Alecensa: Package Insert / Prescribing Info
Package insert / product label
Generic name: alectinib hydrochloride
Dosage form: capsule
Drug class: Multikinase inhibitors
Medically reviewed by Drugs.com. Last updated on May 6, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ALECENSA® (alectinib) capsules, for oral use
Initial U.S. Approval: 2015
Recent Major Changes
Indications and Usage for Alecensa
ALECENSA is a kinase inhibitor indicated for:
- adjuvant treatment in adult patients following tumor resection of anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) (tumors ≥ 4 cm or node positive) as detected by an FDA-approved test. (1.1)
- treatment of adult patients with ALK-positive metastatic NSCLC as detected by an FDA-approved test. (1.2)
Alecensa Dosage and Administration
600 mg orally twice daily. Administer ALECENSA with food. (2.2)
Dosage Forms and Strengths
Capsules: 150 mg (3)
Contraindications
None. (4)
Warnings and Precautions
- Hepatotoxicity: Monitor liver laboratory tests every 2 weeks during the first 3 months of treatment, then once a month and as clinically indicated, with more frequent testing in patients who develop transaminase and bilirubin elevations. In case of severe ALT, AST, or bilirubin elevations, withhold, then reduce dose, or permanently discontinue ALECENSA. (2.4, 5.1)
- Interstitial Lung Disease (ILD)/Pneumonitis: Immediately withhold ALECENSA in patients diagnosed with ILD/pneumonitis and permanently discontinue if no other potential causes of ILD/pneumonitis have been identified. (2.4, 5.2)
- Renal Impairment: Withhold ALECENSA for severe renal impairment, then resume ALECENSA at reduced dose upon recovery or permanently discontinue (2.4, 5.3).
- Bradycardia: Monitor heart rate and blood pressure regularly. If symptomatic, withhold ALECENSA then reduce dose, or permanently discontinue. (2.4, 5.4)
- Severe Myalgia and Creatine Phosphokinase (CPK) Elevation: Assess CPK every 2 weeks during the first month of treatment and in patients reporting unexplained muscle pain, tenderness, or weakness. In case of severe CPK elevations, withhold, then resume or reduce dose. (2.4, 5.5)
- Hemolytic Anemia: If hemolytic anemia is suspected, withhold ALECENSA. If hemolytic anemia is confirmed, consider resuming at a reduced dose upon resolution or permanently discontinue. (5.6)
- Embryo-Fetal Toxicity: ALECENSA can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.7, 8.1, 8.3)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence ≥20%) were hepatotoxicity, constipation, fatigue, myalgia, edema, rash and cough. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2024
Full Prescribing Information
1. Indications and Usage for Alecensa
1.1 Adjuvant Treatment of Resected ALK-Positive Non-Small Cell Lung Cancer (NSCLC)
ALECENSA is indicated as adjuvant treatment in adult patients following tumor resection of anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC) (tumors ≥ 4 cm or node positive), as detected by an FDA-approved test [see Dosage & Administration (2.1)].
1.2 Treatment of Metastatic ALK-Positive NSCLC
ALECENSA is indicated for the treatment of adult patients with ALK-positive metastatic NSCLC as detected by an FDA-approved test [see Dosage & Administration (2.1)].
2. Alecensa Dosage and Administration
2.1 Patient Selection
Select patients with resectable tumors for the adjuvant treatment of NSCLC with ALECENSA based on the presence of ALK positivity in tumor tissue [see Indications and Usage (1.1) and Clinical Studies (14.1)].
Select patients for the treatment of metastatic NSCLC with ALECENSA based on the presence of ALK positivity in tumor tissue or plasma specimens [see Indications and Usage (1.2) and Clinical Studies (14.2)]. If ALK rearrangements are not detected in a plasma specimen, test tumor tissue if feasible.
Information on FDA-approved tests for the detection of ALK rearrangements in NSCLC is available at http://www.fda.gov/CompanionDiagnostics.
2.2 Dosing and Administration
The recommended dosage information for ALECENSA is provided in Table 1.
| Indication | Recommended Dosage of ALECENSA | Duration |
|---|---|---|
| Adjuvant treatment of resected NSCLC | 600 mg orally twice daily with food [see Clinical Pharmacology (12.3)] | For a total of 2 years or until disease recurrence or unacceptable toxicity |
| Metastatic NSCLC | Until disease progression or unacceptable toxicity | |
|
||
2.3 Recommended Dosage for Hepatic Impairment
The recommended dose of ALECENSA in patients with severe hepatic impairment (Child-Pugh C) is 450 mg orally twice daily [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.4 Dose Modifications for Adverse Reactions
The dose reduction schedule for ALECENSA is provided in Table 2.
| Dose Reduction Schedule | Dose Level |
|---|---|
| Starting dose | 600 mg taken orally twice daily |
| First dose reduction | 450 mg taken orally twice daily |
| Second dose reduction | 300 mg taken orally twice daily |
Discontinue if patients are unable to tolerate the 300 mg twice daily dose.
Recommendations for dose modifications of ALECENSA in case of adverse reactions are provided in Table 3.
| Criteria* | ALECENSA Dose Modification |
|---|---|
| ALT or AST elevation of greater than 5 times upper limit of normal (ULN) with total bilirubin less than or equal to 2 times ULN | Temporarily withhold until recovery to baseline or to less than or equal to 3 times ULN, then resume at reduced dose as per Table 2. |
| ALT or AST elevation greater than 3 times ULN with total bilirubin elevation greater than 2 times ULN in the absence of cholestasis or hemolysis | Permanently discontinue ALECENSA. |
| Total bilirubin elevation of greater than 3 times ULN | Temporarily withhold until recovery to baseline or to less than or equal to 1.5 times ULN, then resume at reduced dose as per Table 2. |
| Any grade treatment-related interstitial lung disease (ILD)/pneumonitis | Permanently discontinue ALECENSA. |
| Grade 3 renal impairment | Temporarily withhold until serum creatinine recovers to less than or equal to 1.5 times ULN, then resume at reduced dose. |
| Grade 4 renal impairment | Permanently discontinue ALECENSA. |
| Symptomatic bradycardia | Withhold ALECENSA until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. If contributing concomitant medication is identified and discontinued, or its dose is adjusted, resume ALECENSA at previous dose upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. If no contributing concomitant medication is identified, or if contributing concomitant medications are not discontinued or dose modified, resume ALECENSA at reduced dose (see Table 2) upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. |
| Bradycardia† (life-threatening consequences, urgent intervention indicated) | Permanently discontinue ALECENSA if no contributing concomitant medication is identified. If contributing concomitant medication is identified and discontinued, or its dose is adjusted, resume ALECENSA at reduced dose (see Table 2) upon recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, with frequent monitoring as clinically indicated. Permanently discontinue ALECENSA in case of recurrence. |
| CPK elevation greater than 5 times ULN | Temporarily withhold until recovery to baseline or to less than or equal to 2.5 times ULN, then resume at same dose. |
| CPK elevation greater than 10 times ULN or second occurrence of CPK elevation of greater than 5 times ULN | Temporarily withhold until recovery to baseline or to less than or equal to 2.5 times ULN, then resume at reduced dose as per Table 2. |
| Hemolytic Anemia | Withhold ALECENSA if hemolytic anemia is suspected. Upon resolution, resume at reduced dose or permanently discontinue. |
3. Dosage Forms and Strengths
150 mg hard capsules, white, with "ALE" printed in black ink on the cap and "150 mg" printed in black ink on the body.
5. Warnings and Precautions
5.1 Hepatotoxicity
Severe hepatotoxicity, including drug-induced liver injury, occurred in patients treated with ALECENSA.
In the pooled safety population [see Adverse Reactions (6.1)] of patients who received ALECENSA, hepatotoxicity occurred in 41% of patients and the incidence of Grade ≥ 3 hepatotoxicity was 8%. In the ALINA study, hepatotoxicity occurred in 61% of patients treated with ALECENSA and the incidence of Grade ≥ 3 hepatotoxicity was 4.7%. The majority (72% of 136 patients) of elevated transaminases occurred during the first 3 months of treatment. Treatment discontinuation due to hepatotoxicity occurred in 3.6% of patients who received ALECENSA in the pooled safety population and 1.6% of patients treated in the ALINA study.
In the pooled safety population, concurrent elevations in ALT or AST greater than or equal to 3 times the ULN and total bilirubin greater than or equal to 2 times the ULN, with normal alkaline phosphatase, occurred in less than 1% of patients treated with ALECENSA. Three patients with Grades 3–4 AST/ALT elevations had drug-induced liver injury (documented by liver biopsy in two cases).
Monitor liver function tests including ALT, AST, and total bilirubin every 2 weeks during the first 3 months of treatment, then once a month and as clinically indicated, with more frequent testing in patients who develop transaminase and bilirubin elevations. Based on the severity of the adverse drug reaction, withhold ALECENSA and resume at a reduced dose or permanently discontinue ALECENSA as described in Table 3 [see Dosage and Administration (2.4)].
5.2 Interstitial Lung Disease (ILD)/Pneumonitis
ILD/pneumonitis occurred in patients treated with ALECENSA.
In the pooled safety population [see Adverse Reactions (6.1)], ILD/pneumonitis occurred in 1.3% of patients treated with ALECENSA with 0.4% of patients experiencing Grade 3 ILD/pneumonitis.
Five patients (0.9%) in the pooled safety population discontinued ALECENSA due to ILD/pneumonitis. The median time-to-onset of Grade 3 or higher ILD/pneumonitis was 2.1 months (range: 0.6 months to 3.6 months).
Promptly investigate for ILD/pneumonitis in any patient who presents with worsening of respiratory symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, and fever). Immediately withhold ALECENSA treatment in patients diagnosed with ILD/pneumonitis and permanently discontinue ALECENSA if no other potential causes of ILD/pneumonitis have been identified [see Dosage and Administration (2.4) and Adverse Reactions (6)].
5.3 Renal Impairment
Renal impairment, including fatal cases, occurred in patients treated with ALECENSA.
In the pooled safety population [see Adverse Reactions (6.1)], renal impairment occurred in 12% of patients treated with ALECENSA, including Grade ≥ 3 in 1.7% of patients, of which 0.4% were fatal events. The median time to Grade ≥ 3 renal impairment was 3.7 months (range 0.5 to 31.8 months). Dosage modifications for renal impairment were required in 2.4% of patients.
Permanently discontinue ALECENSA for Grade 4 renal toxicity. Withhold ALECENSA for Grade 3 renal toxicity until recovery to less than or equal to 1.5 times ULN, then resume at reduced dose [see Dosage and Administration (2.4)].
5.4 Bradycardia
Symptomatic bradycardia occurred in patients treated with ALECENSA.
In the pooled safety population [see Adverse Reactions (6.1)], bradycardia occurred in 11% of patients treated with ALECENSA. Twenty percent of 521 patients treated with ALECENSA, for whom serial electrocardiograms (ECGs) were available, had post-dose heart rates of less than 50 beats per minute (bpm).
Monitor heart rate and blood pressure regularly. For asymptomatic bradycardia dose modification is not required. For symptomatic bradycardia that is not life-threatening, withhold ALECENSA until recovery to asymptomatic bradycardia or to a heart rate ≥ 60 bpm and evaluate concomitant medications known to cause bradycardia, as well as anti-hypertensive medications. If bradycardia is attributable to a concomitant medication, resume ALECENSA at a reduced dose (see Table 2) upon recovery to asymptomatic bradycardia or to a heart rate of ≥ 60 bpm, with frequent monitoring as clinically indicated.
Permanently discontinue ALECENSA in cases of life-threatening bradycardia if no contributing concomitant medication is identified [see Dosage and Administration (2.4)]. Permanently discontinue ALECENSA for recurrence of life-threatening bradycardia.
5.5 Severe Myalgia and Creatine Phosphokinase (CPK) Elevation
Severe myalgia and creatine phosphokinase (CPK) elevation occurred in patients treated with ALECENSA.
In the pooled safety population [see Adverse Reactions (6.1)], myalgia (including muscle- and musculoskeletal-related reactions) occurred in 31% of patients treated with ALECENSA, including Grade ≥ 3 in 0.8% of patients. Dosage modifications for myalgia events were required in 2.1% of patients.
In the pooled safety population, of the 491 patients with CPK laboratory data available, elevated CPK occurred in 56% of patients treated with ALECENSA, including 6% Grade ≥ 3. The median time to Grade ≥ 3 CPK elevation was 15 days (interquartile range - 15 –337 days). Dosage modifications for elevation of CPK occurred in 5% of patients.
In the ALINA study, elevated CPK occurred in 77% of 128 patients with CPK laboratory data, including 6% Grade ≥ 3 elevations.
Advise patients to report any unexplained muscle pain, tenderness, or weakness. Assess CPK levels every 2 weeks for the first month of treatment and as clinically indicated in patients reporting symptoms. Based on the severity of the CPK elevation, withhold ALECENSA, then resume or reduce dose [see Dosage and Administration (2.4)].
5.6 Hemolytic Anemia
Hemolytic anemia occurred in patients treated with ALECENSA.
Hemolytic anemia was initially reported with ALECENSA in the postmarketing setting, including cases associated with a negative direct antiglobulin test (DAT) result. Assessments for the determination of hemolytic anemia were subsequently collected in the ALINA study, where hemolytic anemia was observed in 3.1% of patients treated with ALECENSA. If hemolytic anemia is suspected, withhold ALECENSA and initiate appropriate laboratory testing. If hemolytic anemia is confirmed, consider resuming at a reduced dose upon resolution or permanently discontinue ALECENSA [see Dosage and Administration (2.4)].
5.7 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, ALECENSA can cause fetal harm when administered to pregnant women. Oral administration of alectinib to pregnant rats and rabbits during the period of organogenesis resulted in embryo-fetal toxicity and abortion at maternally toxic doses with exposures approximately 2.7-fold those observed in humans with alectinib 600 mg twice daily. Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
Advise females of reproductive potential to use effective contraception during treatment with ALECENSA and for 5 weeks following the last dose [see Use in Specific Populations (8.1 and 8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the label:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.2)]
- Renal Impairment [see Warnings and Precautions (5.3)]
- Bradycardia [see Warnings and Precautions (5.4)]
- Severe Myalgia and Creatine Phosphokinase (CPK) Elevation [see Warnings and Precautions (5.5)]
- Hemolytic Anemia [see Warnings and Precautions (5.6)]
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to ALECENSA as a single agent at 600 mg orally twice daily in 533 patients in Studies NP28761, NP28673, ALEX and ALINA [see Clinical Studies (14)]. Among 533 patients who received ALECENSA, 75% were exposed for 6 months or longer and 64% were exposed for greater than one year. In this pooled safety population, the most common (≥ 20%) adverse reactions were hepatotoxicity (41%), constipation (39%), fatigue (36%), myalgia (31%), edema (29%), rash (23%) and cough (21%). The most common (≥ 2%) Grade 3 or 4 laboratory abnormalities were increased CPK (6%), decreased hemoglobin (4.4%), increased ALT (4.2%), increased bilirubin (4.0%) and increased AST (3.4%).
Adjuvant Treatment of Resected ALK-Positive NSCLC
The safety of ALECENSA was evaluated in ALINA, a multi-center, open-label, randomized trial for the adjuvant treatment of patients with resected ALK-positive NSCLC [see Clinical Studies (14.1)]. At the time of DFS analysis, the median duration of exposure was 23.9 months for ALECENSA and 2.1 months for platinum-based chemotherapy.
Serious adverse reactions occurred in 13% of patients treated with ALECENSA; the most frequent serious adverse reactions (≥ 1%) were pneumonia (3.9%), appendicitis (3.1%), and acute myocardial infarction (1.6%). Permanent discontinuation of ALECENSA due to an adverse event occurred in 5% of patients; the most frequent adverse reactions (≥ 1%) that led to treatment discontinuation were pneumonitis and hepatotoxicity.
Dosage interruptions of ALECENSA due to an adverse reaction occurred in 27% of patients. Adverse reactions which required dosage interruption in ≥ 2% of patients included hepatotoxicity, increased blood CPK, COVID-19, myalgia, abdominal pain, and pneumonia.
Dose reductions of ALECENSA due to an adverse reaction occurred in 26% of patients. Adverse reactions which required dose reductions in ≥ 2% of patients included hepatotoxicity, increased blood CPK, rash, bradycardia and myalgia.
Table 4 and 5 summarize the common adverse reactions and laboratory abnormalities observed in ALINA.
| Adverse Reaction | ALECENSA N=128 | Chemotherapy N=120 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | |
| Based on NCI CTCAE v5.0 | ||||
|
||||
| Hepatobiliary System Disorders | ||||
| Hepatotoxicity* | 61 | 4.7† | 13 | 0 |
| Gastrointestinal Disorders | ||||
| Constipation | 42 | 0.8† | 25 | 0.8† |
| Abdominal pain‡ | 13 | 0 | 10 | 1.7† |
| Diarrhea§ | 13 | 0.8† | 9 | 1.7† |
| Musculoskeletal | ||||
| Myalgia¶ | 34 | 0.8† | 1.7 | 0 |
| Infections and Infestations | ||||
| COVID-19 | 29 | 0 | 0.8 | 0 |
| General Disorders and Administration Site Conditions | ||||
| Fatigue# | 25 | 0.8† | 28 | 4.2† |
| EdemaÞ | 16 | 0 | 1.7 | 0 |
| Skin and Subcutaneous Tissue Disorders | ||||
| Rashß | 23 | 1.6† | 10 | 0 |
| Respiratory System Disorders | ||||
| Coughà | 20 | 0.8† | 3.3 | 0 |
| Dyspneaè | 13 | 0.8† | 2.5 | 0 |
| Renal | ||||
| Renal Impairmentð | 16 | 0.8† | 9 | 0 |
| Nervous System Disorders | ||||
| Dysgeusiaø | 13 | 0 | 3.3 | 0 |
| Headache | 11 | 0 | 7 | 0 |
| Investigations | ||||
| Increased weight | 13 | 0.8† | 0.8 | 0 |
| Cardiac Disorders | ||||
| Bradycardiaý | 12 | 0 | 0 | 0 |
Clinically significant adverse reactions in < 10% of patients who received ALECENSA in ALINA: nausea (8%), vomiting (7%), vision disorders (4.7%; includes blurred vision, visual acuity reduced and photopsia), stomatitis (4.7%; includes stomatitis and mouth ulceration), photosensitivity reaction (3.9%) and pneumonitis (2.3%).
| Parameter | ALECENSA N=128 | Chemotherapy N=120 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | |
| Based on NCI CTCAE v5.0 | ||||
|
||||
| Chemistry | ||||
| Increased CPK | 77 | 8 | 8 | 1.7* |
| Increased AST | 75 | 0.8* | 25 | 0 |
| Increased bilirubin | 68 | 2.3* | 4.2 | 0 |
| Increased alkaline phosphatase | 64 | 0 | 14 | 0 |
| Increased ALT | 57 | 2.3* | 28 | 0 |
| Increased creatinine | 41 | 0 | 23 | 0 |
| Increased uric acid | 30 | 0 | 19 | 0 |
| Hematology | ||||
| Decreased hemoglobin | 69 | 0 | 67 | 0.8* |
Previously Untreated Metastatic ALK-Positive NSCLC
The safety of ALECENSA was evaluated in 152 patients with ALK-positive NSCLC in the ALEX study. The median duration of exposure to ALECENSA was 17.9 months. Patient characteristics of the ALEX study population (n=303) were: median age 56 years, age less than 65 (77%), female (56%), Caucasian (50%), Asian (46%), adenocarcinoma histology (92%), never smoker (63%), and ECOG PS 0 or 1 (93%).
Serious adverse reactions occurred in 28% of patients treated with ALECENSA; serious adverse reactions reported in 2% or more of patients treated with ALECENSA were pneumonia (4.6%), and renal impairment (3.9%). Grade ≥ 3 adverse events were reported for 41% of patients in the ALECENSA arm. Fatal adverse reactions occurred in 3.3% of patients treated with ALECENSA; these were renal impairment (2 patients), sudden death, cardiac arrest, and pneumonia (1 patient each). Permanent discontinuation of ALECENSA for adverse reactions occurred in 11% of patients. Adverse drug reactions that led to discontinuation of ALECENSA in 1% or more of patients were renal impairment (2.0%), hyperbilirubinemia (1.3%), increased ALT (1.3%), and increased AST (1.3%). Dosage interruptions of ALECENSA due to an adverse reaction occurred in 20% of patients. Adverse reactions which required dosage interruption in > 2% of patients included increased ALT, pneumonia. Dose reductions of ALECENSA due to an adverse reaction occurred in 17% of patients. Adverse reactions which required dose reductions in > 2% of patients included hyperbilirubinemia, increased AST and increased ALT.
Tables 6 and 7 summarize the common adverse reactions and laboratory abnormalities observed in ALEX.
| Adverse Reaction | ALECENSA N=152 | Crizotinib N=151 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3-4 (%) | All Grades (%) | Grades 3-4 (%) | |
| NCI CTCAE = National Cancer Institute Common Terminology Criteria for Adverse Events; MedDRA = Medical Dictionary for Regulatory Activities; SOC = System Organ Class. | ||||
|
||||
| Gastrointestinal | ||||
| Constipation | 34 | 0 | 33 | 0 |
| Nausea | 14 | 0.7 | 48 | 3.3 |
| Diarrhea | 12 | 0 | 45 | 2.0 |
| General | ||||
| Fatigue* | 26 | 1.3 | 23 | 0.7 |
| Edema† | 22 | 0.7 | 34 | 0.7 |
| Musculoskeletal | ||||
| Myalgia‡ | 23 | 0 | 4.0 | 0 |
| Skin | ||||
| Rash§ | 15 | 0.7 | 13 | 0 |
| Cardiac | ||||
| Bradycardia¶ | 11 | 0 | 15 | 0 |
| Renal | ||||
| Renal impairment# | 12 | 3.9Þ | 0 | 0 |
The following additional clinically significant adverse drug reactions were observed in patients treated with ALECENSA: weight gain (9.9%), vomiting (7%), photosensitivity reaction (5.3%), vision disorders (4.6%; includes blurred vision, visual impairment, vitreous floaters reduced visual acuity and diplopia), stomatitis (3.3%), dysgeusia (3.3%; includes hypogeusia), interstitial lung disease (1.3%), and drug-induced liver injury (1.3%).
| Parameter | ALECENSA N=152 | Crizotinib N=151 |
||
|---|---|---|---|---|
| All Grades (%) | Grades 3–4 (%) | All Grades (%) | Grades 3–4 (%) | |
| Note: Based on National Cancer Institute Common Terminology Criteria for Adverse Events v4.03. Excludes patients with no post-baseline lab assessments. |
||||
|
||||
| Chemistry | ||||
| Hyperbilirubinemia* | 54 | 5 | 4.7 | 0 |
| Increased AST† | 50 | 6 | 56 | 11 |
| Increased alkaline phosphatase‡ | 50 | 0 | 44 | 0 |
| Increased ALT‡ | 40 | 6 | 62 | 16 |
| Increased creatinine‡,§ | 38 | 4.1 | 23 | 0.7 |
| Increased CPK¶ | 37 | 2.8 | 52 | 1.4 |
| Hypocalcemia* | 29 | 0 | 61 | 1.4 |
| Hyperglycemia# | 22 | 2.2 | 19 | 2.3 |
| HyponatremiaÞ | 18 | 6 | 20 | 4.1 |
| Hypokalemia‡ | 17 | 2 | 12 | 0.7 |
| Hypoalbuminemiaß | 14 | 0 | 57 | 3.4 |
| Hyperkalemia‡ | 12 | 1.4 | 16 | 1.4 |
| Hypophosphatemiaà | 9 | 1.4 | 25 | 2.7 |
| Increased gamma glutamyl transferaseè | 7 | 0.7 | 39 | 4.1 |
| Hematology | ||||
| Anemia‡ | 62 | 7 | 36 | 0.7 |
| Lymphopenia* | 14 | 1.4 | 34 | 4.1 |
| Neutropenia‡ | 14 | 0 | 36 | 7 |
Metastatic ALK-Positive NSCLC Previously Treated with Crizotinib
The safety of ALECENSA was evaluated in 253 patients with ALK-positive non-small cell lung cancer (NSCLC) treated with ALECENSA in two clinical trials, Studies NP28761 and NP28673. The median duration of exposure to ALECENSA was 9.3 months. One hundred sixty-nine patients (67%) were exposed to ALECENSA for more than 6 months, and 100 patients (40%) for more than one year. The population characteristics were: median age 53 years, age less than 65 (86%), female (55%), White (74%), Asian (18%), NSCLC adenocarcinoma histology (96%), never or former smoker (98%), ECOG Performance Status (PS) 0 or 1 (91%), and prior chemotherapy treatment (78%).
Serious adverse reactions occurred in 19% of patients; the most frequently reported serious adverse reactions were pulmonary embolism (1.2%), dyspnea (1.2%), and hyperbilirubinemia (1.2%). Fatal adverse reactions occurred in 2.8% of patients and included hemorrhage (0.8%), intestinal perforation (0.4%), dyspnea (0.4%), pulmonary embolism (0.4%), and endocarditis (0.4%). Permanent discontinuation of ALECENSA for adverse reactions occurred in 6% of patients. The most frequent adverse reactions that led to permanent discontinuation were hyperbilirubinemia (1.6%), increased ALT levels (1.6%), and increased AST levels (1.2%). Overall, 23% of patients initiating treatment at the recommended dose required at least one dose reduction. The median time to first dose reduction was 48 days. The most frequent adverse reactions that led to dose reductions or interruptions were elevations in bilirubin (6%), CPK (4.3%), ALT (4.0%), AST (2.8%), and vomiting (2.8%).
Tables 8 and 9 summarize the common adverse reactions and laboratory abnormalities observed in Studies NP28761 and NP28673.
| Adverse Reactions | ALECENSA N=253 |
|
|---|---|---|
| All Grades (%) | Grades 3–4 (%)* | |
|
||
| Fatigue† | 41 | 1.2 |
| Constipation | 34 | 0 |
| Edema‡ | 30 | 0.8 |
| Myalgia§ | 29 | 1.2 |
| Cough | 19 | 0 |
| Rash¶ | 18 | 0.4 |
| Nausea | 18 | 0 |
| Headache | 17 | 0.8 |
| Diarrhea | 16 | 1.2 |
| Dyspnea | 16 | 3.6# |
| Back pain | 12 | 0 |
| Vomiting | 12 | 0.4 |
| Increased weight | 11 | 0.4 |
| Vision disorderÞ | 10 | 0 |
An additional clinically significant adverse drug reaction was photosensitivity, which occurred in 9.9% of patients exposed to ALECENSA in Studies NP28761 and NP28673. Patients were advised to avoid sun exposure and to use broad-spectrum sunscreen. The incidence of Grade 2 photosensitivity was 0.4%; the remaining events were Grade 1 in severity.
| Parameter | ALECENSA N=250 | |
|---|---|---|
| All Grades (%) | Grades 3–4 (%)* | |
|
||
| Chemistry | ||
| Increased AST | 51 | 3.6 |
| Increased Alkaline Phosphatase | 47 | 1.2 |
| Increased CPK† | 43 | 4.6 |
| Hyperbilirubinemia | 39 | 2.4 |
| Hyperglycemia‡ | 36 | 2.0 |
| Increased ALT | 34 | 4.8 |
| Hypocalcemia | 32 | 0.4 |
| Hypokalemia | 29 | 4.0 |
| Increased Creatinine§ | 28 | 0 |
| Hypophosphatemia | 21 | 2.8 |
| Hyponatremia | 20 | 2.0 |
| Hematology | ||
| Anemia | 56 | 2.0 |
| Lymphopenia¶ | 22 | 4.6 |
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, ALECENSA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on ALECENSA use in pregnant women.
Administration of alectinib to pregnant rats and rabbits by oral gavage during the period of organogenesis resulted in embryo-fetal toxicity and abortion at maternally toxic doses with exposures approximately 2.7-fold those observed in humans treated with alectinib at 600 mg twice daily (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In a preliminary rabbit embryo-fetal study, administration of alectinib by oral gavage during the period of organogenesis resulted in abortion or complete embryo-fetal mortality at a maternally toxic dose of 27 mg/kg/day (approximately 2.9-fold the estimated area under the curve (AUC0-24h,ss) in humans treated with alectinib 600 mg twice daily) in three of six pregnant rabbits. The remaining three pregnant rabbits in this group had few live fetuses, decreased fetal and placental weights, and retroesophageal subclavian artery. In a rat preliminary embryo-fetal development study, administration of alectinib during organogenesis resulted in complete litter loss in all pregnant rats at 27 mg/kg/day (approximately 4.5-fold the estimated AUC0-24h,ss in humans treated with alectinib 600 mg twice daily). Doses greater than or equal to 9 mg/kg/day (approximately 2.7-fold the estimated human AUC0-24h,ss in humans treated with alectinib 600 mg twice daily), resulted in maternal toxicity as well as developmental toxicities including decreased fetal weight, dilated ureter, thymic cord, small ventricle and thin ventricle wall, and reduced number of sacral and caudal vertebrae.
8.2 Lactation
Risk Summary
There are no data on the presence of alectinib or its metabolites in human milk, the effects of alectinib on the breastfed child, or its effects on milk production. Because of the potential for serious adverse reactions in breastfed children from alectinib, advise a lactating woman not to breastfeed during treatment with ALECENSA and for 1 week after the last dose.
8.3 Females and Males of Reproductive Potential
ALECENSA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating ALECENSA [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with ALECENSA and for 5 weeks after the last dose [see Use in Specific Populations (8.1)].
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with ALECENSA and for 3 months following the last dose [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ALECENSA in pediatric patients have not been established.
Animal Data
Juvenile animal studies have not been conducted using alectinib. In general toxicology studies, treatment of rats with doses of alectinib resulting in exposures greater than or equal to approximately 4.5-fold those in humans treated with alectinib at 600 mg twice daily resulted in changes in the growing teeth and bones. Findings in teeth included discoloration and changes in tooth size along with histopathological disarrangement of the ameloblast and odontoblast layers. There were also decreases in the trabecular bone and increased osteoclast activity in the femur and sternum.
8.5 Geriatric Use
Nineteen percent of the 533 patients studied in NP28761, NP28673, ALEX and ALINA were 65 years of age and older (3.2% were 75 years of age and older). No overall differences in effectiveness were observed based on age. Exploratory analysis suggests a higher incidence of serious adverse events (38% vs 25%), more frequent adverse events leading to treatment discontinuations (18% vs 6%) and dose modifications (48% vs 35%) in patients 65 years or older as compared to those younger than 65 years.
8.6 Renal Impairment
No dose adjustment is recommended for patients with mild or moderate renal impairment. The safety of ALECENSA in patients with severe renal impairment (creatinine clearance less than 30 mL/min) or end-stage renal disease has not been studied [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is recommended for patients with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment. Increased exposure of alectinib occurred in patients with severe hepatic impairment (Child-Pugh C). The recommended dose of ALECENSA in patients with severe hepatic impairment (Child-Pugh C) is 450 mg orally twice daily [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
10. Overdosage
No experience with overdose is available. There is no specific antidote for overdose with ALECENSA. Alectinib and its major active metabolite M4 are > 99% bound to plasma proteins; therefore, hemodialysis is likely to be ineffective in the treatment of overdose.
11. Alecensa Description
ALECENSA (alectinib) is a kinase inhibitor for oral administration. The molecular formula for alectinib is C30H34N4O2 ∙ HCl. The molecular weight is 482.62 g/mol (free base form) and 519.08 g/mol (hydrochloride salt). Alectinib is described chemically as 9-ethyl-6, 6-dimethyl-8-[4-(morpholin-4-yl)piperidin-1-yl]-11-oxo-6, 11-dihydro-5H-benzo[b]carbazole-3-carbonitrile hydrochloride. The chemical structure of alectinib is shown below:
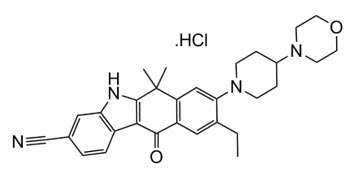
Alectinib HCl is a white to yellow white powder or powder with lumps with a pKa of 7.05 (base).
ALECENSA is supplied as hard capsules containing 150 mg of alectinib (equivalent to 161.33 mg alectinib HCl) and the following inactive ingredients: lactose monohydrate, hydroxypropylcellulose, sodium lauryl sulfate, magnesium stearate, and carboxymethylcellulose calcium. The capsule shell contains hypromellose, carrageenan, potassium chloride, titanium dioxide, corn starch, and carnauba wax. The printing ink contains red iron oxide (E172), yellow iron oxide (E172), FD&C Blue No. 2 aluminum lake (E132), carnauba wax, white shellac, and glyceryl monooleate.
12. Alecensa - Clinical Pharmacology
12.1 Mechanism of Action
Alectinib is a tyrosine kinase inhibitor that targets ALK and RET. In nonclinical studies, alectinib inhibited ALK phosphorylation and ALK-mediated activation of the downstream signaling proteins STAT3 and AKT, and decreased tumor cell viability in multiple cell lines harboring ALK fusions, amplifications, or activating mutations. The major active metabolite of alectinib, M4, showed similar in vitro potency and activity.
Alectinib and M4 demonstrated in vitro and in vivo activity against multiple mutant forms of the ALK enzyme, including some mutations identified in NSCLC tumors in patients who have progressed on crizotinib.
In mouse models implanted with tumors carrying ALK fusions, administration of alectinib resulted in antitumor activity and prolonged survival, including in mouse models implanted intracranially with ALK-driven tumor cell lines.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The ability of alectinib to prolong the QT interval was assessed in 221 patients administered ALECENSA 600 mg twice daily in clinical studies. ALECENSA did not prolong the QTc (QT corrected for heart rate) interval to any clinically relevant extent. One patient had a maximum post-baseline QTcF value of greater than 500 msec, and one patient had a maximum QTcF change from baseline of greater than 60 msec.
12.3 Pharmacokinetics
The pharmacokinetics of alectinib and its major active metabolite M4 have been characterized in patients with ALK-positive NSCLC and healthy subjects.
In patients with ALK-positive NSCLC, the geometric mean (coefficient of variation %) steady-state maximal concentration (Cmax,ss) for alectinib was 665 ng/mL (44%) and for M4 was 246 ng/mL (45%) with peak to trough concentration ratio of 1.2. The geometric mean steady-state area under the curve from 0 to 12 hours (AUC0-12h,ss) for alectinib was 7,430 ng*h/mL (46%) and for M4 was 2,810 ng*h/mL (46%). Alectinib exposure is dose proportional across the dose range of 460 mg to 900 mg (i.e., 0.75 to 1.5 times the approved recommended dosage) under fed conditions. Alectinib and M4 reached steady-state concentrations by day 7. The geometric mean accumulation was approximately 6-fold for both alectinib and M4.
Absorption
Alectinib reached maximal concentrations at 4 hours following administration of ALECENSA 600 mg twice daily under fed conditions in patients with ALK-positive NSCLC.
The absolute bioavailability of alectinib was 37% (90% CI: 34%, 40%) under fed conditions.
A high-fat, high-calorie meal increased the combined exposure (AUC0-inf) of alectinib plus M4 by 3.1-fold (90% CI: 2.7, 3.6) following oral administration of a single 600 mg dose of ALECENSA.
Distribution
The apparent volume of distribution is 4,016 L for alectinib and 10,093 L for M4.
Alectinib and M4 are bound to human plasma proteins greater than 99%, independent of drug concentration.
Alectinib concentrations in the cerebrospinal fluid in patients with ALK-positive NSCLC approximate estimated alectinib free concentrations in the plasma.
In vitro studies suggest that alectinib is not a substrate of P-glycoprotein (P-gp), but M4 is a substrate of P-gp. Alectinib and M4 are not substrates of breast cancer resistance protein (BCRP), organic anion-transporting polypeptide (OATP) 1B1, or OATP1B3.
Elimination
The apparent clearance (CL/F) is 81.9 L/hour for alectinib and 217 L/hour for M4. The geometric mean elimination half-life is 33 hours for alectinib and 31 hours for M4 in patients with ALK-positive NSCLC.
Metabolism
Alectinib is metabolized by CYP3A4 to its major active metabolite M4. The geometric mean metabolite/parent exposure ratio at steady-state is 0.40. M4 is subsequently metabolized by CYP3A4. Alectinib and M4 were the main circulating moieties in plasma, constituting 76% of the total radioactivity.
Excretion
Ninety-eight percent of the radioactivity was excreted in feces following oral administration of a single radiolabeled dose of alectinib under fed conditions. Eighty-four percent of the dose was excreted in the feces as unchanged alectinib, and 6% of the dose was excreted as M4. Excretion of radioactivity in urine was less than 0.5% of administered radiolabeled dose of alectinib.
Specific Populations
Age (21 to 83 years), body weight (38 to 128 kg), mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin 1 to ≤ 1.5 × ULN and AST any value), mild to moderate renal impairment (creatinine clearance 30 to 89 mL/min), race (White, Asian, and Other), and sex had no clinically meaningful effect on the systemic exposure of alectinib and M4. The pharmacokinetics of alectinib have not been studied in patients with severe renal impairment (creatinine clearance < 30 mL/min), or end-stage renal disease.
Hepatic Impairment: Following administration of a single oral dose of 300 mg ALECENSA, the geometric mean ratio [90% confidence interval] for the combined AUCinf of alectinib and M4 in subjects with moderate hepatic impairment (Child-Pugh B) was 1.36 [0.947, 1.96] and in subjects with severe hepatic impairment (Child-Pugh C) was 1.76 [0.984, 3.15] as compared to that in subjects with normal hepatic function. The combined Cmax of alectinib and M4 was comparable among the three groups. No dose adjustment is recommended for patients with mild or moderate hepatic impairment. The recommended dose of ALECENSA in patients with severe hepatic impairment is 450 mg orally twice daily [see Dosage and Administration (2.3) and Use in Specific Populations (8.7)].
Drug Interactions
Effect of Other Drugs on Alectinib
No clinically meaningful effect on the combined exposure of alectinib plus M4 was observed in clinical studies following co-administration of ALECENSA with a strong CYP3A inhibitor (posaconazole), a strong CYP3A inducer (rifampin), or an acid-reducing agent (esomeprazole).
Effect of Alectinib on Other Drugs
No clinically meaningful effect on the exposure of midazolam (sensitive CYP3A substrate) or repaglinide (sensitive CYP2C8 substrate) is expected following co-administration with ALECENSA.
In vitro studies suggest that alectinib and M4 do not inhibit CYP1A2, 2B6, 2C9, 2C19 or 2D6.
In vitro studies suggest that alectinib and M4 inhibit P-gp and BCRP. Alectinib did not inhibit OATP1B1, OATP1B3, OAT1, OAT3, or OCT2 transport activity in vitro.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with alectinib have not been conducted.
Alectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay, but was positive with an increased number of micronuclei in a rat bone marrow micronucleus test. The mechanism of micronucleus induction was abnormal chromosome segregation (aneugenicity) and not a clastogenic effect on chromosomes.
No studies in animals have been performed to evaluate the effect of alectinib on fertility. No adverse effects on male and female reproductive organs were observed in general toxicology studies conducted in rats and monkeys.
14. Clinical Studies
14.1 Adjuvant Treatment of Resected ALK-Positive NSCLC
The efficacy of ALECENSA for the adjuvant treatment of patients with ALK-positive NSCLC following complete tumor resection was evaluated in a global, randomized open-label clinical trial (ALINA: NCT03456076). Eligible patients were required to have resectable ALK-positive NSCLC, Stage IB (tumors ≥ 4 cm) – IIIA per the Union for International Cancer Control/American Joint Committee on Cancer (UICC/AJCC) Staging System, 7th Edition. ALK rearrangements were identified by a locally performed FDA-approved ALK test or by a centrally performed VENTANA ALK (D5F3) CDx assay.
Randomization was stratified by race (Asian vs. other races) and stage of disease (IB vs. II vs. IIIA). Patients were randomized (1:1) to receive ALECENSA 600 mg orally twice daily or platinum-based chemotherapy following tumor resection. Treatment with ALECENSA continued for a total of 2 years, or until disease recurrence or unacceptable toxicity. Platinum-based chemotherapy was administered intravenously for 4 cycles, with each cycle lasting 21 days, according to one of the following regimens:
- Cisplatin 75 mg/m2 on Day 1 plus vinorelbine 25 mg/m2 on Days 1 and 8
- Cisplatin 75 mg/m2 on Day 1 plus gemcitabine 1250 mg/m2 on Days 1 and 8
- Cisplatin 75 mg/m2 on Day 1 plus pemetrexed 500 mg/m2 on Day 1
In the event of intolerance to a cisplatin-based regimen, carboplatin was administered instead of cisplatin in the above combinations at a dose of AUC 5 mg/mL/min or 6 mg/mL/min.
The major efficacy outcome measures were disease-free survival (DFS) in patients with stage II-IIIA NSCLC and DFS in patients with stage IB-IIIA NSCLC (intent-to-treat [ITT] population) as assessed by investigator. DFS was defined as the time from date of randomization to the date of occurrence of any of the following: first documented recurrence of disease, new primary NSCLC, or death due to any cause, whichever occurred first. An additional efficacy outcome measure was overall survival (OS) in the ITT population.
A total of 257 patients were randomized to ALECENSA (N=130) or to chemotherapy (N=127). The median age was 56 years (range: 26 to 87), 24% were ≥ 65 years old; 52% were female; 56% were Asian, 42% were White, 0.4% were Black or African American, 2.3% were race unknown; 0.4% were Hispanic or Latino; 60% were never smokers; 53% had an ECOG PS of 0; 10% of patients had Stage IB, 35% had Stage II and 55% had Stage IIIA disease.
ALINA demonstrated a statistically significant improvement in DFS for patients treated with ALECENSA compared to patients treated with chemotherapy. OS data were not mature at the time of DFS analysis with 2.3% of deaths reported in the ITT population.
The efficacy results from ALINA are summarized in Table 10 and Figure 1.
| Efficacy Parameter | Stage II-IIIA Population | ITT Population | ||
|---|---|---|---|---|
| ALECENSA N=116 | Chemotherapy N=115 | ALECENSA N=130 | Chemotherapy N=127 |
|
| DFS = Disease-Free Survival; ITT = Intent-to-Treat; CI = Confidence Interval; NR = Not Reached; NE = Not Estimable. | ||||
|
||||
| DFS events (%) | 14 (12) | 45 (39) | 15 (12) | 50 (39) |
| Disease recurrence (%) | 14 (12) | 44 (38) | 15 (12) | 49 (38) |
| Death | 0 | 1 (0.9) | 0 | 1 (0.8) |
| Median DFS, months (95% CI)* | NR (NE, NE) | 44.4 (27.8, NE) | NR (NE, NE) | 41.3 (28.5, NE) |
| Hazard Ratio (95% CI)† | 0.24 (0.13, 0.45) | 0.24 (0.13, 0.43) | ||
| p-value‡ | <0.0001 | <0.0001 | ||
Figure 1: Kaplan-Meier Curves of Investigator-Assessed DFS (ITT Population) in ALINA
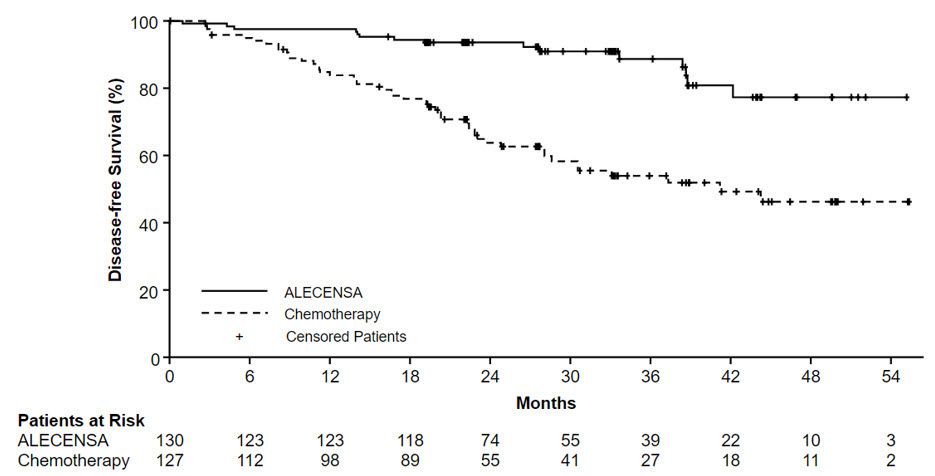
In an exploratory analysis of site(s) of relapse, the proportion of patients with brain involvement at the time of disease recurrence was 4 patients (3.1%) in the ALECENSA arm and 14 patients (11%) in the chemotherapy arm.
14.2 Treatment of Metastatic ALK-Positive NSCLC
Previously Untreated Metastatic ALK-Positive NSCLC
The efficacy of ALECENSA for the treatment of patients with ALK-positive NSCLC who had not received prior systemic therapy for metastatic disease was established in an open-label, randomized, active-controlled, multicenter study (ALEX: NCT02075840). Patients were required to have an ECOG performance status of 0-2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx assay. Neurologically stable patients with treated or untreated central nervous system (CNS) metastases, including leptomeningeal metastases, were eligible; patients with neurologic signs and symptoms due to CNS metastases were required to have completed whole brain radiation or gamma knife irradiation at least 14 days prior to enrollment and be clinically stable. Patients with a baseline QTc > 470 ms were ineligible.
Patients were randomized 1:1 to receive ALECENSA 600 mg orally twice daily or crizotinib 250 mg orally twice daily. Randomization was stratified by ECOG performance status (0/1 vs. 2), race (Asian vs. other races), and the presence or absence of CNS metastases at baseline. Treatment on both arms was continued until disease progression or unacceptable toxicity. The major efficacy outcome measure was progression-free survival (PFS) as determined by investigator assessment according to RECIST v1.1. Additional efficacy outcome measures were PFS as determined by independent review committee (IRC), time to CNS progression by IRC based on RECIST v1.1, objective response rate (ORR) and duration of response (DOR), and OS. Additional exploratory outcome measures were CNS objective response rate (CNS-ORR) and CNS duration of response (CNS-DOR) by IRC in patients with CNS metastases at baseline.
A total of 303 patients were randomized to ALECENSA (n=152) or crizotinib (n=151). The demographic characteristics of the study population were 56% female, median age 56 years (range: 18 to 91 years), 50% White, 46% Asian, 1% Black, and 3% other races. The majority of patients had adenocarcinoma (92%) and never smoked (63%). CNS metastases were present in 40% (n=122) of patients: of these, 43 patients had measurable CNS lesions as determined by an IRC. The ALEX study demonstrated a significant improvement in PFS. The time to cause-specific CNS progression as assessed by IRC was also significantly improved; there was a lower incidence of progression in the CNS as the first site of disease progression, alone or with concurrent systemic progression, in the ALECENSA arm (12%) as compared to the crizotinib arm (45%). Efficacy results from ALEX are summarized in Table 11 and Figure 2.
| ALECENSA N=152 | Crizotinib N=151 |
|
|---|---|---|
| CNS: central nervous system, ORR: overall response rate, IRC: independent review committee, CI: confidence interval, NE: not estimable. | ||
| Progression-Free Survival | ||
| Number of events (%) | 63 (41%) | 92 (61%) |
| Progressive disease (%) | 51 (34%) | 82 (54%) |
| Death (%) | 12 (8%) | 10 (7%) |
| Median in months (95% CI) | 25.7 (19.9, NE) | 10.4 (7.7, 14.6) |
| Hazard ratio (95% CI) * | 0.53 (0.38, 0.73) | |
| P-value * | < 0.0001 | |
| Overall Response Rate | ||
| Overall response rate, % (95% CI) † | 79% (72, 85) | 72% (64, 79) |
| P-value * | 0.1652 | |
| Complete response, % | 13% | 6% |
| Partial response, % | 66% | 66% |
| Duration of Response | ||
| Number of responders | n=120 | n=109 |
| Response duration ≥6 months | 82% | 57% |
| Response duration ≥12 months | 64% | 36% |
| Response duration ≥18 months | 37% | 14% |
Figure 2: Kaplan-Meier Plot of Progression-Free Survival (IRC) in ALEX
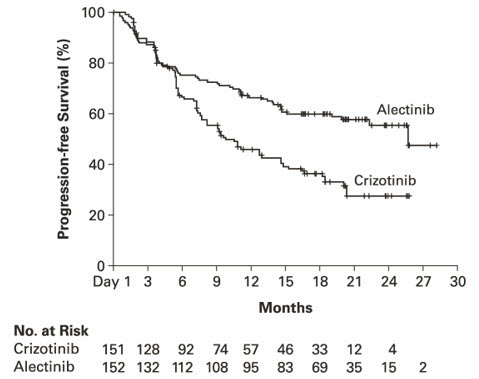
Results for PFS as determined by investigator assessment (HR=0.48 [95% CI: 0.35-0.66], stratified log-rank p<0.0001) were similar to that observed by IRC. At the data cutoff point overall survival data was not mature.
The results of prespecified exploratory analyses of CNS response rate in patients with measurable CNS lesions at baseline are summarized in Table 12.
| ALECENSA | Crizotinib | |
|---|---|---|
| IRC: Independent Review Committee; CI: Confidence Interval; NE: Not Estimable | ||
|
||
| CNS Tumor Response Assessment | N = 21 | N = 22 |
| CNS Objective Response Rate, % (95% CI*) | 81% (58, 95) | 50% (28,72) |
| Complete Response | 38% | 5% |
| Duration of CNS Response | ||
| Number of responders | 17 | 11 |
| CNS response duration ≥ 12 months | 59% | 36% |
Metastatic ALK-Positive NSCLC Previously Treated with Crizotinib
The safety and efficacy of ALECENSA were established in two single-arm, multicenter clinical trials: NP28761 (NCT01588028) and NP28673 (NCT01801111). Patients with locally advanced or metastatic ALK-positive NSCLC, who have progressed on crizotinib, with documented ALK-positive NSCLC based on an FDA-approved test, and ECOG PS of 0-2 were enrolled in both studies. Eligibility criteria permitted enrollment of patients with prior chemotherapy and prior CNS radiotherapy provided that CNS metastases were stable for at least two weeks. All patients received ALECENSA 600 mg orally twice daily. The major efficacy outcome measure in both studies was objective response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) as evaluated per Independent Review Committee (IRC). Additional outcome measures as evaluated by the IRC included duration of response (DOR), CNS ORR, and CNS DOR.
NP28761 was conducted in North America and enrolled 87 patients. Baseline demographic and disease characteristics in NP28761 were median age 54 years old (range 29 to 79, 18% 65 and over), 84% White and 8% Asian, 55% female, 35% ECOG PS 0 and 55% ECOG PS 1, 100% never or former smokers, 99% Stage IV, 94% adenocarcinoma, and 74% prior chemotherapy. The most common sites of extra-thoracic metastasis included 60% CNS (of whom 65% had received CNS radiation), 43% lymph nodes, 36% bone, and 34% liver.
NP28673 was conducted internationally and enrolled 138 patients. Baseline demographic and disease characteristics in NP28673 were median age 52 years old (range 22 to 79, 10% 65 and over), 67% White and 26% Asian, 56% female, 32% ECOG PS 0 and 59% ECOG PS 1, 98% never or former smokers, 99% Stage IV, 96% adenocarcinoma, and 80% prior chemotherapy. The most common sites of extra-thoracic metastasis included 61% CNS (of whom 73% had received CNS radiation), 51% bone, 38% lymph nodes, and 30% liver.
Efficacy results from NP28761 and NP28673 in all treated patients are summarized in Table 13. The median duration of follow-up on Study NP28761 was 4.8 months for both IRC and Investigator assessments and on Study NP28673, 10.9 months for IRC assessment and 7.0 months for Investigator assessment. All responses were partial responses.
| Efficacy Parameter | NP28761 (N=87) | NP28673 (N=138) | ||
|---|---|---|---|---|
| IRC* Assessment | Investigator Assessment | IRC* Assessment | Investigator Assessment | |
|
||||
| Objective Response Rate (95% CI) | 38% (28; 49) | 46% (35; 57) | 44% (36; 53) | 48% (39; 57) |
| Number of Responders | 33 | 40 | 61 | 66 |
| Duration of Response, median in months (95% CI) | 7.5 (4.9, Not Estimable) | NE (4.9, Not Estimable) | 11.2 (9.6, Not Estimable) | 7.8 (7.4, 9.2) |
An assessment of ORR and duration of response for CNS metastases in the subgroup of 51 patients in NP28761 and NP28673 with baseline measurable lesions in the CNS according to RECIST v1.1 are summarized in Table 14. Thirty-five (69%) patients with measurable CNS lesions had received prior brain radiation, including 25 (49%) who completed radiation treatment at least 6 months before starting treatment with ALECENSA. Responses were observed irrespective of prior brain radiation status.
| Efficacy Parameter | N=51 |
|---|---|
| CNS Objective Response Rate (95% CI) | 61% (46, 74) |
| Complete Response | 18% |
| Partial Response | 43% |
| CNS Duration of Response, median in months (95% CI) | 9.1 (5.8, Not Estimable) |
16. How is Alecensa supplied
Hard capsules, white 150 mg capsules with "ALE" printed in black ink on the cap and "150 mg" printed in black ink on the body, available in:
| 240 capsules per bottle: | NDC 50242-130-01 |
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients of the following:
Hepatotoxicity
Inform patients of the signs and symptoms of bilirubin and hepatic transaminase elevations. Advise patients to contact their healthcare provider immediately for signs or symptoms of bilirubin and hepatic transaminase elevations [see Warnings and Precautions (5.1)].
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the risks of severe ILD/pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.2)].
Renal Impairment
Inform patients of the risk of severe and potentially fatal renal impairment. Advise patients to contact their healthcare provider for change in urine color, reduced urine output, or swelling in the legs and feet [see Warnings and Precautions (5.3)].
Bradycardia
Inform patients that symptoms of bradycardia including dizziness, lightheadedness, and syncope can occur while taking ALECENSA. Advise patients to contact their healthcare provider to report these symptoms and to inform their healthcare provider about the use of any heart or blood pressure medications [see Warnings and Precautions (5.4)].
Severe Myalgia/CPK Elevation
Inform patients of signs and symptoms of myalgia, including unexplained and/or persistent muscle pain, tenderness, or weakness. Advise patients to contact their healthcare provider immediately to report new or worsening symptoms of muscle pain or weakness [see Warnings and Precautions (5.5)].
Hemolytic Anemia
Advise patients to contact their healthcare provider if they develop any signs or symptoms of hemolytic anemia [see Warnings and Precautions (5.6)].
Photosensitivity
Inform patients of the signs and symptoms of photosensitivity. Advise patients to avoid prolonged sun exposure while taking ALECENSA and for at least 7 days after study drug discontinuation and to use proper protection from the sun. Advise patients to use a broad spectrum ultraviolet A (UVA)/ultraviolet B (UVB) sunscreen and lip balm (SPF ≥ 50) to help protect against potential sunburn [see Adverse Reactions (6.1)].
Embryo-Fetal Toxicity
ALECENSA can cause fetal harm if taken during pregnancy. Advise a pregnant woman and females of reproductive potential of the potential risk to a fetus [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to use effective contraception during treatment with ALECENSA and for 5 weeks after the last dose of ALECENSA. Advise patients to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1, 8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with ALECENSA and for 3 months after the last dose [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with ALECENSA and for 1 week after the last dose [see Use in Specific Populations (8.2)].
Administration
Instruct patients to take ALECENSA twice a day. Advise patients to take ALECENSA with food and to swallow ALECENSA capsules whole [see Dosage and Administration (2.2)].
Missed Dose
Advise patients that if a dose of ALECENSA is missed or if the patient vomits after taking a dose of ALECENSA, patients should be advised not to take an extra dose, but to take the next dose at the regular time [see Dosage and Administration (2.2)].
Distributed by:
Genentech USA, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
ALECENSA® is a registered trademark of
Chugai Pharmaceutical Co., Ltd., Tokyo, Japan
©2024 Genentech, Inc. All rights reserved.
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 04/2024 | ||
| PATIENT INFORMATION
ALECENSA® (a-le-sen-sah) (alectinib) capsules |
|||
| What is the most important information I should know about ALECENSA? ALECENSA may cause serious side effects, including:
|
|||
|
|
||
|
|||
| What is ALECENSA?
ALECENSA is a prescription medicine used to treat adults with non-small cell lung cancer (NSCLC) that is caused by an abnormal anaplastic lymphoma kinase (ALK) gene:
It is not known if ALECENSA is safe and effective in children. |
|||
Before you take ALECENSA, tell your healthcare provider about all of your medical conditions, including if you:
|
|||
How should I take ALECENSA?
|
|||
| What should I avoid while taking ALECENSA?
Avoid spending time in the sunlight during treatment with ALECENSA and for 7 days after the last dose of ALECENSA. Your skin may be sensitive to the sun (photosensitivity) and you may burn more easily and get severe sunburns. Use sun protecting measures, such as sunscreen and lip balm with an SPF 50 or greater to help protect against sunburn. |
|||
| What are the possible side effects of ALECENSA? ALECENSA may cause serious side effects, including: The most common side effects of ALECENSA include: |
|||
|
|
||
| These are not all of the possible side effects of ALECENSA. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
How should I store ALECENSA?
|
|||
| General information about the safe and effective use of ALECENSA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use ALECENSA for a condition for which it was not prescribed. Do not give ALECENSA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about ALECENSA that is written for health professionals. |
|||
| What are the ingredients in ALECENSA?
Active ingredient: alectinib Inactive ingredients: lactose monohydrate, hydroxypropylcellulose, sodium lauryl sulfate, magnesium stearate and carboxymethylcellulose calcium. Capsule shell contains: hypromellose, carrageenan, potassium chloride, titanium dioxide, corn starch, and carnauba wax. Printing ink contains: red iron oxide (E172), yellow iron oxide (E172), FD&C Blue No. 2 aluminum lake (E132), carnauba wax, white shellac, and glyceryl monooleate. Distributed by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 ALECENSA® is a registered trademark of Chugai Pharmaceutical Co., Ltd., Tokyo, Japan ©2024 Genentech, Inc. For more information, go to www.ALECENSA.com or call 1-800-253-2367. |
|||

| ALECENSA
alectinib hydrochloride capsule |
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||
| Labeler - Genentech, Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche Ltd | 485244961 | ANALYSIS(50242-130) , LABEL(50242-130) , PACK(50242-130) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche AG | 482242971 | ANALYSIS(50242-130) | |
Frequently asked questions
- What is the mechanism of action for Alecensa (alectinib)?
- How long do you take Alecensa for?
- How effective is Alecensa for ALK-positive NSCLC?
- How does Alecensa work?
- What is Alecensa used to treat?
More about Alecensa (alectinib)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- FDA approval history
- Drug class: multikinase inhibitors
- Breastfeeding
- En español
