Synercid: Package Insert / Prescribing Info
Package insert / product label
Generic name: quinupristin and dalfopristin
Dosage form: injection, powder, lyophilized, for solution
Drug class: Streptogramins
Medically reviewed by Drugs.com. Last updated on Jul 10, 2025.
On This Page
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Synercid and other antibacterial drugs, Synercid should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
Synercid Description
Synercid® (quinupristin and dalfopristin powder for injection) I.V., a streptogramin antibacterial agent for intravenous administration, is a sterile lyophilized formulation of two semisynthetic pristinamycin derivatives, quinupristin (derived from pristinamycin I) and dalfopristin (derived from pristinamycin IIA) in the ratio of 30:70 (w/w).
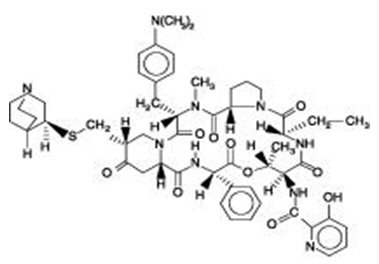
Quinupristin is a white to very slightly yellow, hygroscopic powder. It is a combination of three peptide macrolactones. The main component of quinupristin (> 88.0%) has the following chemical name: N-[(6R,9S,10R,13S,15aS,18R,22S,24aS )-22-[p-(dimethylamino)benzyl]-6-ethyldocosahydro-10,23-dimethyl-5,8,12,15,17,21,24-heptaoxo-13-phenyl-18-[[(3S )-3-quinuclidinylthio] methyl]-12H-pyrido[2,1-f ]pyrrolo-[2,1-l ][1,4,7,10,13,16] oxapentaazacyclononadecin-9-yl]-3-hydroxypicolinamide.
The main component of quinupristin has an empirical formula of C53H67N9O10S, a molecular weight of 1022.24 and the following structural formula:
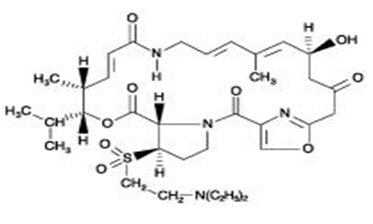
Dalfopristin is a slightly yellow to yellow, hygroscopic, powder. The chemical name for dalfopristin is: (3R,4R,5E,10E,12E,14S,26R,26aS )-26-[[2-(diethylamino)ethyl]sulfonyl]-8,9,14,15,24,25,26,26a-octahydro-14-hydroxy-3-isopropyl-4,12-dimethyl-3H-21,18-nitrilo-1H,22H-pyrrolo[2,1-c ][1,8,4,19]-dioxadiazacyclotetracosine-1,7,16,22(4H,17H )-tetrone.
Dalfopristin has an empirical formula of C34H50N4O9S, a molecular weight of 690.85 and the following structural formula:
Synercid - Clinical Pharmacology
Pharmacokinetics
Quinupristin and dalfopristin are the main active components circulating in plasma in human subjects. Quinupristin and dalfopristin are converted to several active major metabolites: two conjugated metabolites for quinupristin (one with glutathione and one with cysteine) and one non-conjugated metabolite for dalfopristin (formed by drug hydrolysis).
Pharmacokinetic profiles of quinupristin and dalfopristin in combination with their metabolites were determined using a bioassay following multiple 60-minute infusions of Synercid in two groups of healthy young adult male volunteers. Each group received 7.5 mg/kg of Synercid intravenously q12h or q8h for a total of 9 or 10 doses, respectively. The pharmacokinetic parameters were proportional with q12h and q8h dosing; those of the q8h regimen are shown in Table 1:
| Cmax†
(mcg/mL) | AUC‡
(mcg.h/mL) | t 1/2§(hr) | |
|---|---|---|---|
| Quinupristin and metabolites | 3.20 ± 0.67 | 7.20 ± 1.24 | 3.07 ± 0.51 |
| Dalfopristin and metabolite | 7.96 ± 1.30 | 10.57 ± 2.24 | 1.04 ± 0.20 |
The clearances of unchanged quinupristin and dalfopristin are similar (0.72 L/h/kg), and the steady-state volume of distribution for quinupristin is 0.45 L/kg and for dalfopristin is 0.24 L/kg. The elimination half-life of quinupristin and dalfopristin is approximately 0.85 and 0.70 hours, respectively.
The total protein binding of quinupristin is higher than that of dalfopristin. Synercid does not alter the in vitro binding of warfarin to proteins in human serum.
Penetration of unchanged quinupristin and dalfopristin in noninflammatory blister fluid corresponds to about 19% and 11% of that estimated in plasma, respectively. The penetration into blister fluid of quinupristin and dalfopristin in combination with their major metabolites was in total approximately 40% compared to that in plasma.
In vitro, the transformation of the parent drugs into their major active metabolites occurs by non-enzymatic reactions and is not dependent on cytochrome-P450 or glutathione-transferase enzyme activities.
Synercid has been shown to be a major inhibitor (in vitro inhibits 70% cyclosporin A biotransformation at 10 mcg/mL of Synercid) of the activity of cytochrome P450 3A4 isoenzyme. (See WARNINGS.)
Synercid can interfere with the metabolism of other drug products that are associated with QTc prolongation. However, electrophysiologic studies confirm that Synercid does not itself induce QTc prolongation. (See WARNINGS.)
Fecal excretion constitutes the main elimination route for both parent drugs and their metabolites (75 to 77% of dose). Urinary excretion accounts for approximately 15% of the quinupristin and 19% of the dalfopristin dose. Preclinical data in rats have demonstrated that approximately 80% of the dose is excreted in the bile and suggest that in man, biliary excretion is probably the principal route for fecal elimination.
Special Populations
Elderly
The pharmacokinetics of quinupristin and dalfopristin were studied in a population of elderly individuals (range 69 to 74 years). The pharmacokinetics of the drug products were not modified in these subjects.
Renal Insufficiency
In patients with creatinine clearance 6 to 28 mL/min, the AUC of quinupristin and dalfopristin in combination with their major metabolites increased about 40% and 30%, respectively.
In patients undergoing Continuous Ambulatory Peritoneal Dialysis, dialysis clearance for quinupristin, dalfopristin and their metabolites is negligible. The plasma AUC of unchanged quinupristin and dalfopristin increased about 20% and 30%, respectively. The high molecular weight of both components of Synercid suggests that it is unlikely to be removed by hemodialysis.
Hepatic Insufficiency
In patients with hepatic dysfunction (Child-Pugh scores A and B), the terminal half-life of quinupristin and dalfopristin was not modified. However, the AUC of quinupristin and dalfopristin in combination with their major metabolites increased about 180% and 50%, respectively. (See DOSAGE AND ADMINISTRATION and PRECAUTIONS.)
Obesity (body mass index ≥30): In obese patients the Cmax and AUC of quinupristin increased about 30% and those of dalfopristin about 40%.
Microbiology
The streptogramin components of Synercid, quinupristin and dalfopristin, are present in a ratio of 30 parts quinupristin to 70 parts dalfopristin. These two components act synergistically so that Synercid's microbiologic in vitro activity is greater than that of the components individually. Quinupristin's and dalfopristin's metabolites also contribute to the antimicrobial activity of Synercid. In vitro synergism of the major metabolites with the complementary parent compound has been demonstrated.
Mechanism of Action
The site of action of quinupristin and dalfopristin is the bacterial ribosome. Dalfopristin has been shown to inhibit the early phase of protein synthesis while quinupristin inhibits the late phase of protein synthesis. Synercid is bactericidal against isolates of methicillin-susceptible and methicillin-resistant staphylococci. The mode of action of Synercid differs from that of other classes of antibacterial agents such as ß-lactams, aminoglycosides, glycopeptides, quinolones, macrolides, lincosamides and tetracyclines. Therefore, there is no cross resistance between Synercid and these agents when tested by the minimum inhibitory concentration (MIC) method.
Resistance
Resistance to Synercid is associated with resistance to both components (i.e., quinupristin and dalfopristin). In non-comparative studies, emerging resistance to Synercid has occurred.
Interaction with other Antibacterials
In vitro combination testing of Synercid with aztreonam, cefotaxime, ciprofloxacin, and gentamicin against Enterobacteriaceae and Pseudomonas aeruginosa did not show antagonism.
In vitro combination testing of Synercid with prototype drugs of the following classes: aminoglycosides (gentamicin), β-lactams (cefepime, ampicillin, and amoxicillin), glycopeptides (vancomycin), quinolones (ciprofloxacin), tetracyclines (doxycycline) and also chloramphenicol against enterococci and staphylococci did not show antagonism.
Antimicrobial Activity
Synercid has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections, as described in the INDICATIONS AND USAGE section.
Gram-positive bacteria
Staphylococcus aureus (methicillin-susceptible isolates only)
Streptococcus pyogenes
The following in vitro data are available, but their clinical significance is unknown.
At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for quinupristin and dalfopristin (Synercid) against isolates of similar genus or organism group. However, the efficacy of Synercid in treating clinical infections due to these bacteria has not been established in adequate and well-controlled clinical trials.
Gram-positive bacteria
Corynebacterium jeikeium
Staphylococcus aureus (methicillin-resistant isolates)
Staphylococcus epidermidis (including methicillin-resistant isolates)
Streptococcus agalactiae
Indications and Usage for Synercid
To reduce the development of drug-resistant bacteria and maintain the effectiveness of Synercid and other antibacterial drugs, Synercid should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Synercid is indicated in adults for the treatment of the following infections when caused by susceptible strains of the designated microorganisms.
Complicated skin and skin structure infections caused by Staphylococcus aureus (methicillin susceptible) or Streptococcus pyogenes. (See CLINICAL STUDIES.)
Contraindications
Synercid is contraindicated in patients with known hypersensitivity to Synercid, or with prior hypersensitivity to other streptogramins (e.g., pristinamycin or virginiamycin).
Warnings
Drug Interactions
In vitro drug interaction studies have demonstrated that Synercid significantly inhibits cytochrome P450 3A4 metabolism of cyclosporin A, midazolam, nifedipine and terfenadine. In addition, 24 subjects given Synercid 7.5 mg/kg q8h for 2 days and 300 mg of cyclosporine on day 3 showed an increase of 63% in the AUC of cyclosporine, an increase of 30% in the Cmax of cyclosporine, a 77% increase in the t1/2 of cyclosporine, and, a decrease of 34% in the clearance of cyclosporine. Therapeutic level monitoring of cyclosporine should be performed when cyclosporine must be used concomitantly with Synercid.
It is reasonable to expect that the concomitant administration of Synercid and other drugs primarily metabolized by the cytochrome P450 3A4 enzyme system may likely result in increased plasma concentrations of these drugs that could increase or prolong their therapeutic effect and/or increase adverse reactions. (See Table below.) Therefore, coadministration of Synercid with drugs which are cytochrome P450 3A4 substrates and possess a narrow therapeutic window requires caution and monitoring of these drugs (e.g., cyclosporine), whenever possible. Concomitant medications metabolized by the cytochrome P450 3A4 enzyme system that may prolong the QTc interval should be avoided.
Concomitant administration of Synercid and nifedipine (repeated oral doses) and midazolam (intravenous bolus dose) in healthy volunteers led to elevated plasma concentrations of these drugs. The Cmax increased by 18% and 14% (median values) and the AUC increased by 44% and 33% for nifedipine and midazolam, respectively.
|
| Antihistamines: astemizole, terfenadine |
| Anti-HIV (NNRTIs and Protease inhibitors): delavirdine, nevirapine, indinavir, ritonavir |
| Antineoplastic agents: vinca alkaloids (e.g., vinblastine), docetaxel, paclitaxel |
| Benzodiazepines: midazolam, diazepam |
| Calcium channel blockers: dihydropyridines (e.g., nifedipine), verapamil, diltiazem |
| Cholesterol-lowering agents: HMG-CoA reductase inhibitors (e.g., lovastatin) |
| GI motility agents: cisapride |
| Immunosuppressive agents: cyclosporine, tacrolimus |
| Steroids: methylprednisolone |
| Other: carbamazepine, quinidine, lidocaine, disopyramide |
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including Synercid, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
Precautions
General
Prescribing Synercid in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
Venous Irritation
Following completion of a peripheral infusion, the vein should be flushed with 5% Dextrose in Water solution to minimize venous irritation. DO NOT FLUSH with saline or heparin after Synercid administration because of incompatibility concerns.
If moderate to severe venous irritation occurs following peripheral administration of Synercid diluted in 250 mL of Dextrose 5% in water, consideration should be given to increasing the infusion volume to 500 or 750 mL, changing the infusion site, or infusing by a peripherally inserted central catheter (PICC) or a central venous catheter. In clinical trials, concomitant administration of hydrocortisone or diphenhydramine did not appear to alleviate venous pain or inflammation.
Rate of Infusion
In animal studies toxicity was higher when Synercid was administered as a bolus compared to slow infusion. However, the safety of an intravenous bolus of Synercid has not been studied in humans. Clinical trial experience has been exclusively with an intravenous duration of 60 minutes and, thus, other infusion rates cannot be recommended.
Arthralgias/Myalgias
Episodes of arthralgia and myalgia, some severe, have been reported in patients treated with Synercid. In some patients, improvement has been noted with a reduction in dose frequency to q12h. In those patients available for follow-up, treatment discontinuation has been followed by resolution of symptoms. The etiology of these myalgias and arthralgias is under investigation.
Superinfections
The use of antibiotics may promote the overgrowth of nonsusceptible organisms. Should superinfection occur during therapy, appropriate measures should be taken.
Hyperbilirubinemia
Elevations of total bilirubin greater than 5 times the upper limit of normal were noted in approximately 25% of patients in the non-comparative studies. (See ADVERSE REACTIONS: Non-Comparative Trials.) In some patients, isolated hyperbilirubinemia (primarily conjugated) can occur during treatment, possibly resulting from competition between Synercid and bilirubin for excretion. Of note, in the comparative trials, elevations in ALT and AST occurred at a similar frequency in both the Synercid and comparator groups.
Information for Patients
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Patients should be counseled that antibacterial drugs including Synercid should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When Synercid is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by Synercid or other antibacterial drugs in the future.
Drug Interactions
In vitro drug interaction studies have shown that Synercid significantly inhibits cytochrome P450 3A4. (See WARNINGS.)
Synercid does not significantly inhibit human cytochrome P450 1A2, 2A6, 2C9, 2C19, 2D6, or 2E1. Therefore, clinical interactions with drugs metabolized by these cytochrome P450 isoenzymes are not expected.
A drug interaction between Synercid and digoxin cannot be excluded but is unlikely to occur via CYP3A4 enzyme inhibition. Synercid has shown in vitro activity (MICs of 0.25 mcg/mL when tested on two strains) against Eubacterium lentum. Digoxin is metabolized in part by bacteria in the gut and as such, a drug interaction based on Synercid's inhibition of digoxin's gut metabolism (by Eubacterium lentum) may be possible.
In vitro combination testing of Synercid with aztreonam, cefotaxime, ciprofloxacin, and gentamicin, against Enterobacteriaceae and Pseudomonas aeruginosa did not show antagonism.
In vitro combination testing of Synercid with prototype drugs of the following classes: aminoglycosides (gentamicin), β-lactams (cefepime, ampicillin, and amoxicillin), glycopeptides (vancomycin), quinolones (ciprofloxacin), tetracyclines (doxycycline) and also chloramphenicol against enterococci and staphylococci did not show antagonism.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies in animals have not been conducted with Synercid. Five genetic toxicity tests were performed. Synercid, dalfopristin, and quinupristin were tested in the bacterial reverse mutation assay, the Chinese hamster ovary cell HGPRT gene mutation assay, the unscheduled DNA synthesis assay in rat hepatocytes, the Chinese hamster ovary cell chromosome aberration assay, and the mouse micronucleus assay in bone marrow. Dalfopristin was associated with the production of structural chromosome aberrations when tested in the Chinese hamster ovary cell chromosome aberration assay. Synercid and quinupristin were negative in this assay. Synercid, dalfopristin, and quinupristin were all negative in the other four genetic toxicity assays.
No impairment of fertility or perinatal/postnatal development was observed in rats at doses up to 12 to 18 mg/kg (approximately 0.3 to 0.4 times the human dose based on body-surface area).
Pregnancy
Teratogenic Effects
Reproductive studies have been performed in mice at doses up to 40 mg/kg/day (approximately half the human dose based on body-surface area), in rats at doses up to 120 mg/kg/day (approximately 2.5 times the human dose based on body-surface area), and in rabbits at doses up to 12 mg/kg/day (approximately half the human dose based on body-surface area) and have revealed no evidence of impaired fertility or harm to the fetus due to Synercid.
There are, however, no adequate and well-controlled studies with Synercid in pregnant women. Because animal reproduction studies are not always predictive of the human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
In lactating rats, Synercid was excreted in milk. It is not known whether Synercid is excreted in human breast milk. Because many drugs are excreted in human milk, caution should be exercised when Synercid is administered to a nursing woman.
Hepatic Insufficiency
Following a single 1-hour infusion of Synercid (7.5 mg/kg) to patients with hepatic insufficiency, plasma concentrations were significantly increased. (See CLINICAL PHARMACOLOGY: Special Populations.) However, the effect of dose reduction or increase in dosing interval on the pharmacokinetics of Synercid in these patients has not been studied. Therefore, no recommendations can be made at this time regarding the appropriate dose modification.
Pediatric Use
Synercid has been used in a limited number of pediatric patients under emergency-use conditions at a dose of 7.5 mg/kg q8h or q12h. However, the safety and effectiveness of Synercid in patients under 16 years of age have not been established.
Geriatric Use
In phase 3 comparative trials of Synercid, 37% of patients (n=404) were ≥65 years of age, of which 145 were ≥75 years of age. In the phase 3 non-comparative trials, 29% of patients (n=346) were ≥65 years of age, of which 112 were ≥75 years of age. There were no apparent differences in the frequency, type, or severity of related adverse reactions including cardiovascular events between elderly and younger individuals.
Adverse Reactions/Side Effects
The safety of Synercid was evaluated in 1099 patients enrolled in 5 comparative clinical trials. Additionally, 4 non-comparative clinical trials (3 prospective and 1 retrospective in design) were conducted in which 1199 patients received Synercid for infections due to Gram-positive pathogens for which no other treatment option was available. In non-comparative trials, the patients were severely ill, often with multiple co-morbidities or physiological impairments, and may have been intolerant to or failed other antibacterial therapies.
COMPARATIVE TRIALS
ADVERSE REACTION SUMMARY – ALL COMPARATIVE STUDIES
Safety data are available from five comparative clinical studies (n= 1099 Synercid; n= 1095 comparator). One of the deaths in the comparative studies was assessed as possibly related to Synercid. The most frequent reasons for discontinuation due to drug-related adverse reactions were as follows:
| Type | Synercid | Comparator |
|---|---|---|
| Venous | 9.2 | 2.0 |
| Non-venous | 9.6 | 4.3 |
| -Rash | 1.0 | 0.5 |
| -Nausea | 0.9 | 0.6 |
| -Vomiting | 0.5 | 0.5 |
| -Pain | 0.5 | 0.0 |
| -Pruritus | 0.5 | 0.3 |
CLINICAL REACTIONS – ALL COMPARATIVE STUDIES
Adverse reactions with an incidence of ≥1% and possibly or probably related to Synercid administration include:
| Adverse Reactions | % of patients with adverse reactions | |
|---|---|---|
| Synercid | Comparator | |
| Inflammation at infusion site | 42.0 | 25.0 |
| Pain at infusion site | 40.0 | 23.7 |
| Edema at infusion site | 17.3 | 9.5 |
| Infusion site reaction | 13.4 | 10.1 |
| Nausea | 4.6 | 7.2 |
| Thrombophlebitis | 2.4 | 0.3 |
| Diarrhea | 2.7 | 3.2 |
| Vomiting | 2.7 | 3.8 |
| Rash | 2.5 | 1.4 |
| Headache | 1.6 | 0.9 |
| Pruritus | 1.5 | 1.1 |
| Pain | 1.5 | 0.1 |
Additional adverse reactions that were possibly or probably related to Synercid with an incidence less than 1% within each body system are listed below:
Body as a Whole: abdominal pain, worsening of underlying illness, allergic reaction, chest pain, fever, infection;
Cardiovascular: palpitation, phlebitis;
Digestive: constipation, dyspepsia, oral moniliasis, pancreatitis, pseudomembranous enterocolitis, stomatitis;
Metabolic: gout, peripheral edema;
Musculoskeletal: arthralgia, myalgia, myasthenia;
Nervous: anxiety, confusion, dizziness, hypertonia, insomnia, leg cramps, paresthesia, vasodilation;
Respiratory: dyspnea, pleural effusion;
Skin and Appendages: maculopapular rash, sweating, urticaria;
Urogenital: hematuria, vaginitis
CLINICAL REACTIONS – SKIN AND SKIN STRUCTURE STUDIES
In two of the five comparative clinical trials Synercid (n=450) and comparator regimens (e.g., oxacillin/vancomycin or cefazolin/vancomycin; n=443) were studied for safety and efficacy in the treatment of complicated skin and skin structure infections. The adverse event profile seen in the Synercid patients in these two studies differed significantly from that seen in the other comparative studies. What follows is safety data from these two studies.
Discontinuation of therapy was most frequently due to the following drug related events:
| % of patients discontinuing therapy by reaction type | ||
|---|---|---|
| Type | Synercid | Comparator |
| Venous | 12.0 | 2.0 |
| Non-venous | 11.8 | 4.0 |
| -Rash | 2.0 | 0.9 |
| -Nausea | 1.1 | 0.0 |
| -Vomiting | 0.9 | 0.0 |
| -Pain | 0.9 | 0.0 |
| -Pruritus | 0.9 | 0.5 |
Venous adverse events were seen predominately in patients who had peripheral infusions. The most frequently reported venous and non-venous adverse reactions possibly or probably related to study drug were:
| % of patients with adverse reactions | ||
|---|---|---|
| Synercid | Comparator | |
| Venous | 68.0 | 32.7 |
| -Pain at infusion site | 44.7 | 17.8 |
| -Inflammation at infusion site | 38.2 | 14.7 |
| -Edema at infusion site | 18.0 | 7.2 |
| -Infusion site reaction | 11.6 | 3.6 |
| Non-venous | 24.7 | 13.1 |
| -Nausea | 4.0 | 2.0 |
| -Vomiting | 3.7 | 1.0 |
| -Rash | 3.1 | 1.3 |
| -Pain | 3.1 | 0.2 |
There were eight (1.7%) episodes of thrombus or thrombophlebitis in the Synercid arms and none in the comparator arms.
LABORATORY EVENTS-ALL COMPARATIVE STUDIES
Table 7 shows the number (%) of patients exhibiting laboratory values above or below the clinically relevant "critical" values during treatment phase (with an incidence of 0.1% or greater in either treatment group).
| Parameter | Critically High or Low Value | Synercid Critically High or Low | Comparator Critically High or Low |
|---|---|---|---|
| AST | > 10 × ULN | 9 (0.9) | 2 (0.2) |
| ALT | > 10 × ULN | 4 (0.4) | 4 (0.4) |
| Total Bilirubin | > 5 × ULN | 9 (0.9) | 2 (0.2) |
| Conjugated Bilirubin | > 5 × ULN | 29 (3.1) | 12 (1.3) |
| LDH | > 5 × ULN | 10 (2.6) | 8 (2.1) |
| Alk Phosphatase | > 5 × ULN | 3 (0.3) | 7 (0.7) |
| Gamma-GT | > 10 × ULN | 19 (1.9) | 10 (1.0) |
| CPK | > 10 × ULN | 6 (1.6) | 5 (1.4) |
| Creatinine | ≥ 440 μmoL/L | 1 (0.1) | 1 (0.1) |
| BUN | ≥ 35.5 mmoL/L | 2 (0.3) | 9 (1.2) |
| Blood Glucose | > 22.2 mmoL/L | 11 (1.3) | 11 (1.3) |
| < 2.2 mmoL/L | 1 (0.1) | 1 (0.1) | |
| Bicarbonates | > 40 mmoL/L | 2 (0.3) | 3 (0.5) |
| < 10 mmoL/L | 3 (0.5) | 3 (0.5) | |
| CO2 | > 50 mmoL/L | 0 (0.0) | 0 (0.0) |
| < 15 mmoL/L | 1 (0.2) | 0 (0.0) | |
| Sodium | > 160 mmoL/L | 0 (0.0) | 0 (0.0) |
| < 120 mmoL/L | 5 (0.5) | 3 (0.3) | |
| Potassium | > 6.0 mmoL/L | 3 (0.3) | 6 (0.6) |
| < 2.0 mmoL/L | 0 (0.0) | 1 (0.1) | |
| Hemoglobin | < 8 g/dL | 25 (2.6) | 16 (1.6) |
| Hematocrit | > 60% | 2 (0.2) | 0 (0.0) |
| Platelets | > 1,000,000/mm3 | 2 (0.2) | 2 (0.2) |
| < 50,000/mm3 | 6 (0.6) | 7 (0.7) |
NON-COMPARATIVE TRIALS
CLINICAL ADVERSE REACTIONS
Approximately one-third of patients discontinued therapy in these trials due to adverse events. However, the discontinuation rate due to adverse reactions assessed by the investigator as possibly or probably related to Synercid therapy was approximately 5.0%.
There were three prospectively designed non-comparative clinical trials in patients (n = 972) treated with Synercid. One of these studies (301), had more complete documentation than the other two (398A and 398B). The most common events probably or possibly related to therapy are presented in Table 8:
| Adverse Reactions | % of patients with adverse reaction | ||
|---|---|---|---|
| Study 301 | Study 398A | Study 398B | |
| Arthralgia | 7.8 | 5.2 | 4.3 |
| Myalgia | 5.1 | 0.95 | 3.1 |
| Arthralgia and Myalgia | 7.4 | 3.3 | 6.8 |
| Nausea | 3.8 | 2.8 | 4.9 |
The percentage of patients who experienced severe related arthralgia and myalgia was 3.3% and 3.1%, respectively. The percentage of patients who discontinued treatment due to related arthralgia and myalgia was 2.3% and 1.8%, respectively.
LABORATORY EVENTS
The most frequently observed abnormalities in laboratory studies were in total and conjugated bilirubin, with increases greater than 5 times upper limit of normal, irrespective of relationship to Synercid, reported in 25.0% and 34.6% of patients, respectively. The percentage of patients who discontinued treatment due to increased total and conjugated bilirubin was 2.7% and 2.3%, respectively. Of note, 46.5% and 59.0% of patients had high baseline total and conjugated bilirubin levels before study entry.
OTHER
Serious adverse reactions in clinical trials, including non-comparative studies, considered possibly or probably related to Synercid administration with an incidence < 0.1% include: acidosis, anaphylactoid reaction, apnea, arrhythmia, bone pain, cerebral hemorrhage, cerebrovascular accident, coagulation disorder, convulsion, dysautonomia, encephalopathy, grand mal convulsion, hemolysis, hemolytic anemia, heart arrest, hepatitis, hypoglycemia, hyponatremia, hypoplastic anemia, hypoventilation, hypovolemia, hypoxia, jaundice, mesenteric arterial occlusion, neck rigidity, neuropathy, pancytopenia, paraplegia, pericardial effusion, pericarditis, respiratory distress syndrome, shock, skin ulcer, supraventricular tachycardia, syncope, tremor, ventricular extrasystoles and ventricular fibrillation. Cases of hypotension and gastrointestinal hemorrhage were reported in less than 0.2% of patients.
Related/similar drugs
Overdosage
There are four reports of patients receiving Synercid doses at up to three times that recommended (7.5 mg/kg). No adverse events were considered possibly or probably related to Synercid overdose. Signs of acute overdosage may include dyspnea, emesis, tremors, and ataxia as seen in animals given extremely high doses (50 mg/kg) of Synercid. Patients who receive an overdose should be carefully observed and given supportive treatment. Synercid is not removed by peritoneal dialysis or by hemodialysis.
Synercid Dosage and Administration
Synercid should be administered by intravenous infusion in 5% Dextrose in Water solution over a 60-minute period. (See WARNINGS.) An infusion pump or device may be used to control the rate of infusion. If necessary, central venous access (e.g., PICC) can be used to administer Synercid to decrease the incidence of venous irritation. The recommended dosage for the treatment of complicated skin and skin structure infections is 7.5 mg/kg q12h. The minimum recommended treatment duration for complicated skin and skin structure infections is seven days.
Special Populations
Elderly
No dosage adjustment of Synercid is required for use in the elderly. (See CLINICAL PHARMACOLOGY: Pharmacokinetics and PRECAUTIONS: Geriatric Use.)
Renal Insufficiency
No dosage adjustment of Synercid is required for use in patients with renal impairment or patients undergoing peritoneal dialysis. (See CLINICAL PHARMACOLOGY: Pharmacokinetics.)
Hepatic Insufficiency
Data from clinical trials of Synercid suggest that the incidence of adverse effects in patients with chronic liver insufficiency or cirrhosis was comparable to that in patients with normal hepatic function. Pharmacokinetic data in patients with hepatic cirrhosis (Child Pugh A or B) suggest that dosage reduction may be necessary but exact recommendations cannot be made at this time. (See CLINICAL PHARMACOLOGY: Special Populations and PRECAUTIONS: General: Hepatic Insufficiency sections.)
Pediatric Patients
The recommended dose of Synercid for pediatric patients (12 to < 18 years of age) is 7.5 mg/kg q12h. No dosing recommendations are available in pediatric patients less than 12 years of age. (See PRECAUTIONS: Pediatric Use.)
Preparation and administration of solution
- Reconstitute the 500 mg single dose vial by slowly adding 5 mL of 5% Dextrose in Water or Sterile Water for injection.
- GENTLY swirl the vial by manual rotation without shaking to ensure dissolution of contents while LIMITING FOAM FORMATION.
- Allow the solution to sit for a few minutes until all the foam has disappeared. The resulting solution should be clear. Vials reconstituted in this manner will give a solution of 100 mg/mL. CAUTION: FURTHER DILUTION REQUIRED BEFORE INFUSION.
- According to the patient's weight, the reconstituted Synercid solution should be added to 250 mL of 5% Dextrose solution. An infusion volume of 100 mL may be used for central line infusions.
- If moderate to severe venous irritation occurs following peripheral administration of Synercid diluted in 250 mL of Dextrose 5% in water, consideration should be given to increasing the infusion volume to 500 or 750 mL, changing the infusion site, or infusing by a peripherally inserted central catheter (PICC) or a central venous catheter.
- The desired dose should be administered by intravenous infusion over 60 minutes.
NOTE: As for other parenteral drug products, Synercid should be inspected visually for particulate matter prior to administration.
COMPATIBILITY
DO NOT DILUTE WITH SALINE SOLUTIONS BECAUSE SYNERCID IS NOT COMPATIBLE WITH THESE AGENTS. Synercid should not be mixed with, or physically added to, other drugs except for the following drugs where compatibility by Y-site injection has been established:
| Admixture and Concentration | IV Infusion Solutions for Admixture |
|---|---|
| Aztreonam 20 mg/mL | D5W |
| Ciprofloxacin 1 mg/mL | D5W |
| Fluconazole 2 mg/mL | Used as the undiluted solution |
| Haloperidol 0.2 mg/mL | D5W |
| Metoclopramide 5 mg/mL | D5W |
| Potassium Chloride 40 mEq/L | D5W |
| D5W = 5% Dextrose Injection |
If Synercid is to be given concomitantly with another drug, each drug should be given separately in accordance with the recommended dosage and route of administration for each drug.
With intermittent infusion of Synercid and other drugs through a common intravenous line, the line should be flushed before and after administration with 5% Dextrose in Water solution.
Storage and Handling
Before Reconstitution: The unopened vials should be stored in a refrigerator at 2 to 8°C (36 to 46°F).
Reconstituted and Infusions Solutions
Because Synercid contains no antibacterial preservative, it should be reconstituted under strict aseptic conditions (e.g., Laminar Air Flow Hood). The reconstituted solution should be diluted within 30 minutes. Vials are for single use. The storage time of the diluted solution should be as short as possible to minimize the risk of microbial contamination. Stability of the diluted solution prior to the infusion is established as 5 hours at room temperature or 54 hours if stored under refrigeration 2 to 8°C (36 to 46°F). The solution should not be frozen.
How is Synercid supplied
Synercid is supplied as a sterile lyophilized pyrogen-free preparation in single-dose 10 mL type I glass vials with gray elastomeric closure, and aluminum seal with a dark blue flip-off cap for the 500 mg vial.
| NDC 61570-260-10 | Synercid IV 500 mg | 150 mg quinupristin and 350 mg dalfopristin | 10 vials |
Clinical Studies
COMPARATIVE TRIALS
Complicated Skin and Skin Structure Infections
Two randomized, open-label, controlled clinical trials of Synercid (7.5 mg/kg q12h intravenously [iv]) in the treatment of complicated skin and skin structure infections were performed. The comparator drug was oxacillin (2g q6h iv) in the first study (JRV 304) and cefazolin (1g q8h iv) in the second study (JRV 305); however, in both studies vancomycin (1g q12h iv) could be substituted for the specified comparator if the causative pathogen was suspected or confirmed methicillin-resistant staphylococcus or if the patient was allergic to penicillins, cephalosporins or carbapenems. Study JRV 304 enrolled 450 patients (n = 229 Synercid; n= 221 Comparator) and Study JRV 305 enrolled 443 patients (n = 221 Synercid; n = 222 Comparator).
In the first study, 105 patients (45.9%) and 106 patients (48.0%) in the Synercid and Comparator arms, respectively, were found to be clinically evaluable. For the second study, these values were 113 (51.1%) and 120 (54.1%) patients in the Synercid and Comparator arms, respectively. Patients were found not to be clinically evaluable for reasons such as: wrong diagnosis, lower extremity infection in patients with diabetes or peripheral vascular disease since these infections were assumed to include aerobic gram-negative and anaerobic organisms, no specimen for culture obtained, insufficient therapy, no test of cure assessment, etc.
For the patients found to be clinically evaluable, in Study JRV 304 the success rate was 49.5% in the Synercid arm and 51.9% in the Comparator arm. In Study JRV 305, the success rates were 66.4% and 64.2% in the Synercid and Comparator arms, respectively.
Table 10 shows the clinical success rate (combined results from two clinical trials) in the clinically evaluable population. Due to the small numbers of patients in the subsets, statistical conclusions could not be reached.
| Cured or Improved | ||||
|---|---|---|---|---|
| Infection Type | Synercid | Comparator | ||
| (n/N) | (%) | (n/N) | (%) | |
| Erysipelas (cellulitis) | 52/82 | (63.4) | 43/77 | (55.8) |
| Post-operative infections | 14/38 | (36.8) | 24/42 | (57.1) |
| Traumatic wound infection | 33/55 | (60.0) | 33/55 | (60.0) |
SAFETY
Discontinuations of therapy because of adverse reactions which were probably or possibly due to drug therapy occurred more than four times as often in the Synercid group than in the comparator group. Approximately half of the discontinuations in the Synercid arm were due to venous adverse events. (See ADVERSE REACTIONS: Clinical Reactions: Skin and Skin Structure Studies.)
Keep out of the reach of children.
This product's label may have been updated. For current full prescribing information, please visit www.pfizer.com

PRINCIPAL DISPLAY PANEL - 500 mg Vial Label
NDC 61570-260-01
Synercid® I.V.
(quinupristin 150mg and dalfopristin 350mg)
for Injection
500 mg
Single Dose Vial, For I.V. Use Only-Not For Direct Infusion
1 Sterile Vial
Pfizer
Injectables
Rx only

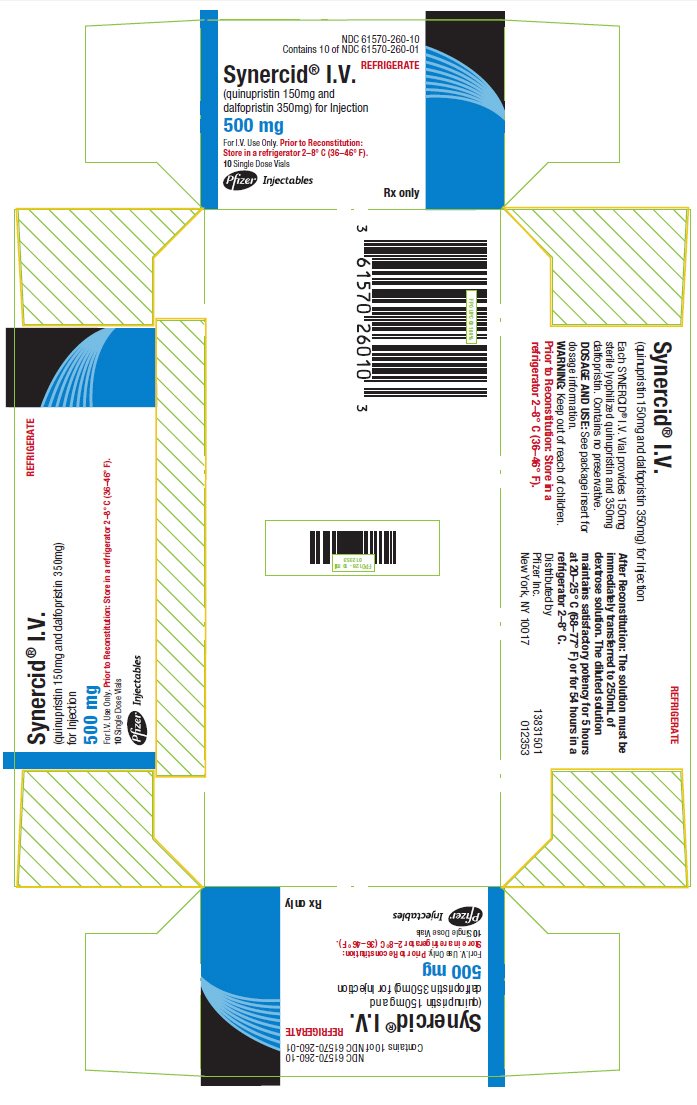
PRINCIPAL DISPLAY PANEL - 10 Vial Carton
NDC 61570-260-10
Contains 10 of NDC 61570-260-01
REFRIGERATE
Synercid® I.V.
(quinupristin 150mg and
dalfopristin 350mg) for Injection
500 mg
For I.V. Use Only. Prior to Reconstitution:
Store in a refrigerator 2–8° C (36–46° F).
10 Single Dose Vials
Pfizer
Injectables
Rx only
| SYNERCID
quinupristin and dalfopristin injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Pfizer Laboratories Div Pfizer Inc (134489525) |
More about Synercid (dalfopristin / quinupristin)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- Drug class: streptogramins