Sickle cell anemia
Medically reviewed by Drugs.com. Last updated on Aug 6, 2025.
What is sickle cell anemia?
Sickle cell anemia is an inherited blood disorder.
It causes:
- chronic destruction of red blood cells, causing severe anemia
- episodes of intense pain
- vulnerability to infections
- organ damage
- in some cases, early death.
Hemoglobin is a protein in red blood cells that carries oxygen. People with sickle cell anemia inherit a defective type of hemoglobin. When oxygen levels inside a red blood cell get low, the defective hemoglobin forms long rods. These rods stretch the red blood cells into long, abnormal sickle shapes. In contrast, normal red blood cells are disc-shaped.
|
|
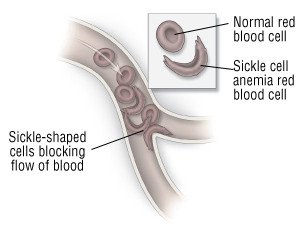
Sickle-shaped red blood cells cannot easily pass through the body's blood vessels. Instead, they clog blood vessels. They block the flow of blood and cut off the oxygen supply to tissues and organs. This lack of oxygen can damage the body's organs and limbs. It causes severe pain in any affected area.
A normal red cell's lifespan is 120 days. In contrast, sickled blood cells last only 10 to 20 days. As a result, patients with sickle cell disease have chronic anemia, an abnormally low level of red blood cells.
The spleen is an organ that helps clear infections. Sickle cell disease damages the spleen. By the time a child with sickle cell anemia is 4 years old, the spleen has usually stopped functioning. As a result, people with sickle cell disease have an increased risk of developing life-threatening infections.
To have fully developed sickle cell anemia, you must inherit one gene for the illness from each parent. Sometimes a person inherits only one sickle cell gene from one parent. This person is said to have sickle cell trait rather than sickle cell anemia. People with sickle cell trait usually have no symptoms, though they can pass the gene to their children.
Symptoms of sickle cell anemia
Symptoms of sickle cell anemia include:
- fatigue, shortness of breath, pale skin and fingernails due to anemia
- recurrent bouts of pain in the abdomen, chest, back, arms, or legs
- a yellowing of the skin and whites of the eyes
- slowed growth and delayed puberty in children
- frequent infections
- eye problems, including blindness
- stroke.
When sickled red cells block blood vessels, the oxygen supply to body cells is obstructed. This causes painful episodes called crises. Painful sickle cell crises can affect many different joints and organs. The back, chest, extremities, and abdomen are affected most commonly. The level of pain varies, from trivial to excruciating. The episodes typically last from two to seven days.
In about half the cases, the pain crisis is accompanied by
- fever
- nausea
- vomiting
- high blood pressure
- fast heart rate.
Triggers for these painful episodes include:
- infection
- stress
- alcohol consumption
- dehydration.
Women are more likely to experience a painful episode during their menstrual period. But the majority of sickle cell crises have no identifiable cause.
Diagnosing sickle cell anemia
Your doctor will ask you about
- a history of painful crises
- neurological problems
- chest pain
- history of infections.
He or she then will perform a physical examination. It will focus on your heart, lungs, joints, eyes, and neurological system.
Routine blood tests include a complete blood count (CBC), which can detect anemia. A microscopic examination of the blood may reveal the characteristic sickled cells. A blood test called hemoglobin electrophoresis confirms the diagnosis by identifying the abnormal form of hemoglobin that is produced in people with the disease.
In families with a history of sickle cell anemia, a doctor may screen for the disorder whenever a new baby is born in your family. Prenatal screening also can be done.
Expected duration of sickle cell anemia
As a genetic disease, sickle cell anemia is generally considered a lifelong disease. However, a few people have been cured after having a blood and bone marrow (stem cell) transplant or gene therapy.
Preventing sickle cell anemia
The only way to prevent sickle cell anemia is for two people who carry the sickle cell gene to avoid having a child together.
If sickle cell anemia or sickle cell trait runs in your family, you and your spouse may wish to speak with a genetic counselor. He or she can explain your chances of passing the condition to your children.
Drugs used to treat this and similar conditions
Treatment options
The following list of medications are related to or used in the treatment of this condition.
Treating sickle cell anemia
Treatment of sickle cell anemia may include the following:
- Folic acid supplements: to ensure that enough of this nutrient is available to make new red blood cells
- Vaccinations: to prevent the infections that are more likely to occur in people with sickle cell anemia, a person should be vaccinated for
- pneumococcal pneumonia
- Haemophilus influenza
- meningitis
- hepatitis A and B
- varicella
- rotavirus
- tetanus
- diphtheria
- pertussis
- influenza
- Human papillomavirus (HPV)
- COVID-19
- poliovirus (in those parts of the world where the virus persists)
- Antibiotic therapy: an emergency supply of antibiotics commonly prescribed for future use if infection is suspected. In some cases, daily preventive antibiotics are recommended to protect people with sickle cell anemia from serious bacterial infections, especially if the spleen is not functioning normally.
- Fluids, oxygen, and pain-killing medications: these can be helpful to manage painful sickle crises
- Blood transfusions: these can be helpful to treat anemia and painful crises
- Hydroxyurea (Hydrea): this medication is the mainstay of treatment in children and adults. Hydroxyurea
- reduces the number of painful crises
- reduces the need for blood transfusions
- decreases rates of hospital admission
- may prolong survival.
- L-glutamine (Endari, Glutasolve): this drug may reduce the number and severity of painful crises. It is an amino acid that can be used in adult and pediatric patients 5 years of age and older.
- Crizanlizumab (Adakveo): this is an antibody treatment that can help prevent blockage of blood vessels. It’s FDA-approved for those with sickle cell anemia who are 16 years of age and older.
- Laser coagulation: this treatment can prevent vision loss in people with retinopathy (disease of the retina) due to sickle cell anemia. Routine eye examinations are recommended for early detection of this complication.
As noted above, a blood and bone marrow (stem cell) transplant may cure the illness. The child has to have a suitable donor such as a genetically similar sibling, parent, or child. However, this is a high-risk treatment that is primarily reserved for those with severe and uncontrolled symptoms.
When to call a professional
People with sickle cell disease must see their doctor regularly and receive comprehensive care.
Call the doctor immediately when anyone with sickle cell disease:
- develops a fever or any other sign of infection
- has severe pain in any part of their body
- develops breathing problems
- develops any neurological symptoms.
Prognosis
Sickle cell anemia affects different people differently.
For example, some patients have only mild symptoms with less than one crisis episode per year. Others have more severe symptoms with frequent crises.
While there is not yet a widely available cure for sickle cell anemia, life expectancy for those with the illness has dramatically increased over the past 30 years.
Additional info
National Heart, Lung, and Blood Institute (NHLBI)
https://www.nhlbi.nih.gov/
March of Dimes Birth Defects Foundation
https://www.marchofdimes.org/
Sickle Cell Disease Association Of America
https://www.sicklecelldisease.org/
Further information
Always consult your healthcare provider to ensure the information displayed on this page applies to your personal circumstances.