KEVZARA on CYP450 enzyme activity may persist for several weeks after stopping therapy [see Clinical Pharmacology (12.3)].
7.3 Live Vaccines
Avoid concurrent use of live vaccines during treatment with KEVZARA [see Warnings and Precautions (5.7)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
The limited human data with KEVZARA in pregnant women are not sufficient to inform drug-associated risk for major birth defects and miscarriage. Monoclonal antibodies, such as sarilumab, are actively transported across the placenta during the third trimester of pregnancy and may affect immune response in the in utero exposed infant (see Clinical Considerations). From animal data, and consistent with the mechanism of action, levels of IgG, in response to antigen challenge, may be reduced in the fetus/infant of treated mothers (see Clinical Considerations and Data). In an animal reproduction study, consisting of a combined embryo-fetal and pre- and postnatal development study with monkeys that received intravenous administration of sarilumab, there was no evidence of embryotoxicity or fetal malformations with exposures up to approximately 84 times the maximum recommended human dose (MRHD) (see Data). The literature suggests that inhibition of IL-6 signaling may interfere with cervical ripening and dilatation and myometrial contractile activity leading to potential delays of parturition (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. KEVZARA should be used in pregnancy only if the potential benefit justifies the potential risk to the fetus.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Risks and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to KEVZARA in utero [see Warnings and Precautions (5.7)]. From the animal data, and consistent with the mechanism of action, levels of IgG, in response to antigen challenge, may be reduced in the fetus/infant of treated mothers (see Data).
Data
Animal Data
In a combined embryo-fetal and pre- and postnatal development study, pregnant cynomolgus monkeys received sarilumab at intravenous doses of 0, 5, 15, or 50 mg/kg/week from confirmation of pregnancy at gestation day (GD) 20, throughout the period of organogenesis (up to approximately GD 50), and continuing to natural birth of infants at around GD 165. Maintenance of pregnancy was not affected at any doses. Sarilumab was not embryotoxic or teratogenic with exposures up to approximately 84 times the MRHD (based on AUC with maternal intravenous doses up to 50 mg/kg/week). Sarilumab had no effect on neonatal growth and development evaluated up to one month after birth. Sarilumab was detected in the serum of neonates up to one month after birth, suggesting that the antibody had crossed the placenta.
Following antigen challenge, decreased IgG titers attributed to the immunosuppressive action of sarilumab were evident in studies with older monkeys, with exposures up to approximately 80 times the MRHD (based on AUC with intravenous doses up to 50 mg/kg/week) and juvenile mice treated with an analogous antibody, which binds to murine IL-6Rα to inhibit IL-6 mediated signaling, at subcutaneous doses up to 200 mg/kg/week. These findings suggest the potential for decreased IgG titers, following antigen challenge, in infants of mothers treated with KEVZARA.
Parturition is associated with significant increases of IL-6 in the cervix and myometrium. The literature suggests that inhibition of IL-6 signaling may interfere with cervical ripening and dilatation and myometrial contractile activity leading to potential delays of parturition. For mice deficient in IL-6 (ll6-/- null mice), parturition was delayed relative to wild-type (ll6+/+) mice. Administration of recombinant IL-6 to ll6-/- null mice restored the normal timing of delivery.
8.2 Lactation
Risk Summary
No information is available on the presence of sarilumab in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. Maternal IgG is present in human milk. If sarilumab is transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to sarilumab are unknown. The lack of clinical data during lactation precludes clear determination of the risk of KEVZARA to an infant during lactation; therefore, the developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for KEVZARA and the potential adverse effects on the breastfed child from KEVZARA or from the underlying maternal condition.
8.4 Pediatric Use
KEVZARA is approved for active polyarticular juvenile idiopathic arthritis (pJIA) in pediatric patients who weigh 63 kg or greater. Use of KEVZARA in this patient population is supported by evidence from adequate and well-controlled studies of KEVZARA in adults with RA, pharmacokinetic data from adult patients with RA, and a pharmacokinetic, pharmacodynamic, dose-finding, and safety study in pediatric patients with pJIA 2 years of age and older. KEVZARA is not approved in pediatric patients weighing less than 63 kg because of the lack of an appropriate dosage form [see Adverse Reactions (6.1), Pharmacokinetics (12.3), and Clinical Studies (14.1, 14.3)].
The safety and effectiveness of KEVZARA have not been established in pediatric patients with pJIA below the age of 2 years.
8.5 Geriatric Use
Of the total number of patients with RA exposed to KEVZARA in clinical studies [see Clinical Studies (14.1)], 450 patients (15%) were 65 years of age and over, while 48 patients (1.6%) were 75 years and over. In clinical studies, no overall differences in safety and efficacy were observed between older and younger patients. The frequency of serious infections among KEVZARA and placebo-treated patients 65 years of age and older was higher than those under the age of 65.
Of the total number of patients with PMR exposed to KEVZARA in the clinical study (Study 3) [see Clinical Studies (14.2)], 16 patients (27.1%) were under 65 years of age, 33 patients (55.9%) were 65 to 75 years of age, and 10 patients (17.0%) were 75 years and over. The median age in the PMR study was 69.0 years and all patients were on baseline corticosteroid. There were no differences in the incidence of serious infections between the KEVZARA group and placebo group. In Study 3, no overall differences in safety were observed between older and younger patients.
As there is a higher incidence of infections in the elderly population in general, caution should be used when treating the elderly.
8.6 Hepatic Impairment
The safety and efficacy of KEVZARA have not been studied in patients with hepatic impairment, including patients with positive HBV or HCV serology [see Warnings and Precautions (5.6)].
8.7 Renal Impairment
No dose adjustment is required in patients with mild to moderate renal impairment. KEVZARA has not been studied in patients with severe renal impairment [see Clinical Pharmacology (12.3)].
11. Kevzara Description
Sarilumab is a human recombinant monoclonal antibody of the IgG1 subclass that binds to the IL-6 receptor and has an approximate molecular weight of 150 kDa. Sarilumab is produced by recombinant DNA technology in Chinese Hamster Ovary cell suspension culture.
KEVZARA (sarilumab) injection for subcutaneous administration is supplied as a sterile, clear and colorless to pale yellow, preservative-free solution of approximately pH 6.0. KEVZARA is supplied in a single-dose pre-filled syringe and pre-filled pen.
Each syringe or pen delivers 1.14 mL of solution containing 150 mg or 200 mg of sarilumab, arginine (8.94 mg), histidine (3.71 mg), polysorbate 20 (2.28 mg), sucrose (57 mg) and Water for Injection, USP.
12. Kevzara - Clinical Pharmacology
12.1 Mechanism of Action
Sarilumab binds to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R), and has been shown to inhibit IL-6-mediated signaling through these receptors. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types including T- and B-cells, lymphocytes, monocytes, and fibroblasts. IL-6 has been shown to be involved in diverse physiological processes such as T-cell activation, induction of immunoglobulin secretion, initiation of hepatic acute phase protein synthesis, and stimulation of hematopoietic precursor cell proliferation and differentiation. IL-6 is also produced by synovial and endothelial cells leading to local production of IL-6 in joints affected by inflammatory processes such as rheumatoid arthritis.
12.2 Pharmacodynamics
Following single-dose subcutaneous administration of sarilumab 200-mg and 150-mg in patients with RA, rapid reduction of CRP levels was observed. Levels were reduced to normal within 2 weeks after treatment initiation. Following single-dose sarilumab administration, in patients with RA, absolute neutrophil counts decreased to the nadir between 3 to 4 days and thereafter recovered towards baseline [see Warnings and Precautions (5.2)]. Treatment with sarilumab resulted in decreases in fibrinogen and serum amyloid A, and increases in hemoglobin and serum albumin. In patients with pJIA, decreases in CRP, erythrocyte sedimentation rate (ESR) and neutrophil count were observed after KEVZARA administration.
12.3 Pharmacokinetics
Rheumatoid Arthritis
Absorption
The pharmacokinetics of sarilumab were characterized in 1770 adult patients with rheumatoid arthritis (RA) treated with sarilumab which included 631 patients treated with 150 mg and 682 patients treated with 200 mg doses by subcutaneous injection every two weeks for up to 52 weeks. The median tmax was observed in 2 to 4 days.
At steady state, exposure over the dosing interval measured by area under curve (AUC) increased 2-fold with an increase in dose from 150 to 200 mg every two weeks. Steady state was reached in 14 to 16 weeks with a 2- to 3-fold accumulation compared to single dose exposure.
For the 150 mg every two weeks dose regimen, the estimated mean (± SD) steady-state AUC, Cmin and Cmax of sarilumab were 202 ± 120 mg.day/L, 6.35 ± 7.54 mg/L, and 20.0 ± 9.20 mg/L, respectively.
For the 200 mg every two weeks dose regimen, the estimated mean (± SD) steady-state AUC, Cmin and Cmax of sarilumab were 395 ± 207 mg.day/L, 16.5 ± 14.1 mg/L, and 35.6 ± 15.2 mg/L, respectively.
Elimination
Sarilumab is eliminated by parallel linear and non-linear pathways. At higher concentrations, the elimination is predominantly through the linear, non-saturable proteolytic pathway, while at lower concentrations, non-linear saturable target-mediated elimination predominates. The half-life of sarilumab is concentration-dependent. At 200 mg every 2 weeks, the concentration-dependent half-life is up to 10 days in patients with RA at steady state. At 150 mg every 2 weeks, the concentration-dependent half-life is up to 8 days in patients with RA at steady state.
After the last steady state dose of 150 mg and 200 mg sarilumab, the median times to non-detectable concentration are 28 and 43 days, respectively.
Population pharmacokinetic analyses in patients with RA revealed that there was a trend toward higher apparent clearance of sarilumab in the presence of anti-sarilumab antibodies.
Polymyalgia Rheumatica
The pharmacokinetic profile of subcutaneous sarilumab in PMR patients was determined using a population pharmacokinetic analysis on a data set including 58 PMR patients treated with repeated subcutaneous administration of sarilumab 200 mg every two weeks. In general, pharmacokinetic exposures were higher in patients with PMR when compared to patients with RA. For this dose regimen, the estimated mean (± SD) steady-state AUC, Cmin and Cmax of sarilumab were 551 ± 321 mg.day/L, 27.0 ± 21.5 mg/L, and 46.5 ± 23.0 mg/L, respectively. The median time to steady state in PMR patients was estimated to be 28 weeks. There was accumulation following subcutaneous administration of sarilumab 200 mg, with an accumulation ratio of approximately 6-fold based on the mean trough concentrations.
Polyarticular juvenile idiopathic arthritis (pJIA)
The pharmacokinetics of sarilumab in pJIA patients was characterized by a population pharmacokinetic analysis which included 101 pediatric patients 2 to 17 years of age who were treated with repeated subcutaneous doses of sarilumab.
For 3 mg/kg sarilumab (patients with a body weight ≥30 kg) given every 2 weeks, the estimated mean (±SD) steady-state AUC, Cmin, and Cmax of sarilumab were 276 ± 121 mg.day/L, 9.57 ± 5.84 mg/L, and 27.1 ± 11.6 mg/L, respectively.
For 4 mg/kg sarilumab (patients with a body weight 10 to <30 kg) given every 2 weeks, the estimated mean (± SD) steady-state AUC, Cmin, and Cmax of sarilumab were 395 ± 101 mg.day/L, 14.4 ± 9.81 mg/L, and 40.4 ± 7.77 mg/L, respectively.
Steady state was reached in 12 to 28 weeks with a 2- to 4-fold accumulation compared to single dose exposure for 3 and 4 mg/kg q2w. Steady state concentrations were within the range of exposures in adult RA patients following 150 mg/200 mg every 2 weeks.
Specific Populations
Population pharmacokinetic analyses in adult patients showed that age, gender and race did not meaningfully influence the pharmacokinetics of sarilumab. Although body weight influenced the pharmacokinetics of sarilumab, no dose adjustments are recommended for any of these demographics in adult patients.
Hepatic Impairment
No formal study of the effect of hepatic impairment on the pharmacokinetics of sarilumab was conducted.
Renal Impairment
No formal study of the effect of renal impairment on the pharmacokinetics of sarilumab was conducted. Based on population pharmacokinetic analysis of data from 1770 patients with RA, including patients with mild (creatinine clearance (CLcr): 60 to 90 mL/min; N=471 at baseline) or moderate (CLcr: 30 to 60 mL/min; N=74 at baseline) renal impairment, CLcr was correlated with sarilumab exposure. However, the effect of CLcr on exposure is not sufficient to warrant a dose adjustment [see Use in Specific Populations (8.7)]. Patients with severe renal impairment were not studied.
Drug-Drug Interactions
CYP450 Substrates
Simvastatin is a CYP3A4 and OATP1B1 substrate. In 17 patients with RA, one week following a single 200-mg subcutaneous administration of sarilumab, exposure of simvastatin and simvastatin acid decreased by 45% and 36%, respectively [see Drug Interactions (7.2)].
12.6 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to sarilumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In the RA pre-rescue population, 4.0% of patients treated with KEVZARA 200 mg + DMARD, 5.7% of patients treated with KEVZARA 150 mg + DMARD and 1.9% of patients treated with placebo + DMARD, exhibited an anti-drug antibody (ADA) response. Neutralizing antibodies (NAb) were detected in 1.0% of patients on KEVZARA 200 mg + DMARD, 1.6% of patients on KEVZARA 150 mg + DMARD, and 0.2% of patients on placebo + DMARD.
In RA patients treated with KEVZARA monotherapy, 9.2% of patients exhibited an ADA response with 6.9% of patients also exhibiting NAbs. Prior to administration of KEVZARA, 2.3% of patients exhibited an ADA response.
No correlation was observed between ADA development and either loss of efficacy or adverse reactions in RA patients.
In the PMR population, 1 patient (1.8%) in the KEVZARA 200 mg + 14-week corticosteroid taper group exhibited an ADA response. None of the patients in the placebo + 52-week corticosteroid taper group exhibited an ADA response. Neutralizing antibodies were detected in the PMR patient with ADA response on KEVZARA 200 mg; the patient did not demonstrate a clinical response. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the safety, and/or effectiveness of sarilumab is unknown.
In the pJIA population, 3 (4.3%) patients treated with the recommended dose exhibited an anti-drug antibody (ADA) response. Neutralizing antibodies were detected in one pJIA patient with ADA response. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the safety, and/or effectiveness of sarilumab is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term animal studies have been performed to establish the carcinogenicity potential of sarilumab. Literature indicates that the IL-6 pathway can mediate anti-tumor responses by promoting increased immune cell surveillance of the tumor microenvironment. However, available published evidence also supports that IL-6 signaling through the IL-6 receptor may be involved in pathways that lead to tumorigenesis. The malignancy risk in humans from an antibody that disrupts signaling through the IL-6 receptor, such as sarilumab, is presently unknown.
Fertility and reproductive performance were unaffected in male and female mice treated with an analogous antibody, which binds to murine IL-6Rα to inhibit IL-6 mediated signaling, at subcutaneous doses of 10, 25, and 100 mg/kg twice per week.
14. Clinical Studies
14.1 Rheumatoid Arthritis
Design of Clinical Studies in Adults with Moderately to Severely Active RA
The efficacy and safety of KEVZARA in RA were assessed in two randomized, double-blind, placebo-controlled multicenter studies (Study 1 and Study 2) in patients older than 18 years with moderately to severely active rheumatoid arthritis (RA) diagnosed according to American College of Rheumatology (ACR) criteria. Patients had at least 8 tender and 6 swollen joints at baseline.
Study 1 (NCT01061736) evaluated 1197 patients with moderately to severely active rheumatoid arthritis who had inadequate clinical response to methotrexate (MTX). Patients received subcutaneous KEVZARA 200 mg, KEVZARA 150 mg, or placebo every two weeks with concomitant MTX. After Week 16 in Study 1, patients with an inadequate response could have been rescued with KEVZARA 200 mg every two weeks.
Study 2 (NCT01146652) evaluated 546 patients with moderately to severely active rheumatoid arthritis who had an inadequate clinical response or were intolerant to one or more TNF-α antagonists. Patients received subcutaneous KEVZARA 200 mg, KEVZARA 150 mg, or placebo every two weeks with concomitant conventional DMARDs (MTX, sulfasalazine, leflunomide, and/or hydroxychloroquine). After Week 12 in Study 2, patients with an inadequate response could have been rescued with KEVZARA 200 mg every two weeks.
In Studies 1 and 2, the primary endpoint was the proportion of patients who achieved an ACR20 response at Week 24. Other key endpoints evaluated included change from baseline in HAQ-DI at Week 16 in Study 1 and at Week 12 in Study 2, and change from baseline in van der Heijde-modified Total Sharp Score (mTSS) at Week 52 in Study 1.
Clinical Response
The percentages of KEVZARA every two weeks + MTX/DMARD-treated patients achieving ACR20, ACR50 and ACR70 responses in Studies 1 and 2 are shown Table 4. In both studies, patients treated with either 200 mg or 150 mg of KEVZARA every two weeks + MTX/DMARD had higher ACR20, ACR50, and ACR70 response rates versus placebo + MTX/DMARD-treated patients at Week 24.
In Studies 1 and 2, a greater proportion of patients treated with KEVZARA 200 mg or 150 mg every two weeks plus MTX/DMARD achieved a low level of disease activity as measured by a Disease Activity Score 28-C-Reactive Protein (DAS28-CRP) <2.6 compared with placebo + MTX/DMARD at the end of the studies (Table 4). In Study 1, the proportion of patients achieving DAS28-CRP <2.6 who had at least 3 or more active joints at the end of Week 24 was 33.1%, 37.8% and 20%, in the KEVZARA 200 mg + MTX/DMARD arm, KEVZARA 150 mg + MTX/DMARD arm, and placebo arm respectively.
| Percentage of Patients | ||||||
|---|---|---|---|---|---|---|
| Study 1 | Study 2 | |||||
| Placebo + MTX N=398 | KEVZARA 150 mg + MTX N=400 | KEVZARA 200 mg + MTX N=399 | Placebo + DMARD(s)†
N=181 | KEVZARA 150 mg + DMARD(s)†
N=181 | KEVZARA 200 mg + DMARD(s)†
N=184 |
|
|
||||||
| ACR20 | ||||||
| Week 12 | 34.7% | 54.0% | 64.9% | 37.6% | 54.1% | 62.5% |
| Difference from placebo (95% CI)‡ | 19.4% (12.6%, 26.1%) | 30.2% (23.6%, 36.8%) | 16.6% (6.7%, 26.5%) | 25.3% (15.7%, 34.8%) |
||
| Week 24§ | 33.4% | 58.0% | 66.4% | 33.7% | 55.8% | 60.9% |
| Difference from placebo (95% CI)‡ | 24.6% (18.0%, 31.3%) | 33.0% (26.5%, 39.5%) | 22.1% (12.6%, 31.6%) | 27.4% (17.7%, 37.0%) |
||
| Week 52 | 31.7% | 53.5% | 58.6% | |||
| Difference from placebo (95% CI)‡ | 21.9% (15.2%, 28.5%) | 27.0% (20.5%, 33.6%) | NA¶ | NA¶ | NA¶ | |
| ACR50 | ||||||
| Week 12 | 12.3% | 26.5% | 36.3% | 13.3% | 30.4% | 33.2% |
| Difference from placebo (95% CI)‡ | 14.2% (8.9%, 19.6%) | 24.1% (18.4%, 29.8%) | 17.1% (9.2%, 25.1%) | 20.1% (12.0%, 28.3%) |
||
| Week 24 | 16.6% | 37.0% | 45.6% | 18.2% | 37.0% | 40.8% |
| Difference from placebo (95% CI)‡ | 20.4% (14.5%, 26.3%) | 29.1% (23.0%, 35.1%) | 18.8% (10.2%, 27.4%) | 22.8% (14.0%, 31.6%) |
||
| Week 52 | 18.1% | 40.0% | 42.9% | |||
| Difference from placebo (95% CI)‡ | 21.9% (15.8%, 28.0%) | 24.8% (18.7%, 30.9%) | NA¶ | NA¶ | NA¶ | |
| ACR70 | ||||||
| Week 12 | 4.0% | 11.0% | 17.5% | 2.2% | 13.8% | 14.7% |
| Difference from placebo (95% CI)‡ | 7.0% (3.4%, 10.6%) | 13.5% (9.4%, 17.7%) | 11.6% (6.2%, 17.0%) | 12.5% (7.1%, 17.9%) |
||
| Week 24 | 7.3% | 19.8% | 24.8% | 7.2% | 19.9% | 16.3% |
| Difference from placebo (95% CI)‡ | 12.5% (7.8%, 17.1%) | 17.5% (12.6%, 22.5%) | 12.7% (6.1%, 19.3%) | 9.2% (2.8%, 15.7%) |
||
| Week 52 | 9.0% | 24.8% | 26.8% | |||
| Difference from placebo (95% CI)‡ | 15.7% (10.6%, 20.8%) | 17.8% (12.6%, 23.0%) | NA¶ | NA¶ | NA¶ | |
| Major clinical response# | ||||||
| Responders | 3.0% | 12.8% | 14.8% | NA¶ | NA¶ | NA¶ |
| Difference from placebo (95% CI)‡ | 9.7% (6.1%, 13.4%) | 11.8% (7.9%, 15.6%) | ||||
| DAS28-CRP < 2.6Þ | ||||||
| Week 12 | ||||||
| Percentage of patients | 4.8% | 18.0% | 23.1% | 3.9% | 17.1% | 17.9% |
| Difference from placebo (95% CI)‡ | 13.3% (9.0%, 17.5%) | 18.3% (13.7%, 23.0%) | 13.3% (7.3%, 19.3%) | 14.1% (8.0%, 20.3%) |
||
| Week 24 | ||||||
| Percentage of patients | 10.1% | 27.8% | 34.1% | 7.2% | 24.9% | 28.8% |
| Difference from placebo (95% CI)‡ | 17.7% (12.5%, 23.0%) | 24.0% (18.5%, 29.5%) | 17.7% (10.5%, 24.9%) | 21.7% (14.3%, 29.1%) |
||
The percent ACR20 response by visit in Study 1 is shown in Figure 1. A similar response curve was observed in Study 2.
| Figure 1: Percent of ACR20 Response by Visit for Study 1 (Adults with Moderately to Severely Active RA) |
|---|
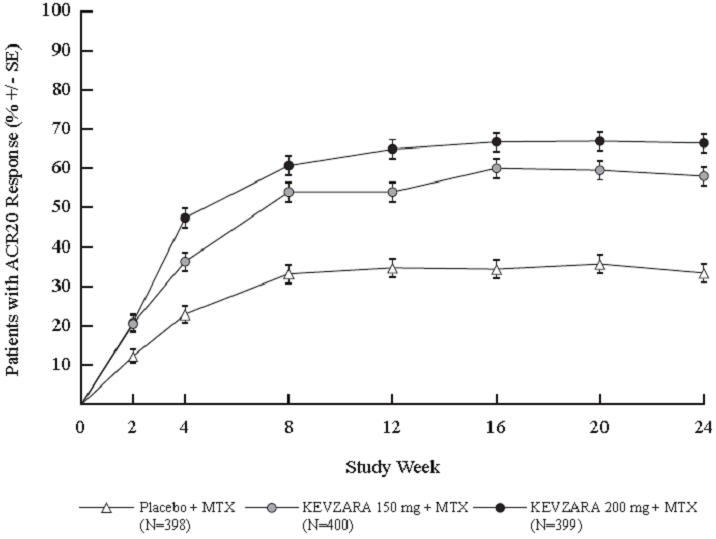 |
The results of the components of the ACR response criteria at Week 12 for Studies 1 and 2 are shown in Table 5.
| Study 1 | Study 2 | |||||
|---|---|---|---|---|---|---|
| Component means (range/units) | Placebo + MTX (N=398) | KEVZARA 150 mg + MTX (N=400) | KEVZARA 200 mg + MTX (N=399) | Placebo + DMARD(s) (N=181) | KEVZARA 150 mg + DMARD(s) (N=181) | KEVZARA 200 mg + DMARD(s) (N=184) |
|
||||||
| Tender Joints (0–68) | ||||||
| Baseline | 26.80 | 27.21 | 26.50 | 29.42 | 27.66 | 29.55 |
| Week 12 | 16.25 | 12.88 | 11.78 | 19.18 | 13.38 | 13.10 |
| Change from baseline | -10.51 | -14.42 | -14.94 | -9.79 | -14.11 | -15.92 |
| Swollen Joints (0–66) | ||||||
| Baseline | 16.68 | 16.60 | 16.77 | 20.21 | 19.60 | 19.97 |
| Week 12 | 9.66 | 7.50 | 6.79 | 12.50 | 8.82 | 8.28 |
| Change from baseline | -7.02 | -9.03 | -10.12 | -7.25 | -10.77 | -10.89 |
| Pain VAS* (0–100 mm) | ||||||
| Baseline | 63.71 | 65.48 | 66.71 | 71.57 | 71.02 | 74.86 |
| Week 12 | 49.25 | 41.47 | 36.93 | 54.77 | 43.45 | 41.66 |
| Change from baseline | -14.45 | -23.73 | -29.77 | -16.12 | -27.95 | -32.77 |
| Physician global VAS* (0–100 mm) | ||||||
| Baseline | 62.86 | 63.43 | 63.59 | 68.39 | 68.10 | 67.76 |
| Week 12 | 39.25 | 31.32 | 28.47 | 43.73 | 33.65 | 30.18 |
| Change from baseline | -23.63 | -31.85 | -34.84 | -24.60 | -34.92 | -36.92 |
| Patient global VAS* (0–100 mm) | ||||||
| Baseline | 63.70 | 64.43 | 66.49 | 68.77 | 67.71 | 70.89 |
| Week 12 | 49.37 | 41.52 | 38.05 | 53.67 | 41.99 | 41.74 |
| Change from baseline | -13.92 | -22.88 | -28.39 | -15.05 | -26.05 | -28.83 |
| HAQ-DI (0–3) | ||||||
| Baseline | 1.61 | 1.63 | 1.69 | 1.80 | 1.72 | 1.82 |
| Week 12 | 1.34 | 1.15 | 1.13 | 1.49 | 1.23 | 1.33 |
| Change from baseline | -0.27 | -0.47 | -0.57 | -0.29 | -0.50 | -0.49 |
| CRP (mg/L) | ||||||
| Baseline | 20.46 | 22.57 | 22.23 | 26.02 | 23.60 | 30.77 |
| Week 12 | 19.61 | 9.24 | 3.30 | 21.72 | 9.21 | 4.58 |
| Change from baseline | -0.58 | -13.59 | -18.31 | -3.39 | -14.24 | -25.91 |
Radiographic Response
In Study 1, structural joint damage was assessed radiographically and expressed as the van der Heijde-modified Total Sharp Score (mTSS) and its components, the erosion score and joint space narrowing score. Radiographs of hands and feet were obtained at baseline, 24 weeks, and 52 weeks and scored independently by at least two well-trained readers who were blinded to treatment group and visit number.
Both doses of KEVZARA + MTX were superior to placebo + MTX in the change from baseline in mTSS over 52 weeks (see Table 6). Less progression of both erosion and joint space narrowing scores over 52 weeks was reported in the KEVZARA + MTX treatment groups compared to the placebo + MTX group.
Treatment with KEVZARA + MTX was associated with significantly less radiographic progression of structural damage as compared with placebo + MTX. At Week 52, 55.6% of patients receiving KEVZARA 200 mg + MTX and 47.8% of patients receiving KEVZARA 150 mg + MTX had no progression of structural damage (as defined by a change in the Total Sharp Score of zero or less) compared with 38.7% of patients receiving placebo.
| Study 1 | |||
|---|---|---|---|
| Placebo + MTX (N=398) | KEVZARA 150 mg + MTX (N=400) | KEVZARA 200 mg + MTX (N=399) |
|
| Modified Total Sharp Score (mTSS) | |||
| Mean change | 2.78 | 0.90 | 0.25 |
| LS† mean difference (95% CI‡) | -1.88 (-2.74, -1.01) | -2.52 (-3.38, -1.66) | |
| Erosion score | |||
| Mean change | 1.46 | 0.42 | 0.05 |
| LS† mean difference (95% CI‡) | -1.03 (-1.53, -0.53) | -1.40 (-1.90, -0.90) | |
| Joint space narrowing score | |||
| Mean change | 1.32 | 0.47 | 0.20 |
| LS† mean difference (95% CI‡) | -0.85 (-1.34, -0.35) | -1.12 (-1.61, -0.63) | |
Physical Function Response
In Studies 1 and 2, physical function and disability were assessed by the Health Assessment Questionnaire Disability Index (HAQ-DI). Patients receiving KEVZARA 200 mg every two weeks + MTX/DMARD and KEVZARA 150 mg every two weeks + MTX/DMARD demonstrated greater improvement from baseline in physical function compared to placebo + MTX/DMARD at Week 16 and Week 12 in Studies 1 and 2, respectively (Table 7).
| Study 1 | Study 2 | |||||
|---|---|---|---|---|---|---|
| Week 16 | Week 12 | |||||
| Placebo + MTX (N=398) | KEVZARA 150 mg + MTX (N=400) | KEVZARA 200 mg + MTX (N=399) | Placebo + DMARD(s) (N=181) | KEVZARA 150 mg + DMARD(s) (N=181) | KEVZARA 200 mg + DMARD(s) (N=184) |
|
| HAQ-DI | ||||||
| Change from baseline | -0.30 | -0.54 | -0.58 | -0.29 | -0.50 | -0.49 |
| Difference from placebo (95% CI)* | -0.24 (-0.31, -0.16) | -0.26 (-0.34, -0.18) | -0.20 (-0.32, -0.09) | -0.21 (-0.33, -0.10) |
||
| % of patients with clinically meaningful improvement† | 42.5% | 53.8% | 57.4% | 35.9% | 47% | 51.1% |
Other Health Related Outcomes
General health status was assessed by the Short Form health survey (SF-36) in Studies 1 and 2. Patients receiving KEVZARA 200 mg every two weeks + MTX/DMARD demonstrated greater improvement from baseline compared to placebo + MTX/DMARD in the physical component summary (PCS) at Week 24, but there was no evidence of a difference between the treatment groups in the mental component summary (MCS) at Week 24. Patients receiving KEVZARA 200 mg + MTX/DMARD reported greater improvement relative to placebo in the domains of Physical Functioning, Role Physical, Bodily Pain, General Health Perception, Vitality, Social Functioning and Mental Health, but not in the Role Emotional domain.
14.2 Polymyalgia Rheumatica
The efficacy and safety of KEVZARA in PMR were assessed in a randomized, double-blind, placebo-controlled, 52-week, multicenter study (Study 3) (NCT03600818) in adults with PMR diagnosed according to American College of Rheumatology/European Union League against Rheumatism (ACR/EULAR) classification criteria. Patients had at least one episode of unequivocal PMR flare while attempting to taper corticosteroids.
In Study 3, patients with active PMR were randomized to receive KEVZARA 200 mg every two weeks with a pre-defined 14-week taper of prednisone (n= 60) or placebo every two weeks with a pre-defined 52-week taper of prednisone (n=58). One participant was randomized but not treated in the KEVZARA 200 mg arm. Patients experiencing a disease flare or unable to adhere to the assigned prednisone tapering schedule could receive corticosteroids as rescue therapy.
The primary endpoint was the proportion of patients with sustained remission at Week 52. Sustained remission was defined as achievement of disease remission no later than Week 12, absence of disease flare from Week 12 through Week 52, sustained reduction of CRP (to <10 mg/L) from Week 12 through Week 52, and successful adherence to prednisone taper from Week 12 through Week 52. An additional endpoint was total cumulative corticosteroid dose over 52 weeks.
Clinical Response
The proportion of participants achieving sustained remission at Week 52 was higher in the KEVZARA arm compared to the placebo arm; this difference was statistically significant. At 52 weeks, a higher proportion of patients in the KEVZARA arm achieved each component of the sustained remission endpoint compared to the placebo. An analysis was conducted that removed all acute phase reactants (CRP and ESR) criteria from the definition of the sustained remission, given sarilumab's direct impact on acute phase reactants. The results of this analysis were consistent with the primary analysis (see Table 8).
| Placebo (N=58) | KEVZARA (N=60) |
||
|---|---|---|---|
|
|||
| Sustained remission at Week 52 | |||
| Number of patients with sustained remission, n (%) | 6 (10.3) | 17 (28.3) | |
| Proportion difference (95% CI) vs. placebo | 18.0 (4.2, 31.8; p=0.0193) | ||
| Components of sustained remission at Week 52 | |||
| Absence of signs and symptoms and CRP < 10 mg/L (disease remission*) no later than Week 12, n (%) | 22 (37.9) | 28 (46.7) | |
| Absence of disease flare† from Week 12 through Week 52, n (%) | 19 (32.8) | 33 (55.0) | |
| Sustained reduction of CRP (<10 mg/L) from Week 12 through Week 52, n (%) | 26 (44.8) | 40 (66.7) | |
| Successful adherence to prednisone taper from Week 12 through Week 52, n (%) | 14 (24.1) | 30 (50.0) | |
| Sensitivity analysis removing acute phase reactants (CRP and ESR) from sustained remission at Week 52 | |||
| Number of patients with sustained remission, n (%) | 8 (13.8) | 19 (31.7) | |
| Proportion difference (95% CI) for sarilumab vs. placebo | 17.9 (3.1, 32.6) | ||
Effect on Concomitant Corticosteroid Use
The total actual cumulative corticosteroid dose included all corticosteroids taken during the study (i.e., prednisone taper regimen per protocol, add-on prednisone prior to Week 12, corticosteroid use due to rescue, or corticosteroid use during the treatment period to manage an adverse reaction not related to PMR). The total actual cumulative prednisone equivalent corticosteroid dose was lower in the KEVZARA arm (mean [SD] 1039.5 [612.2] mg and median 777 mg) relative to the placebo arm (mean [SD] 2235.8 [839.4] mg and median 2044 mg).
14.3 Polyarticular Juvenile Idiopathic Arthritis (pJIA)
Supportive efficacy and safety data were provided from Study 4, which was a multicenter, open-label, two-phase study in patients aged 2 to 17 years of age with polyarticular juvenile idiopathic arthritis (pJIA) diagnosed according to American College Rheumatology (ACR) classification criteria who had an inadequate response to current therapy. This study was divided into a dose range finding portion and a confirmatory portion. Three doses were investigated in the 12-week core treatment phase of the dose range finding portion. Following the dose selection, patients were enrolled to receive the recommended dose [3 mg/kg every 2 weeks (q2w) in 42 patients weighing ≥30 kg (Group A) and 4 mg/kg q2w in 31 patients weighing ≥10 kg and <30 kg (Group B)]. A total of 101 patients were treated, including 73 patients who received the recommended dose regimen from baseline and 20 patients who had their dose switched to the recommended dose during the study. Of the 73 patients who received the recommended dose throughout treatment, baseline mean disease duration was 2.48 years and a JADAS-27 score of 22.73. At baseline, 84.9% of patients had received at least one conventional DMARD (mainly methotrexate), 13.7% received systemic glucocorticoids, and 19.2% had prior treatment with biological DMARDs (mainly TNF antagonists). The patients treated had subtypes of JIA that, at disease onset, included rheumatoid factor positive (17.8%), negative polyarticular JIA (65.8%), or extended oligoarticular JIA (16.4%).
Use of KEVZARA in pediatric patients with pJIA is supported by evidence from adequate and well-controlled studies of KEVZARA in adults with RA, pharmacokinetic data from adult patients with RA, and pharmacokinetic (PK) comparability from Study 4 [see Clinical Pharmacology (12.3)].
16. How is Kevzara supplied
KEVZARA (sarilumab) injection is supplied as a clear and colorless to pale yellow solution in single-dose pre-filled syringes, and single-dose pre-filled pens.
| Strength | Package Size | NDC Number |
|---|---|---|
| 150 mg/1.14 mL | 2 syringes per pack | 0024-5908-01 |
| 200 mg/1.14 mL | 2 syringes per pack | 0024-5910-01 |
| Strength | Package Size | NDC Number |
|---|---|---|
| 150 mg/1.14 mL | 2 pre-filled pens per pack | 0024-5920-01 |
| 200 mg/1.14 mL | 2 pre-filled pens per pack | 0024-5922-01 |
Storage and Stability
Refrigerate at 36°F to 46°F (2°C to 8°C) in original carton to protect from light. Do not freeze. Do not shake.
If needed, patients/caregivers may store KEVZARA at room temperature up to 77°F (25°C) up to 14 days in the outer carton. Do not store above 77°F (25°C). After removal from the refrigerator, use KEVZARA within 14 days or discard.
17. Patient Counseling Information
Advise the patients to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Infections
Inform patients that KEVZARA may lower their resistance to infections. Instruct patients to contact their physician immediately when symptoms suggesting infection appear, to ensure rapid evaluation and appropriate treatment [see Warning and Precautions (5.1)].
Gastrointestinal Perforation
Inform patients that some patients, particularly those also taking NSAIDS, and/or steroids, have had tears (perforations) of the stomach or intestines. Inform patients that gastrointestinal perforations have been reported in KEVZARA-treated patients in clinical studies, primarily as a complication of diverticulitis. Instruct patients to contact their physician immediately when symptoms of severe, persistent abdominal pain appear to ensure rapid evaluation and appropriate treatment.
Hypersensitivity and Serious Allergic Reaction
Assess patient suitability for home use for SC injection. Inform patients that some patients who have been treated with KEVZARA have developed serious allergic reactions. Advise patients to seek immediate medical attention if they experience any symptom of serious allergic reactions.
Instruction on Injection Technique
Instruct patients and caregivers to read the Instructions for Use before the patient starts using KEVZARA, and each time the patient gets a refill as there may be new information they need to know.
Provide guidance to patients and caregivers on proper subcutaneous injection technique, including aseptic technique, and how to use the pre-filled syringe or pre-filled pen correctly (see Instructions for Use).
The pre-filled syringe or pre-filled pen should be left at room temperature for 30 minutes or 60 minutes respectively (see the Instructions for Use) prior to use. The syringe or pen should be used within 14 days after being taken out of the refrigerator. A puncture-resistant container should be used to dispose the used pre-filled syringes or pre-filled pens and should be kept out of the reach of children. Instruct patients or caregivers in the technique as well as proper pre-filled syringe or pre-filled pen disposal, and caution against reuse of these items.
REGENERON
sanofi
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License No. 1752
Marketed by:
sanofi-aventis U.S. LLC (Morristown, NJ 07960) and
Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
For patent information: www.kevzara-patents.com
KEVZARA® is a registered trademark of Sanofi Biotechnology
©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC
| MEDICATION GUIDE KEVZARA® (KEV-za-ra) (sarilumab) injection, for subcutaneous use |
|||
|---|---|---|---|
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: May 2025 | ||
| What is the most important information I should know about KEVZARA? KEVZARA can cause serious side effects including:
|
|||
|
|
||
See "What are the possible side effects of KEVZARA?" for more information about side effects. |
|||
| What is KEVZARA?
KEVZARA is an injectable prescription medicine called an interleukin-6 (IL-6) receptor blocker. KEVZARA is used:
It is not known if KEVZARA is safe and effective in children with pJIA under 2 years of age. |
|||
| Who should not use KEVZARA?
Do not use KEVZARA if you or your child are allergic to sarilumab or any of the ingredients in KEVZARA. See the end of this Medication Guide for a complete list of ingredients in KEVZARA. |
|||
Before you or your child use KEVZARA, talk to your healthcare provider about all of your medical conditions, including if you or your child:
Tell your healthcare provider about all the medicines you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show to your healthcare provider and pharmacist when you get a new medicine. |
|||
How should I use KEVZARA?
|
|||
| What are the possible side effects of KEVZARA? KEVZARA can cause serious side effects, including:
|
|||
|
|
||
Common side effects of KEVZARA include:
These are not all of the possible side effects of KEVZARA. |
|||
How should I store KEVZARA?
|
|||
| General Information about the safe and effective use of KEVZARA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use KEVZARA for a condition for which it was not prescribed. Do not give KEVZARA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about KEVZARA that is written for health professionals. |
|||
| What are the ingredients in KEVZARA? Active Ingredient: sarilumab Inactive Ingredients: arginine, histidine, polysorbate 20, sucrose, and Water for Injection, USP. REGENERON sanofi Manufactured by: sanofi-aventis U.S. LLC Morristown, NJ 07960, A SANOFI COMPANY U.S. License # 1752. Marketed by: sanofi-aventis U.S. LLC (Morristown, NJ 07960) and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591) KEVZARA® is a registered trademark of Sanofi Biotechnology ©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC For more information, go to www.KEVZARA.com or call 1-844-KEVZARA (1-844-538-9272). |
|||
Instructions For UseKEVZARA® (KEV-za-ra)(sarilumab)injection, for subcutaneous useSingle-dose Pre-filled Syringe (150 mg/1.14 mL)
Important information:
KEVZARA is available as a single-dose pre-filled syringe (called "syringe" in these instructions). The pre-filled syringe contains 150 mg of KEVZARA for injection under the skin (subcutaneous injection) 1 time every 2 weeks.
Do not try to inject KEVZARA until you have been shown the right way to give the injections by your healthcare provider.
| Do | Do not |
|---|---|
|
|
Keep these instructions for future use.
If you have any further questions, ask your healthcare provider or call 1-844-KEVZARA (1-844-538-9272).
KEVZARA pre-filled syringe parts
What you will need for your injection:
- A new KEVZARA 150 mg/1.14 mL pre-filled syringe
- Alcohol wipe
- Cotton ball or gauze
- Sharps disposal container. See "How should I dispose of (throw away) KEVZARA pre-filled syringes?" at the end of this Instructions for Use.
Step A: Get ready for an injection
| 1. Prepare all of the equipment you will need on a clean, flat working surface. | |
|  |
| 2. Look at the label. | |
|  |
| 3. Look at the medicine. | |
|  |
| 4. Lay the syringe on a flat surface and allow it to warm up at room temperature for at least 30 minutes. | |
| 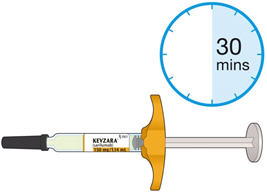 |
| 5. Select the injection site. | |
| 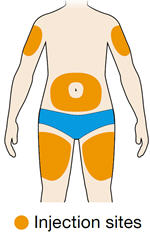 |
| 6. Prepare the injection site. | |
|
Step B: Give the injection
Complete Step B after completing all steps in Step A "Get ready for an injection".
| 1. Pull off the needle cap. | |
|  |
| 2. Pinch the skin. | |
|  |
| 3. Insert the needle into the fold of skin at about a 45 degree angle. | 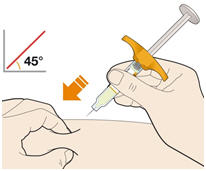 |
| 4. Push the plunger down. | |
| 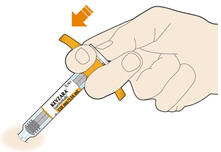 |
| 5. Before you remove the needle, check that the syringe is empty. | |
| 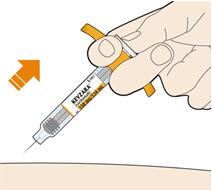 |
| 6. Dispose of the syringe. | |
| 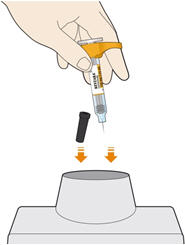 |
How should I dispose of (throw away) KEVZARA pre-filled syringes?
- Put the used syringe in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) the syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not reuse the syringe.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Important: Always keep the sharps disposal container out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
REGENERON
sanofi
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License # 1752
Marketed by: sanofi-aventis U.S. LLC (Morristown, NJ 07960)
and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
KEVZARA® is a registered trademark of Sanofi Biotechnology
©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC
Issued: May 2025
Instructions For UseKEVZARA® (KEV-za-ra)(sarilumab)injection, for subcutaneous useSingle-dose Pre-filled Syringe (200 mg/1.14 mL)
Important information:
KEVZARA is available as a single-dose pre-filled syringe (called "syringe" in these instructions). The pre-filled syringe contains 200 mg of KEVZARA for injection under the skin (subcutaneous injection) 1 time every 2 weeks.
Do not try to inject KEVZARA until you have been shown the right way to give the injections by your healthcare provider.
| Do | Do not |
|---|---|
|
|
Keep these instructions for future use.
If you have any further questions, ask your healthcare provider or call 1-844-KEVZARA (1-844-538-9272).
KEVZARA pre-filled syringe parts
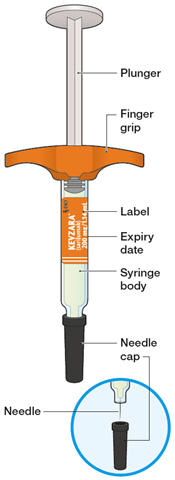
What you will need for your injection:
- A new KEVZARA 200 mg/1.14 mL pre-filled syringe
- Alcohol wipe
- Cotton ball or gauze
- Sharps disposal container. See "How should I dispose of (throw away) KEVZARA pre-filled syringes?" at the end of this Instructions for Use.
Step A: Get ready for an injection
| 1. Prepare all of the equipment you will need on a clean, flat working surface. | |
|  |
| 2. Look at the label. | |
|  |
| 3. Look at the medicine. | |
|  |
| 4. Lay the syringe on a flat surface and allow it to warm up at room temperature for at least 30 minutes. | |
|  |
| 5. Select the injection site. | |
| 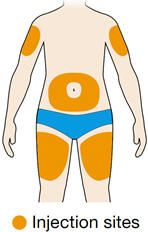 |
| 6. Prepare the injection site. | |
|
Step B: Give the injection
Complete Step B after completing all steps in Step A "Get ready for an injection"
| 1. Pull off the needle cap. | |
| 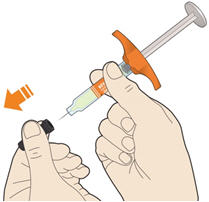 |
| 2. Pinch the skin. | |
| 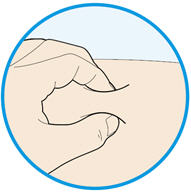 |
| 3. Insert the needle into the fold of skin at about a 45 degree angle. | |
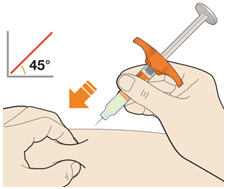 |
|
| 4. Push the plunger down. | |
|  |
| 5. Before you remove the needle, check that the syringe is empty. | |
| 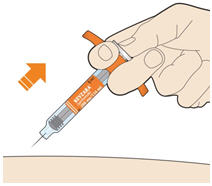 |
| 6. Dispose of the syringe. | |
| 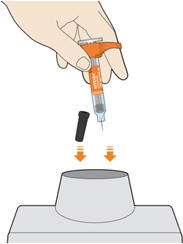 |
How should I dispose of (throw away) KEVZARA pre-filled syringes?
- Put the used syringe in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) the syringe in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not reuse the syringe.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Important: Always keep the sharps disposal container out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
REGENERON
sanofi
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License # 1752
Marketed by: sanofi-aventis U.S. LLC, (Morristown, NJ 07960)
and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
KEVZARA® is a registered trademark of Sanofi Biotechnology
©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC
Issued: May 2025
Instructions For UseKEVZARA® (KEV-za-ra)(sarilumab)injection, for subcutaneous useSingle-dose pre-filled pen (150 mg/1.14 mL)
Important information:
KEVZARA is available as a single-dose pre-filled pen (called "pen" in these instructions). The pre-filled pen contains 150 mg of KEVZARA for injection under the skin (subcutaneous injection) 1 time every 2 weeks.
Do not try to inject KEVZARA until you have been shown the right way to give the injections by your healthcare provider.
| Do | Do not |
|---|---|
|
|
Keep these instructions for future use.
If you have any further questions, ask your healthcare provider or call 1-844-KEVZARA (1-844-538-9272).
KEVZARA pre-filled pen parts
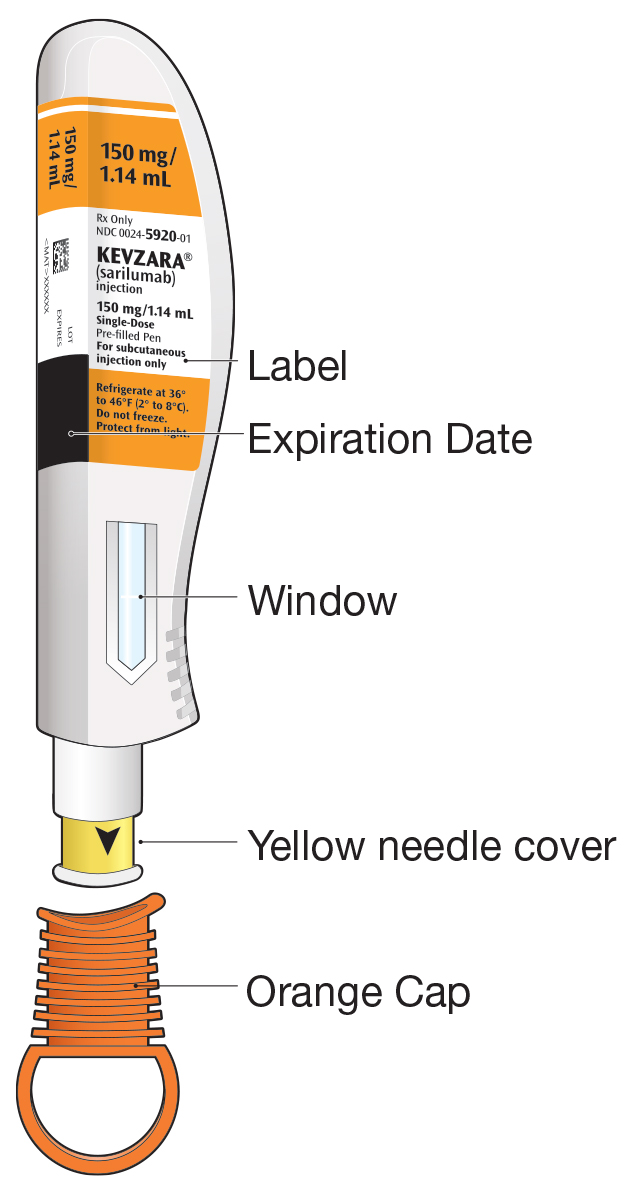
What you will need for your injection:
- An unused KEVZARA 150 mg/1.14 mL pre-filled pen
- Alcohol wipe
- Cotton ball or gauze
- Sharps disposal container. See "How should I throw away (dispose of) KEVZARA pre-filled pens?" at the end of this Instructions for Use.
Step A: Get ready for an injection
| 1. Prepare all of the equipment you will need on a clean, flat working surface. | |
|
|
| 2. Look at the label. | |
|  |
| 3. Look at the window. | |
| 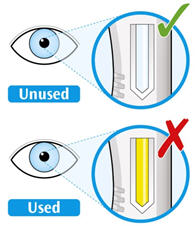 |
| 4. Lay the pen on a flat surface and allow it to warm up at room temperature for at least 60 minutes. | |
| 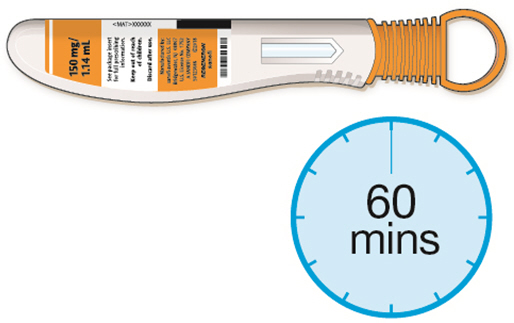 |
| 5. Select the injection site. | |
| 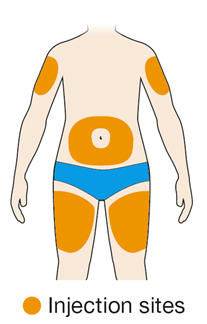 |
| 6. Prepare the injection site. | |
|
|
Step B: Give the injection
Complete Step B after completing all steps in Step A "Get ready for an injection"
| 1. Twist or pull off the orange cap. | |
|  |
| 2. Put the yellow needle cover on your skin at about a 90 degree angle. | |
| 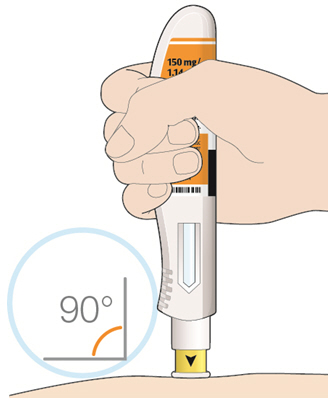 |
| 3. Press down and hold the pen firmly against your skin. | |
|  |
| 4. Keep holding the pen firmly against your skin. | |
| 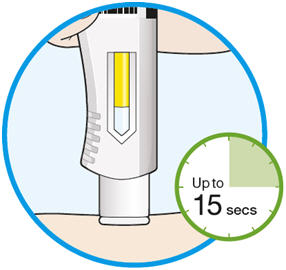 |
| 5. There will be a second click when the injection is complete. Check to see if the entire window has turned solid yellow before you remove the pen. | |
|  |
| 6. Remove the pen from your skin. | |
|  |
| 7. Dispose of the pen | |
| 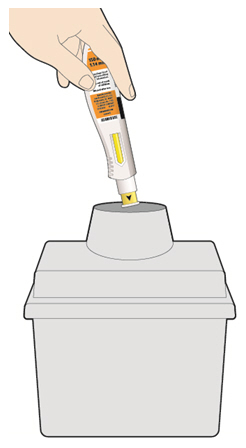 |
How should I throw away (dispose of) KEVZARA pre-filled pens?
- Put the used pen in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) the pen in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not reuse the pen.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Important: Always keep the sharps disposal container out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
REGENERON
sanofi
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License # 1752
Marketed by: sanofi-aventis U.S. LLC (Morristown, NJ 07960)
and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
KEVZARA® is a registered trademark of Sanofi Biotechnology
©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC
Issued: May 2025
Instructions For UseKEVZARA® (KEV-za-ra)(sarilumab)injection, for subcutaneous useSingle-dose pre-filled pen (200 mg/1.14 mL)
Important information:
KEVZARA is available as a single-dose pre-filled pen (called "pen" in these instructions). The pre-filled pen contains 200 mg of KEVZARA for injection under the skin (subcutaneous injection) 1 time every 2 weeks.
Do not try to inject KEVZARA until you have been shown the right way to give the injections by your healthcare provider.
| Do | Do not |
|---|---|
|
|
Keep these instructions for future use.
If you have any further questions, ask your healthcare provider or call 1-844-KEVZARA (1-844-538-9272).
KEVZARA pre-filled pen parts

What you will need for your injection:
- An unused KEVZARA 200 mg/1.14 mL pre-filled pen
- Alcohol wipe
- Cotton ball or gauze
- Sharps disposal container. See "How should I throw away (dispose of) KEVZARA pre-filled pens?" at the end of this Instructions for Use.
Step A: Get ready for an injection
| 1. Prepare all of the equipment you will need on a clean, flat working surface. | |
|
|
| 2. Look at the label. | |
|  |
| 3. Look at the window. | |
| 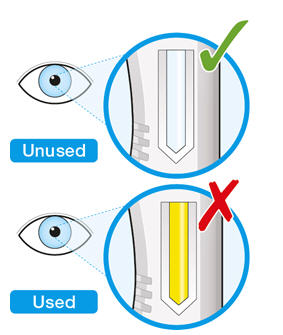 |
| 4. Lay the pen on a flat surface and allow it to warm up at room temperature for at least 60 minutes. | |
|  |
| 5. Select the injection site. | |
| 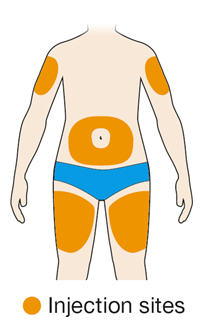 |
| 6. Prepare the injection site. | |
|
|
Step B: Give the injection
Complete Step B after completing all steps in Step A "Get ready for an injection"
| 1. Twist or pull off the orange cap. | |
| 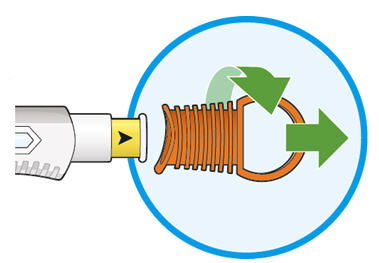 |
| 2. Put the yellow needle cover on your skin at about a 90 degree angle. | |
| 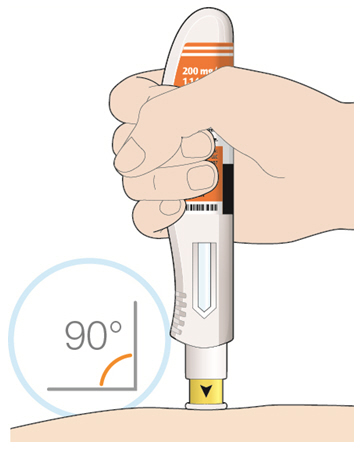 |
| 3. Press down and hold the pen firmly against your skin. | |
| 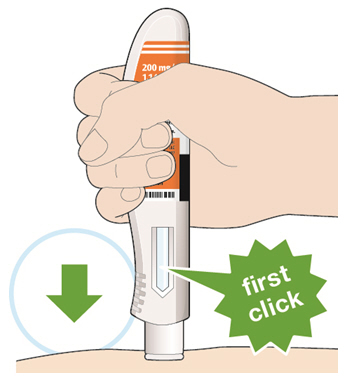 |
| 4. Keep holding the pen firmly against your skin. | |
|  |
| 5. There will be a second click when the injection is complete. Check to see if the entire window has turned solid yellow before you remove the pen. | |
| 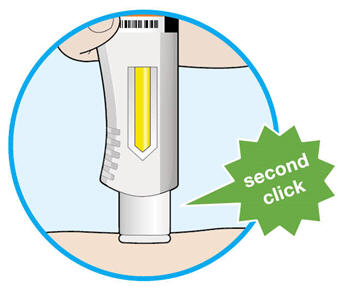 |
| 6. Remove the pen from your skin. | |
| 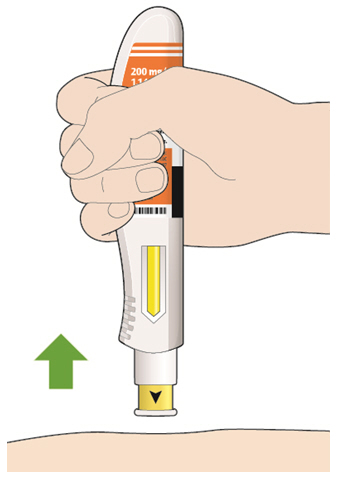 |
| 7. Dispose of the pen | |
| 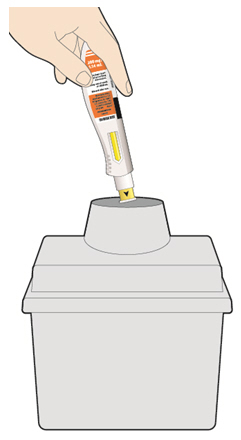 |
How should I throw away (dispose of) KEVZARA pre-filled pens?
- Put the used pen in an FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) the pen in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not reuse the pen.
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
Important: Always keep the sharps disposal container out of the reach of children.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
REGENERON
sanofi
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License # 1752
Marketed by: sanofi-aventis U.S. LLC (Morristown, NJ 07960)
and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY 10591)
KEVZARA® is a registered trademark of Sanofi Biotechnology
©2025 Regeneron Pharmaceuticals, Inc. / sanofi-aventis U.S. LLC
Issued: May 2025
PRINCIPAL DISPLAY PANEL - 150 mg/1.14 mL Syringe Carton
NDC 0024-5908-01
Rx Only
KEVZARA®
(sarilumab)
injection
150 mg/1.14 mL (131.6 mg/mL)
2 Single-Dose Pre-filled Syringes
For Subcutaneous Injection Use Only
REGENERON
sanofi
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original
carton to protect from light. Do NOT freeze. Do NOT shake.
If needed, patients/caregivers may store KEVZARA at room
temperature up to 77°F (25°C) up to 14 days in the outer carton.
Do not store above 77°F (25°C). After removal from the
refrigerator, use KEVZARA within 14 days or discard.
Date removed from the refrigerator ____/____/____
ATTENTION: Enclosed Medication Guide is required for each patient
150 mg/1.14 mL

PRINCIPAL DISPLAY PANEL - 200 mg/1.14 mL Syringe Carton
NDC 0024-5910-01
Rx Only
KEVZARA®
(sarilumab)
injection
200 mg/1.14 mL (175.4 mg/mL)
2 Single-Dose Pre-filled Syringes
For Subcutaneous Injection Use Only
REGENERON
sanofi
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original
carton to protect from light. Do NOT freeze. Do NOT shake.
If needed, patients/caregivers may store KEVZARA at room
temperature up to 77°F (25°C) up to 14 days in the outer carton.
Do not store above 77°F (25°C). After removal from the
refrigerator, use KEVZARA within 14 days or discard.
Date removed from the refrigerator ____/____/____
ATTENTION: Enclosed Medication Guide is required for each patient
200 mg/1.14 mL

PRINCIPAL DISPLAY PANEL - 150 mg/1.14 mL Pen Carton
NDC 0024-5920-01
Rx Only
KEVZARA®
(sarilumab)
injection
150 mg/1.14 mL (131.6 mg/mL)
2 Single-Dose Pre-filled Pens
For Subcutaneous Injection Use Only
REGENERON
sanofi
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original
carton to protect from light. Do NOT freeze. Do NOT shake.
If needed, patients/caregivers may store KEVZARA at room
temperature up to 77°F (25°C) up to 14 days in the outer carton.
Do not store above 77°F (25°C). After removal from the
refrigerator, use KEVZARA within 14 days or discard.
Date removed from the refrigerator ____/____/____
ATTENTION: Enclosed Medication Guide is required for each patient
150 mg/1.14 mL
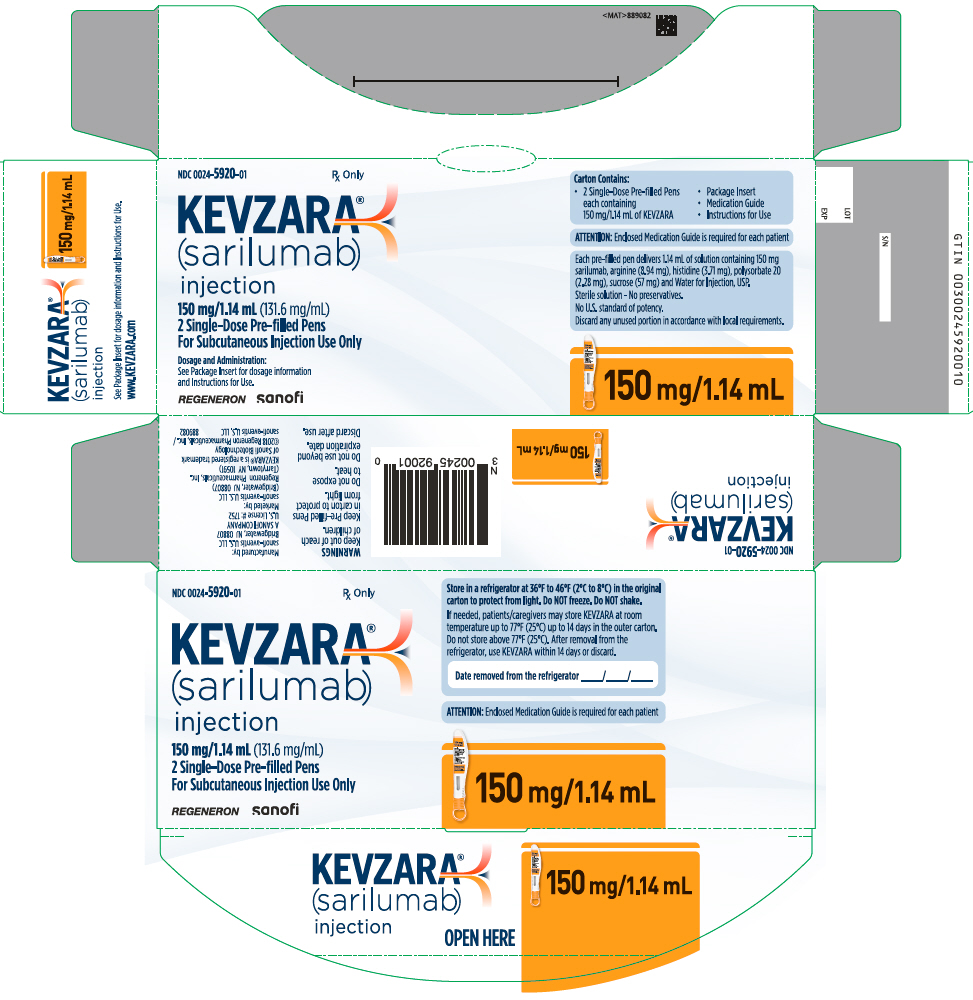
PRINCIPAL DISPLAY PANEL - 200 mg/1.14 mL Pen Carton
NDC 0024-5922-01
Rx Only
KEVZARA®
(sarilumab)
injection
200 mg/1.14 mL (175.4 mg/mL)
2 Single-Dose Pre-filled Pens
For Subcutaneous Injection Use Only
REGENERON
sanofi
Store in a refrigerator at 36°F to 46°F (2°C to 8°C) in the original
carton to protect from light. Do NOT freeze. Do NOT shake.
If needed, patients/caregivers may store KEVZARA at room
temperature up to 77°F (25°C) up to 14 days in the outer carton.
Do not store above 77°F (25°C). After removal from the
refrigerator, use KEVZARA within 14 days or discard.
Date removed from the refrigerator ____/____/____
ATTENTION: Enclosed Medication Guide is required for each patient
200 mg/1.14 mL
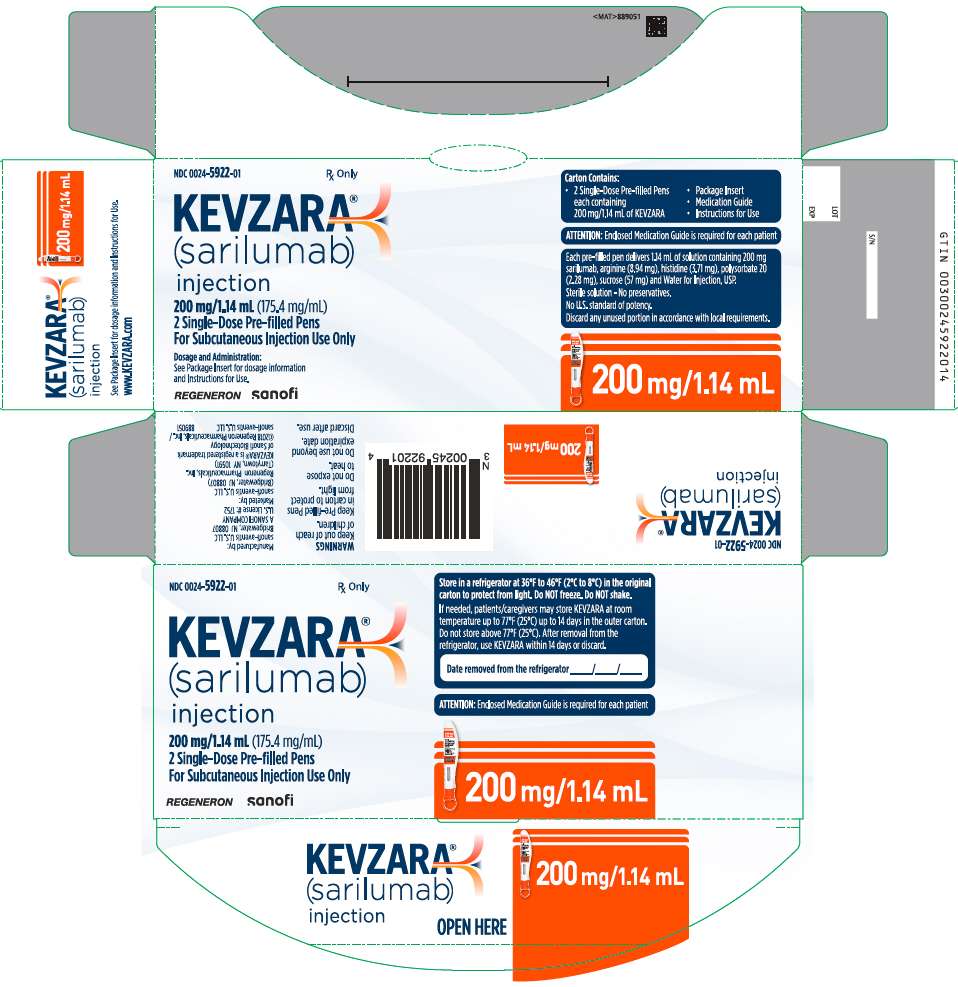
| KEVZARA
sarilumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| KEVZARA
sarilumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| KEVZARA
sarilumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| KEVZARA
sarilumab injection, solution |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Sanofi-Aventis U.S. LLC (824676584) |
| Registrant - Sanofi Winthrop Industrie (297730488) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi Winthrop Industrie | 297730488 | ANALYSIS(0024-5908, 0024-5910, 0024-5920, 0024-5922) , MANUFACTURE(0024-5908, 0024-5910, 0024-5920, 0024-5922) , PACK(0024-5908, 0024-5910, 0024-5920, 0024-5922) , LABEL(0024-5908, 0024-5910, 0024-5920, 0024-5922) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi-Aventis Deutschland GmbH | 313218430 | ANALYSIS(0024-5920, 0024-5922) , MANUFACTURE(0024-5920, 0024-5922) , PACK(0024-5920, 0024-5922) , LABEL(0024-5920, 0024-5922) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Regeneron Pharmaceuticals, Inc. | 945589711 | API MANUFACTURE(0024-5908, 0024-5910, 0024-5920, 0024-5922) , ANALYSIS(0024-5908, 0024-5910, 0024-5920, 0024-5922) , MANUFACTURE(0024-5908, 0024-5910, 0024-5920, 0024-5922) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 050424395 | PACK(0024-5908, 0024-5910, 0024-5920, 0024-5922) , LABEL(0024-5908, 0024-5910, 0024-5920, 0024-5922) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Ireland Limited | 985127419 | ANALYSIS(0024-5908, 0024-5910) , MANUFACTURE(0024-5908, 0024-5910) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Nelson Laboratories, LLC | 151663234 | ANALYSIS(0024-5908, 0024-5910) | |
Biological Products Related to Kevzara
Find detailed information on biosimilars for this medication.
Frequently asked questions
- What are the new drugs for rheumatoid arthritis (RA)?
- How long does it take for Kevzara to work?
- Does Kevzara cause weight gain?
- How do you use Kevzara (sarilumab) for the treatment of rheumatoid arthritis?
More about Kevzara (sarilumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (31)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: antirheumatics
- Breastfeeding
- En español
