Venofer Injection: Package Insert / Prescribing Info
Package insert / product label
Generic name: iron sucrose
Dosage form: injection, solution
Drug class: Iron products
J Code (medical billing code): J1756 (1 mg, intravenous)
Medically reviewed by Drugs.com. Last updated on Jan 1, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
VENOFER ® safely and effectively. See full prescribing information for VENOFER.
Venofer (iron sucrose) injection, for intravenous use
Initial U.S. Approval: 2000
Indications and Usage for Venofer Injection
Venofer is an iron replacement product indicated for the treatment of iron deficiency anemia (IDA) in patients with chronic kidney disease (CKD). (1)
Venofer Injection Dosage and Administration
| Population | Dose | |
| Adult patients | Hemodialysis Dependent-Chronic Kidney Disease (HDD-CKD) (2.2) | 100 mg slow intravenous injection or infusion |
| Non-Dialysis Dependent-Chronic Kidney Disease (NDD-CKD) (2.3) | 200 mg slow intravenous injection or infusion | |
| Peritoneal Dialysis Dependent-Chronic Kidney Disease (PDD-CKD) (2.4) | 300 mg or 400 mg intravenous infusion | |
| Pediatric patients | HDD-CKD (2.5), PDD-CKD or NDD-CKD (2.6) | 0.5 mg/kg slow intravenous injection or infusion |
Dosage Forms and Strengths
Injection: 100 mg/5 mL (20 mg/mL) in single-dose vials. (3)
Contraindications
- Known hypersensitivity to Venofer (4)
Warnings and Precautions
- Hypersensitivity Reactions: Observe for signs and symptoms of hypersensitivity during and after Venofer administration for at least 30 minutes and until clinically stable following completion of each administration. Only administer Venofer when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions. (5.1)
- Hypotension: May cause hypotension. Monitor for signs and symptoms of hypotension during and following each administration. (5.2)
- Iron Overload: Regularly monitor hematologic responses during therapy. Do not administer to patients with iron overload. (5.3)
Adverse Reactions/Side Effects
- Adult patients: The most common adverse reactions (≥2%) are diarrhea, nausea, vomiting, headache, dizziness, hypotension, pruritus, pain in extremity, arthralgia, back pain, muscle cramp, injection site reactions, chest pain, and peripheral edema. (6.1)
- Pediatric patients: The most common adverse reactions (≥2%) are headache, respiratory tract viral infection, peritonitis, vomiting, pyrexia, dizziness, cough, nausea, arteriovenous fistula thrombosis, hypotension, and hypertension. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact American Regent, Inc. at 1-800-734-9236 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2022
Full Prescribing Information
1. Indications and Usage for Venofer Injection
Venofer is indicated for the treatment of iron deficiency anemia (IDA) in patients with chronic kidney disease (CKD).
2. Venofer Injection Dosage and Administration
Venofer must only be administered intravenously either by slow injection or by infusion. The dosage of Venofer is expressed in mg of elemental iron. Each mL contains 20 mg of elemental iron.
2.1 Mode of Administration
Administer Venofer only intravenously by slow injection or by infusion. The dosage of Venofer is expressed in mg of elemental iron. Each mL contains 20 mg of elemental iron.
2.2 Adult Patients with Hemodialysis Dependent-Chronic Kidney Disease (HDD-CKD)
Administer Venofer 100 mg undiluted as a slow intravenous injection over 2 to 5 minutes, or as an infusion of 100 mg diluted in a maximum of 100 mL of 0.9% NaCl over a period of at least 15 minutes, per consecutive hemodialysis session [see How Supplied/Storage and Handling (16.2).] Administer Venofer early during the dialysis session (generally within the first hour). The usual total treatment course of Venofer is 1000 mg. Venofer treatment may be repeated if iron deficiency reoccurs.
2.3 Adult Patients with Non-Dialysis Dependent-Chronic Kidney Disease (NDD-CKD)
Administer Venofer 200 mg undiluted as a slow intravenous injection over 2 to 5 minutes or as an infusion of 200 mg in a maximum of 100 mL of 0.9% NaCl over a period of 15 minutes. Administer on 5 different occasions over a 14 day period. There is limited experience with administration of an infusion of 500 mg of Venofer, diluted in a maximum of 250 mL of 0.9% NaCl, over a period of 3.5 to 4 hours on Day 1 and Day 14 [see How Supplied/Storage and Handling (16.2).]Venofer treatment may be repeated if iron deficiency reoccurs.
2.4 Adult Patients with Peritoneal Dialysis Dependent-Chronic Kidney Disease (PDD-CKD)
Administer Venofer in 3 divided doses, given by slow intravenous infusion, within a 28 day period: 2 infusions each of 300 mg over 1.5 hours 14 days apart followed by one 400 mg infusion over 2.5 hours 14 days later. Dilute Venofer in a maximum of 250 mL of 0.9% NaCl [see How Supplied/Storage and Handling (16.2).] Venofer treatment may be repeated if iron deficiency reoccurs.
2.5 Pediatric Patients (2 Years of Age and Older) with HDD-CKD for Iron Maintenance Treatment
For iron maintenance treatment: Administer Venofer at a dose of 0.5 mg/kg, not to exceed 100 mg per dose, every two weeks for 12 weeks given undiluted by slow intravenous injection over 5 minutes or diluted in 0.9% NaCl at a concentration of 1 to 2 mg/mL and administered over 5 to 60 minutes. Do not dilute to concentrations below 1 mg/mL [see How Supplied/Storage and Handling [16.2].) Venofer treatment may be repeated if necessary.
The dosing for iron replacement treatment in pediatric patients with HDD-CKD has not been established.
2.6 Pediatric Patients (2 Years of Age and Older) with NDD-CKD or PDD-CKD who are on Erythropoietin Therapy for Iron Maintenance Treatment
For iron maintenance treatment: Administer Venofer at a dose of 0.5 mg/kg, not to exceed 100 mg per dose, every four weeks for 12 weeks given undiluted by slow intravenous injection over 5 minutes or diluted in 0.9% NaCl at a concentration of 1 to 2 mg/mL and administered over 5 to 60 minutes. Do not dilute to concentrations below 1 mg/mL [see How Supplied/Storage and Handling (16.2).] Venofer treatment may be repeated if necessary.
The dosing for iron replacement treatment in pediatric patients with NDD-CKD or PDD-CKD has not been established.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and fatal, have been reported in patients receiving Venofer. Patients may present with shock, clinically significant hypotension, loss of consciousness, and/or collapse. If hypersensitivity reactions or signs of intolerance occur during administration, stop Venofer immediately. Monitor patients for signs and symptoms of hypersensitivity during and after Venofer administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Venofer when personnel and therapies are immediately available for the treatment of serious hypersensitivity reactions. Most reactions associated with intravenous iron preparations occur within 30 minutes of the completion of the infusion [see Adverse Reactions (6.1 and 6.2)].
5.2 Hypotension
Venofer may cause clinically significant hypotension. Monitor for signs and symptoms of hypotension following each administration of Venofer. Hypotension following administration of Venofer may be related to the rate of administration and/or total dose administered [see Dosage and Administration (2), Warnings and Precautions (5.1), and Adverse Reactions (6.2)].
5.3 Iron Overload
Excessive therapy with parenteral iron can lead to excess storage of iron with the possibility of iatrogenic hemosiderosis. All adult and pediatric patients receiving Venofer require periodic monitoring of hematologic and iron parameters (hemoglobin, hematocrit, serum ferritin and transferrin saturation). Do not administer Venofer to patients with evidence of iron overload. Transferrin saturation (TSAT) values increase rapidly after intravenous administration of iron sucrose; do not perform serum iron measurements for at least 48 hours after intravenous dosing [see Dosage and Administration (2) and Overdosage (10)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
-
Hypersensitivity Reactions [see Warnings and Precautions (5.1)]
-
Hypotension [see Warnings and Precautions (5.2)]
-
Iron Overload [see Warnings and Precautions (5.3)]
6.1 Adverse Reactions in Clinical Trials
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug may not reflect the rates observed in practice.
Adverse Reactions in Adults Patients with CKD
The frequency of adverse reactions associated with the use of Venofer has been documented in six clinical trials involving 231 patients with HDD-CKD, 139 patients with NDD-CKD and 75 patients with PDD-CKD. Adverse reactions reported by ≥ 2% of treated patients in the six clinical trials for which the rate for Venofer exceeds the rate for comparator are listed by indication in Table 1. Patients with HDD-CKD received 100 mg doses at 10 consecutive dialysis sessions until a cumulative dose of 1000 mg was administered. Patients with NDD-CKD received either 5 doses of 200 mg over 2 weeks or 2 doses of 500 mg separated by fourteen days, and patients with PDD-CKD received 2 doses of 300 mg followed by a dose of 400 mg over a period of 4 weeks.
|
* EPO=Erythropoietin |
|||||
| Body System/Adverse Reactions
| HDD-CKD | NDD-CKD | PDD-CKD | ||
| Venofer | Venofer | Oral Iron | Venofer | EPO* Only | |
| (N=231) | (N=139) | (N=139) | (N=75) | (N=46) | |
| % | % | % | % | % | |
| Subjects with any adverse reaction | 78.8 | 76.3 | 73.4 | 72.0 | 65.2 |
| Ear and Labyrinth Disorders | |||||
| Ear Pain | 0 | 2.2 | 0.7 | 0 | 0 |
| Eye Disorders | |||||
| Conjunctivitis | 0.4 | 0 | 0 | 2.7 | 0 |
| Gastrointestinal Disorders | |||||
| Abdominal pain | 3.5 | 1.4 | 2.9 | 4.0 | 6.5 |
| Diarrhea | 5.2 | 7.2 | 10.1 | 8.0 | 4.3 |
| Dysgeusia | 0.9 | 7.9 | 0 | 0 | 0 |
| Nausea | 14.7 | 8.6 | 12.2 | 5.3 | 4.3 |
| Vomiting | 9.1 | 5.0 | 8.6 | 8.0 | 2.2 |
| General Disorders and | |||||
| Administration Site Conditions | |||||
| Asthenia | 2.2 | 0.7 | 2.2 | 2.7 | 0 |
| Chest pain | 6.1 | 1.4 | 0 | 2.7 | 0 |
| Feeling abnormal | 3.0 | 0 | 0 | 0 | 0 |
| Infusion site pain or burning | 0 | 5.8 | 0 | 0 | 0 |
| Injection site extravasation | 0 | 2.2 | 0 | 0 | 0 |
| Peripheral edema | 2.6 | 7.2 | 5.0 | 5.3 | 10.9 |
| Pyrexia | 3.0 | 0.7 | 0.7 | 1.3 | 0 |
| Infections and Infestations | |||||
| Nasopharyngitis, Sinusitis, Upper respiratory tract infections, Pharyngitis | 2.6 | 2.2 | 4.3 | 16.0 | 4.3 |
| Injury, Poisoning and Procedural | |||||
| Complications | |||||
| Graft complication | 9.5 | 1.4 | 0 | 0 | 0 |
| Metabolism and Nutrition Disorders | |||||
| Fluid overload | 3.0 | 1.4 | 0.7 | 1.3 | 0 |
| Gout | 0 | 2.9 | 1.4 | 0 | 0 |
| Hyperglycemia | 0 | 2.9 | 0 | 0 | 2.2 |
| Hypoglycemia | 0.4 | 0.7 | 0.7 | 4.0 | 0 |
| Musculoskeletal and Connective | |||||
| Tissue Disorders | |||||
| Arthralgia | 3.5 | 1.4 | 2.2 | 4.0 | 4.3 |
| Back pain | 2.2 | 2.2 | 3.6 | 1.3 | 4.3 |
| Muscle cramp | 29.4 | 0.7 | 0.7 | 2.7 | 0 |
| Myalgia | 0 | 3.6 | 0 | 1.3 | 0 |
| Pain in extremity | 5.6 | 4.3 | 0 | 2.7 | 6.5 |
| Nervous System Disorders | |||||
| Dizziness | 6.5 | 6.5 | 1.4 | 1.3 | 4.3 |
| Headache | 12.6 | 2.9 | 0.7 | 4.0 | 0 |
| Respiratory, Thoracic and | |||||
| Mediastinal Disorders | |||||
| Cough | 3.0 | 2.2 | 0.7 | 1.3 | 0 |
| Dyspnea | 3.5 | 5.8 | 1.4 | 1.3 | 2.2 |
| Nasal congestion | 0 | 1.4 | 2.2 | 1.3 | 0 |
| Skin and Subcutaneous | |||||
| Tissue Disorders | |||||
| Pruritus | 3.9 | 2.2 | 4.3 | 2.7 | 0 |
| Vascular Disorders | |||||
| Hypertension | 6.5 | 6.5 | 4.3 | 8.0 | 6.5 |
| Hypotension | 39.4 | 2.2 | 0.7 | 2.7 | 2.2 |
One hundred thirty (11%) of the 1,151 patients evaluated in the 4 U.S. trials in HDD-CKD patients (studies A, B and the two post marketing studies) had prior other intravenous iron therapy and were reported to be intolerant (defined as precluding further use of that iron product). When these patients were treated with Venofer there were no occurrences of adverse reactions that precluded further use of Venofer [see Warning and Precautions (5)].
Adverse Reactions in Pediatric Patients with CKD (ages 2 years and older)
In a randomized, open-label, dose-ranging trial for iron maintenance treatment with Venofer in pediatric patients with CKD on stable erythropoietin therapy [see Clinical Studies (14.7)], at least one adverse reaction was experienced by 57% (27/47) of the patients receiving Venofer 0.5 mg/kg, 53% (25/47) of the patients receiving Venofer 1 mg/kg, and 55% (26/47) of the patients receiving Venofer 2 mg/kg.
A total of 5 (11%) subjects in the Venofer 0.5 mg/kg group, 10 (21%) patients in the Venofer 1 mg/kg group, and 10 (21%) patients in the Venofer 2 mg/kg group experienced at least 1 serious adverse reaction during the study. The most common adverse reactions (>2% of patients) in all patients were headache (6%), respiratory tract viral infection (4%), peritonitis (4%), vomiting (4%), pyrexia (4%), dizziness (4%), cough (4%), nausea (3%), arteriovenous fistula thrombosis (2%), hypotension (2%), and hypertension (2.1%).
6.2 Adverse Reactions from Post-Marketing Experience
The following adverse reactions have been identified during post-approval use of Venofer. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In the post-marketing safety studies in 1,051 treated patients with HDD-CKD, the adverse reactions reported by >1% were cardiac failure congestive, sepsis and dysgeusia.
-
Immune system disorders: anaphylactic-type reactions, angioedema
-
Psychiatric disorders: confusion
-
Nervous system disorders: convulsions, collapse, light-headedness, loss-of-consciousness
-
Cardiovascular System: bradycardia, shock, acute myocardial ischemia with or without myocardial infarction or with in-stent thrombosis in the context of a hypersensitivity reaction.
-
Respiratory, thoracic and mediastinal disorders: bronchospasm, dyspnea
-
Musculoskeletal and connective tissue disorders: back pain, swelling of the joints
-
Renal and urinary disorders: chromaturia
-
General disorders and administration site conditions: hyperhidrosis
Symptoms associated with Venofer total dosage or infusing too rapidly included hypotension, dyspnea, headache, vomiting, nausea, dizziness, joint aches, paresthesia, abdominal and muscle pain, edema, and cardiovascular collapse. These adverse reactions have occurred up to 30 minutes after the administration of Venofer injection. Reactions have occurred following the first dose or subsequent doses of Venofer. Symptoms may respond to intravenous fluids, hydrocortisone, and/or antihistamines. Slowing the infusion rate may alleviate symptoms.
Injection site discoloration has been reported following extravasation. Assure stable intravenous access to avoid extravasation.
Related/similar drugs
7. Drug Interactions
Venofer may reduce the absorption of concomitantly administered oral iron preparations.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Published studies on intravenous iron sucrose treatment after the first trimester of pregnancy have not shown adverse maternal or fetal outcomes (see Data). Available reports of intravenous iron sucrose use in pregnant women during the first trimester are insufficient to assess the risk of major birth defects and miscarriage. There are risks to the mother and fetus associated with untreated IDA in pregnancy as well as risks to the fetus associated with maternal severe hypersensitivity reactions (see Clinical Considerations). Animal reproduction studies of iron sucrose administered to rats and rabbits during the period of organogenesis at elemental iron doses equivalent to the maximum recommended human dose based on body surface area revealed no evidence of harm to the fetus (see Data). The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically-recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Iron deficiency anemia during pregnancy should be treated. Untreated IDA in pregnancy is associated with adverse maternal outcomes such as post-partum anemia. Adverse pregnancy outcomes associated with IDA include increased risk for preterm delivery and low birth weight.
Fetal/Neonatal adverse reactions
Severe adverse reactions including circulatory failure (severe hypotension, shock including in the context of anaphylactic reaction) may occur in pregnant women with parenteral iron products (such as Venofer) which may cause fetal bradycardia, especially during the second and third trimester.
Data
Human Data
Published data from randomized controlled studies and prospective observational studies on the use of Venofer in pregnant women have not reported an association of Venofer and adverse developmental outcomes. However, these studies did not include women exposed during the first trimester of pregnancy and were not designed to assess the risk of major birth defects. Maternal adverse events reported in these studies are similar to those reported during clinical trials in adult males and non-pregnant females [seeAdverse Reactions (6.1)].
Animal Data
Iron sucrose was administered intravenously to rats and rabbits during the period of organogenesis at elemental iron doses up to 13 mg/kg/day (0.25 times or equivalent to the maximum recommended human dose based on body surface area, respectively) and revealed no evidence of harm to the fetus.
8.2 Lactation
Risk Summary
Iron sucrose is present in human milk, and available published reports following exposure to 100-300 mg intravenous iron sucrose have not reported adverse reactions in breastfed infants (see Data). There are no data on the effects on milk production. The developmental and health benefits of breastfeeding should be considered, along with the mother’s clinical need for Venofer and any potential adverse effects on the breastfed child from Venofer or from the underlying maternal condition.
Data
A published study showed no difference in iron concentration in the colostrum of 10 iron deficient breastfeeding women who were 2 to 3 days postpartum and received a single dose of 100 mg of intravenous iron sucrose compared to 5 breastfeeding women who received no iron. These results may underestimate the amount of iron in breastmilk following the standard dose of Venofer.
A published report of 78 breastfeeding women who received 300 mg of intravenous iron sucrose over 3 days (infant age not reported) did not report on the safety of iron sucrose in breastfed infants; however adverse reactions in breastfed infants were not reported.
Clinical Considerations
Monitor breastfed infants for gastrointestinal toxicity (constipation, diarrhea).
8.4 Pediatric Use
Safety and effectiveness of Venofer for iron replacement treatment in pediatric patients with dialysis-dependent or non-dialysis-dependent CKD have not been established.
Safety and effectiveness of Venofer for iron maintenance treatment in pediatric patients 2 years of age and older with dialysis-dependent or non-dialysis-dependent CKD receiving erythropoietin therapy were studied. Venofer at doses of 0.5 mg/kg, 1 mg/kg, and 2 mg/kg was administered. All three doses maintained hemoglobin between 10.5 g/dL and 14.0 g/dL in about 50% of subjects over the 12-week treatment period with stable EPO dosing [see Clinical Studies (14.7)].
Venofer has not been studied in patients younger than 2 years of age.
In a country where Venofer is available for use in children, at a single site, five premature infants (weight less than 1,250 g) developed necrotizing enterocolitis and two of the five died during or following a period when they received Venofer, several other medications and erythropoietin. Necrotizing enterocolitis may be a complication of prematurity in very low birth weight infants. No causal relationship to Venofer or any other drugs could be established.
8.5 Geriatric Use
Of the 1,051 patients in two post-marketing safety studies of Venofer, 40% were 65 years and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. In general, dose administration to an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10. Overdosage
No data are available regarding overdosage of Venofer in humans. Excessive dosages of Venofer may lead to accumulation of iron in storage sites potentially leading to hemosiderosis. Do not administer Venofer to patients with iron overload [see Warnings and Precautions (5.3)]. Venofer is not dialyzable through CA210 (Baxter) High Efficiency or Fresenius F80A High Flux dialysis membranes.
Toxicities in single-dose studies in mice and rats, at intravenous iron sucrose doses up to 8 times the maximum recommended human dose based on body surface area, included sedation, hypoactivity, pale eyes, bleeding in the gastrointestinal tract and lungs, and mortality.
11. Venofer Injection Description
Venofer (iron sucrose injection, USP), an iron replacement product, is a brown, sterile, aqueous, complex of polynuclear iron (III)-hydroxide in sucrose for intravenous use. Iron sucrose injection has a molecular weight of approximately 34,000 to 60,000 daltons and a proposed structural formula:
[Na2Fe5O8(OH) ·3(H2O)]n ·m(C12H22O11)
where: n is the degree of iron polymerization and m is the number of sucrose molecules associated with the iron (III)-hydroxide.
Each mL contains 20 mg elemental iron as iron sucrose in water for injection. Venofer is available in 5 mL single-dose vials (100 mg elemental iron per 5 mL). The drug product contains approximately 30% sucrose w/v (300 mg/mL). Sodium hydroxide may be added to adjust pH to 10.5 to 11.1. The product contains no preservatives. The osmolarity of the injection is 1,250 mOsmol/L.
12. Venofer Injection - Clinical Pharmacology
12.1 Mechanism of Action
Venofer is an aqueous complex of poly-nuclear iron (III)-hydroxide in sucrose. Following intravenous administration, Venofer is dissociated into iron and sucrose and the iron is transported as a complex with transferrin to target cells including erythroid precursor cells. The iron in the precursor cells is incorporated into hemoglobin as the cells mature into red blood cells.
12.2 Pharmacodynamics
Following intravenous administration, Venofer is dissociated into iron and sucrose. In 22 patients undergoing hemodialysis and receiving erythropoietin (recombinant human erythropoietin) therapy treated with iron sucrose containing 100 mg of iron, three times weekly for three weeks, significant increases in serum iron and serum ferritin and significant decreases in total iron binding capacity occurred four weeks from the initiation of iron sucrose treatment.
12.3 Pharmacokinetics
In healthy adults administered intravenous doses of Venofer, its iron component exhibited first order kinetics with an elimination half-life of 6 h, total clearance of 1.2 L/h, and steady state apparent volume of distribution of 7.9 L. The iron component appeared to distribute mainly in blood and to some extent in extravascular fluid. A study evaluating Venofer containing 100 mg of iron labeled with 52Fe/59Fe in patients with iron deficiency showed that a significant amount of the administered iron is distributed to the liver, spleen and bone marrow and that the bone marrow is an irreversible iron trapping compartment.
Following intravenous administration of Venofer, iron sucrose is dissociated into iron and sucrose. The sucrose component is eliminated mainly by urinary excretion. In a study evaluating a single intravenous dose of Venofer containing 1,510 mg of sucrose and 100 mg of iron in 12 healthy adults (9 female, 3 male: age range 32 to 52), 68.3% of the sucrose was eliminated in urine in 4 h and 75.4% in 24 h. Some iron was also eliminated in the urine. Neither transferrin nor transferrin receptor levels changed immediately after the dose administration. In this study and another study evaluating a single intravenous dose of iron sucrose containing 500 to 700 mg of iron in 26 patients with anemia on erythropoietin therapy (23 female, 3 male; age range 16 to 60), approximately 5% of the iron was eliminated in urine in 24 h at each dose level. The effects of age and gender on the pharmacokinetics of Venofer have not been studied.
Pharmacokinetics in Pediatric Patients
In a single-dose PK study of Venofer, patients with NDD-CKD ages 12 to 16 (N=11) received intravenous bolus doses of Venofer at 7 mg/kg (maximum 200 mg) administered over 5 minutes. Following a single dose of Venofer, the half-life of total serum iron was 8 hours. The mean Cmax and AUC values were 8545 μg/dl and 31305 hr•μg/dL, respectively, which were 1.42- and 1.67-fold higher than dose adjusted adult Cmax and AUC values.
Venofer is not dialyzable through CA210 (Baxter) High Efficiency or Fresenius F80A High Flux dialysis membranes. In in vitro studies, the amount of iron sucrose in the dialysate fluid was below the levels of detection of the assay (less than 2 parts per million).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with iron sucrose.
Iron sucrose was not mutagenic in vitro in the bacterial reverse mutation assay (Ames test) or the mouse lymphoma assay. Iron sucrose was not clastogenic in the in vitro chromosome aberration assay using human lymphocytes or in the in vivo mouse micronucleus assay.
Iron sucrose at intravenous doses up to 15 mg/kg/day of elemental iron (1.2 times the maximum recommended human dose based on body surface area) had no effect on fertility and reproductive function of male and female rats.
14. Clinical Studies
Five clinical trials involving 647 adult patients and one clinical trial involving 131 pediatric patients were conducted to assess the safety and efficacy of Venofer.
14.1 Clinical Studies Overview
Five clinical trials involving 647 adult patients and one clinical trial involving 131 pediatric patients were conducted to assess the safety and efficacy of Venofer.
14.2 Study A: Hemodialysis Dependent-Chronic Kidney Disease (HDD–CKD)
Study A was a multicenter, open-label, historically-controlled study in 101 patients with HDD-CKD (77 patients with Venofer treatment and 24 in the historical control group) with IDA. Eligibility criteria for Venofer treatment included patients undergoing chronic hemodialysis, receiving erythropoietin, hemoglobin level between 8.0 and 11.0 g/dL, transferrin saturation <20%, and serum ferritin <300 ng/mL. The mean age of the patients was 65 years with the age range of 31 to 85 years. Of the 77 patients, 44 (57%) were male and 33 (43%) were female.
Venofer 100 mg was administered at 10 consecutive dialysis sessions either as slow injection or a slow infusion. The historical control population consisted of 24 patients with similar ferritin levels as patients treated with Venofer, who were off intravenous iron for at least 2 weeks and who had received erythropoietin therapy with hematocrit averaging 31 to 36 for at least two months prior to study entry. The mean age of patients in the historical control group was 56 years, with an age range of 29 to 80 years. Patient age and serum ferritin level were similar between treatment and historical control patients.
Patients in the Venofer treated population showed a greater increase in hemoglobin and hematocrit than did patients in the historical control population. See Table 2.
| **p < 0.01 and *p < 0.05 compared to historical control from ANCOVA analysis with baseline hemoglobin, serum ferritin and erythropoietin dose as covariates. |
||||||
| Efficacy parameters | End of treatment | 2 week follow-up | 5 week follow-up | |||
| Venofer (n=69) | Historical Control (n=18) | Venofer
(n=73) | Historical Control
(n=18) | Venofer
(n=71) | Historical
Control (n=15) |
|
| Hemoglobin (g/dL) | 1.0 ± 0.12** | 0.0 ± 0.21 | 1.3 ± 0.14** | -0.6 ± 0.24 | 1.2 ± 0.17* | -0.1 ± 0.23 |
| Hematocrit (%) | 3.1 ± 0.37** | -0.3 ± 0.65 | 3.6 ± 0.44** | -1.2 ± 0.76 | 3.3 ± 0.54 | 0.2 ± 0.86 |
Serum ferritin increased at endpoint of study from baseline in the Venofer-treated population (165.3 ± 24.2 ng/mL) compared to the historical control population (-27.6 ± 9.5 ng/mL). Transferrin saturation also increased at endpoint of study from baseline in the Venofer-treated population (8.8 ± 1.6%) compared to this historical control population (-5.1 ± 4.3%).
14.3 Study B: Hemodialysis Dependent-Chronic Kidney Disease (HDD-CKD)
Study B was a multicenter, open label study of Venofer in 23 patients with iron deficiency and HDD-CKD who had been discontinued from iron dextran due to intolerance. Eligibility criteria were otherwise identical to Study A. The mean age of the patients in this study was 53 years, with ages ranging from 21 to 79 years. Of the 23 patients enrolled in the study, 10 (44%) were male and 13 (56%) were female.
All 23 enrolled patients were evaluated for efficacy. Increases in mean hemoglobin (1.1 ± 0.2 g/dL), hematocrit (3.6 ± 0.6%), serum ferritin (266.3 ± 30.3 ng/mL) and transferrin saturation (8.7 ± 2.0%) were observed from baseline to end of treatment.
14.4 Study C: Hemodialysis Dependent-Chronic Kidney Disease (HDD-CKD)
Study C was a multicenter, open-label study in patients with HDD-CKD. This study enrolled patients with a hemoglobin ≤ 10 g/dL, a serum transferrin saturation ≤ 20%, and a serum ferritin ≤ 200 ng/mL, who were undergoing maintenance hemodialysis 2 to 3 times weekly. The mean age of the patients enrolled in this study was 41 years, with ages ranging from 16 to 70 years. Of 130 patients evaluated for efficacy in this study, 68 (52%) were male and 62 (48%) were female. Forty-eight percent of the patients had previously been treated with oral iron. Exclusion criteria were similar to those in studies A and B. Venofer was administered in doses of 100 mg during sequential dialysis sessions until a pre-determined (calculated) total dose of iron was administered. A 50 mg dose (2.5 mL) was given to patients within two weeks of study entry as a test dose. Twenty-seven patients (20%) were receiving erythropoietin treatment at study entry and they continued to receive the same erythropoietin dose for the duration of the study.
The modified intention-to-treat (mITT) population consisted of 131 patients. Increases from baseline in mean hemoglobin (1.7 g/dL), hematocrit (5%), serum ferritin (434.6 ng/mL), and serum transferrin saturation (14%) were observed at week 2 of the observation period and these values remained increased at week 4 of the observation period.
14.5 Study D: Non-Dialysis Dependent-Chronic Kidney Disease (NDD-CKD)
Study D (NCT00236977) was a randomized, open-label, multicenter, active-controlled study of the safety and efficacy of oral iron versus Venofer in patients with NDD-CKD with or without erythropoietin therapy. Erythropoietin therapy was stable for 8 weeks prior to randomization. In the study 188 patients with NDD-CKD, hemoglobin of ≤ 11.0 g/dL, transferrin saturation ≤ 25%, ferritin ≤ 300 ng/mL were randomized to receive oral iron (325 mg ferrous sulfate three times daily for 56 days); or Venofer (either 200 mg over 2 to 5 minutes 5 times within 14 days or two 500 mg infusions on Day 1 and Day 14, administered over 3.5 to 4 hours). The mean age of the 91 treated patients in the Venofer group was 61.6 years (range 25 to 86 years) and 64 years (range 21 to 86 years) for the 91 patients in the oral iron group.
A statistically significantly greater proportion of Venofer subjects (35/79; 44.3%) compared to oral iron subjects (23/82; 28%) had an increase in hemoglobin ≥ 1 g/dL at anytime during the study (p = 0.03).
14.6 Study E: Peritoneal Dialysis Dependent-Chronic Kidney Disease (PDD-CKD)
Study E (NCT00236938) was a randomized, open-label, multicenter study comparing patients with PDD-CKD receiving an erythropoietin and intravenous iron to patients with PDD-CKD receiving an erythropoietin alone without iron supplementation. Patients with PDD-CKD, stable erythropoietin for 8 weeks, hemoglobin of ≤ 11.5 g/dL, TSAT ≤ 25%, ferritin ≤ 500 ng/mL were randomized to receive either no iron or Venofer (300 mg in 250 mL 0.9% NaCl over 1.5 hours on Day 1 and 15 and 400 mg in 250 mL 0.9% NaCl over 2.5 hours on Day 29). The mean age of the 75 treated patients in the Venofer / erythropoietin group was 51.9 years (range 21 to 81 years) vs. 52.8 years (range 23 to 77 years) for 46 patients in the erythropoietin alone group.
Patients in the Venofer / erythropoietin group had statistically significantly greater mean change from baseline to the highest hemoglobin value (1.3 g/dL), compared to subjects who received erythropoietin alone (0.6 g/dL) (p < 0.01). A greater proportion of subjects treated with Venofer / erythropoietin (59.1 %) had an increase in hemoglobin of ≥ 1 g/dL at any time during the study compared to the subjects who received erythropoietin only (33.3%).
14.7 Study F: Iron Maintenance Treatment Dosing in Pediatric Patients Ages 2 Years and Older with Chronic Kidney Disease
Study F (NCT00239642) was a randomized, open-label, dose-ranging study for iron maintenance treatment in pediatric patients with dialysis-dependent or non-dialysis-dependent CKD on stable erythropoietin therapy. The study randomized patients to one of three doses of Venofer (0.5 mg/kg, 1 mg/kg or 2 mg/kg). The mean age was 13 years (range 2 to 20 years). Over 70% of patients were 12 years or older in all three groups. There were 84 males and 61 females. About 60% of patients underwent hemodialysis and 25% underwent peritoneal dialysis in all three dose groups. At baseline, the mean hemoglobin was 12 g/dL, the mean TSAT was 33% and the mean ferritin was 300 ng/mL. Patients with HDD-CKD received Venofer once every other week for 6 doses. Patients with PDD-CKD or NDD-CKD received Venofer once every 4 weeks for 3 doses. Among 131 evaluable patients with stable erythropoietin dosing, the proportion of patients who maintained hemoglobin between 10.5 g/dL and 14.0 g/dL during the 12-week treatment period was 58.7%, 46.7%, and 45.0% in the Venofer 0.5 mg/kg, 1 mg/kg, and 2 mg/kg groups, respectively. A dose-response relationship was not demonstrated.
16. How is Venofer Injection supplied
16.1 How Supplied
Venofer is supplied sterile in 5 mL single-dose vials. Each 5 mL vial contains 100 mg elemental iron (20 mg/mL).
NDC-0517-2340-99 100 mg/5 mL Single-Dose Vial Packages of 10
16.2 Stability and Storage
Contains no preservatives. Store in original carton at 20°C to 25°C (68° F to 77° F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Do not freeze.
Syringe Stability: Venofer, when diluted with 0.9% NaCl at concentrations ranging from 2 mg to 10 mg of elemental iron per mL, or undiluted (20 mg elemental iron per mL) and stored in a plastic syringe, was found to be physically and chemically stable for 7 days at controlled room temperature (25°C ± 2°C) and under refrigeration (4°C ± 2°C).
Intravenous Admixture Stability: Venofer, when added to intravenous infusion bags (PVC or non-PVC) containing 0.9% NaCl at concentrations ranging from 1 mg to 2 mg of elemental iron per mL, has been found to be physically and chemically stable for 7 days at controlled room temperature (25°C ± 2°C).
Do not dilute to concentrations below 1 mg/mL.
Do not mix Venofer with other medications or add to parenteral nutrition solutions for intravenous infusion.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to infusion.
17. Patient Counseling Information
Prior History of Reactions to Parenteral Iron Products
Question patients regarding any prior history of reactions to parenteral iron products [see Warnings and Precautions (5)].
Serious Hypersensitivity Reactions
Advise patients to report any symptoms of hypersensitivity that may develop during and following Venofer administration, such as rash, itching, dizziness, light-headedness, swelling, and breathing problems [see Warnings and Precautions (5)].
AMERICAN REGENT, INC.
Shirley, NY USA 11967
Venofer is manufactured under license from Vifor (International) Inc., Switzerland.
PremierProRx® is a registered trademark of Premier Healthcare Alliance, L.P., used under license.

IN2340-99
MG #44828
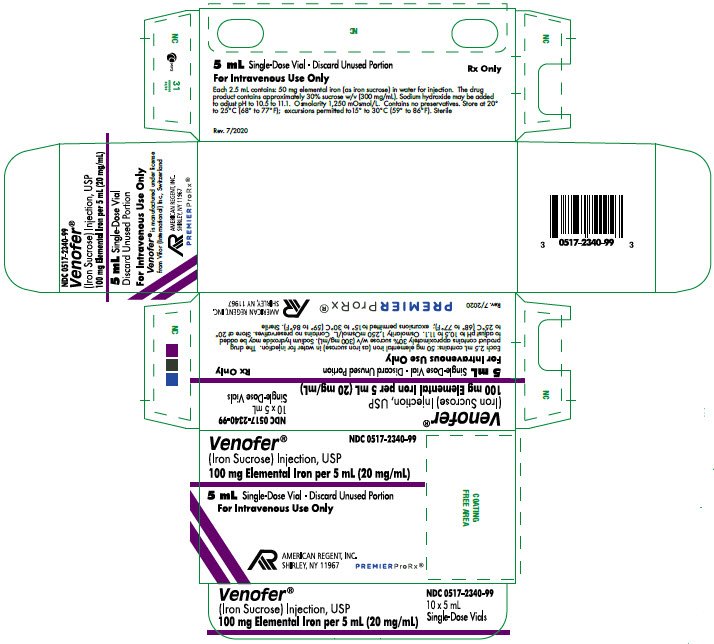
PRINCIPAL DISPLAY PANEL - Container Label
0517-2340-99
Venofer®
(Iron Sucrose) Injection, USP
100 Elemental Iron per 5 mL (20 mg/mL)
5 mL Single-Dose Vial
Discard Unused Portion
For Intravenous Use Only
Rx Only

PRINCIPAL DISPLAY PANEL - Carton Labeling
NDC 0517-2340-99
10 x 5 mL Single-Use Vials
VENOFER®
(Iron Sucrose) Injection, USP
100 mg Elemental Iron per 5 mL (20 mg/mL)
5 mL Single-Dose Vial - Discard Unused Portion
For Intravenous Use Only
Rx Only
Each 5 mL contains: 100 mg elemental iron (as iron sucrose) in water for injection. The drug product contains approximately 30% sucrose w/v (300 mg/mL). Sodium Hydroxide may be added to adjust pH to 10.5 to 11.1. Osmolarity 1,250 mOsmol/L. Contains no preservatives. Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F). Sterile.
AMERICAN
REGENT, INC.
SHIRLEY, NY 11967

Rev. 7/2020

| VENOFER
iron sucrose injection, solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - American Regent, Inc. (002033710) |
| Registrant - American Regent, Inc. (002033710) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| American Regent, Inc. | 002033710 | analysis(0517-2340) , manufacture(0517-2340) | |
More about Venofer (iron sucrose)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (41)
- Side effects
- Dosage information
- During pregnancy
- Drug class: iron products
- Breastfeeding
- En español