Tavneos: Package Insert / Prescribing Info
Package insert / product label
Generic name: avacopan
Dosage form: capsule, liquid filled
Drug class: Selective immunosuppressants
Medically reviewed by Drugs.com. Last updated on Jul 2, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
TAVNEOS® (avacopan) capsules, for oral use
Initial U.S. Approval: 2021
Indications and Usage for Tavneos
TAVNEOS is a complement 5a receptor (C5aR) antagonist indicated as an adjunctive treatment of adult patients with severe active anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. TAVNEOS does not eliminate glucocorticoid use. (1)
Tavneos Dosage and Administration
The recommended dosage is 30 mg (three 10 mg capsules) twice daily, with food. (2)
Dosage Forms and Strengths
Capsules: 10 mg (3)
Contraindications
Serious hypersensitivity to avacopan or to any of the excipients. (4)
Warnings and Precautions
- Hepatotoxicity: Increase in liver function tests occurred in clinical trials. Obtain liver function tests before initiation of therapy and monitor as clinically indicated. (5.1)
- Serious Hypersensitivity Reactions: Cases of angioedema occurred in a clinical trial. Observe for signs and symptoms of angioedema and manage accordingly. (5.2)
- Hepatitis B Virus (HBV) Reactivation: Cases of HBV reactivation occurred in a clinical trial. Withhold TAVNEOS and institute appropriate anti-infective therapy. (5.3)
- Serious Infections: Avoid use of TAVNEOS in patients with active, serious infection, including localized infections. (5.4)
Adverse Reactions/Side Effects
The most common adverse reactions (≥5%) are: nausea, headache, hypertension, diarrhea, vomiting, rash, fatigue, upper abdominal pain, dizziness, blood creatinine increased, and paresthesia. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Inc. at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
Drug Interactions
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2024
Full Prescribing Information
1. Indications and Usage for Tavneos
TAVNEOS is indicated as an adjunctive treatment of adult patients with severe active anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (granulomatosis with polyangiitis [GPA] and microscopic polyangiitis [MPA]) in combination with standard therapy including glucocorticoids. TAVNEOS does not eliminate glucocorticoid use.
2. Tavneos Dosage and Administration
2.1 Recommended Evaluations Prior to Treatment Initiation
Before initiating TAVNEOS, perform the following evaluations:
- Liver Function Tests: Obtain liver test panel (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin) before initiating TAVNEOS. TAVNEOS is not recommended for use in patients with cirrhosis, especially those with severe hepatic impairment (Child-Pugh C) [see Warnings and Precautions (5.1) and Use in Specific Populations (8.7)].
- Hepatitis B (HBV) Serology: Screen patients for HBV infection by measuring HBsAg and anti-HBc. For patients with evidence of prior or current HBV infection, consult with a physician with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy before or during treatment with TAVNEOS [see Warnings and Precautions (5.3)].
2.2 Recommended Dosage and Administration
The recommended dose of TAVNEOS is 30 mg (three 10 mg capsules) twice daily, with food.
Advise patients that TAVNEOS capsules should not be crushed, chewed or opened.
If a dose is missed, instruct the patient to wait until the usual scheduled time to take the next regular dose. Instruct the patient not to double the next dose.
3. Dosage Forms and Strengths
Capsules: 10 mg, opaque, yellow and light orange with CCX168 printed in black.
4. Contraindications
TAVNEOS is contraindicated in patients with serious hypersensitivity reaction to avacopan or to any of the excipients [see Warnings and Precautions (5.2)].
5. Warnings and Precautions
5.1 Hepatotoxicity
Serious cases of hepatic injury have been observed in patients taking TAVNEOS. During controlled trials, the TAVNEOS treatment group had a higher incidence of transaminase elevations and hepatobiliary events, including serious and life-threatening events [see Adverse Reactions 6.1].
Obtain liver test panel (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], alkaline phosphatase, and total bilirubin) before initiating TAVNEOS, every 4 weeks after start of therapy for the first 6 months of treatment and as clinically indicated thereafter.
If a patient receiving treatment with TAVNEOS presents with an elevation in ALT or AST to >3 times the upper limit of normal, evaluate promptly and consider pausing treatment as clinically indicated.
If AST or ALT is >5 times the upper limit of normal, or if a patient develops transaminases >3 times the upper limit of normal with elevation of bilirubin to >2 times the upper limit of normal, discontinue TAVNEOS until TAVNEOS-induced liver injury is ruled out [see Adverse Reactions (6.1)].
TAVNEOS is not recommended for patients with active, untreated and/or uncontrolled chronic liver disease (e.g., chronic active hepatitis B, untreated hepatitis C, uncontrolled autoimmune hepatitis) and cirrhosis. Consider the risk and benefit before administering TAVNEOS to a patient with liver disease. Monitor patients closely for hepatic adverse reactions [see Use in Specific Populations (8.7)].
5.2 Hypersensitivity Reactions
TAVNEOS may cause angioedema [see Adverse Reactions (6.1)]. In clinical trials, two cases of angioedema occurred, including one serious event requiring hospitalization. If angioedema occurs, discontinue TAVNEOS immediately, provide appropriate therapy, and monitor for airway compromise. TAVNEOS must not be re-administered unless another cause has been established. Educate patients on recognizing the signs and symptoms of a hypersensitivity reaction and to seek immediate medical care should they develop.
5.3 Hepatitis B Virus (HBV) Reactivation
Hepatitis B virus (HBV) reactivation, including life threatening hepatitis B, was observed in the clinical program.
HBV reactivation is defined as an abrupt increase in HBV replication, manifesting as a rapid increase in serum HBV DNA levels or detection of HBsAg, in a person who was previously HBsAg negative and anti-HBc positive. Reactivation of HBV replication is often followed by hepatitis, i.e., increase in transaminase levels. In severe cases, increase in bilirubin levels, liver failure, and death can occur.
Screen patients for HBV infection by measuring HBsAg and anti-HBc before initiating treatment with TAVNEOS. For patients who show evidence of prior hepatitis B infection (HBsAg positive [regardless of antibody status] or HBsAg negative but anti-HBc positive), consult physicians with expertise in managing hepatitis B regarding monitoring and consideration for HBV antiviral therapy before and/or during TAVNEOS treatment.
Monitor patients with evidence of current or prior HBV infection for clinical and laboratory signs of hepatitis, or HBV reactivation during and for six months following TAVNEOS therapy.
In patients who develop reactivation of HBV while on TAVNEOS, immediately discontinue TAVNEOS and any concomitant therapy associated with HBV reactivation, and institute appropriate treatment. Insufficient data exist regarding the safety of resuming TAVNEOS treatment in patients who develop HBV reactivation. Resumption of TAVNEOS treatment in patients whose HBV reactivation resolves should be discussed with physicians with expertise in managing HBV.
5.4 Serious Infections
Serious infections, including fatal infections, have been reported in patients receiving TAVNEOS. The most common serious infections reported in the TAVNEOS group were pneumonia and urinary tract infections.
Avoid use of TAVNEOS in patients with an active, serious infection, including localized infections. Consider the risks and benefits of treatment prior to initiating TAVNEOS in patients:
- with chronic or recurrent infection
- who have been exposed to tuberculosis
- with a history of a serious or an opportunistic infection
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses; or
- with underlying conditions that may predispose them to infection.
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with TAVNEOS. Interrupt TAVNEOS if a patient develops a serious or opportunistic infection. A patient who develops a new infection during treatment with TAVNEOS should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient; appropriate antimicrobial therapy should be initiated, the patient should be closely monitored, and TAVNEOS should be interrupted if the patient is not responding to antimicrobial therapy. TAVNEOS may be resumed once the infection is controlled.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Hepatitis B Virus (HBV) Reactivation [see Warnings and Precautions (5.3)]
- Serious Infections [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because the clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The identification of potential adverse drug reactions was based on safety data from the phase 3 clinical trial in which 330 patients with ANCA-associated vasculitis were randomized 1:1 to either TAVNEOS or prednisone [see Clinical Studies (14)]. The mean age of patients was 60.9 years (range of 13 to 88 years), with a predominance of men (56.4%) and Caucasians (84.2%). The cumulative exposure to TAVNEOS was 138.7 patient-years. Additionally, two phase 2 trials were conducted in ANCA-associated vasculitis. The cumulative clinical trial exposure from the phase 2 and 3 trials equals 212.3 patient-years.
The most frequent serious adverse reactions reported more frequently in patients treated with TAVNEOS than with prednisone were pneumonia (4.8% TAVNEOS vs. 3.7% prednisone), GPA (3.0% TAVNEOS vs. 0.6% prednisone), acute kidney injury (1.8% TAVNEOS vs. 0.6% prednisone), and urinary tract infection (1.8% TAVNEOS vs. 1.2% prednisone). Within 52 weeks, 4 patients in the prednisone treatment group (2.4%) and 2 patients in the TAVNEOS group (1.2%) died. There were no deaths in the phase 2 trials.
In the phase 3 trial, seven patients (4.2%) in the TAVNEOS treatment group and 2 patients (1.2%) in the prednisone treatment group discontinued treatment due to hepatic-related adverse reactions, including hepatobiliary adverse reactions and liver enzymes abnormalities. The most frequent adverse reaction that led to drug discontinuation reported by > 1 patient and more frequently reported in patients treated with TAVNEOS was hepatic function abnormal (1.8%).
The most common adverse reactions that occurred in ≥5% of patients and higher in the TAVNEOS group as compared with the prednisone group are listed in Table 1.
| Adverse Reaction | Prednisone (N = 164) n (%) | TAVNEOS (N = 166) n (%) |
|---|---|---|
| N = number of patients randomized to treatment group in the Safety Population; n = number of patients in specified category. | ||
| Nausea | 34 (20.7) | 39 (23.5) |
| Headache | 23 (14.0) | 34 (20.5) |
| Hypertension | 29 (17.7) | 30 (18.1) |
| Diarrhea | 24 (14.6) | 25 (15.1) |
| Vomiting | 21 (12.8) | 25 (15.1) |
| Rash | 13 (7.9) | 19 (11.4) |
| Fatigue | 15 (9.1) | 17 (10.2) |
| Upper abdominal pain | 10 (6.1) | 11 (6.6) |
| Dizziness | 10 (6.1) | 11 (6.6) |
| Blood creatinine increased | 8 (4.9) | 10 (6.0) |
| Paresthesia | 7 (4.3) | 9 (5.4) |
Hepatotoxicity and Elevated Liver Function Tests
In the phase 3 trial, a total of 19 patients (11.6%) in the prednisone group and 22 patients (13.3%) in the TAVNEOS group had hepatic-related adverse reactions, including hepatobiliary adverse reactions and liver enzyme abnormalities. Study medication was paused or discontinued permanently due to hepatic-related adverse reactions in 5 patients (3.0%) in the prednisone group and 9 patients (5.4%) in the TAVNEOS group. Serious hepatic-related adverse reactions were reported in 6 patients (3.7%) in the prednisone group and 9 patients (5.4%) in the TAVNEOS group. A serious hepatic-related adverse reaction was reported in 1 patient in the TAVNEOS group in the phase 2 studies.
Related/similar drugs
7. Drug Interactions
7.1 CYP3A4 Inducers
Avacopan exposure is decreased when co-administered with strong CYP3A4 enzyme inducers such as rifampin [see Clinical Pharmacology (12.3)]. Avoid coadministration of strong and moderate CYP3A4 inducers with TAVNEOS.
7.2 CYP3A4 Inhibitors
Avacopan exposure is increased when co-administered with strong CYP3A4 enzyme inhibitors such as itraconazole [see Clinical Pharmacology (12.3)]. Administer TAVNEOS 30 mg once daily when coadministered with strong CYP3A4 inhibitors.
7.3 CYP3A4 Substrates
Avacopan is a moderate CYP3A4 inhibitor. Co-administration of avacopan and 40 mg simvastatin increases the systemic exposure of simvastatin. While taking TAVNEOS, limit simvastatin dosage to 10 mg daily (or 20 mg daily for patients who have previously tolerated simvastatin 80 mg daily for at least one year without evidence of muscle toxicity). Consider dose reduction of CYP3A4 substrates when co-administering TAVNEOS with CYP3A4 substrates. Consult the concomitant CYP3A4 substrate product information when considering administration of such products together with TAVNEOS [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies with TAVNEOS in pregnant women to inform a drug-associated risk. In animal reproduction studies, oral administration of avacopan to pregnant hamsters and rabbits during the period of organogenesis produced no evidence of fetal harm with exposures up to approximately 5 and 0.6 times, respectively, the exposure at the maximum recommended human dose (MRHD) of 30 mg twice daily (on an area under the curve [AUC] basis). Avacopan caused an increase in the number of abortions in rabbits at an exposure 0.6 times the MRHD (see Animal Data).
The background risk of major birth defects and miscarriage for the indicated population are unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
In an embryo-fetal development study with pregnant hamsters dosed by the oral route during the period of organogenesis from gestation days 6 to 12, avacopan produced an increase in the incidence of a skeletal variation, described as supernumerary ribs, at an exposure that was 5 times the MRHD (on an AUC basis with a maternal oral dose of 1000 mg/kg/day). No structural abnormalities were noted with exposures up to 5 times the MRHD (on an AUC basis with maternal oral doses up to 1000 mg/kg/day).
In an embryo-fetal development study with pregnant rabbits dosed by the oral route during the period of organogenesis from gestation days 6 to 18, avacopan caused an increase in the number of abortions at an exposure 0.6 times the MRHD (on an AUC basis with a maternal oral dose of 200 mg/kg/day), however, no evidence of fetal harm was observed with such exposures. Maternal toxicity, as evidenced by decreased body weight gains, was observed at exposures 0.6 times and higher than the MRHD (on an AUC basis with maternal oral doses of 30 mg/kg/day and higher).
In a prenatal and postnatal development study with pregnant hamsters dosed by the oral route during the periods of gestation and lactation from gestation day 6 to lactation day 20, avacopan had no effects on the growth and development of offspring with exposures up to approximately 5 times the MRHD (on an AUC basis with maternal oral doses up to 1000 mg/kg/day).
8.2 Lactation
Risk Summary
There are no available data on the effects of avacopan on the breastfed child or on milk production. It is unknown whether avacopan is secreted in human milk. Avacopan was detected in the plasma of undosed hamster pups nursing from drug-treated dams (see Animal Data). The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TAVNEOS and any potential adverse effects on the breast-fed infant from TAVNEOS or from the underlying maternal condition.
Animal Data
Avacopan has not been measured in the milk of lactating animals; however, it was detected in the plasma of nursing offspring in a pre- and post-natal development study with hamsters at a pup to maternal plasma ratio of 0.37. This finding suggests that avacopan is secreted into the milk of lactating hamsters. [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of TAVNEOS in pediatric patients have not been established.
8.5 Geriatric Use
Of the 86 geriatric patients who received TAVNEOS in the phase 3 randomized clinical trial for ANCA-associated vasculitis [see Clinical Studies (14)], 62 patients were between 65-74 years and 24 were 75 years or older. No overall differences in safety or effectiveness were observed between geriatric patients and younger patients.
8.6 Patients with Renal Impairment
No dose adjustment is required for patients with mild, moderate, or severe renal impairment [see Clinical Pharmacology (12.3)]. TAVNEOS has not been studied in patients with ANCA-associated vasculitis who are on dialysis.
11. Tavneos Description
TAVNEOS (avacopan) capsules contain avacopan, a C5aR antagonist. Avacopan is a chiral molecule containing two stereocenters and has a chemical name of (2R,3S)-2-[4-(cyclopentylamino)phenyl]-1-(2-fluoro-6-methylbenzoyl)-N-[4-methyl-3-(trifluoromethyl)phenyl]piperidine-3-carboxamide. It has a molecular formula of C33H35F4N3O2 and a molecular weight of 582 g/mol. Avacopan has the following structural formula:
Avacopan is a white to pale yellow crystalline solid that is soluble in organic solvents and practically insoluble in water.
TAVNEOS is available as a 10 mg capsule for oral administration. The capsules include the following inactive ingredients: Polyethylene glycol 4000 (PEG-4000), Polyoxyl-40 hydrogenated castor oil. The capsules are a light orange and yellow opaque bicolor gelatin capsule with a clear gelatin sealing band. The top half of the capsule is printed with "CCX168" in black ink. The capsule shell contains gelatin, red iron oxide, yellow iron oxide, and titanium dioxide, and the capsule sealing band contains gelatin and polysorbate 80.
12. Tavneos - Clinical Pharmacology
12.1 Mechanism of Action
Avacopan is a complement 5a receptor (C5aR) antagonist that inhibits the interaction between C5aR and the anaphylatoxin C5a. Avacopan blocks C5a-mediated neutrophil activation and migration. The precise mechanism by which avacopan exerts a therapeutic effect in patients with ANCA-associated vasculitis has not been definitively established.
12.2 Pharmacodynamics
Avacopan blocks the C5a-induced upregulation of CD11b (integrin alpha M) on neutrophils taken from humans dosed with avacopan. The clinical significance of the pharmacodynamic effect is unclear.
12.3 Pharmacokinetics
Based on population pharmacokinetic analysis, the mean steady state plasma exposure estimates of avacopan are 3466 ± 1921 ng∙h/mL for the 12-hour area under the plasma drug concentration over time curve (AUC0-12hr) and 349 ± 169 ng/mL for the maximum plasma concentration (Cmax) in patients with ANCA-associated vasculitis receiving 30 mg avacopan twice daily. Steady state plasma levels of avacopan are reached by 13 weeks and the accumulation is approximately 4-fold.
Absorption
Co-administration of 30 mg in capsule formulation with a high-fat, high-calorie meal increases AUC and Cmax of avacopan by approximately 72% and 8%, respectively, and delays tmax by approximately 4 hours (from 2.0 hours to 6.0 hours).
Distribution
The plasma protein binding (e.g., to albumin and α1-acid glycoprotein) of avacopan and metabolite M1 is greater than 99.9%. The apparent volume of distribution of avacopan is estimated to be 345 L.
Elimination
Based on population pharmacokinetic analysis, the estimated total apparent body clearance (CL/F) of avacopan is 16.3 L/h. Following a single dose of 30 mg avacopan with food, the mean elimination half-lives of avacopan and M1 are 97.6 hours and 55.6 hours, respectively, in healthy subjects.
Metabolism
CYP3A4 is the major enzyme responsible for the clearance of avacopan and for the formation and clearance of the major circulating metabolite M1, a mono-hydroxylated product of avacopan. M1 was present at ~12% of the total drug-related materials in plasma and has approximately the same activity as avacopan on the C5aR.
Excretion
The main route of clearance of avacopan is metabolism followed by biliary excretion of the metabolites into feces. Following oral administration of radiolabelled avacopan, about 77% and 10% of the radioactivity was recovered in feces and urine, respectively, and 7% and <0.1% of the radioactive dose was recovered as unchanged avacopan in feces and urine, respectively.
Specific Populations
No clinically significant differences in plasma exposure of avacopan and metabolite M1 were observed based on race (White, Asian, Black), gender (female 31%), age (18 to 83 years), body weight (40.3-174 kg), and renal function (eGFR 14-170 mL/min/1.73 m2 at baseline).
Patients with Hepatic Impairment
Mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment had no clinically relevant effect on avacopan and M1 plasma exposure. In subjects with mild or moderate hepatic impairment, avacopan AUC increased by 12% and 12%, respectively, Cmax decreased by 13% and 17%, respectively, compared to subjects with normal liver function. In subjects with mild or moderate hepatic impairment, M1 AUC increased by 11% and 18%, respectively, Cmax decreased by 5% and 16%, respectively, compared to subjects with normal liver function.
TAVNEOS has not been studied in subjects with severe hepatic impairment (Child-Pugh Class C).
Drug Interaction Studies
Effects of Other Drugs on TAVNEOS
Avacopan is primarily metabolized by CYP3A4. In vitro studies indicate that avacopan is not a substrate of BCRP and P-gp efflux, and OATP1B1 and OATP1B3 uptake transporters. M1 is a substrate of P-gp but is not a substrate of BCRP efflux, and OATP1B1 and OATP1B3 uptake transporters. Summary of results from a clinical study which evaluated the effect of co-administered drugs on avacopan and M1 plasma exposures is shown in Table 2.
| Co-administered Drug | Regimen of Co-administered Drug | Ratio (90% CI)* | ||
|---|---|---|---|---|
| Cmax | AUC | |||
| CI: Confidence interval | ||||
|
||||
| Strong CYP3A4 inhibitor: itraconazole | 200 mg once daily for 4 days | Avacopan | 1.87 (1.70, 2.06) | 2.19 (2.00, 2.41) |
| M1 | 1.03 (0.95, 1.11) | 1.19 (1.11, 1.28) |
||
| Strong CYP3A4 inducer: rifampin | 600 mg once daily for 11 days | Avacopan | 0.21 (0.18, 0.25) | 0.07 (0.06, 0.10) |
| M1 | 0.27 (0.23, 0.31) | 0.07 (0.06, 0.09) |
||
Proton-pump inhibitors such as omeprazole are not expected to have a clinically relevant effect on avacopan plasma exposure.
Effect of TAVNEOS on Other Drug Substances
In vitro studies indicate that avacopan does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, and CYP2D6, and does not induce CYP1A2 and CYP2B6, but shows induction and time-dependent inhibition of CYP3A4. In vitro studies indicate that M1 does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C19, and CYP2D6, and has a low potential to induce CYP3A4, CYP1A2 and CYP2B6, but may inhibit CYP2C9 and CYP3A4.
In vitro studies indicate that avacopan and M1 do not inhibit the transporters P-gp, BCRP, OATP1B1, OATP1B3, OCT2, OAT1, OAT3, MATE1, and MATE2K at clinically relevant concentrations.
Summary of results from clinical studies which evaluated the effect of avacopan on other drugs is shown in Table 3.
| Co-administered Drug | Regimen of Avacopan | Ratio (90% CI)* | |
|---|---|---|---|
| Cmax | AUC | ||
| CI: Confidence interval | |||
|
|||
| Sensitive CYP3A4 substrate: simvastatin | 60 mg twice daily for 7 days (fed)† | 3.20 (2.49, 4.10) | 3.53 (3.23, 3.85) |
| Sensitive CYP3A4 substrate: midazolam | 30 mg twice daily for 11 days (fasted)‡ | 1.55 (1.41, 1.69) | 1.81 (1.65, 1.98) |
| Sensitive CYP2C9 substrate: celecoxib | 30 mg twice daily for 11 days (fasted)‡ | 1.64 (1.34, 2.00) | 1.15 (1.03, 1.28) |
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies in Sprague-Dawley rats and Syrian hamsters were conducted to assess the carcinogenic potential of avacopan. Avacopan demonstrated no tumorigenic potential in a study with rats that received oral doses up to 100 mg/kg/day (approximately 3 times the MRHD in adults on an AUC basis) and a study in hamsters that received oral doses up to 100 mg/kg/day (approximately 6 times the MRHD in adults on an AUC basis).
14. Clinical Studies
The efficacy and safety of TAVNEOS was evaluated in a double-blind, active-controlled, phase 3 clinical trial (NCT02994927) in 330 patients with newly diagnosed or relapsed ANCA-associated vasculitis who were randomized 1:1 to one of the following treatment groups:
- TAVNEOS group (N = 166): Patients received 30 mg avacopan twice daily for 52 weeks plus prednisone-matching placebo for 20 weeks
- Prednisone group (N = 164): Patients received avacopan-matched placebo twice daily for 52 weeks plus prednisone (tapered from 60 mg/day to 0 over 20 weeks)
All patients in both groups received one of the following standard immunosuppressive regimens:
- IV cyclophosphamide 15 mg/kg IV up to 1.2 g maximum every 2 to 3 weeks for 13 weeks followed by oral azathioprine 1 mg/kg/day with titration up to 2 mg/kg/day (or mycophenolate mofetil at a target dose of 2 g/day if azathioprine was contraindicated) from Week 15 onwards
- Oral cyclophosphamide 2 mg/kg/day (maximum 200 mg/day) for 14 weeks followed by azathioprine 1 mg/kg/day with titration up to 2 mg/kg/day (or mycophenolate mofetil at a target dose of 2 g/day if azathioprine was contraindicated) from Week 15 onwards
- IV rituximab 375 mg/m2 once weekly for 4 weeks without azathioprine or mycophenolate mofetil
Glucocorticoids were allowed as pre-medication for rituximab to reduce hypersensitivity reactions, taper after glucocorticoids given during the Screening period, treatment of persistent vasculitis, worsening of vasculitis, or relapses, as well as for non-vasculitis reasons such as adrenal insufficiency.
Randomization was stratified based on 3 factors: newly-diagnosed or relapsing ANCA-associated vasculitis, proteinase 3 positive or myeloperoxidase positive ANCA-associated vasculitis, and standard immunosuppressive regimen. The primary endpoints of the study were disease remission at Week 26 and sustained disease remission at Week 52. Disease remission was defined as achieving a Birmingham Vasculitis Activity Score (BVAS) of 0 and no use of glucocorticoids for treatment of ANCA-associated vasculitis from Week 22 to Week 26. Sustained remission was defined as remission at Week 26 and remission at Week 52, without relapse between Week 26 and Week 52. Remission at Week 52 was defined as BVAS of 0 and no use of glucocorticoids for treatment of ANCA-associated vasculitis from Week 48 to Week 52. Relapse was defined as occurrence of one major item, at least 3 non-major items, or 1 or 2 non-major items for at least 2 consecutive visits on the BVAS after remission (BVAS of 0) had been achieved.
The two treatment groups were well balanced regarding baseline demographics and disease characteristics of patients in this trial. The mean patient age was 60.9 years. Most patients were male (56.4%), Caucasian (84.2%), and had newly diagnosed disease (69.4%). Patients had either GPA (54.8%) or MPA (45.2%) and had presence of anti-PR3 (43.0%) or anti-MPO (57.0%) antibodies. Mean baseline BVAS was 16.2; patients most commonly had manifestations within the renal component (81.2%), general component (68.2%), ear/nose/throat component (43.6%), and chest component (43.0%). Approximately 65% of patients received rituximab, 31% received IV cyclophosphamide, and 4% received oral cyclophosphamide.
Remission at Week 26 and Sustained Remission at Week 52
Remission was achieved by 72.3% of patients in the TAVNEOS group and 70.1% of patients in the prednisone group at Week 26 (treatment difference: 3.4%, 95% CI [-6.0%, 12.8%]). At Week 52, a significantly higher percentage of patients had sustained remission in TAVNEOS group (65.7%) compared to the prednisone group (54.9%), as presented in Table 4.
| Prednisone (N = 164) n (%) | TAVNEOS (N = 166) n (%) | Estimate of Treatment Difference | P-value* | |
|---|---|---|---|---|
| CI = confidence interval; N = number of patients in the analysis population for the specified treatment group; n = number of patients with disease remission; % = 100*n/N | ||||
|
||||
| Sustained Remission at Week 52 | 90 (54.9%) | 109 (65.7%) | 12.5%† | 0.013 |
| 95% CI | (46.9, 62.6)‡ | (57.9, 72.8)‡ | (2.6, 22.3)§ | |
In pre-specified subgroup efficacy analyses, sustained remission at 52 weeks in patients was examined based on stratification factors and GPA/MPA disease. The results are displayed in Figure 1 below.
Figure 1. Forest Plot of Sustained Remission at Week 52 Based on Disease Related Variables
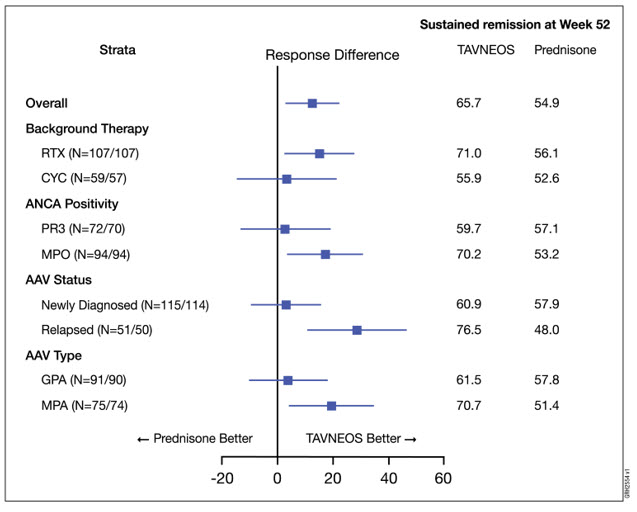
AAV = ANCA-associated vasculitis, CYC = cyclophosphamide, GPA = granulomatosis with polyangiitis, MPA = microscopic polyangiitis; MPO = myeloperoxidase positive, PR3 = anti-proteinase 3 positive, and RTX = rituximab. The treatment difference between TAVNEOS and prednisone groups is presented with point estimate and 95% confidence interval using normal approximation. The notation N = XXX/YYY indicates the number of patients randomized who received at least one dose of drug in TAVNEOS arm and prednisone arm, respectively. Subgroup findings should be interpreted with caution due to small sample sizes and overlapping subgroups.
16. How is Tavneos supplied
TAVNEOS (avacopan) capsule is supplied as a 10 mg, hard, opaque yellow and light orange capsule with "CCX168" printed in black.
- Bottle containing 180 capsules with child resistant induction seal closure (NDC 73556-168-01)
- Bottle containing 30 capsules with child resistant induction seal closure (NDC 73556-168-02)
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
- Dosage and Administration: Instruct the patient that TAVNEOS should be swallowed whole. TAVNEOS should not be chewed or crushed. If a dose is missed, instruct the patient to take the next scheduled dose [see Dosage and Administration (2.2)].
- Hypersensitivity Reactions: Advise patients to seek immediate medical attention when experiencing any signs or symptoms suggesting angioedema (swelling of face, extremities, eyes, lips, tongue, and difficulty in swallowing or breathing) and to discontinue the drug until they have consulted with the prescribing physician [see Warnings and Precautions (5.2)].
- Hepatotoxicity: Inform patients of the signs and symptoms of hepatic adverse reactions. Advise patients to contact their healthcare provider immediately for signs or symptoms of liver problems; yellowing of the skin or the white part of the eyes (jaundice), dark or brown (tea colored) urine, pain on the upper right side of the stomach area (abdomen), bleeding or bruising [see Warnings and Precautions (5.1)].
- Infections: Inform patients that serious infections have been reported in patients receiving TAVNEOS, including reactivation of hepatitis B infection. Instruct patients to contact their healthcare provider immediately if they develop any signs or symptoms of an infection [see Warnings and Precautions (5.4)].
- Lactation: Consider benefits/risk during lactation [see Use in Specific Populations (8.2)].
TAVNEOS® (avacopan)
Manufactured for:
ChemoCentryx, Inc.
One Amgen Center Drive
Thousand Oaks, CA 91320-1799
Patent: https://pat.amgen.com/tavneos
© 2021, 2024 ChemoCentryx, Inc. All rights reserved.
1xxxxxx – v3
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 8/2023 | |||
| MEDICATION GUIDE TAVNEOS® (tav' nee ose) (avacopan) capsules, for oral use |
||||
|
What is the most important information I should know about TAVNEOS? TAVNEOS may cause serious side effects, including:
|
||||
|
|
|||
| Your healthcare provider will do blood tests to check how well your liver is working before starting and during your treatment with TAVNEOS. | ||||
What is TAVNEOS?
|
||||
Do not take TAVNEOS:
|
||||
Before taking TAVNEOS, tell your healthcare provider about all your medical conditions including if you:
|
||||
How should I take TAVNEOS?
|
||||
| What are the possible side effects of TAVNEOS?
TAVNEOS may cause serious side effects, including:
|
||||
|
|
|
||
| These are not all the possible side effects of TAVNEOS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||||
| How should I store TAVNEOS?
Store TAVNEOS capsules at room temperature between 68°F to 77°F (20°C to 25°C). Keep TAVNEOS and all medicines out of reach of children. |
||||
| General information about the safe and effective use of TAVNEOS.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TAVNEOS for a condition for which it was not prescribed. Do not give TAVNEOS to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information that is written for healthcare professionals. |
||||
| What are the ingredients in TAVNEOS? Active ingredient: avacopan Inactive ingredients: Polyethylene glycol 4000 (PEG-4000), Polyoxyl-40 hydrogenated castor oil. |
||||
| Manufactured for: ChemoCentryx, Inc., One Amgen Center Drive, Thousand Oaks, CA 91320-1799 Patent: https://pat.amgen.com/tavneos © 2021, 2023 ChemoCentryx, Inc. All rights reserved. 1xxxxxx - v2 For more information, go to www.TAVNEOS.com or call Amgen at 1-800-772-6436. |
||||


| TAVNEOS
avacopan capsule |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - ChemoCentryx, Inc. (002356900) |
More about Tavneos (avacopan)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (4)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: selective immunosuppressants
- Breastfeeding
- En español
