Atovaquone Suspension: Package Insert / Prescribing Info
Package insert / product label
Dosage form: oral suspension
Drug class: Miscellaneous antibiotics
Medically reviewed by Drugs.com. Last updated on Aug 17, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ATOVAQUONE oral suspension
Initial U.S. Approval: 1992
Indications and Usage for Atovaquone Suspension
Atovaquone oral suspension is a quinone antimicrobial drug indicated for:
- Prevention of Pneumocystis jirovecii pneumonia (PCP) in adults and adolescents aged 13 years and older who cannot tolerate trimethoprim-sulfamethoxazole (TMP-SMX). (1.1)
- Treatment of mild-to-moderate PCP in adults and adolescents aged 13 years and older who cannot tolerate TMP-SMX. (1.2)
Limitations of Use (1.3):
- Treatment of severe PCP (alveolar arterial oxygen diffusion gradient [(A-a)DO2] >45 mm Hg) with atovaquone has not been studied.
- The efficacy of atovaquone in subjects who are failing therapy with TMP-SMX has also not been studied.
Atovaquone Suspension Dosage and Administration
• Prevention of PCP: 1,500 mg (10 mL) once daily with food (2.1)
• Treatment of PCP: 750 mg (5 mL) twice daily with food for 21 days (2.2)
• Supplied in a unit dose cup
Shake gently before use. (2.3)
Dosage Forms and Strengths
Oral suspension: 750 m L per 5 mL (3)
Contraindications
Known serious allergic/hypersensitivity reaction (e.g., angioedema, bronchospasm, throat tightness, urticaria) to atovaquone or any of the components of atovaquone. (4)
Warnings and Precautions
- Failure to administer atovaquone oral suspension with food may result in lower plasma atovaquone concentrations and may limit response to therapy. Patients with gastrointestinal disorders may have limited absorption resulting in suboptimal atovaquone concentrations. (5.1)
- Hepatotoxicity: Elevated liver chemistry tests and cases of hepatitis and fatal liver failure have been reported. (5.2)
Adverse Reactions/Side Effects
• PCP Prevention: The most frequent adverse reactions (≥25% that required discontinuation) were diarrhea, rash, headache, nausea, and fever. (6.1)
• PCP Treatment: The most frequent adverse reactions (≥ 14% that required discontinuation) were rash
(including maculopapular), nausea, diarrhea, headache, vomiting, and fever. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact PAI Pharma at 1-800-845-8210 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Concomitant administration of rifampin or rifabutin reduces atovaquone concentrations; concomitant use with atovaquone oral suspension is not recommended. (7.1)
- Concomitant administration of tetracycline reduces atovaquone concentrations; use caution when coadministering. Monitor patients for potential loss of efficacy of atovaquone if coadministration of tetracycline is necessary. (7.2)
- Concomitant administration with metoclopramide reduces atovaquone concentrations; administer concomitantly only if other antiemetics are not available. (7.3)
- Concomitant administration of indinavir reduces indinavir trough concentrations; use caution when coadministering. Monitor patients for potential loss of efficacy of indinavir if coadministration is necessary. (7.4)
Use In Specific Populations
Lactation: Breastfeeding is not recommended in mothers with HIV-1 infection due to the potential for HIV-1 transmission. (8.2)
See 17 for Patient Counseling Information
Revised:01/2024
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 8/2024
Full Prescribing Information
1. Indications and Usage for Atovaquone Suspension
1.1 Prevention of Pneumocystis jirovecii Pneumonia
Atovaquone oral suspension is indicated for the prevention of Pneumocystis jirovecii pneumonia (PCP) in adults and adolescents (aged 13 years and older) who cannot tolerate trimethoprim-sulfamethoxazole (TMP-SMX).
1.2 Treatment of Mild-to-Moderate Pneumocystis jirovecii Pneumonia
Atovaquone oral suspension is indicated for the acute oral treatment of mild-to-moderate PCP in adults and adolescents (aged 13 years and older) who cannot tolerate TMP-SMX.
1.3 Limitations of Use
Clinical experience with atovaquone for the treatment of PCP has been limited to subjects with mild-to-moderate PCP (alveolar-arterial oxygen diffusion gradient [(A-a)DO2] ≤45 mm Hg). Treatment of more severe episodes of PCP with atovaquone has not been studied. The efficacy of atovaquone in subjects who are failing therapy with TMP-SMX has also not been studied.
2. Atovaquone Suspension Dosage and Administration
2.1 Dosage for the Prevention of P. jirovecii Pneumonia
The recommended oral dosage is 1,500 mg (10 mL) once daily administered with food.
2.2 Dosage for the Treatment of Mild-to-Moderate P. jirovecii Pneumonia
The recommended oral dosage is 750 mg (5 mL) twice daily (total daily dose = 1,500 mg) administered with food for 21 days.
2.3 Important Administration Instructions
Administer atovaquone oral suspension with food to avoid lower plasma atovaquone concentrations that may limit response to therapy [see Warnings and Precautions (5.1), Clinical Pharmacology (12.3)].
Atovaquone Oral Suspension
Shake Unit dose cup gently before administering the recommended dosage.
3. Dosage Forms and Strengths
Atovaquone oral suspension USP is a bright yellow, tutti frutti flavored, oral suspension containing 750 mg of atovaquone per 5 mL. Atovaquone is supplied in in a 5 mL unit dose cup.
4. Contraindications
Atovaquone oral suspension is contraindicated in patients who develop or have a history of hypersensitivity reactions (e.g., angioedema, bronchospasm, throat tightness, urticaria) to atovaquone or any of the components of atovaquone oral solution.
5. Warnings and Precautions
5.1 Risk of Limited Oral Absorption
Absorption of orally administered atovaquone oral suspension is limited but can be significantly increased when the drug is taken with food. Failure to administer atovaquone oral suspension with food may result in lower plasma atovaquone concentrations and may limit response to therapy.
Consider therapy with other agents in patients who have difficulty taking atovaquone oral suspension with food or in patients who have gastrointestinal disorders that may limit absorption of oral medications [see Clinical Pharmacology (12.3)].
5.2 Hepatotoxicity
Cases of cholestatic hepatitis, elevated liver enzymes, and fatal liver failure have been reported in patients treated with atovaquone [see Adverse Reactions (6.2)].
If treating patients with severe hepatic impairment, closely monitor patients following administration of atovaquone.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in other sections of the labeling:
• Hepatotoxicity [see Warnings and Precautions (5.2)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Additionally, because many subjects who participated in clinical trials with atovaquone had complications of advanced human immunodeficiency virus (HIV) disease, it was often difficult to distinguish adverse reactions caused by atovaquone from those caused by underlying medical conditions.
PCP Prevention Trials
In 2 clinical trials, atovaquone oral suspension was compared with dapsone or aerosolized pentamidine in HIV-1-infected adolescent (13 to 18 years) and adult subjects at risk of PCP (CD4 count <200 cells/mm3 or a prior episode of PCP) and unable to tolerate TMP-SMX.
Dapsone Comparative Trial: In the dapsone comparative trial (n = 1,057), the majority of subjects were white (64%), male (88%), and receiving prophylaxis for PCP at randomization (73%); the mean age was 38 years. Subjects received atovaquone oral suspension 1,500 mg once daily (n = 536) or dapsone 100 mg once daily (n = 521); median durations of exposure were 6.7 and 6.5 months, respectively. Adverse reaction data were collected only for adverse reactions requiring discontinuation of treatment, which occurred at similar frequencies in subjects treated with atovaquone oral suspension or dapsone (Table 1). Among subjects taking neither dapsone nor atovaquone at enrollment (n = 487), adverse reactions requiring discontinuation of treatment occurred in 43% of subjects treated with dapsone and 20% of subjects treated with atovaquone oral suspension. Gastrointestinal adverse reactions (nausea, diarrhea, and vomiting) were more frequently reported in subjects treated with atovaquone oral suspension (Table 1).
Table 1. Percentage (>2%) of Subjects with Selected Adverse Reactions Requiring Discontinuation of Treatment in the Dapsone Comparative PCP Prevention Trial
| Adverse Reaction | All Subjects | |
|
Atovaquone Oral Suspension | Dapsone 100 mg/day (n=521) % |
|
| Rash | 6.3 | 8.8 |
| Nausea | 4.1 | 0.6 |
| Diarrhea | 3.2 | 0.2 |
| Vomiting | 2.2 | 0.6 |
Aerosolized Pentamidine Comparative Trial: In the aerosolized pentamidine comparative trial (n = 549), the majority of subjects were white (79%), male (92%), and were primary prophylaxis patients at enrollment (58%); the mean age was 38 years. Subjects received atovaquone oral suspension once daily at a dose of 750 mg (n = 188) or 1,500 mg (n = 175) or received aerosolized pentamidine 300 mg every 4 weeks (n = 186); the median durations of exposure were 6.2, 6.0, and 7.8 months, respectively. Table 2 summarizes the clinical adverse reactions reported by ≥20% of the subjects receiving either the 1,500-mg dose of atovaquone oral suspension or aerosolized pentamidine.
Rash occurred more often in subjects treated with atovaquone oral suspension (46%) than in subjects treated with aerosolized pentamidine (28%). Treatment-limiting adverse reactions occurred in 25% of subjects treated with atovaquone oral suspension 1,500 mg once daily and in 7% of subjects treated with aerosolized pentamidine. The most frequent adverse reactions requiring discontinuation of dosing in the group receiving atovaquone oral suspension 1,500 mg once daily were rash (6%), diarrhea (4%), and nausea (3%). The most frequent adverse reaction requiring discontinuation of dosing in the group receiving aerosolized pentamidine was bronchospasm (2%).
Table 2. Percentage (≥20%) of Subjects with Selected Adverse Reactions in the Aerosolized Pentamidine Comparative PCP Prevention Trial
| Adverse Reaction | All Subjects | |
|
Atovaquone Oral Suspension | Aerosolized Pentamidine (N=186) % |
|
| Diarrhea | 42 | 35 |
| Rash | 39 | 28 |
| Headache | 28 | 22 |
| Nausea | 26 | 23 |
| Fever | 25 | 18 |
| Rhinitis | 24 | 17 |
Other reactions occurring in ≥10% of subjects receiving the recommended dose of atovaquone oral suspension (1,500 mg once daily) included vomiting, sweating, flu syndrome, sinusitis, pruritus, insomnia, depression, and myalgia.
PCP Treatment Trials
Safety information is presented from 2 clinical efficacy trials of the atovaquone tablet formulation: 1) a randomized, double-blind trial comparing atovaquone tablets with TMP-SMX in subjects with acquired immunodeficiency syndrome (AIDS) and mild-to-moderate PCP [(A-a)DO2] ≤45 mm Hg and PaO2 ≥60 mm Hg on room air; 2) a randomized, open-label trial comparing atovaquone tablets with intravenous (IV) pentamidine isethionate in subjects with mild-to-moderate PCP who could not tolerate trimethoprim or sulfa antimicrobials.
TMP-SMX Comparative Trial: In the TMP-SMX comparative trial (n = 408), the majority of subjects were white (66%) and male (95%); the mean age was 36 years. Subjects received atovaquone 750 mg (three 250-mg tablets) 3 times daily for 21 days or TMP 320 mg plus SMX 1,600 mg 3 times daily for 21 days; median durations of exposure were 21 and 15 days, respectively.
Table 3 summarizes all clinical adverse reactions reported by ≥10% of the trial population regardless of attribution. Nine percent of subjects who received atovaquone and 24% of subjects who received TMP-SMX discontinued therapy due to an adverse reaction. Among the subjects who discontinued, 4% of subjects receiving atovaquone and 8% of subjects in the TMP-SMX group discontinued therapy due to rash.
The incidence of adverse reactions with atovaquone oral suspension at the recommended dose (750 mg twice daily) was similar to that seen with the tablet formulation.
Table 3. Percentage (≥10%) of Subjects with Selected Adverse Reactions in the TMP-SMX Comparative PCP Treatment Trial
| Adverse Reaction | Atovaquone Tablets (n=203) % | TMP-SMX (n=205) % |
| Rash (including maculopapular | 23 | 34 |
| Nausea | 21 | 44 |
| Diarrhea | 19 | 7 |
| Headache | 16 | 22 |
| Vomiting | 14 | 35 |
| Fever | 14 | 25 |
| Insomnia | 10 | 9 |
Two percent of subjects treated with atovaquone and 7% of subjects treated with TMP-SMX had therapy prematurely discontinued due to elevations in ALT/AST.
Pentamidine Comparative Trial: In the pentamidine comparative trial (n = 174), the majority of subjects in the primary therapy trial population (n = 145) were white (72%) and male (97%); the mean age was 37 years. Subjects received atovaquone 750 mg (three 250-mg tablets) 3 times daily for 21 days or a 3- to 4-mg/kg single pentamidine isethionate IV infusion daily for 21 days; the median durations of exposure were 21 and 14 days, respectively.
Table 4 summarizes the clinical adverse reactions reported by ≥10% of the primary therapy trial population regardless of attribution. Fewer subjects who received atovaquone reported adverse reactions than subjects who received pentamidine (63% vs. 72%). However, only 7% of subjects discontinued treatment with atovaquone due to adverse reactions, while 41% of subjects who received pentamidine discontinued treatment for this reason. Of the 5 subjects who discontinued therapy with atovaquone, 3 reported rash (4%). Rash was not severe in any subject. The most frequently cited reasons for discontinuation of pentamidine therapy were hypoglycemia (11%) and vomiting (9%).
Table 4. Percentage (≥10%) of Subjects with Selected Adverse Reactions in the Pentamidine Comparative PCP Treatment Trial (Primary Therapy Group)
| Adverse Reaction | Atovaquone Tablets (n=73) % | Pentamidine (n=71) % |
| Fever | 40 | 25 |
| Nausea | 22 | 37 |
| Rash | 22 | 13 |
| Diarrhea | 21 | 31 |
| Insomnia | 19 | 14 |
| Headache | 18 | 28 |
| Vomiting | 14 | 17 |
| Cough | 14 | 1 |
| Sweat | 10 | 3 |
| Monilia, oral | 10 | 3 |
Laboratory abnormality was reported as the reason for discontinuation of treatment in 2 of 73 subjects (3%) who received atovaquone, and in 14 of 71 subjects (20%) who received pentamidine. One subject (1%) receiving atovaquone had elevated creatinine and BUN levels and 1 subject (1%) had elevated amylase levels. In this trial, elevated levels of amylase occurred in subjects (8% versus 4%) receiving atovaquone tablets or pentamidine, respectively.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of atovaquone oral suspension. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders
Methemoglobinemia, thrombocytopenia.
Immune System Disorders
Hypersensitivity reactions including angioedema, bronchospasm, throat tightness, and urticaria.
Eye Disorders
Vortex keratopathy.
Gastrointestinal Disorders
Pancreatitis.
Hepatobiliary Disorders
Hepatitis, fatal liver failure.
Skin and Subcutaneous Tissue Disorders
Erythema multiforme, Stevens-Johnson syndrome, and skin desquamation.
Renal and Urinary Disorders
Acute renal impairment.
Related/similar drugs
7. Drug Interactions
7.1 Rifampin/Rifabutin
Concomitant administration of rifampin or rifabutin and atovaquone oral suspension is known to reduce atovaquone concentrations [see Clinical Pharmacology (12.3)]. Concomitant administration of atovaquone oral suspension and rifampin or rifabutin is not recommended.
7.2 Tetracycline
Concomitant administration of tetracycline and atovaquone oral suspension has been associated with a reduction in plasma concentrations of atovaquone [see Clinical Pharmacology (12.3)]. Caution should be used when prescribing tetracycline concomitantly with atovaquone oral suspension. Monitor patients for potential loss of efficacy of atovaquone if coadministration is necessary.
7.3 Metoclopramide
Metoclopramide may reduce the bioavailability of atovaquone and should be used only if other antiemetics are not available [see Clinical Pharmacology (12.3)].
7.4 Indinavir
Concomitant administration of atovaquone and indinavir did not result in any change in the steady-state AUC and Cmax of indinavir but resulted in a decrease in the Ctrough of indinavir [see Clinical Pharmacology (12.3)]. Caution should be exercised when prescribing atovaquone oral suspension with indinavir due to the decrease in trough concentrations of indinavir. Monitor patients for potential loss of efficacy of indinavir if coadministration with atovaquone oral suspension is necessary.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Available data from postmarketing experience with use of atovaquone in pregnant women are insufficient to identify a drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. Pregnant women with HIV who are infected with PCP are at increased risk of adverse pregnancy outcomes (see Clinical Considerations). Atovaquone given orally by gavage to pregnant rats and rabbits during organogenesis did not cause fetal malformations at plasma concentrations up to 3 times and 0.5 times, respectively, the estimated human exposure based on steady-state plasma concentrations (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk: Pregnant women with HIV who are infected with PCP are at increased risk of severe illness and maternal death associated with PCP compared with non-pregnant women.
Data
Animal Data: Atovaquone administered in oral doses of 250, 500, and 1,000 mg/kg/day to pregnant rats during organogenesis (Gestation Day [GD] 6 to GD15) did not cause maternal or embryo-fetal toxicity at doses up to 1,000 mg/kg/day corresponding to maternal plasma concentrations approximately 3 times the estimated human exposure during the treatment of PCP based on steady-state plasma concentrations. In pregnant rabbits, atovaquone administered in oral doses of 300, 600, and 1,200 mg/kg/day during organogenesis (GD6 to GD18) caused decreased fetal body length at a maternally toxic dose of 1,200 mg/kg/day corresponding to a plasma concentration that is approximately 0.5 times the estimated human exposure based on steady-state plasma concentrations. In a pre- and post-natal study in rats, atovaquone administered in oral doses of 250, 500, and 1,000 mg/kg/day from GD15 until Lactation Day (LD) 20 did not impair the growth or developmental effects in first generation offspring at doses up to 1,000 mg/kg/day corresponding to approximately 3 times the estimated human exposure based on steady-state plasma concentrations during the treatment of PCP. Atovaquone crossed the placenta and was present in fetal rat and rabbit tissue.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1–infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1. There are no data on the presence of atovaquone in human milk, the effects on the breastfed child, or the effects on milk production. Atovaquone was detected in rat milk when lactating rats were administered oral atovaquone (see Data). When a drug is present in animal milk, it is likely the drug will be present in human milk. Because of the potential for HIV-1 transmission to HIV-negative infants, instruct mothers with HIV-1 not to breastfeed if they are taking atovaquone for the prevention or treatment of PCP.
Data
In a rat study with doses of 10 and 250 mg/kg given orally by gavage on postpartum Day 11, atovaquone concentrations in the milk were 30% of the concurrent atovaquone concentrations in the maternal plasma at both doses. The concentration of drug in animal milk does not necessarily predict the concentration of drug in human milk.
8.4 Pediatric Use
Evidence of safety and effectiveness in pediatric patients (aged 12 years and younger) has not been established. In a trial of atovaquone oral suspension administered once daily with food for 12 days to 27 HIV-1-infected, asymptomatic infants and children aged between 1 month and 13 years, the pharmacokinetics of atovaquone were age-dependent. The average steady-state plasma atovaquone concentrations in the 24 subjects with available concentration data are shown in Table 5.
Table 5. Average Steady-state Plasma Atovaquone Concentrations in Pediatric Subjects
| Age | Dose of Atovaquone Oral Suspension | ||
| 10 mg/kg | 30 mg/kg | 45 mg/kg | |
| Average Css in mcg/mL (mean ± SD) | |||
| 1-3 months | 5.9 (n=1) | 27.8 ± 5.8 (n=4) | — |
| >3-24 months | 5.7 ± 5.1 (n=4) | 9.8 ± 3.2 (n=4) | 15.4 ± 6.6 (n=4) |
| >2-13 years | 16.8 ± 6.4 (n=4) | 37.1 ± 10.9 (n=3) | — |
| Css = Concentration at steady state. | |||
10. Overdosage
Overdoses up to 31,500 mg of atovaquone have been reported. In one such patient who also took an unspecified dose of dapsone, methemoglobinemia occurred. Rash has also been reported after overdose. There is no known antidote for atovaquone, and it is currently unknown if atovaquone is dialyzable.
11. Atovaquone Suspension Description
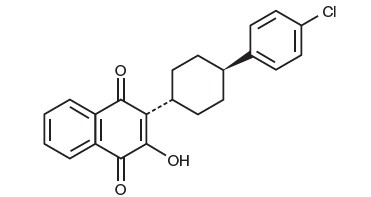
Atovaquone oral suspension USP is a quinone antimicrobial drug. The chemical name of atovaquone is trans-2-[4-(4-chlorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthalenedione. Atovaquone is a yellow crystalline solid that is practically insoluble in water. It has a molecular weight of 366.84 and the molecular formula C22H19ClO3. The compound has the following structural formula:
Atovaquone oral suspension USP is a formulation of micro-fine particles of atovaquone, USP.
Each 5 mL of atovaquone oral suspension USP contains 750 mg of atovaquone and the inactive ingredients benzyl alcohol, tutti frutti flavor, poloxamer 188, purified water, saccharin sodium, and xanthan gum.
12. Atovaquone Suspension - Clinical Pharmacology
Atovaquone is a quinone antimicrobial drug [see Microbiology (12.4)].
12.2 Pharmacodynamics
Relationship between Plasma Atovaquone Concentrations and Clinical Outcome
In a comparative clinical trial, HIV/AIDS subjects received atovaquone tablets 750 mg 3 times daily or TMP-SMX for treatment of mild-to-moderate PCP for 21 days [see Clinical Studies (14.2)]; the relationship between atovaquone plasma concentrations and successful treatment outcome from 113 of these subjects for whom both steady-state drug concentrations and outcome data were available is shown in Table 6.
Table 6. Relationship between Plasma Atovaquone Concentrations and Successful Treatment Outcome.
| Steady-state Plasma Atovaquone Concentrations (mg/mL) | Successful Treatmenta
No. Successes/No. in Group (%) |
| 0 to <5 | 0/6 (0%) |
| 5 to <10 | 18/26 (69%) |
| 10 to <15 | 30/38 (79%) |
| 15 to <20 | 18/19 (95%) |
| ≥20 | 24/24 (100%) |
a Successful treatment outcome was defined as improvement in clinical and respiratory measures persisting
at least 4 weeks after cessation of therapy. Improvement in clinical and respiratory measures was assessed
using a composite of parameters that included oral body temperature, respiratory rate, and severity scores
for cough, dyspnea, and chest pain/tightness.
Cardiac Effects
The effect of atovaquone oral suspension on the QT interval is unknown in humans.
12.3 Pharmacokinetics
Plasma atovaquone concentrations do not increase proportionally with dose following ascending repeat-dose administration of atovaquone oral suspension in healthy subjects. When atovaquone oral suspension was administered with food at dosage regimens of 500 mg once daily, 750 mg once daily, and 1,000 mg once daily, mean (±SD) steady-state plasma atovaquone concentrations were 11.7 ± 4.8, 12.5 ± 5.8, and 13.5 ± 5.1 mcg/mL, respectively. The corresponding mean (±SD) Cmax concentrations were 15.1 ± 6.1,
15.3 ± 7.6, and 16.8 ± 6.4 mcg/mL.
Absorption
Atovaquone is a highly lipophilic compound with low aqueous solubility. The mean (±SD) absolute bioavailability of atovaquone from a 750-mg dose of atovaquone oral suspension administered under fed conditions in 9 HIV-1–infected (CD4 >100 cells/mm3) volunteers was 47% ± 15%.
Effect of Food: Administering atovaquone oral suspension with food enhances atovaquone bioavailability. Sixteen healthy subjects received a single 750-mg dose of atovaquone oral suspension after an overnight fast and following a meal (23 g fat: 610 kCal). The mean (±SD) atovaquone AUC under fasting and fed conditions were 324 ± 115 and 801 ± 320 h•mcg/mL, respectively, representing a 2.6 ± 1.0-fold increase.
Distribution
Following IV administration of atovaquone, the mean (±SD) volume of distribution at steady state (Vdss) was 0.60 ± 0.17 L/kg (n = 9). Atovaquone is extensively bound to plasma proteins (99.9%) over the concentration range of 1 to 90 mcg/mL. In 3 HIV-1-infected children who received 750 mg atovaquone as the tablet formulation 4 times daily for 2 weeks, the cerebrospinal fluid concentrations of atovaquone were 0.04, 0.14, and 0.26 mcg/mL, representing less than 1% of the plasma concentration.
Elimination
The mean (±SD) half-life of atovaquone was 62.5 ± 35.3 hours after IV administration and ranged from 67.0 ± 33.4 to 77.6 ± 23.1 hours following administration of atovaquone oral suspension.
Metabolism: The metabolism of atovaquone is unknown.
Excretion: Following oral administration of 14C-labelled atovaquone to healthy subjects, greater than 94% of the dose was recovered as unchanged atovaquone in the feces over 21 days.
Specific Populations
Patients with Hepatic or Renal Impairment: The pharmacokinetics of atovaquone have not been studied in patients with hepatic or renal impairment.
HIV-Infected Subjects: When atovaquone oral suspension was administered to 5 HIV-1–infected subjects at a dose of 750 mg twice daily, the mean (±SD) steady-state plasma atovaquone concentration was21.0 ± 4.9 mcg/mL and mean (±SD) Cmax was 24.0 ± 5.7 mcg/mL. The mean (±SD) minimum plasma atovaquone concentration (Cmin) associated with the 750-mg twice-daily regimen was 16.7 ± 4.6 mcg/mL.
In an open-label PCP trial in 18 HIV-1–infected subjects, administration of atovaquone oral suspension 750 mg twice daily with meals resulted in a mean (±SD) steady-state plasma atovaquone concentration of 22.0 ± 10.1 mcg/mL.
The mean (±SD) plasma clearance of atovaquone following IV administration in 9 HIV-1–infected subjects was 10.4 ± 5.5 mL/min (0.15 ± 0.09 mL/min/kg).
Drug Interaction Studies
Rifampin/Rifabutin: In a trial with 13 HIV-1–infected volunteers, the oral administration of rifampin 600 mg every 24 hours with atovaquone oral suspension 750 mg every 12 hours resulted in a 52% ± 13% decrease in the mean (±SD) steady-state plasma atovaquone concentration and a 37% ± 42% increase in the mean (±SD) steady-state plasma rifampin concentration. The half-life of atovaquone decreased from 82 ± 36 hours when administered without rifampin to 50 ± 16 hours with rifampin. In a trial of 24 healthy volunteers, the oral administration of rifabutin 300 mg once daily with atovaquone oral suspension 750 mg twice daily resulted in a 34% decrease in the mean steady-state plasma atovaquone concentration and a 19% decrease in the mean steady-state plasma rifabutin concentration.
Tetracycline: Concomitant treatment with tetracycline has been associated with a 40% reduction in plasma concentrations of atovaquone.
Metoclopramide: Concomitant treatment with metoclopramide has been associated with a 50% reduction in steady-state atovaquone plasma concentrations.
Indinavir: Concomitant administration of atovaquone (750 mg twice daily with food for 14 days) and indinavir (800 mg three times daily without food for 14 days) did not result in any change in the steady-state AUC and Cmax of indinavir, but resulted in a decrease in the Ctrough of indinavir (23% decrease [90% CI: 8%, 35%]).
Trimethoprim/Sulfamethoxazole (TMP-SMX): Concomitant administration of atovaquone oral suspension 500 mg once daily (not the approved dosage) and TMP-SMX in 6 HIV-infected adult subjects did not result in significant changes in either atovaquone or TMP-SMX exposure.
Zidovudine: The administration of atovaquone tablets 750 mg every 12 hours with zidovudine 200 mg every 8 hours to 14 HIV-1 infected subjects resulted in a 24% ± 12% decrease in zidovudine apparent oral clearance, leading to a 35% ± 23% increase in plasma zidovudine AUC. The glucuronide metabolite:parent ratio decreased from a mean of 4.5 when zidovudine was administered alone to 3.1 when zidovudine was administered with atovaquone tablets. This effect is minor and would not be expected to produce clinically significant events. Zidovudine had no effect on atovaquone pharmacokinetics.
12.4 Microbiology
Mechanism of Action
Atovaquone is a hydroxy-1,4-naphthoquinone, an analog of ubiquinone, with antipneumocystis activity. The mechanism of action against Pneumocystis jirovecii has not been fully elucidated. In Plasmodium species, the site of action appears to be the cytochrome bc1 complex (Complex III). Several metabolic enzymes are linked to the mitochondrial electron transport chain via ubiquinone. Inhibition of electron transport by atovaquone results in indirect inhibition of these enzymes. The ultimate metabolic effects of such blockade may include inhibition of nucleic acid and adenosine triphosphate (ATP) synthesis.
Antimicrobial Activity
Atovaquone is active against P. jirovecii[see Clinical Studies (14)].
Resistance
Phenotypic resistance to atovaquone in vitro has not been demonstrated for P. jirovecii. However, in 2 subjects who developed PCP after prophylaxis with atovaquone, DNA sequence analysis identified mutations in the predicted amino acid sequence of P. jirovecii cytochrome b (a likely target site for atovaquone). The clinical significance of this is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis and Mutagenesis and Impairment of Fertility
Carcinogenicity studies in rats were negative; 24-month studies in mice (dosed with 50, 100, or 200 mg/kg/day), showed treatment-related increases in incidence of hepatocellular adenoma and hepatocellular carcinoma at all doses tested, which correlated with 1.4 to 3.6 times the average steady-state plasma concentrations in humans during acute treatment of PCP. Atovaquone was negative with or without metabolic activation in the Ames Salmonella mutagenicity assay, the mouse lymphoma mutagenesis assay, and the cultured human lymphocyte cytogenetic assay. No evidence of genotoxicity was observed in the in vivo mouse micronucleus assay.
Impairment of Fertility
Atovaquone administered by oral gavage in doses of 100, 300, or 1,000 mg/kg/day to adult male rats from 73 days prior to mating until 20 days after mating and to adult female rats from 14 days prior to mating until LD20 did not impair male or female fertility or early embryonic development at doses up to 1,000 mg/kg/day corresponding to plasma exposures of approximately 3 times the estimated human exposure based on steady-state plasma concentrations.
14. Clinical Studies
14.1 Prevention of PCP
The indication for prevention of PCP is based on the results of 2 clinical trials comparing atovaquone oral suspension with dapsone or aerosolized pentamidine in HIV-1-infected adolescent (aged 13 to 18 years) and adult subjects at risk of PCP (CD4 count <200 cells/mm3 or a prior episode of PCP) and unable to tolerate TMP-SMX.
Dapsone Comparative Trial
This open-label trial enrolled 1,057 subjects, randomized to receive atovaquone oral suspension 1,500 mg once daily (n = 536) or dapsone 100 mg once daily (n = 521). The majority of subjects were white (64%), male (88%), and receiving prophylaxis for PCP at randomization (73%); the mean age was 38 years. Median follow-up was 24 months. Subjects randomized to the dapsone arm who were seropositive for Toxoplasmagondii and had a CD4 count <100 cells/mm3 also received pyrimethamine and folinic acid. PCP event rates are shown in Table 7. Mortality rates were similar.
Aerosolized Pentamidine Comparative Trial
This open-label trial enrolled 549 subjects, randomized to receive atovaquone oral suspension 1,500 mg once daily (n = 175), atovaquone oral suspension 750 mg once daily
(n = 188), or aerosolized pentamidine 300 mg once monthly (n = 186). The majority of subjects were white (79%), male (92%), and were primary prophylaxis patients at enrollment (58%); the mean age was 38 years. Median follow-up was 11.3 months. The results of the PCP event rates appear in Table 7. Mortality rates were similar among the groups.
Table 7. Confirmed or Presumed/Probable PCP Events (As-Treated Analysis)a
| Trial 1 |
Trial 2 |
||||
| Assessment | Atovaquone Oral Suspension 1,500 mg/day (n=527) | Dapsone 100 mg/day (n=510) | Atovaquone Oral Suspension 750 mg/day (n=188) | Atovaquone Oral Suspension 1,500 mg/day (n=172) | Aerosolozed Pentamidine 300 mg/ month (n=169) |
| % | 15 | 19 | 23 | 18 | 17 |
| Relative Riskb
(CI)c | 0.77 (0.57, 1.04) | 1.47 (0.86, 2.50) | 1.14 (0.63, 2.06) | ||
a Those events occurring during or within 30 days of stopping assigned treatment.
b Relative risk <1 favors atovaquone and values >1 favor comparator. Trials results did not show superiority of atovaquone to the comparator.
c The confidence level of the interval for the dapsone comparative trial was 95% and for the pentamidine comparator trial was 97.5%.
An analysis of all PCP events (intent-to-treat analysis) for both trials showed results similar to those shown in Table 7.
14.2 Treatment of PCP
The indication for treatment of mild-to-moderate PCP is based on the results of 2 efficacy trials: a randomized, double-blind trial comparing atovaquone tablets with TMP-SMX in subjects with HIV/AIDS and mild-to-moderate PCP (defined in the protocol as [(A-a)DO2] ≤45 mm Hg and PaO2 ≥60 mm Hg on room air) and a randomized open-label trial comparing atovaquone tablets with IV pentamidine isethionate in subjects with mild-to-moderate PCP who could not tolerate trimethoprim or sulfa antimicrobials. Both trials were
conducted with the tablet formulation using 750 mg three times daily. Results from these efficacy trials established a relationship between plasma atovaquone concentration and successful outcome. Successful outcome was defined as improvement in clinical and respiratory measures persisting at least 4 weeks after cessation of therapy. [see Clinical Pharmacology (12.2)].
TMP-SMX Comparative Trial
This double-blind, randomized trial compared the safety and efficacy of atovaquone tablets with that of TMP-SMX for the treatment of subjects with HIV/AIDS and histologically confirmed PCP. Only subjects with mild-to-moderate PCP were eligible for enrollment.
A total of 408 subjects were enrolled into the trial. The majority of subjects were white (66%) and male (95%); the mean age was 36 years. Eighty-six subjects without histologic confirmation of PCP were excluded from the efficacy analyses. Of the 322 subjects with histologically confirmed PCP, 160 were randomized to receive 750 mg atovaquone (three 250-mg tablets) 3 times daily for 21 days and 162 were randomized to receive 320 mg TMP plus 1,600 mg SMX 3 times daily for 21 days. Therapy success was defined as improvement in clinical and respiratory measures persisting at least 4 weeks after cessation of therapy. Improvement in clinical and respiratory measures was assessed using a composite of parameters that included oral body temperature, respiratory rate, severity scores for cough, dyspnea, and chest pain/tightness. Therapy failures included lack of response, treatment discontinuation due to an adverse experience, and unevaluable.
There was a significant difference (P = 0.03) in mortality rates between the treatment groups favoring TMP-SMX. Among the 322 subjects with confirmed PCP, 13 of 160 (8%) subjects treated with atovaquone and 4 of 162 (2.5%) subjects receiving TMP-SMX died during the 21-day treatment course or 8-week follow-up period. In the intent-to-treat analysis for all 408 randomized subjects, there were 16 (8%) deaths among subjects treated with atovaquone and 7 (3.4%) deaths among subjects treated with TMP-SMX (P = 0.051). Of the 13 subjects with confirmed PCP and treated with atovaquone who died, 4 died of PCP and 5 died with a combination of bacterial infections and PCP; bacterial infections did not appear to be a factor in any of the 4 deaths among TMP-SMX-treated subjects.
A correlation between plasma atovaquone concentrations and death demonstrated that subjects with lower plasma concentrations were more likely to die. For those subjects for whom Day 4 plasma atovaquone concentration data are available, 5 (63%) of 8 subjects with concentrations <5 mcg/mL died during participation in the trial. However, only 1 (2.0%) of the 49 subjects with Day 4 plasma atovaquone concentrations ≥5 mcg/mL died. Sixty-two percent of subjects on atovaquone and 64% of subjects on TMP-SMX were classified as protocol-defined therapy successes (Table 8).
Table 8. Outcome for PCP-positive Subjects Enrolled in the TMP-SMX Comparative Trial
| Number of Subjects (%) | ||||
| Outcome of Therapya | Atovaquone Tablets (n=160) | TMP-SMX (n=162) | ||
| Therapy success | 99 | 62% | 103 | 64% |
|
Therapy failure due to: | 28 11 22 | 17% 7% 14% | 10 33 16 | 6% 20% 10% |
| Required alternate PCP therapy during trial | 55 | 34% | 55 | 34% |
a As defined by the protocol and described in trial description above.
The failure rate due to lack of response was significantly higher for subjects receiving atovaquone, while the failure rate due to an adverse reaction was significantly higher for subjects receiving TMP-SMX.
Pentamidine Comparative Trial
This unblinded, randomized trial was designed to compare the safety and efficacy of atovaquone with that of pentamidine for the treatment of histologically-confirmed mild or moderate PCP in subjects with HIV/AIDS. Approximately 80% of the subjects either had a history of intolerance to trimethoprim or sulfa antimicrobials (the primary therapy group) or were experiencing intolerance to TMP-SMX with treatment of an episode of PCP at the time of enrollment in the trial (the salvage treatment group). A total of 174 subjects were enrolled into the trial. Subjects were randomized to receive atovaquone 750 mg (three 250-mg tablets) 3 times daily for 21 days or pentamidine isethionate 3- to 4-mg/kg single IV infusion daily for 21 days. The majority of subjects were white (72%) and male (97%); the mean age was approximately 37 years. Thirty-nine subjects without histologic confirmation of PCP were excluded from the efficacy analyses. Of the 135 subjects with histologically-confirmed PCP, 70 were randomized to receive atovaquone and 65 to pentamidine. One hundred and ten (110) of these were in the primary therapy group and 25 were in the salvage therapy group. One subject in the primary therapy group randomized to receive pentamidine did not receive trial medication.
There was no difference in mortality rates between the treatment groups. Among the 135 subjects with confirmed PCP, 10 of 70 (14%) subjects receiving atovaquone and 9 of 65 (14%) subjects receiving pentamidine died during the 21-day treatment course or 8-week follow-up period. In the intent-to-treat analysis for all subjects, there were 11 (12.5%) deaths among those treated with atovaquone and 12 (14%) deaths among those treated with pentamidine. Among subjects for whom Day 4 plasma atovaquone concentrations were available, 3 of 5 (60%) subjects with concentrations <5 mcg/mL died during participation in the trial. However, only 2 of 21 (9%) subjects with Day 4 plasma concentrations ≥5 mcg/mL died. The therapeutic outcomes for the 134 subjects who received trial medication in this trial are presented in Table 9.
Table 9. Outcome of Treatment for PCP-positive Subjects (%) Enrolled in the Pentamidine Comparative Trial
| Primary Treatment | Salvage Treatment | |||||||
| Outcome of Therapy | Atovaquone (n=56) | Pentamidine (n=53) | Atovaquone (n=14) | Pentamidine (n=11) |
||||
| Therapy success | 32 | 57% | 21 | 40% | 13 | 93% | 7 | 64% |
| Therapy failure due to: -Lack of response -Adverse reaction -Unevaluable | 16 2 6 | 29% 3.6% 11% | 9 19 4 | 17% 36% 8% | 0 0 1 | 7% | 0 3 1 | 27% 9% |
| Required alternate PCP therapy during trial | 19 | 34% | 29 | 55% | 0 | 4 | 36% | |
16. How is Atovaquone Suspension supplied
Atovaquone oral suspension USP (bright yellow, tutti frutti flavored) containing 750 mg atovaquone per 5 mL.
- NDC 0904-7459-41 5 mL unit dose cup.
- Case contains 18 unit dose cups of 5 mL (0904-7459-25), package in 3 trays of 6 unit dose cups each,
- 42 unit dose cups of 5 mL (NDC 0904-7459-53), package in 7 trays of 6 unit dose cups each.
Store at 15ºC to 25ºC (59ºF to 77ºF).
Do not freeze. Dispense in tight container as defined in USP.
17. Patient Counseling Information
Administration Instructions
Instruct patients to:
• Ensure the prescribed dose of atovaquone oral suspension is taken as directed.
• Take their daily doses of atovaquone oral suspension with food, as food will significantly improve the
absorption of the drug.
• Shake atovaquone oral suspension gently before use each time.
Lactation
Instruct mothers with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in the breast
milk [see Use in Specific Populations (8.2)].
Distributed by:
Major® Pharmaceuticals
Indianapolis, IN 46268
R01/24


| ATOVAQUONE
atovaquone oral suspension suspension |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Major Pharmaceuticals (191427277) |
| Registrant - PAI Holdings, LLC (044940096) |
More about atovaquone
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (6)
- Latest FDA alerts (4)
- Side effects
- Dosage information
- During pregnancy
- Drug class: miscellaneous antibiotics
- Breastfeeding
- En español