Caprelsa: Package Insert / Prescribing Info
Package insert / product label
Generic name: vandetanib
Dosage form: tablet, film coated
Drug classes: EGFR inhibitors, Multikinase inhibitors, VEGF/VEGFR inhibitors
Medically reviewed by Drugs.com. Last updated on May 26, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
CAPRELSA® (vandetanib) tablets, for oral use
Initial U.S. Approval: 2011
WARNING: QT PROLONGATION, TORSADES DE POINTES, AND SUDDEN DEATH
See full prescribing information for complete boxed warning.
CAPRELSA can prolong the QT interval. Torsades de pointes and sudden death have occurred in patients receiving CAPRELSA. Do not use CAPRELSA in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome. Correct hypocalcemia, hypokalemia and/or hypomagnesemia prior to CAPRELSA administration. Monitor electrolytes periodically. Avoid drugs known to prolong the QT interval. Only prescribers and pharmacies certified with the restricted distribution program are able to prescribe and dispense CAPRELSA (5.1, 5.16).
Recent Major Changes
| Warnings and Precautions (5.17) | 05/2025 |
Indications and Usage for Caprelsa
CAPRELSA is a kinase inhibitor indicated for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease. (1)
Use CAPRELSA in patients with indolent, asymptomatic or slowly progressing disease only after careful consideration of the treatment related risks of CAPRELSA. (1)
Caprelsa Dosage and Administration
Dosage Forms and Strengths
100 mg and 300 mg tablets (3)
Contraindications
Do not use in patients with congenital long QT syndrome. (4)
Warnings and Precautions
- Prolonged QT interval, torsades de pointes, and sudden death: Monitor electrocardiograms and levels of serum potassium, calcium, magnesium and TSH. Reduce CAPRELSA dose as appropriate. (2.1, 5.1)
- Severe skin reactions, including toxic epidermal necrolysis and Stevens-Johnson syndrome, some fatal. Discontinue CAPRELSA for severe skin reactions. (2.1, 5.2)
- Interstitial lung disease (ILD), including fatalities: investigate unexplained non-specific respiratory signs and symptoms. Discontinue CAPRELSA for confirmed ILD. (2.1, 5.3)
- Ischemic cerebrovascular events, hemorrhage, heart failure, diarrhea, hypertension, and reversible posterior leukoencephalopathy syndrome: Discontinue or interrupt CAPRELSA. (2.1, 5.4, 5.5, 5.6, 5.7, 5.9, 5.10)
- Impaired wound healing: Withhold for at least 1 month prior to elective surgery. Do not administer CAPRELSA for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment with CAPRELSA after resolution of wound healing complications has not been established. (5.14)
- Embryo-fetal toxicity: Can cause fetal harm. Advise women of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.15, 8.1)
- REMS: CAPRELSA is available only through a restricted distribution program called the CAPRELSA REMS Program. (5.16)
- Osteonecrosis, including osteonecrosis of the jaw: Withhold CAPRELSA for 1 month prior to scheduled dental surgery and permanently discontinue if osteonecrosis occurs (5.17, 6.2).
Adverse Reactions/Side Effects
The most common adverse drug reactions (>20%) seen with CAPRELSA and with a between-arm difference of ≥5 % have been diarrhea/colitis, rash, acneiform dermatitis, hypertension, nausea, headache, upper respiratory tract infections, decreased appetite and abdominal pain. (6)
To report SUSPECTED ADVERSE REACTIONS, Contact Sanofi Genzyme at 1-800-817-2722 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 5/2025
Full Prescribing Information
WARNING: QT PROLONGATION, TORSADES DE POINTES, AND SUDDEN DEATH
CAPRELSA can prolong the QT interval. Torsades de pointes and sudden death have occurred in patients receiving CAPRELSA. Do not use CAPRELSA in patients with hypocalcemia, hypokalemia, hypomagnesemia, or long QT syndrome. Correct hypocalcemia, hypokalemia and/or hypomagnesemia prior to CAPRELSA administration. Monitor electrolytes periodically. Avoid drugs known to prolong the QT interval. Only prescribers and pharmacies certified with the restricted distribution program are able to prescribe and dispense CAPRELSA [see Warnings and Precautions (5.1, 5.16)].
1. Indications and Usage for Caprelsa
CAPRELSA is indicated for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease.
Use CAPRELSA in patients with indolent, asymptomatic or slowly progressing disease only after careful consideration of the treatment related risks of CAPRELSA.
2. Caprelsa Dosage and Administration
The recommended dose of CAPRELSA is 300 mg taken orally once daily until disease progression or unacceptable toxicity occurs.
CAPRELSA may be taken with or without food.
Do not take a missed dose within 12 hours of the next dose.
Do not crush CAPRELSA tablets. The tablets can be dispersed in 2 ounces of water by stirring for approximately 10 minutes (will not completely dissolve). Do not use other liquids for dispersion. Swallow immediately after dispersion. Mix any remaining residue with 4 additional ounces of water and swallow.
The dispersion can also be administered through nasogastric or gastrostomy tubes.
2.1 Dosage Adjustment
For Adverse Reactions
The 300 mg daily dose can be reduced to 200 mg (two 100 mg tablets) and then to 100 mg for Common Terminology Criteria for Adverse Events (CTCAE) Grade 3 or greater toxicities.
Interrupt CAPRELSA for the following:
- Corrected QT interval, Fridericia (QTcF) greater than 500 ms: Resume at a reduced dose when the QTcF returns to less than 450 ms.
- CTCAE Grade 3 or greater toxicity: Resume at a reduced dose when the toxicity resolves or improves to CTCAE Grade 1.
For recurrent toxicities, reduce the dose of CAPRELSA to 100 mg after resolution or improvement to CTCAE Grade 1 severity, if continued treatment is warranted.
Adverse events including QT interval prolongation should be monitored closely as they may not resolve fully until approximately three plasma half-lives of the drug. Monitor appropriately [see Warnings and Precautions (5.1), (5.2), (5.3), (5.4), (5.5), (5.6), (5.7), and (5.9)].
For Patients with Renal Impairment
Reduce the starting dose to 200 mg in patients with moderate (creatinine clearance ≥30 to <50 mL/min) renal impairment [see Warnings and Precautions (5.12) and Use in Specific Populations (8.6)].
For Patients with Hepatic Impairment
CAPRELSA is not recommended for use in patients with moderate and severe hepatic impairment [see Use in Specific Populations (8.7)].
3. Dosage Forms and Strengths
CAPRELSA 100 mg tablets are white, round, biconvex, film-coated, and intagliated with 'Z 100' on one side and plain on the reverse side.
CAPRELSA 300 mg tablets are white, oval, biconvex, film-coated, and intagliated with 'Z 300' on one side and plain on the reverse side.
5. Warnings and Precautions
5.1 QT Prolongation and Torsades de Pointes
CAPRELSA can prolong the QT interval in a concentration-dependent manner [see Clinical Pharmacology (12.2)]. Torsades de pointes, ventricular tachycardia and sudden deaths have occurred in patients treated with CAPRELSA.
Do not start CAPRELSA treatment in patients whose QTcF interval is greater than 450 ms. Do not administer CAPRELSA to patients who have a history of Torsades de pointes, congenital long QT syndrome, bradyarrhythmias or uncompensated heart failure. CAPRELSA has not been studied in patients with ventricular arrhythmias or recent myocardial infarction. Vandetanib exposure is increased in patients with impaired renal function. Reduce the starting dose to 200 mg in patients with moderate renal impairment and monitor QT interval frequently.
Obtain an ECG and serum potassium, calcium, magnesium and TSH at baseline, 2 to 4 weeks and 8 to 12 weeks after starting treatment with CAPRELSA, and every 3 months thereafter. Monitor electrolytes and ECGs more frequently in patients who experience diarrhea. Following any dose reduction for QT prolongation or any dose interruption greater than 2 weeks, conduct QT assessments as described above. Maintain serum potassium levels of 4 mEq/L or higher (within normal range) and maintain serum magnesium and calcium levels within normal ranges to reduce the risk of QT prolongation.
Avoid using CAPRELSA with drugs known to prolong the QT interval [see Warnings and Precautions (5.11) and Drug Interactions (7.4)]. If such drugs are given to patients already receiving CAPRELSA and no alternative therapy exists, perform ECG monitoring of the QT interval more frequently.
Stop CAPRELSA in patients who develop a QTcF greater than 500 ms until the QTcF returns to less than 450 ms. Dosing of CAPRELSA can then be resumed at a reduced dose [see Dosage and Administration (2.1)].
5.2 Severe Skin Reactions
Severe and sometimes fatal skin reactions, including toxic epidermal necrolysis (TEN) and Stevens-Johnson syndrome, have occurred in patients treated with CAPRELSA. Permanently discontinue CAPRELSA for severe skin reactions and refer the patient for urgent medical evaluation. Systemic therapies such as corticosteroids may be required.
Photosensitivity reactions can occur during CAPRELSA treatment and up to 4 months after treatment discontinuation.
5.3 Interstitial Lung Disease
Interstitial Lung Disease (ILD) or pneumonitis, including fatalities, has occurred in patients treated with CAPRELSA. Consider a diagnosis of ILD in patients presenting with non-specific respiratory signs and symptoms.
Interrupt CAPRELSA for acute or worsening pulmonary symptoms. Discontinue CAPRELSA if ILD is confirmed.
5.4 Ischemic Cerebrovascular Events
Ischemic cerebrovascular events, including fatalities, occurred in patients treated with CAPRELSA. In the randomized medullary thyroid cancer (MTC) study, ischemic cerebrovascular events occurred more frequently with CAPRELSA compared to placebo (1.3% compared to 0%). The safety of resumption of CAPRELSA therapy after resolution of an ischemic cerebrovascular event has not been studied. Discontinue CAPRELSA in patients who experience a severe ischemic cerebrovascular event.
5.5 Hemorrhage
Serious hemorrhagic events, including fatalities, occurred in patients treated with CAPRELSA. Do not administer CAPRELSA to patients with a recent history of hemoptysis of ≥1/2 teaspoon of red blood. Discontinue CAPRELSA in patients with severe hemorrhage.
5.6 Heart Failure
Heart failure, including fatalities, occurred in patients treated with CAPRELSA. Monitor for signs and symptoms of heart failure. Consider discontinuation of CAPRELSA in patients with heart failure. Heart failure may not be reversible upon stopping CAPRELSA.
5.7 Diarrhea
Diarrhea of Grade 3 or greater severity occurred in 11% of patients receiving CAPRELSA in the randomized MTC study. If diarrhea occurs, carefully monitor serum electrolytes and ECGs to reduce the risk and enable early detection of QT prolongation resulting from dehydration [see Warnings and Precautions (5.1)]. Interrupt CAPRELSA for severe diarrhea. Upon improvement, resume CAPRELSA at a reduced dose [see Dosage and Administration (2.1)].
5.8 Hypothyroidism
In the randomized MTC study in which 90% of the patients enrolled had prior thyroidectomy, increased dosing of thyroid replacement therapy was required in 49% of CAPRELSA-treated patients compared to 17% of placebo-treated patients. Obtain Thyroid-stimulating hormone (TSH) at baseline, at 2 to 4 weeks and 8 to 12 weeks after starting treatment with CAPRELSA, and every 3 months thereafter. If signs or symptoms of hypothyroidism occur, examine thyroid hormone levels and adjust thyroid replacement therapy accordingly.
5.9 Hypertension
Hypertension, including hypertensive crisis, has occurred in patients treated with CAPRELSA. Monitor all patients for hypertension. Dose reduction or interruption for hypertension may be necessary. If hypertension cannot be controlled, do not resume CAPRELSA [see Dosage and Administration (2.1)].
5.10 Reversible Posterior Leukoencephalopathy Syndrome
Reversible posterior leukoencephalopathy syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by an MRI of the brain, has occurred in patients treated with CAPRELSA. Consider this syndrome in any patient presenting with seizures, headache, visual disturbances, confusion or altered mental function. In clinical studies, three of four patients who developed RPLS while taking CAPRELSA also had hypertension. Discontinue CAPRELSA treatment in patients with RPLS.
5.11 Drug Interactions
Avoid administration of CAPRELSA with anti-arrhythmic drugs (including but not limited to amiodarone, disopyramide, procainamide, sotalol, dofetilide) and other drugs that may prolong the QT interval (including but not limited to chloroquine, clarithromycin, dolasetron, granisetron, haloperidol, methadone, moxifloxacin, and pimozide) [see Drug Interactions (7.4) and Clinical Pharmacology (12.2)].
5.12 Renal Failure
Renal failure occurred in patients treated with CAPRELSA [see Adverse Reactions (6.1)]. Withhold, reduce the dose or permanently discontinue based on severity [see Dosage and Administration (2.1)].
Vandetanib exposure is increased in patients with impaired renal function. Reduce the starting dose to 200 mg in patients with moderate renal impairment and monitor the QT interval closely [see Dosage and Administration (2.1)]. Vandetanib is not recommended for use in patients with severe renal impairment (clearance below 30 mL/min). There is no information available for patients with end-stage renal disease requiring dialysis [see Boxed Warning, Dosage and Administration (2.1), Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
5.13 Hepatic Impairment
CAPRELSA is not recommended for use in patients with moderate and severe hepatic impairment, as safety and efficacy have not been established [see Dosage and Administration (2.1)].
5.14 Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Impaired wound healing has occurred in patients treated with CAPRELSA.
Withhold CAPRELSA for at least 1 month prior to elective surgery. Do not administer CAPRELSA for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment with CAPRELSA after resolution of wound healing complications has not been established.
5.15 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, CAPRELSA can cause fetal harm when administered to a pregnant woman. In rats, vandetanib was embryotoxic, fetotoxic, and induced fetal malformations at exposures equivalent to or lower than those expected at the 300 mg clinical dose and had adverse effects on female fertility, embryofetal development, and postnatal development of pups.
Advise patients of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CAPRELSA and for 4 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with CAPRELSA and for 4 months after the last dose [see Use in Specific Populations (8.1), (8.3)].
5.16 CAPRELSA REMS (Risk Evaluation and Mitigation Strategy) Program
Because of the risk of QT prolongation, Torsades de pointes, and sudden death, CAPRELSA is available only through a restricted distribution program called the CAPRELSA REMS Program. Only prescribers and pharmacies certified with the program are able to prescribe and dispense CAPRELSA.
To learn about the specific REMS requirements and to enroll in the CAPRELSA REMS Program, call 1-800-817-2722 or visit www.caprelsarems.com.
5.17 Osteonecrosis
Osteonecrosis, including osteonecrosis of the jaw (ONJ), has occurred during treatment with CAPRELSA. Concomitant exposure to other risk factors, such as bisphosphonates, denosumab, dental disease or invasive dental procedures, may increase the risk of ONJ. Perform an oral examination prior to and periodically during treatment with CAPRELSA. Advise patients regarding good oral hygiene practices. Avoid invasive dental procedures while on CAPRELSA treatment, particularly in patients at higher risk. Withhold CAPRELSA for at least one month prior to scheduled dental surgery or invasive dental procedures. Permanently discontinue CAPRELSA if ONJ develops. [see Adverse Reactions (6.2)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed elsewhere in the label:
- QT Prolongation and Torsades de Pointes [see Boxed Warning, Warnings and Precautions (5.1)]
- Severe Skin Reactions [see Warnings and Precautions (5.2)]
- Interstitial Lung Disease [see Warnings and Precautions (5.3)]
- Ischemic Cerebrovascular Events [see Warnings and Precautions (5.4)]
- Hemorrhage [see Warnings and Precautions (5.5)]
- Heart Failure [see Warnings and Precautions (5.6)]
- Diarrhea [see Warnings and Precautions (5.7)]
- Hypothyroidism [see Warnings and Precautions (5.8)]
- Hypertension [see Warnings and Precautions (5.9)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
- Renal Failure [see Warnings and Precautions (5.12)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.15)]
- Osteonecrosis [see Warnings and Precautions (5.17)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Patients with unresectable locally advanced or metastatic medullary thyroid cancer were treated with CAPRELSA 300 mg (n=231) or Placebo (n=99). The population exposed to CAPRELSA was 58% male, 94% white, and had a median age of 50 years. The data described below reflect a median exposure to CAPRELSA for 607 days.
The most commonly reported adverse drug reactions which occurred in >20% of CAPRELSA-treated patients and with a between-arm difference of ≥5% included, in order of decreasing frequency: diarrhea/colitis, rash, acneiform dermatitis, hypertension, nausea, headache, upper respiratory tract infection, decreased appetite, and abdominal pain.
Among CAPRELSA-treated patients, dose interruption occurred in 109 (47%) and dose reduction occurred in 83 (36%). Adverse reactions led to study treatment discontinuation in 28 of 231 patients (12%) receiving CAPRELSA and in 3 of 99 patients (3.0%) receiving placebo. Adverse reactions leading to permanent discontinuation in 2 or more (≥0.9%) patients treated with CAPRELSA were: asthenia (1.7%), rash (1.7%), diarrhea (0.9%), fatigue (0.9%), pyrexia (0.9%), elevated creatinine (0.9%), QT prolongation (0.9%), and hypertension (0.9%).
| System Organ Class | CAPRELSA 300 mg | Placebo | ||
|---|---|---|---|---|
| Preferred Term | N=231 | N=99 | ||
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
|
||||
| Gastrointestinal Disorders | ||||
| Diarrhea/Colitis | 57 | 11 | 27 | 2 |
| Nausea | 33 | 1 | 16 | 0 |
| Abdominal Pain† | 21 | 3 | 11 | 0 |
| Vomiting | 15 | 1 | 7 | 0 |
| Dyspepsia | 11 | 0 | 4 | 0 |
| Dry Mouth | 9 | 0 | 3 | 0 |
| Skin and Cutaneous Disorders | ||||
| Rash‡ | 53 | 5 | 12 | 0 |
| Dermatitis Acneiform/Acne | 35 | 1 | 7 | 0 |
| Dry Skin | 15 | 0 | 5 | 0 |
| Photosensitivity Reaction | 13 | 2 | 0 | 0 |
| Pruritus | 11 | 1 | 4 | 0 |
| Nail abnormalities§ | 9 | 0 | 0 | 0 |
| Alopecia | 8 | N/A | 0 | N/A |
| Vascular Disorders | ||||
| Hypertension/Hypertensive Crisis/Accelerated Hypertension | 33 | 9 | 5 | 1 |
| Nervous System Disorders | ||||
| Headache | 26 | 1 | 9 | 0 |
| Dysgeusia | 8 | 0 | 3 | 0 |
| General Disorders | ||||
| Fatigue¶ | 24 | 6 | 23 | 1 |
| Infections | ||||
| Upper Respiratory Tract Infections# | 23 | 0 | 16 | 0 |
| Metabolic and Nutritional Disorders | ||||
| Decreased Appetite | 21 | 4 | 12 | 0 |
| Hypocalcemia | 11 | 2 | 3 | 0 |
| Investigations | ||||
| ECG QT ProlongedÞ | 14 | 8 | 1 | 1 |
| Eye Disorders | ||||
| Corneal Abnormalitiesß | 13 | 0 | 1 | 0 |
| Blurred Vision | 9 | 0 | 1 | 0 |
| Renal Disorders | ||||
| Proteinuria | 10 | 0 | 2 | 0 |
| Psychiatric Disorders | ||||
| Depression | 10 | 2 | 3 | 0 |
| Endocrine Disorders | ||||
| Hypothyroidism | 6 | 0 | 0 | 0 |
| Musculoskeletal Disorders | ||||
| Muscle Spasms | 6 | 0 | 1 | 0 |
Other Clinically Relevant Adverse Effects
In patients with medullary thyroid cancer treated with CAPRELSA or placebo (NCT00410761), clinically important uncommon adverse drug reactions included pancreatitis (0.4% vs 0%), intestinal perforation (0.4% vs 0%), and heart failure (0.9% vs 0%).
Blurred vision was commonly reported (9% vs 1%) in this trial. Scheduled slit lamp examinations revealed corneal opacities (vortex keratopathies) in treated patients, which can lead to halos and decreased visual acuity. Perform ophthalmologic examination, including slit lamp examination, in patients who report visual changes.
Grade 1 to 2 bleeding events were also more common in patients receiving CAPRELSA compared to placebo (14% vs 7%).
| Laboratory Abnormalities | CAPRELSA 300 mg N=231 | Placebo N=99 |
||
|---|---|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) |
|
|
||||
| Chemistries | ||||
| Hypocalcemia | 57 | 6 | 25 | 3 |
| ALT Increased | 51 | 2 | 19 | 0 |
| Hypoglycemia | 24 | 0 | 7 | 1 |
| Creatinine Increased | 16 | 0 | 1 | 0 |
| Hypomagnesemia | 7 | <1 | 2 | 0 |
| Hematologic | ||||
| Neutropenia | 10 | <1 | 5 | 2 |
| Thrombocytopenia | 9 | 0 | 3 | 0 |
No patient with a Grade 3 to 4 ALT elevation had a concomitant increase in bilirubin in the MTC study.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of CAPRELSA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Vascular disorders: Arterial (including aortic) aneurysms, dissections, and rupture
Musculoskeletal and connective tissue disorders: Osteonecrosis
General disorders: Impaired wound healing
Related/similar drugs
7. Drug Interactions
7.1 Effect of CYP3A4 Inducers on CAPRELSA
Rifampicin, a strong CYP3A4 inducer, decreased vandetanib plasma concentrations. Avoid concomitant use of known strong CYP3A4 inducers during CAPRELSA therapy. Avoid concomitant use of St. John's wort because it can decrease vandetanib exposure unpredictably [see Clinical Pharmacology (12.3)].
7.2 Effect of CAPRELSA on OCT2 Transporter
CAPRELSA increased plasma concentrations of metformin that is transported by the organic cation transporter type 2 (OCT2). Use caution and closely monitor for toxicities when administering CAPRELSA with drugs that are transported by OCT2 [see Clinical Pharmacology (12.3)].
7.3 Effect of CAPRELSA on Digoxin
CAPRELSA increased plasma concentrations of digoxin. Use caution and closely monitor for toxicities when administering CAPRELSA with digoxin [see Clinical Pharmacology (12.3)].
7.4 Drugs that Prolong the QT Interval
Avoid concomitant use of CAPRELSA with agents that may prolong the QT interval [see Warnings and Precautions (5.11)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action and findings in animals, CAPRELSA can cause fetal harm when administered to a pregnant woman. There are no available human data on CAPRELSA use in pregnant women to inform a drug-associated risk. Vandetanib is embryotoxic, fetotoxic, and induced fetal malformations in rats at exposures less than or equal to those expected at the recommended human dose of 300 mg/day. Advise patients of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
In reproductive toxicity studies, administration of vandetanib to female rats prior to mating and through the first week of pregnancy at a dose of 25 mg/kg/day (approximately equal to the human exposure at the 300 mg clinical dose based on Cmax), there were increases in pre-implantation loss and post-implantation loss resulting in a reduction in the number of live embryos.
During organogenesis, vandetanib caused an increase in post-implantation loss, including occasional total litter loss at a dose of 25 mg/kg/day. At doses greater than 10 mg/kg/day (approximately 0.4 times the human Cmax at the 300 mg clinical dose) treatment with vandetanib resulted in increases in late embryofetal death and decreases in fetal birth weight. A no-effect level for malformations was not identified in this study. Administration of vandetanib at doses greater than or equal to 1 mg/kg/day (approximately 0.03 times the human Cmax at the 300 mg clinical dose) resulted in dose dependent increases in both malformations of the heart vessels and skeletal variations including delayed ossification of the skull, vertebrae, and sternum, indicating delayed fetal development.
In a rat prenatal and postnatal development study, at doses (1 and 10 mg/kg/day) producing mild maternal toxicity during gestation and/or lactation, vandetanib decreased pup survival and reduced postnatal pup growth. Reduced postnatal pup growth was associated with a delay in physical development.
8.2 Lactation
Risk Summary
There are no data on the presence of vandetanib or its metabolites in human milk, the effects on the breastfed child or on milk production. Vandetanib was present in the milk of lactating rats (see Data). Because of the potential for serious adverse reactions from CAPRELSA in breastfed children, advise lactating women not to breastfeed during treatment with CAPRELSA and for 4 months after the last dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with CAPRELSA [see Use in Specific Populations (8.1)].
Contraception
CAPRELSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Infertility
There are no data on the effect of CAPRELSA on human fertility. Results from animal studies indicate that vandetanib can impair male and female fertility [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
The MTC study of CAPRELSA did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently compared to younger patients.
8.6 Renal Impairment
Vandetanib exposure is increased in patients with impaired renal function. Reduce the starting dose to 200 mg in patients with moderate (creatinine clearance ≥30 to <50 mL/min) renal impairment [see Dosage and Administration (2.1), Warnings and Precautions (5.12), and Clinical Pharmacology (12.3)].
Vandetanib is not recommended for use in patients with severe renal impairment (clearance below 30 mL/min) [see Warnings and Precautions (5.12)]. Patients with end-stage renal disease requiring dialysis were not studied [see Adverse Reactions (6.1)].
8.7 Hepatic Impairment
The pharmacokinetics of CAPRELSA were evaluated after a single dose of 800 mg in subjects with mild (n=8), moderate (n=7), and severe (n=6) hepatic impairment and normal hepatic function (n=5). Subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe (Child-Pugh class C) hepatic impairment had comparable mean AUC and clearance values to those with normal hepatic function.
There are limited data in patients with liver impairment (serum bilirubin greater than 1.5 times the upper limit of normal). CAPRELSA is not recommended for use in patients with moderate and severe hepatic impairment, as safety and efficacy have not been established [see Dosage and Administration (2.1) and Warnings and Precautions (5.13)].
10. Overdosage
In the event of an overdose, monitor patients closely for QTc prolongation. Adverse events including QT interval prolongation should be monitored closely as they may not resolve fully until approximately three plasma half-lives of the drug.
11. Caprelsa Description
Vandetanib has the chemical name N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl) methoxy]quinazolin-4-amine.
The structural and molecular formulas are:
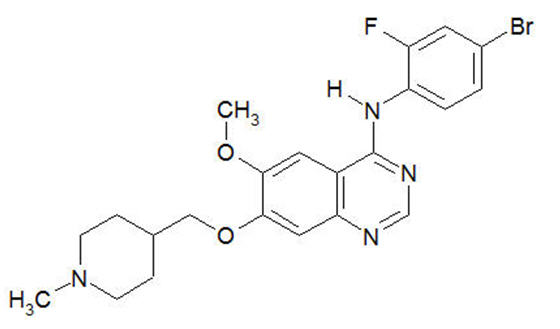
C22H24BrFN4O2
Vandetanib has a molecular weight of 475.36 g/mol. Vandetanib exhibits pH-dependent solubility, with increased solubility at lower pH. Vandetanib is practically insoluble in water with a value of 0.008 mg/mL at 25°C (77°F).
CAPRELSA tablets for daily oral administration are available in two dosage strengths containing either 100 mg or 300 mg of vandetanib. The tablet cores contain the following inactive ingredients: calcium hydrogen phosphate dihydrate, crospovidone, magnesium stearate, microcrystalline cellulose, and povidone. The tablet film-coat contains the following inactive ingredients: hypromellose 2910, macrogol 300, and titanium dioxide E171.
12. Caprelsa - Clinical Pharmacology
12.1 Mechanism of Action
In vitro studies have shown that vandetanib inhibits the tyrosine kinase activity of the EGFR and VEGFR families, RET, BRK, TIE2, and members of the EPH receptor and Src kinase families. These receptor tyrosine kinases are involved in both normal cellular function and pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, and maintenance of the tumor microenvironment. In addition, the N-desmethyl metabolite of the drug, representing 7 to 17.1% of vandetanib exposure, has similar inhibitory activity to the parent compound for VEGF receptors (KDR and Flt-1) and EGFR.
In vitro, vandetanib inhibited epidermal growth factor (EGF)-stimulated receptor tyrosine kinase phosphorylation in tumor cells and endothelial cells and VEGF-stimulated tyrosine kinase phosphorylation in endothelial cells.
In vivo, vandetanib administration reduced tumor cell-induced angiogenesis, tumor vessel permeability, and inhibited tumor growth and metastasis in mouse models of cancer.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In 231 patients with medullary thyroid cancer randomized to receive CAPRELSA 300 mg once daily in the phase 3 clinical trial. CAPRELSA was associated with sustained plasma concentration-dependent QT prolongation. Based on the exposure-response relationship, the mean (90% CI) QTcF change from baseline (ΔQTcF) was 35 (33–36) ms for the 300 mg dose. The ΔQTcF remained above 30 ms for the duration of the trial (up to 2 years). In addition, 36% of patients experienced greater than 60 ms increase in ΔQTcF and 4.3% of patients had QTcF greater than 500 ms. Cases of Torsades de pointes and sudden death have occurred [see Boxed Warning and Warnings and Precautions (5.1), (5.11)].
12.3 Pharmacokinetics
A population pharmacokinetic analysis of CAPRELSA was conducted in 231 patients with MTC following oral administration of 300 mg daily doses. The pharmacokinetics of CAPRELSA at the 300 mg dose in MTC patients are characterized by a mean clearance of approximately 13.2 L/h, a mean volume of distribution of approximately 7450 L, and a median plasma half-life of 19 days.
Absorption
Following oral administration of CAPRELSA, absorption is slow with peak plasma concentrations typically achieved at a median of 6 hours, range 4 to 10 hours, after dosing. Vandetanib accumulates approximately 8-fold on multiple dosing with steady state achieved in approximately 3 months.
Exposure to vandetanib is unaffected by food.
Distribution
Vandetanib binds to human serum albumin and α1-acid-glycoprotein with in vitro protein binding being approximately 90%. In ex vivo plasma samples from colorectal cancer patients at steady state exposure after 300 mg once daily, the mean percentage protein binding was 94%.
Metabolism
Following oral dosing of 14C-vandetanib, unchanged vandetanib and metabolites vandetanib N-oxide and N-desmethyl vandetanib were detected in plasma, urine and feces. A glucuronide conjugate was seen as a minor metabolite in excreta only. N-desmethyl-vandetanib is primarily produced by CYP3A4 and vandetanib-N-oxide by flavin-containing monooxygenase enzymes FMO1 and FMO3. N-desmethyl-vandetanib and vandetanib-N-oxide circulate at concentrations of approximately 7–17% and 1.4–2.2%, respectively, of those of vandetanib.
Excretion
Within a 21-day collection period after a single dose of 14C-vandetanib, approximately 69% was recovered with 44% in feces and 25% in urine. Excretion of the dose was slow and further excretion beyond 21 days would be expected based on the plasma half-life.
Vandetanib was not a substrate of hOCT2 expressed in HEK293 cells. Vandetanib inhibits the uptake of the selective OCT2 marker substrate 14C-creatinine by HEK-OCT2 cells, with a mean IC50 of 2.1 μg/mL. This is higher than vandetanib plasma concentrations (0.81 μg/mL) observed after multiple dosing at 300 mg. Inhibition of renal excretion of creatinine by vandetanib provides an explanation for increases in plasma creatinine seen in human subjects receiving vandetanib.
Specific Populations
Effects of age and gender
In a population pharmacokinetic evaluation in cancer patients, no relationship was apparent between oral clearance of vandetanib and patient age or gender.
Ethnicity
Based on a cross-study comparison in a limited number of patients, Japanese (N=3) and Chinese (N=7) patients had average exposures of vandetanib that were higher than Caucasian (N=7) patients receiving the same dose of CAPRELSA.
Effect of renal impairment
The pharmacokinetics of vandetanib were evaluated after a single CAPRELSA dose of 800 mg in six subjects with mild (creatinine clearance = 50 to <80 mL/min), eight subjects with moderate (creatinine clearance ≥30 to <50 mL/min), six subjects with severe (creatinine clearance <30 mL/min) renal impairment and ten subjects with normal (creatinine clearance >80 mL/min) renal function. Subjects with mild renal impairment had a comparable mean AUC of vandetanib to that with normal renal function. In subjects with moderate or severe renal impairment, the average AUC of vandetanib increased by 39% and 41%, respectively, compared to patients with normal renal function [see Dosage and Administration (2.1), Warnings and Precautions (5.12), and Use in Specific Populations (8.6)].
Drug Interactions
Effect of other drugs on CAPRELSA
Strong CYP3A4 inducers: In a cross-over study in 12 healthy volunteers, a single oral 300 mg dose of CAPRELSA was administered alone on day 1 and on day 10 in combination with daily doses of 600 mg of rifampicin (a strong CYP3A4 inducer) given on days 1 to 31. The coadministration of rifampicin with CAPRELSA decreased the geometric mean AUC0–504h of vandetanib by 40% (90% confidence interval (CI): 56%, 63%) compared to vandetanib alone. No clinically meaningful change in the mean Cmax of vandetanib was observed. The geometric mean AUC0–504h and Cmax of N-desmethylvandetanib increased by 266% and 414%, respectively, in the presence of rifampicin compared with vandetanib alone [see Drug Interactions (7.1)].
Strong CYP3A4 inhibitors: In a cross-over study in 14 healthy volunteers, a single oral 300 mg dose of CAPRELSA was administered alone and on day 4 in combination with daily doses of 200 mg of itraconazole (a strong CYP3A4 inhibitor) given on days 1 to 24. No change was observed in the geometric mean AUC0–504h or Cmax of vandetanib when itraconazole was coadministered with CAPRELSA.
Gastric pH elevating agents: In a cross-over study of 14 healthy volunteers, a single oral 300 mg dose of CAPRELSA was administered alone and in combination with five daily doses of 40 mg omeprazole (a proton pump inhibitor). No clinically meaningful change was observed in the geometric mean AUC0–504h and Cmax of vandetanib when omeprazole was coadministered with CAPRELSA.
In a cross-over study of 16 healthy volunteers, a single 300 mg oral dose of CAPRELSA was administered alone and after two oral doses of 150 mg of ranitidine (a H2 receptor antagonist) administered about 12 hours apart. No change was observed in the geometric mean AUC0–504h and Cmax of vandetanib when ranitidine was coadministered with CAPRELSA.
Effect of CAPRELSA on other drugs
Sensitive CYP3A4 substrates: In a cross-over study of 16 healthy volunteers, a single oral 7.5 mg dose of midazolam (as 2 mg/mL oral syrup), a sensitive CYP3A4 substrate, was administered alone and 8 days after receiving a single 800 mg oral dose of CAPRELSA. No change was observed in the geometric mean Cmax and AUCinf of midazolam when CAPRELSA was coadministered with midazolam.
Substrates of OCT2 transporter: In a cross-over study of 13 healthy volunteers, a single 1000 mg oral dose of metformin, a substrate of OCT2, was administered alone and 3 hours after receiving a single 800 mg oral dose of CAPRELSA. The coadministration of CAPRELSA with metformin increased the geometric mean AUCinf of metformin by 74% (90% CI: 58%, 92%) and geometric mean Cmax of metformin by 50% (90% CI: 34%, 67%) compared to metformin alone [see Drug Interactions (7.2)].
Substrates of P-glycoprotein transporter: In a cross-over study of 14 healthy volunteers, a single oral 0.25 mg dose of digoxin, a substrate of P-glycoprotein, was administered alone and in combination with a single 300 mg oral dose of CAPRELSA. The coadministration of CAPRELSA increased the geometric mean Cmax digoxin by 29% (90% CI: 10%, 52%) and the geometric mean of AUC0–t of digoxin by 23% (90% CI: 12%, 34%) compared to digoxin alone [see Drug Interactions (7.3)].
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Vandetanib was not carcinogenic in a 2-year study in rats when administered by daily oral gavage at doses of up to 10 mg/kg (0.7 times the human Cmax at the 300 mg clinical dose), or in the Tg∙RasH2 mouse when administered by daily oral gavage at doses of up to 30 mg/kg (~5 times the human Cmax at the clinical dose of 300 mg) for 26 weeks. Vandetanib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay and was not clastogenic in either the in vitro cytogenetic assay using human lymphocytes or in the in vivo rat micronucleus assay.
Based on nonclinical findings, male and female fertility may be impaired by treatment with CAPRELSA. In a fertility study of male rats, vandetanib had no effect on copulation or fertility rate when untreated females were mated with males administered 1, 5, or 20 mg/kg/day of vandetanib (approximately 0.03, 0.22, or 0.40 times, respectively, the human exposure based on area under the curve (AUC) in patients with cancer at the 300 mg clinical dose); however, in the same study there was a slight decrease in the number of live embryos in females mated with males treated at the 20 mg/kg/day dose level and an increase in preimplantation loss in females mated with males administered vandetanib at doses of ≥5 mg/kg/day. In a female fertility study, there was a trend towards increased estrus cycle irregularity, a slight reduction in pregnancy incidence and an increase in implantation loss. In a one-month repeat-dose toxicity study in rats, there was a decrease in the number of corpora lutea in the ovaries of rats administered 75 mg/kg/day vandetanib (approximately 1.8 times the human exposure based on AUC at the 300 mg clinical dose).
13.2 Animal Toxicology and/or Pharmacology
In an animal model of wound-healing, mice dosed with vandetanib had reduced skin-breaking strength compared with controls. This suggests that CAPRELSA slows but does not prevent wound healing. The appropriate interval between discontinuation of CAPRELSA and subsequent elective surgery required to avoid the risks of impaired wound healing has not been determined.
14. Clinical Studies
14.1 Clinical Trial in Patients with Medullary Thyroid Cancer
A double-blind, placebo-controlled study (Study D4200C00058, NCT00410761) randomized patients with unresectable locally advanced or metastatic medullary thyroid cancer to CAPRELSA 300 mg (n=231) versus placebo (n=100).
The major efficacy outcome measure was progression-free survival (PFS) with CAPRELSA compared to placebo. Other efficacy outcome measures included evaluation of overall survival (OS) and overall objective response rate (ORR). Centralized, independent blinded review of the imaging data was used in the assessment of PFS and ORR. Upon objective disease progression based on the investigator's assessment, patients were discontinued from blinded study treatment and given the option to receive open-label CAPRELSA. Forty-seven percent (109/231) of the patients initially randomized to CAPRELSA opted to receive open-label CAPRELSA after disease progression, and 79% (79/100) of the patients initially randomized to placebo opted to receive open-label CAPRELSA after disease progression.
The result of the PFS analysis, based on the central review RECIST assessment, showed a statistically significant improvement in PFS for patients randomized to CAPRELSA (Hazard Ratio (HR) = 0.35; 95% Confidence Interval (CI) = 0.24-0.53; p<0.001). Analyses in the subgroups of patients who were symptomatic or had progressed within 6 months prior to their enrollment showed similar PFS results (HR = 0.31 95% CI: 0.19, 0.53 for symptomatic patients; HR = 0.41 95% CI: 0.25, 0.66 for patients who had progressed within 6 months prior to enrollment). Median final OS were similar across both treatment arms.
The overall objective response rate (ORR) for patients randomized to CAPRELSA was 44% compared to 1% for patients randomized to placebo. All objective responses were partial responses.
| Figure 1: Kaplan-Meier Curves for Progression Free Survival in Study D4200C00058 |
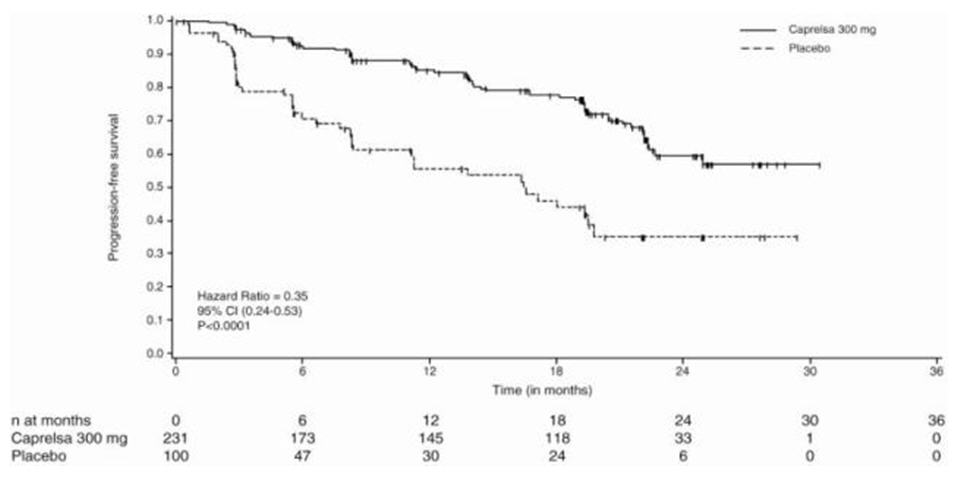 |
| Vandetanib 300 mg (N=231) | Placebo (N=100) |
|
|---|---|---|
| Progression Free Survival | ||
| Events (%) | 59 (26.0) | 41 (41.0) |
| Median, months | NR* | 16.4 |
| (95% CI) | (22.6, NE†) | (8.3, 19.7) |
| Hazard Ratio (95% CI) | 0.35 (0.24, 0.53) | |
| p-value | <0.001 | |
| Overall Survival | ||
| Deaths (%) | 116 (50.2) | 52 (52.0) |
| Median, months | 81.6 | 80.4 |
| (95% CI) | (64.6, 98.5) | (52.5, NE) |
| Hazard Ratio (95% CI) | 0.99 (0.72, 1.38) | |
| p-value | 0.975 | |
16. How is Caprelsa supplied
100 mg Tablets available in bottles containing 30 tablets (NDC 58468-7820-3).
300 mg Tablets available in bottles containing 30 tablets (NDC 58468-7840-3).
16.1 Storage and Handling
CAPRELSA tablets should be stored at room temperature between 68°F and 77°F (20°C and 25°C); excursions permitted to 59°F–86°F (15°C-30°C) [See USP controlled room temperature].
Procedures for proper handling and disposal of anticancer drugs should be considered. A guideline on this subject has been published.1 Do not crush CAPRELSA tablets.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
QT Prolongation and Torsades de Pointes
Advise patients to contact their healthcare provider in the event of syncope, pre-syncopal symptoms, and cardiac palpitations. Advise patients that their healthcare provider will monitor their electrolytes and ECGs during treatment [see Warnings and Precautions (5.1)].
Severe Skin Reactions
Advise patients to contact their healthcare provider in the event of skin reactions or rash [see Warnings and Precautions (5.2)].
Interstitial Lung Disease (ILD)
Advise patients to contact their health care provider in the event of sudden onset or worsening of breathlessness, persistent cough or fever [see Warnings and Precautions (5.3)].
Diarrhea
Advise patients to contact their healthcare provider in the event of diarrhea [see Warnings and Precautions (5.7)].
Reversible Posterior Leukoencephalopathy Syndrome (RPLS)
Advise patients to contact their healthcare provider in the event of seizures, headaches, visual disturbances, confusion or difficulty thinking [see Warnings and Precautions (5.10)].
Impaired Wound Healing
Advise patients that CAPRELSA may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.14)].
Osteonecrosis
Advise patients regarding good oral hygiene practices and to have preventive dentistry performed prior to treatment with CAPRELSA and throughout treatment with CAPRELSA and to inform their healthcare provider of any planned dental procedures.
Advise patients to immediately contact their healthcare provider for signs or symptoms associated with osteonecrosis [see Warnings and Precautions (5.17)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with CAPRELSA [see Use in Specific Populations (8.1), (8.3)].
Advise females of reproductive potential to use effective contraception during treatment with CAPRELSA and for 4 months after the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with CAPRELSA and for 4 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with CAPRELSA and for 4 months after the last dose [see Use in Specific Populations (8.2)].
Photosensitivity
Advise patients to use appropriate sun protection due to the increased susceptibility to sunburn while taking CAPRELSA and for at least 4 months after drug discontinuation [see Warnings and Precautions (5.2)].
Administration
Advise patients that CAPRELSA can be taken with or without food and not to crush CAPRELSA tablets [see Clinical Pharmacology (12.3)].
Manufactured for:
Genzyme Corporation
Cambridge, MA 02141
A SANOFI COMPANY
CAPRELSA is a registered trademark of Genzyme Corporation.
©2025 Genzyme Corporation.
For patent information: https://www.sanofi.us/en/products-and-resources/patents
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: 05/2025 | |
| MEDICATION GUIDE
CAPRELSA® (kap-rel-sah) (vandetanib) tablets |
||
| Read this Medication Guide before you start taking CAPRELSA and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your healthcare provider about your medical condition or treatment. | ||
| What is the most important information I should know about CAPRELSA? CAPRELSA can cause a change in the electrical activity of your heart called QT prolongation, which can cause irregular heartbeats and that may lead to death. You should not take CAPRELSA if you have had a condition called long QT syndrome since birth. Your healthcare provider should perform tests to check the levels of your blood potassium, calcium, magnesium, and thyroid-stimulating hormone (TSH), as well as the electrical activity of your heart with a test called an electrocardiogram (ECG). You should have these tests:
Call your healthcare provider right away if you feel faint, light-headed, or feel your heart beating irregularly during treatment with CAPRELSA. These may be symptoms related to QT prolongation. CAPRELSA is only available through a restricted program called the CAPRELSA Risk Evaluation and Mitigation Strategy (REMS) Program. For more information about the CAPRELSA REMS Program, call 1-800-817-2722 or go to www.caprelsa.com. See "What are the possible side effects of CAPRELSA?" for more information about side effects. |
||
| What is CAPRELSA?
CAPRELSA is a prescription medicine used to treat medullary thyroid cancer that cannot be removed by surgery or that has spread to other parts of the body. It takes a long time to get rid of CAPRELSA from your body and you may be at risk for side effects related to CAPRELSA after you have stopped your treatment. It is not known if CAPRELSA is safe and effective in children. |
||
| Who should not take CAPRELSA? Do not take CAPRELSA if you have QT prolongation. |
||
Before you take CAPRELSA, tell your healthcare provider about all of your medical conditions, including if you:
Especially tell your healthcare provider if you take:
Do not take other medicines during treatment with CAPRELSA until you have talked with your healthcare provider or pharmacist. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
||
How should I take CAPRELSA?
|
||
What should I avoid while taking CAPRELSA?
|
||
| What are the possible side effects of CAPRELSA? CAPRELSA may cause serious side effects, including:
|
||
|
|
|
|
||
|
|
|
|
||
| The most common side effects of CAPRELSA include: | ||
|
|
|
| Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of CAPRELSA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
How should I store CAPRELSA?
|
||
| General information about the safe and effective use of CAPRELSA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use CAPRELSA for a condition for which it was not prescribed. Do not give CAPRELSA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about CAPRELSA that is written for health professionals. |
||
| What are the ingredients in CAPRELSA? Active ingredient: vandetanib Inactive ingredients:
CAPRELSA is a registered trademark of Genzyme Corporation. ©2025 Genzyme Corporation. For more information, go to www.caprelsa.com or call 1-800-817-2722. |
||
| CAPRELSA
vandetanib tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| CAPRELSA
vandetanib tablet, film coated |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genzyme Corporation (025322157) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Lonza AG | 480007517 | API MANUFACTURE(58468-7820, 58468-7840) , ANALYSIS(58468-7820, 58468-7840) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| IPR Pharmaceuticals, Inc | 156931248 | MANUFACTURE(58468-7820, 58468-7840) , ANALYSIS(58468-7820, 58468-7840) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Penn Pharmaceutical Services Limited | 226277259 | ANALYSIS(58468-7820, 58468-7840) , MANUFACTURE(58468-7820, 58468-7840) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AstraZeneca UK Ltd | 232235408 | ANALYSIS(58468-7820, 58468-7840) , MANUFACTURE(58468-7820, 58468-7840) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| AstraZeneca Pharmaceuticals LP | 054743190 | LABEL(58468-7820, 58468-7840) , PACK(58468-7820, 58468-7840) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| EUROAPI UK LIMITED | 229522842 | PACK(58468-7820, 58468-7840) , LABEL(58468-7820, 58468-7840) | |
More about Caprelsa (vandetanib)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: EGFR inhibitors
- Breastfeeding
- En español
