Opdivo: Package Insert / Prescribing Info
Package insert / product label
Generic name: nivolumab
Dosage form: injection
Drug class: Anti-PD-1 and PD-L1 monoclonal antibodies (immune checkpoint inhibitors)
J Code (medical billing code): J9299 (1 mg, intravenous)
Medically reviewed by Drugs.com. Last updated on Jul 20, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
OPDIVO® (nivolumab) injection, for intravenous use
Initial U.S. Approval: 2014
Indications and Usage for Opdivo
OPDIVO is a programmed death receptor-1 (PD-1)-blocking antibody indicated for the treatment of:
Melanoma
- •
- adult and pediatric (12 years and older) patients with unresectable or metastatic melanoma, as a single agent or in combination with ipilimumab. (1.1)
- •
- for the adjuvant treatment of adult and pediatric patients 12 years and older with completely resected Stage IIB, Stage IIC, Stage III, or Stage IV melanoma. (1.2)
Non-Small Cell Lung Cancer (NSCLC)
- •
- adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer in the neoadjuvant setting, in combination with platinum-doublet chemotherapy. (1.3)
- •
- adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer and no known EGFR mutations or ALK rearrangements, for neoadjuvant treatment, in combination with platinum-doublet chemotherapy, followed by single-agent OPDIVO as adjuvant treatment after surgery. (1.4)
- •
- adult patients with metastatic non-small cell lung cancer expressing PD‑L1 (≥1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, as first-line treatment in combination with ipilimumab. (1.5)
- •
- adult patients with metastatic or recurrent non-small cell lung cancer with no EGFR or ALK genomic tumor aberrations as first-line treatment, in combination with ipilimumab and 2 cycles of platinum-doublet chemotherapy. (1.5)
- •
- adult patients with metastatic non-small cell lung cancer and progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO. (1.5)
Malignant Pleural Mesothelioma
- •
- adult patients with unresectable malignant pleural mesothelioma, as first-line treatment in combination with ipilimumab. (1.6)
Renal Cell Carcinoma (RCC)
- •
- adult patients with intermediate or poor risk advanced renal cell carcinoma, as a first-line treatment in combination with ipilimumab. (1.7)
- •
- adult patients with advanced renal cell carcinoma, as a first-line treatment in combination with cabozantinib. (1.7)
- •
- adult patients with advanced renal cell carcinoma who have received prior anti-angiogenic therapy. (1.7)
Classical Hodgkin Lymphoma (cHL)
- •
- adult patients with classical Hodgkin lymphoma that has relapsed or progressed aftera: (1.8)
- •
- autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin, or
- •
- 3 or more lines of systemic therapy that includes autologous HSCT.
Squamous Cell Carcinoma of the Head and Neck (SCCHN)
- •
- adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck with disease progression on or after a platinum-based therapy. (1.9)
Urothelial Carcinoma
- •
- adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC. (1.10)
- •
- adult patients with unresectable or metastatic urothelial carcinoma, as first-line treatment in combination with cisplatin and gemcitabine. (1.10)
- •
- adult patients with locally advanced or metastatic urothelial carcinoma who:
- •
- have disease progression during or following platinum-containing chemotherapy.
- •
- have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. (1.10)
Colorectal Cancer
- •
- adult and pediatric (12 years and older) patients with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer (CRC) in combination with ipilimumab. (1.11)
- •
- adult and pediatric (12 years and older) patients with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. (1.11)
Hepatocellular Carcinoma (HCC)
- •
- adult patients with unresectable or metastatic hepatocellular carcinoma (HCC), as a first-line treatment in combination with ipilimumab. (1.12)
- •
- in combination with ipilimumab in adult patients with unresectable or metastatic HCC who have been previously treated with sorafenib. (1.12)
Esophageal Cancer
- •
- adult patients with completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease, who have received neoadjuvant chemoradiotherapy (CRT). (1.13)
- •
- adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma as first-line treatment in combination with fluoropyrimidine- and platinum‑containing chemotherapy whose tumors express PD-L1 (≥1). (1.13)
- •
- adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma as first-line treatment in combination with ipilimumab whose tumors express PD-L1 (≥1). (1.13)
- •
- adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy. (1.13)
Gastric Cancer, Gastroesophageal Junction Cancer, and Esophageal Adenocarcinoma
- •
- adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma whose tumors express PD-L1 (≥1) in combination with fluoropyrimidine- and platinum-containing chemotherapy. (1.14)
a This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
Opdivo Dosage and Administration
- •
- Administer by intravenous infusion after dilution based upon recommended infusion rate for each indication. (2)
- •
- Unresectable or metastatic melanoma
- •
- Adult and pediatric patients weighing 40 kg or greater: 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Pediatric patients weighing less than 40 kg: 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks. (2.2)
- •
- Adult and pediatric patients weighing 40 kg or greater: 1 mg/kg followed by ipilimumab 3 mg/kg on the same day every 3 weeks for 4 doses, then 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Pediatric patients weighing less than 40 kg: 1 mg/kg followed by ipilimumab 3 mg/kg on the same day every 3 weeks for 4 doses, then 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks. (2.2)
- •
- Adjuvant treatment of melanoma
- •
- Neoadjuvant treatment of resectable (tumors ≥4 cm or node positive) non-small cell lung cancer
- •
- 360 mg with platinum-doublet chemotherapy on the same day every 3 weeks for 3 cycles. (2.2)
- •
- Neoadjuvant and adjuvant treatment of resectable non-small cell lung cancer
- •
- 360 mg with platinum-doublet chemotherapy on the same day every 3 weeks for up to 4 cycles, then continued as single-agent OPDIVO 480 mg every 4 weeks after surgery for up to 13 cycles (~1 year). (2.2)
- •
- Metastatic non-small cell lung cancer
- •
- Malignant pleural mesothelioma
- •
- 360 mg every 3 weeks with ipilimumab 1 mg/kg every 6 weeks. (2.2)
- •
- Advanced renal cell carcinoma
- •
- 3 mg/kg followed by ipilimumab 1 mg/kg on the same day every 3 weeks for 4 doses, then 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks administered in combination with cabozantinib 40 mg once daily without food. (2.2)
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Classical Hodgkin lymphoma
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Recurrent or metastatic squamous cell carcinoma of the head and neck
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Adjuvant treatment of urothelial carcinoma
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- First-line unresectable or metastatic urothelial carcinoma
- •
- 360 mg every 3 weeks with cisplatin and gemcitabine on the same day for up to 6 cycles, then 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Previously treated locally advanced or metastatic urothelial carcinoma
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer
- •
- Adult and pediatric patients weighing 40 kg or greater: 240 mg followed by ipilimumab 1 mg/kg on the same day every 3 weeks for a maximum of 4 doses, then 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Pediatric patients weighing less than 40 kg: 3 mg/kg followed by ipilimumab 1 mg/kg on the same day every 3 weeks for a maximum of 4 doses, then 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks. (2.2)
- •
- Microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed following prior treatment for metastatic disease.
- •
- Hepatocellular carcinoma
- •
- 1 mg/kg followed by ipilimumab 3 mg/kg on the same day every 3 weeks for 4 doses, then 240 mg every 2 weeks or 480 mg every 4 weeks. (2.2)
- •
- Adjuvant treatment of resected esophageal or gastroesophageal cancer
- •
- 240 mg every 2 weeks or 480 mg every 4 weeks for total treatment duration of 1 year. (2.2)
- •
- Esophageal squamous cell carcinoma
- •
- Gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma (GC, GEJC, or EAC)
- •
- See full Prescribing Information for preparation and administration instructions and dosage modifications for adverse reactions.
Dosage Forms and Strengths
- •
- Injection: 40 mg/4 mL (10 mg/mL), 100 mg/10 mL (10 mg/mL), 120 mg/12 mL (10 mg/mL), and 240 mg/24 mL (10 mg/mL) solution in a single-dose vial. (3)
Contraindications
- •
- None. (4)
Warnings and Precautions
- •
-
Immune-Mediated Adverse Reactions: (5.1)
- •
- Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue, including the following: immune-mediated pneumonitis, immune-mediated colitis, immune-mediated hepatitis and hepatotoxicity, immune-mediated endocrinopathies, immune-mediated dermatologic adverse reactions, and immune-mediated nephritis and renal dysfunction.
- •
- Monitor for early identification and management. Evaluate liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment.
- •
- Withhold or permanently discontinue based on severity and type of reaction. (2.3)
- •
- Infusion-related reactions: Interrupt, slow the rate of infusion, or permanently discontinue OPDIVO based on severity of reaction. (5.2)
- •
- Complications of allogeneic HSCT: Fatal and other serious complications can occur in patients who receive allogeneic HSCT before or after being treated with a PD-1/PD-L1 blocking antibody. (5.3)
- •
- Embryo-Fetal toxicity: Can cause fetal harm. Advise females of reproductive potential of potential risk to a fetus and to use effective contraception. (5.4, 8.1, 8.3)
- •
- Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials. (5.5)
Adverse Reactions/Side Effects
Most common adverse reactions (incidence ≥20%) in patients were:
- •
- As a single agent: fatigue, rash, musculoskeletal pain, pruritus, diarrhea, nausea, asthenia, cough, dyspnea, constipation, decreased appetite, back pain, arthralgia, upper respiratory tract infection, pyrexia, headache, abdominal pain, vomiting, and urinary tract infection. (6.1)
- •
- In combination with ipilimumab: fatigue, diarrhea, rash, pruritus, nausea, musculoskeletal pain, pyrexia, cough, decreased appetite, vomiting, abdominal pain, dyspnea, upper respiratory tract infection, arthralgia, headache, hypothyroidism, constipation, decreased weight, and dizziness. (6.1)
- •
- In combination with platinum-doublet chemotherapy: nausea, fatigue, musculoskeletal pain, constipation, decreased appetite, rash, vomiting, and peripheral neuropathy. (6.1)
- •
- In combination with ipilimumab and platinum-doublet chemotherapy: fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus. (6.1)
- •
- In combination with cabozantinib: diarrhea, fatigue, hepatotoxicity, palmar-plantar erythrodysesthesia syndrome, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection. (6.1)
- •
- In combination with fluoropyrimidine- and platinum-containing chemotherapy: nausea, peripheral neuropathy, decreased appetite, fatigue, constipation, stomatitis, diarrhea, vomiting, abdominal pain, and musculoskeletal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Opdivo
1.1 Unresectable or Metastatic Melanoma
OPDIVO, as a single agent or in combination with ipilimumab, is indicated for the treatment of adult and pediatric patients 12 years and older with unresectable or metastatic melanoma.
1.2 Adjuvant Treatment of Melanoma
OPDIVO is indicated for the adjuvant treatment of adult and pediatric patients 12 years and older with completely resected Stage IIB, Stage IIC, Stage III, or Stage IV melanoma.
1.3 Neoadjuvant Treatment of Resectable Non-Small Cell Lung Cancer
OPDIVO, in combination with platinum-doublet chemotherapy, is indicated as neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) non-small cell lung cancer (NSCLC).
1.4 Neoadjuvant and Adjuvant Treatment of Resectable Non-Small Cell Lung Cancer
OPDIVO, in combination with platinum-doublet chemotherapy, is indicated for the neoadjuvant treatment of adult patients with resectable (tumors ≥4 cm or node positive) NSCLC and no known epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK) rearrangements, followed by single-agent OPDIVO as adjuvant treatment after surgery.
1.5 Metastatic Non-Small Cell Lung Cancer
- •
- OPDIVO, in combination with ipilimumab, is indicated for the first-line treatment of adult patients with metastatic NSCLC whose tumors express PD-L1 (≥1%) as determined by an FDA-approved test [see Dosage and Administration (2.1)], with no EGFR or ALK genomic tumor aberrations.
- •
- OPDIVO, in combination with ipilimumab and 2 cycles of platinum-doublet chemotherapy, is indicated for the first-line treatment of adult patients with metastatic or recurrent NSCLC, with no EGFR or ALK genomic tumor aberrations.
- •
- OPDIVO is indicated for the treatment of adult patients with metastatic NSCLC with progression on or after platinum-based chemotherapy. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approved therapy for these aberrations prior to receiving OPDIVO.
1.6 Malignant Pleural Mesothelioma
OPDIVO, in combination with ipilimumab, is indicated for the first-line treatment of adult patients with unresectable malignant pleural mesothelioma.
1.7 Advanced Renal Cell Carcinoma
- •
- OPDIVO, in combination with ipilimumab, is indicated for the first-line treatment of adult patients with intermediate or poor risk advanced RCC.
- •
- OPDIVO, in combination with cabozantinib, is indicated for the first-line treatment of adult patients with advanced RCC.
- •
- OPDIVO, as a single agent, is indicated for the treatment of adult patients with advanced renal cell carcinoma (RCC) who have received prior anti-angiogenic therapy.
1.8 Classical Hodgkin Lymphoma
OPDIVO is indicated for the treatment of adult patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after:
- •
- autologous hematopoietic stem cell transplantation (HSCT) and brentuximab vedotin, or
- •
- 3 or more lines of systemic therapy that includes autologous HSCT.
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14.8)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
1.9 Squamous Cell Carcinoma of the Head and Neck
OPDIVO is indicated for the treatment of adult patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after platinum-based therapy.
1.10 Urothelial Carcinoma
OPDIVO is indicated for the adjuvant treatment of adult patients with urothelial carcinoma (UC) who are at high risk of recurrence after undergoing radical resection of UC [see Clinical Studies (14.10)].
OPDIVO, in combination with cisplatin and gemcitabine, is indicated for the first-line treatment of adult patients with unresectable or metastatic urothelial carcinoma.
OPDIVO is indicated for the treatment of adult patients with locally advanced or metastatic urothelial carcinoma who:
- •
- have disease progression during or following platinum-containing chemotherapy.
- •
- have disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy.
1.11 Microsatellite Instability-High or Mismatch Repair Deficient Metastatic Colorectal Cancer
- •
- OPDIVO, in combination with ipilimumab, is indicated for the treatment of adult and pediatric patients 12 years and older with unresectable or metastatic microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) colorectal cancer (CRC).
- •
- OPDIVO, as a single agent, is indicated for the treatment of adult and pediatric patients 12 years and older with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer (CRC) that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan.
1.12 Hepatocellular Carcinoma
- •
- OPDIVO, in combination with ipilimumab, is indicated for the first-line treatment of adult patients with unresectable or metastatic hepatocellular carcinoma (HCC).
- •
- OPDIVO, in combination with ipilimumab, is indicated for the treatment of adult patients with unresectable or metastatic HCC who have been previously treated with sorafenib.
1.13 Esophageal Cancer
- •
- OPDIVO is indicated for the adjuvant treatment of completely resected esophageal or gastroesophageal junction cancer with residual pathologic disease in adult patients who have received neoadjuvant chemoradiotherapy (CRT).
- •
- OPDIVO, in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC) whose tumors express PD-L1 (≥1) [see Dosage and Administration (2.1)].
- •
- OPDIVO, in combination with ipilimumab, is indicated for the first-line treatment of adult patients with unresectable advanced or metastatic esophageal squamous cell carcinoma (ESCC) whose tumors express PD-L1 (≥1) [see Dosage and Administration (2.1)].
- •
- OPDIVO is indicated for the treatment of adult patients with unresectable advanced, recurrent or metastatic esophageal squamous cell carcinoma (ESCC) after prior fluoropyrimidine- and platinum-based chemotherapy.
1.14 Gastric Cancer, Gastroesophageal Junction Cancer, and Esophageal Adenocarcinoma
OPDIVO, in combination with fluoropyrimidine- and platinum-containing chemotherapy, is indicated for the treatment of adult patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma whose tumors express PD-L1 (≥1) [see Dosage and Administration (2.1)].
2. Opdivo Dosage and Administration
2.1 Patient Selection
Information on FDA-approved tests for patient selection is available at:
https://www.fda.gov/CompanionDiagnostics
Non-Small Cell Lung Cancer
- •
- Select patients with metastatic NSCLC for treatment with OPDIVO in combination with ipilimumab based on PD-L1 expression [see Clinical Studies (14.5)].
Esophageal Cancer
- •
- Select patients with unresectable advanced or metastatic ESCC for treatment with OPDIVO in combination with fluoropyrimidine- and platinum-containing chemotherapy based on PD-L1 expression [see Clinical Studies (14.13)].
- •
-
Select patients with unresectable advanced or metastatic ESCC for treatment with OPDIVO in combination with ipilimumab based on PD-L1 expression [see Clinical Studies (14.13)].
- •
- An FDA-approved companion diagnostic for the detection of PD-L1 expression in patients with advanced or metastatic ESCC is not available.
Gastric Cancer, Gastroesophageal Junction Cancer, and Esophageal Adenocarcinoma
- •
-
Select patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma for treatment with OPDIVO in combination with fluoropyrimidine- and platinum-containing chemotherapy based on PD-L1 expression [see Clinical Studies (14.14)].
- •
- An FDA-approved companion diagnostic for the detection of PD-L1 expression in patients with advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma is not available.
2.2 Recommended Dosage
The recommended dosages of intravenous OPDIVO as a single agent are presented in Table 1.
Administer OPDIVO as a 30-minute intravenous infusion [see Dosage and Administration (2.4)].
|
Indication |
Recommended OPDIVO Dosage |
Duration of Therapy |
|
Metastatic non-small cell lung cancer |
240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease progression or unacceptable toxicity |
|
Advanced renal cell carcinoma |
||
|
Classical Hodgkin lymphoma |
||
|
Squamous cell carcinoma of the head and neck |
||
|
Locally advanced or metastatic urothelial carcinoma |
||
|
Esophageal squamous cell carcinoma |
||
|
Unresectable or metastatic melanoma |
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more: 240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease progression or unacceptable toxicity |
|
Pediatric patients age 12 years and older and weighing less than 40 kg: 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks |
||
|
Adjuvant treatment of melanoma |
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more: 240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease recurrence or unacceptable toxicity for up to 1 year |
|
Pediatric patients age 12 years and older and weighing less than 40 kg: 3 mg/kg every 2 weeks or 6 mg/kg every 4 weeks |
||
|
Adjuvant treatment of urothelial carcinoma (UC) |
240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease recurrence or unacceptable toxicity for up to 1 year |
|
Microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) metastatic colorectal cancer that has progressed following prior treatment for metastatic disease |
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more: 240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease progression or unacceptable toxicity |
|
Pediatric patients age 12 years and older and weighing less than 40 kg: 3 mg/kg every 2 weeks |
||
|
Adjuvant treatment of resected esophageal or gastroesophageal junction cancer |
240 mg every 2 weeks or 480 mg every 4 weeks |
Until disease progression or unacceptable toxicity for a total treatment duration of 1 year |
The recommended dosages of OPDIVO in combination with other therapeutic agents are presented in Table 2. Administer OPDIVO on the same day as other therapeutic agents.
Refer to the respective Prescribing Information for each therapeutic agent administered in combination with OPDIVO for the recommended dosage information, as appropriate.
|
Indication |
Recommended OPDIVO Dosage |
Duration of Therapy |
|
Unresectable or metastatic melanoma |
1 mg/kg every 3 weeks |
In combination with ipilimumab for a maximum of 4 doses or until unacceptable toxicity, whichever occurs earlier |
|
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more: 240 mg every 2 weeks |
After completing 4 doses of combination therapy, administer as single agent until disease progression or unacceptable toxicity |
|
|
Pediatric patients age 12 years and older and weighing less than 40 kg: 3 mg/kg every 2 weeks |
||
|
Neoadjuvant treatment of resectable non-small cell lung cancer |
360 mg every 3 weeks |
In combination with platinum-doublet chemotherapy for 3 cycles |
|
Neoadjuvant and adjuvant treatment of resectable non-small cell lung cancer |
Neoadjuvant: 360 mg every 3 weeks |
Neoadjuvant treatment in combination with chemotherapy for up to 4 cycles or until disease progression or unacceptable toxicity, followed by adjuvant treatment with OPDIVO as a single agent after surgery for up to 13 cycles (approximately 1 year) or until disease recurrence or unacceptable toxicity |
|
Adjuvant: 480 mg every 4 weeks |
||
|
Metastatic non-small cell lung cancer expressing PD‑L1 |
360 mg every 3 weeks |
In combination with ipilimumab until disease progression, unacceptable toxicity, or up to 2 years in patients without disease progression |
|
Metastatic or recurrent non-small cell lung cancer |
360 mg every 3 weeks |
In combination with ipilimumab until disease progression, unacceptable toxicity, or up to 2 years in patients without disease progression |
|
2 cycles of histology-based platinum‑doublet chemotherapy |
||
|
Malignant pleural mesothelioma |
360 mg every 3 weeks |
In combination with ipilimumab until disease progression, unacceptable toxicity, or up to 2 years in patients without disease progression |
|
Advanced renal cell carcinoma |
3 mg/kg every 3 weeks |
In combination with ipilimumab |
|
240 mg every 2 weeks |
After completing 4 doses of combination therapy with ipilimumab, administer as single agent until disease progression or unacceptable toxicity |
|
|
240 mg every 2 weeks Administer OPDIVO in combination with cabozantinib 40 mg orally once daily without food |
OPDIVO: Until disease progression, unacceptable toxicity, or up to 2 years |
|
|
Cabozantinib: Until disease progression or unacceptable toxicity |
||
|
First-line unresectable or metastatic urothelial carcinoma |
360 mg every 3 weeks |
In combination with cisplatin and gemcitabine |
|
240 mg every 2 weeks |
After completing up to 6 cycles of combination therapy, administer as single agent until disease progression, unacceptable toxicity, or up to 2 years from first dose |
|
|
Microsatellite instability-high (MSI‑H) or mismatch repair deficient (dMMR) metastatic colorectal cancer |
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more:
240 mg every 3 weeks |
In combination with ipilimumab for a maximum of 4 doses |
|
Pediatric patients age 12 years and older and weighing less than 40 kg: |
||
|
Adult patients and pediatric patients age 12 years and older and weighing 40 kg or more: 240 mg every 2 weeks |
After completing a maximum of 4 doses of combination therapy, administer as single agent until disease progression or unacceptable toxicity, or up to 2 years |
|
|
Pediatric patients age 12 years and older and weighing less than 40 kg: 3 mg/kg every 2 weeks |
||
|
Hepatocellular carcinoma |
1 mg/kg every 3 weeks |
In combination with ipilimumab |
|
240 mg every 2 weeks |
After completing a maximum of 4 doses of combination therapy, administer as single agent until disease progression, unacceptable toxicity, or up to 2 years |
|
|
Esophageal squamous cell carcinoma |
240 mg every 2 weeks Administer OPDIVO in combination with fluoropyrimidine- and platinum‑containing chemotherapy |
OPDIVO: Until disease progression, unacceptable toxicity, or up to 2 years |
|
Chemotherapy: Until disease progression or unacceptable toxicity |
||
|
3 mg/kg every 2 weeks |
In combination with ipilimumab until disease progression, unacceptable toxicity, or up to 2 years |
|
|
Gastric cancer, Gastroesophageal junction cancer, and Esophageal adenocarcinoma |
240 mg every 2 weeks |
Until disease progression, unacceptable toxicity, or up to 2 years |
2.3 Dosage Modifications
No dose reduction for OPDIVO is recommended. In general, withhold OPDIVO for severe (Grade 3) immune-mediated adverse reactions. Permanently discontinue OPDIVO for life-threatening (Grade 4) immune-mediated adverse reactions, recurrent severe (Grade 3) immune-mediated reactions that require systemic immunosuppressive treatment, or an inability to reduce corticosteroid dose to 10 mg or less of prednisone or equivalent per day within 12 weeks of initiating steroids.
Dosage modifications for OPDIVO or OPDIVO in combination for adverse reactions that require management different from these general guidelines are summarized in Table 3 and Table 4.
When OPDIVO is administered in combination with ipilimumab, withhold or permanently discontinue both ipilimumab and OPDIVO for an adverse reaction meeting these dose modification guidelines.
| a Resume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of last dose or inability to reduce prednisone to 10 mg per day (or equivalent) or less within 12 weeks of initiating steroids. b If AST and ALT are less than or equal to ULN at baseline, withhold or permanently discontinue OPDIVO based on recommendations for hepatitis with no liver involvement. c Depending on clinical severity, consider withholding for Grade 2 endocrinopathy until symptom improvement with hormone replacement. Resume once acute symptoms have resolved. ALT = alanine aminotransferase, AST = aspartate aminotransferase, DRESS = Drug Rash with Eosinophilia and Systemic Symptoms, SJS = Stevens Johnson Syndrome, TEN = toxic epidermal necrolysis, ULN = upper limit normal |
||
|
Adverse Reaction |
Severity |
Dosage Modification |
|
Immune-Mediated Adverse Reactions [see Warnings and Precautions (5.1)] |
||
|
Pneumonitis |
Grade 2 |
Withholda |
|
Grades 3 or 4 |
Permanently discontinue |
|
|
Colitis For colitis in patients treated with combination therapy with ipilimumab, see Table 4. |
Grade 2 or 3 |
Withholda |
|
Grade 4 |
Permanently discontinue |
|
|
Hepatitis with no tumor involvement of the liver For liver enzyme elevations in patients treated with combination therapy with ipilimumab, see Table 4. |
AST/ALT increases to >3 and ≤8 times ULN or Total bilirubin increases to >1.5 and ≤3 times ULN. |
Withholda |
|
AST or ALT increases to >8 times ULN or Total bilirubin increases to >3 times ULN. |
Permanently discontinue |
|
|
Hepatitis with tumor involvement of the liverb For liver enzyme elevations in patients treated with combination therapy with ipilimumab, see Table 4. |
Baseline AST/ALT is >1 and ≤3 times ULN and increases to >5 and ≤10 times ULN or Baseline AST/ALT is >3 and ≤5 times ULN and increases to >8 and ≤10 times ULN. |
Withholda |
|
AST/ALT increases to >10 times ULN or Total bilirubin increases to >3 times ULN. |
Permanently discontinue |
|
|
Endocrinopathiesc |
Grade 3 or 4 |
Withhold until clinically stable or permanently discontinue depending on severity |
|
Nephritis with Renal Dysfunction |
Grade 2 or 3 increased blood creatinine |
Withholda |
|
Grade 4 increased blood creatinine |
Permanently discontinue |
|
|
Exfoliative Dermatologic Conditions |
Suspected SJS, TEN, or DRESS |
Withhold |
|
Confirmed SJS, TEN, or DRESS |
Permanently discontinue |
|
|
Myocarditis |
Grades 2, 3, or 4 |
Permanently discontinue |
|
Neurological Toxicities |
Grade 2 |
Withholda |
|
Grade 3 or 4 |
Permanently discontinue |
|
|
Other Adverse Reactions |
||
|
Infusion-Related Reactions |
Grade 1 or 2 |
Interrupt or slow the rate of infusion |
|
Grade 3 or 4 |
Permanently discontinue |
|
| a Resume in patients with complete or partial resolution (Grade 0 to 1) after corticosteroid taper. Permanently discontinue if no complete or partial resolution within 12 weeks of last dose or inability to reduce prednisone to 10 mg per day (or equivalent) or less within 12 weeks of initiating steroids. b If AST and ALT are less than or equal to ULN at baseline, withhold or permanently discontinue OPDIVO in combination with ipilimumab based on recommendations for hepatitis with no liver involvement. c Consider corticosteroid therapy for hepatic adverse reactions if OPDIVO is withheld or discontinued when administered in combination with cabozantinib. d After recovery, rechallenge with one or both of OPDIVO and cabozantinib may be considered. If rechallenging with cabozantinib with or without OPDIVO, refer to cabozantinib Prescribing Information. |
|||
|
Treatment |
Adverse Reaction |
Severity |
Dosage Modification |
|
OPDIVO in combination with ipilimumab |
Colitis |
Grade 2 |
Withholda |
|
Grade 3 or 4 |
Permanently discontinue |
||
|
Hepatitis with no tumor involvement of the liver or Hepatitis with tumor involvement of the liver/non-HCC |
AST/ALT increases to >3 times ULN and ≤5 times ULN or Total bilirubin increases to ≥1.5 and ≤3 times ULN. |
Withholda |
|
|
AST or ALT >5 times ULN or Total bilirubin >3 times ULN. |
Permanently discontinue |
||
|
Hepatitis with tumor involvement of the liverb/HCC |
Baseline AST/ALT is >1 and ≤3 times ULN and increases to >5 and ≤10 times ULN or Baseline AST/ALT is >3 and ≤5 times ULN and increases to >8 and ≤10 times ULN. |
Withholda |
|
|
AST/ALT increases to >10 times ULN or Total bilirubin increases to >3 times ULN. |
Permanently discontinue |
||
|
OPDIVO in combination with cabozantinib |
Liver enzyme elevations |
ALT or AST >3 times ULN but ≤10 times ULN with concurrent total bilirubin <2 times ULN |
Withholdc both OPDIVO and cabozantinib until adverse reactions recoverd to Grades 0‑1 |
|
ALT or AST >10 times ULN or >3 times ULN with concurrent total bilirubin ≥2 times ULN |
Permanently discontinuec both OPDIVO and cabozantinib |
||
2.4 Preparation and Administration
Visually inspect for particulate matter and discoloration. OPDIVO is a clear to opalescent, colorless to pale-yellow solution. Discard if cloudy, discolored, or contains extraneous particulate matter other than a few translucent-to-white, proteinaceous particles. Do not shake.
Preparation
- •
- Withdraw the required volume of OPDIVO and transfer into an intravenous container.
- •
- Dilute OPDIVO with either 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP to prepare an infusion with a final concentration ranging from 1 mg/mL to 10 mg/mL. The total volume of infusion must not exceed 160 mL.
- •
- For adult and pediatric patients with body weight 40 kg or greater, do not exceed a total volume of infusion of 160 mL.
- •
- For adult and pediatric patients with body weight less than 40 kg, do not exceed a total volume of infusion of 4 mL/kg of body weight.
- •
- Mix diluted solution by gentle inversion. Do not shake.
- •
- Discard partially used vials or empty vials of OPDIVO.
- •
- The product does not contain a preservative.
- •
- After preparation, store the diluted solution either:
- •
- at room temperature at 20°C to 25°C (68°F to 77°F) and room light for no more than 8 hours from the time of preparation to end of the infusion. Discard diluted solution if not used within 8 hours from the time of preparation; or
- •
- under refrigeration at 2°C to 8°C (36°F to 46°F) and protected from light for no more than 7 days from the time of preparation to end of infusion. Discard diluted solution if not used within 7 days from the time of preparation.
- •
- Do not freeze.
Administration
- •
- Administer the infusion, after dilution, over 30 minutes through an intravenous line containing a sterile, nonpyrogenic, low protein binding in-line filter (pore size of 0.2 micrometer to 1.2 micrometer).
- •
- Administer OPDIVO in combination with other therapeutic agents as follows:
|
Combination Therapy |
|
|
Ipilimumab |
Administer OPDIVO first, followed by the other therapeutic agent(s). |
|
Platinum-Doublet Chemotherapy |
|
|
Ipilimumab and Platinum-Doublet Chemotherapy |
|
|
Fluoropyrimidine- and Platinum- Containing Chemotherapy |
|
- •
- Use separate infusion bags and filters for each infusion.
- •
- Flush the intravenous line at end of infusion.
- •
- Do not co-administer other drugs through the same intravenous line.
3. Dosage Forms and Strengths
Injection: 40 mg/4 mL (10 mg/mL), 100 mg/10 mL (10 mg/mL), 120 mg/12 mL (10 mg/mL), and 240 mg/24 mL (10 mg/mL) clear to opalescent, colorless to pale-yellow solution in a single-dose vial.
5. Warnings and Precautions
5.1 Severe and Fatal Immune-Mediated Adverse Reactions
OPDIVO is a monoclonal antibody that belongs to a class of drugs that bind to either the programmed death-receptor 1 (PD-1) or the PD-ligand 1 (PD-L1), blocking the PD-1/PD-L1 pathway, thereby removing inhibition of the immune response, potentially breaking peripheral tolerance and inducing immune-mediated adverse reactions. Important immune-mediated adverse reactions listed under Warnings and Precautions may not include all possible severe and fatal immune-mediated reactions.
Immune-mediated adverse reactions, which may be severe or fatal, can occur in any organ system or tissue. Immune-mediated adverse reactions can occur at any time after starting treatment with a PD-1/PD-L1 blocking antibody. While immune-mediated adverse reactions usually manifest during treatment with PD‑1/PD-L1 blocking antibodies, immune-mediated adverse reactions can also manifest after discontinuation of PD-1/PD-L1 blocking antibodies.
Early identification and management of immune-mediated adverse reactions are essential to ensure safe use of PD-1/PD-L1 blocking antibodies. Monitor patients closely for symptoms and signs that may be clinical manifestations of underlying immune-mediated adverse reactions. Evaluate liver enzymes, creatinine, and thyroid function at baseline and periodically during treatment. In cases of suspected immune-mediated adverse reactions, initiate appropriate workup to exclude alternative etiologies, including infection. Institute medical management promptly, including specialty consultation as appropriate.
Withhold or permanently discontinue OPDIVO depending on severity [see Dosage and Administration (2.3)]. In general, if OPDIVO requires interruption or discontinuation, administer systemic corticosteroid therapy (1 to 2 mg/kg/day prednisone or equivalent) until improvement to Grade 1 or less. Upon improvement to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Consider administration of other systemic immunosuppressants in patients whose immune-mediated adverse reactions are not controlled with corticosteroid therapy.
Toxicity management guidelines for adverse reactions that do not necessarily require systemic steroids (e.g., endocrinopathies and dermatologic reactions) are discussed below.
Immune-Mediated Pneumonitis
OPDIVO can cause immune-mediated pneumonitis, which is defined as requiring use of steroids and no clear alternate etiology. In patients treated with other PD-1/PD-L1 blocking antibodies, the incidence of pneumonitis is higher in patients who have received prior thoracic radiation.
OPDIVO as a Single Agent
Immune-mediated pneumonitis occurred in 3.1% (61/1994) of patients receiving OPDIVO as a single agent, including Grade 4 (<0.1%), Grade 3 (0.9%), and Grade 2 (2.1%) adverse reactions. Pneumonitis led to permanent discontinuation of OPDIVO in 1.1% and withholding of OPDIVO in 0.8% of patients.
Systemic corticosteroids were required in 100% (61/61) of patients with pneumonitis. Pneumonitis resolved in 84% of the 61 patients. Of the 15 patients in whom OPDIVO was withheld for pneumonitis, 14 reinitiated OPDIVO after symptom improvement; of these, 4 (29%) had recurrence of pneumonitis.
OPDIVO with Ipilimumab
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: In NSCLC, immune-mediated pneumonitis occurred in 9% (50/576) of patients receiving OPDIVO 3 mg/kg every 2 weeks with ipilimumab 1 mg/kg every 6 weeks, including Grade 4 (0.5%), Grade 3 (3.5%), and Grade 2 (4%) immune-mediated pneumonitis. Four patients (0.7%) died due to pneumonitis. Immune-mediated pneumonitis led to permanent discontinuation of OPDIVO with ipilimumab in 5% of patients and withholding of OPDIVO with ipilimumab in 3.6% of patients.
Systemic corticosteroids were required in 100% of patients with pneumonitis. Pneumonitis resolved in 72% of the patients. Approximately 13% (2/16) of patients had recurrence of pneumonitis after reinitiation of OPDIVO with ipilimumab.
Immune-Mediated Colitis
OPDIVO can cause immune-mediated colitis, defined as requiring use of corticosteroids and no clear alternate etiology. A common symptom included in the definition of colitis was diarrhea. Cytomegalovirus (CMV) infection/reactivation has been reported in patients with corticosteroid-refractory immune-mediated colitis. In cases of corticosteroid-refractory colitis, consider repeating infectious workup to exclude alternative etiologies.
OPDIVO as a Single Agent
Immune-mediated colitis occurred in 2.9% (58/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (1.7%) and Grade 2 (1%) adverse reactions. Colitis led to permanent discontinuation of OPDIVO in 0.7% and withholding of OPDIVO in 0.9% of patients.
Systemic corticosteroids were required in 100% (58/58) of patients with colitis. Four patients required addition of infliximab to high-dose corticosteroids. Colitis resolved in 86% of the 58 patients. Of the 18 patients in whom OPDIVO was withheld for colitis, 16 reinitiated OPDIVO after symptom improvement; of these, 12 (75%) had recurrence of colitis.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Immune-mediated colitis occurred in 25% (115/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 4 (0.4%), Grade 3 (14%), and Grade 2 (8%) adverse reactions. Colitis led to permanent discontinuation of OPDIVO with ipilimumab in 14% and withholding of OPDIVO with ipilimumab in 4.4% of patients.
Systemic corticosteroids were required in 100% (115/115) of patients with colitis. Approximately 23% of patients required addition of infliximab to high-dose corticosteroids. Colitis resolved in 93% of the 115 patients. Of the 20 patients in whom OPDIVO with ipilimumab was withheld for colitis, 16 reinitiated treatment after symptom improvement; of these, 9 (56%) had recurrence of colitis.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Immune-mediated colitis occurred in 9% (60/666) of patients with RCC or CRC receiving OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 3 (4.4%) and Grade 2 (3.7%) adverse reactions. Colitis led to permanent discontinuation of OPDIVO with ipilimumab in 3.2% and withholding of OPDIVO with ipilimumab in 2.7% of patients with RCC or CRC.
Systemic corticosteroids were required in 100% (60/60) of patients with colitis. Approximately 23% of patients with immune-mediated colitis required addition of infliximab to high-dose corticosteroids. Colitis resolved in 95% of the 60 patients. Of the 18 patients in whom OPDIVO with ipilimumab was withheld for colitis, 16 reinitiated treatment after symptom improvement; of these, 10 (63%) had recurrence of colitis.
Immune-Mediated Hepatitis and Hepatotoxicity
OPDIVO can cause immune-mediated hepatitis, defined as requiring the use of corticosteroids and no clear alternate etiology.
OPDIVO as a Single Agent
Immune-mediated hepatitis occurred in 1.8% (35/1994) of patients receiving OPDIVO as a single agent, including Grade 4 (0.2%), Grade 3 (1.3%), and Grade 2 (0.4%) adverse reactions. Hepatitis led to permanent discontinuation of OPDIVO in 0.7% and withholding of OPDIVO in 0.6% of patients.
Systemic corticosteroids were required in 100% (35/35) of patients with hepatitis. Two patients required the addition of mycophenolic acid to high-dose corticosteroids. Hepatitis resolved in 91% of the 35 patients. Of the 12 patients in whom OPDIVO was withheld for hepatitis, 11 reinitiated OPDIVO after symptom improvement; of these, 9 (82%) had recurrence of hepatitis.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Immune-mediated hepatitis occurred in 15% (70/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 4 (2.4%), Grade 3 (11%), and Grade 2 (1.8%) adverse reactions. Immune-mediated hepatitis led to permanent discontinuation of OPDIVO with ipilimumab in 8% or withholding of OPDIVO with ipilimumab in 3.5% of patients.
Systemic corticosteroids were required in 100% (70/70) of patients with hepatitis. Approximately 9% of patients with immune-mediated hepatitis required the addition of mycophenolic acid to high-dose corticosteroids. Hepatitis resolved in 91% of the 70 patients. Of the 16 patients in whom OPDIVO with ipilimumab was withheld for hepatitis, 14 reinitiated treatment after symptom improvement; of these, 8 (57%) had recurrence of hepatitis.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Immune-mediated hepatitis occurred in 7% (48/666) of patients with RCC or CRC receiving OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 4 (1.2%), Grade 3 (4.9%), and Grade 2 (0.4%) adverse reactions. Immune-mediated hepatitis led to permanent discontinuation of OPDIVO with ipilimumab in 3.6% and withholding of OPDIVO with ipilimumab in 2.6% of patients with RCC or CRC.
Systemic corticosteroids were required in 100% (48/48) of patients with hepatitis. Approximately 19% of patients with immune-mediated hepatitis required addition of mycophenolic acid to high-dose corticosteroids. Hepatitis resolved in 88% of the 48 patients. Of the 17 patients in whom OPDIVO with ipilimumab was withheld for hepatitis, 14 reinitiated treatment after symptom improvement; of these, 10 (71%) had recurrence of hepatitis.
OPDIVO with Cabozantinib
OPDIVO in combination with cabozantinib can cause hepatic toxicity with higher frequencies of Grade 3 and 4 ALT and AST elevations compared to OPDIVO alone. Monitor liver enzymes before initiation of and periodically throughout treatment. Consider more frequent monitoring of liver enzymes as compared to when the drugs are administered as single agents. For elevated liver enzymes, interrupt OPDIVO and cabozantinib and consider administering corticosteroids [see Dosage and Administration (2.3)].
With the combination of OPDIVO and cabozantinib, Grades 3 and 4 increased ALT or AST were seen in 11% of patients [see Adverse Reactions (6.1)]. ALT or AST >3 times ULN (Grade ≥2) was reported in 83 patients, of whom 23 (28%) received systemic corticosteroids; ALT or AST resolved to Grades 0-1 in 74 (89%). Among the 44 patients with Grade ≥2 increased ALT or AST who were rechallenged with either OPDIVO (n=11) or cabozantinib (n=9) administered as a single agent or with both (n=24), recurrence of Grade ≥2 increased ALT or AST was observed in 2 patients receiving OPDIVO, 2 patients receiving cabozantinib, and 7 patients receiving both OPDIVO and cabozantinib.
Immune-Mediated Endocrinopathies
Adrenal Insufficiency
OPDIVO can cause primary or secondary adrenal insufficiency. For grade 2 or higher adrenal insufficiency, initiate symptomatic treatment, including hormone replacement as clinically indicated. Withhold OPDIVO depending on severity [see Dosage and Administration (2.3)].
OPDIVO as a Single Agent
Adrenal insufficiency occurred in 1% (20/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (0.4%) and Grade 2 (0.6%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of OPDIVO in 0.1% and withholding of OPDIVO in 0.4% of patients.
Approximately 85% of patients with adrenal insufficiency received hormone replacement therapy. Systemic corticosteroids were required in 90% (18/20) of patients with adrenal insufficiency. Adrenal insufficiency resolved in 35% of the 20 patients. Of the 8 patients in whom OPDIVO was withheld for adrenal insufficiency, 4 reinitiated OPDIVO after symptom improvement and all required hormone replacement therapy for their ongoing adrenal insufficiency.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Adrenal insufficiency occurred in 8% (35/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 4 (0.2%), Grade 3 (2.4%), and Grade 2 (4.2%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of OPDIVO with ipilimumab in 0.4% and withholding of OPDIVO with ipilimumab in 2% of patients.
Approximately 71% (25/35) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 37% of the 35 patients. Of the 9 patients in whom OPDIVO with ipilimumab was withheld for adrenal insufficiency, 7 reinitiated treatment after symptom improvement and all required hormone replacement therapy for their ongoing adrenal insufficiency.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Adrenal insufficiency occurred in 7% (48/666) of patients with RCC or CRC who received OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 4 (0.3%), Grade 3 (2.5%), and Grade 2 (4.1%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of OPDIVO with ipilimumab in 1.2% and withholding of OPDIVO with ipilimumab in 2.1% of patients with RCC or CRC.
Approximately 94% (45/48) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 29% of the 48 patients. Of the 14 patients in whom OPDIVO with ipilimumab was withheld for adrenal insufficiency, 11 reinitiated treatment after symptom improvement; of these, all received hormone replacement therapy and 2 (18%) had recurrence of adrenal insufficiency.
OPDIVO with Cabozantinib
Adrenal insufficiency occurred in 4.7% (15/320) of patients with RCC who received OPDIVO with cabozantinib, including Grade 3 (2.2%), and Grade 2 (1.9%) adverse reactions. Adrenal insufficiency led to permanent discontinuation of OPDIVO and cabozantinib in 0.9% and withholding of OPDIVO and cabozantinib in 2.8% of patients with RCC.
Approximately 80% (12/15) of patients with adrenal insufficiency received hormone replacement therapy, including systemic corticosteroids. Adrenal insufficiency resolved in 27% (n=4) of the 15 patients. Of the 9 patients in whom OPDIVO with cabozantinib was withheld for adrenal insufficiency, 6 reinstated treatment after symptom improvement; of these, all (n=6) received hormone replacement therapy and 2 had recurrence of adrenal insufficiency.
Hypophysitis
OPDIVO can cause immune-mediated hypophysitis. Hypophysitis can present with acute symptoms associated with mass effect such as headache, photophobia, or visual field defects. Hypophysitis can cause hypopituitarism. Initiate hormone replacement as clinically indicated. Withhold or permanently discontinue OPDIVO depending on severity [see Dosage and Administration (2.3)].
OPDIVO as a Single Agent
Hypophysitis occurred in 0.6% (12/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (0.2%) and Grade 2 (0.3%) adverse reactions. Hypophysitis led to permanent discontinuation of OPDIVO in <0.1% and withholding of OPDIVO in 0.2% of patients.
Approximately 67% (8/12) of patients with hypophysitis received hormone replacement therapy, including systemic corticosteroids. Hypophysitis resolved in 42% of the 12 patients. Of the 3 patients in whom OPDIVO was withheld for hypophysitis, 2 reinitiated OPDIVO after symptom improvement; of these, none had recurrence of hypophysitis.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Hypophysitis occurred in 9% (42/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 3 (2.4%) and Grade 2 (6%) adverse reactions. Hypophysitis led to permanent discontinuation of OPDIVO with ipilimumab in 0.9% and withholding of OPDIVO with ipilimumab in 4.2% of patients.
Approximately 86% of patients with hypophysitis received hormone replacement therapy. Systemic corticosteroids were required in 88% (37/42) of patients with hypophysitis. Hypophysitis resolved in 38% of the 42 patients. Of the 19 patients in whom OPDIVO with ipilimumab was withheld for hypophysitis, 9 reinitiated treatment after symptom improvement; of these, 1 (11%) had recurrence of hypophysitis.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Hypophysitis occurred in 4.4% (29/666) of patients with RCC or CRC receiving OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 4 (0.3%), Grade 3 (2.4%), and Grade 2 (0.9%) adverse reactions. Hypophysitis led to permanent discontinuation of OPDIVO with ipilimumab in 1.2% and withholding of OPDIVO with ipilimumab in 2.1% of patients with RCC or CRC.
Approximately 72% (21/29) of patients with hypophysitis received hormone replacement therapy, including systemic corticosteroids. Hypophysitis resolved in 59% of the 29 patients. Of the 14 patients in whom OPDIVO with ipilimumab was withheld for hypophysitis, 11 reinitiated treatment after symptom improvement; of these, 2 (18%) had recurrence of hypophysitis.
Thyroid Disorders
OPDIVO can cause immune-mediated thyroid disorders. Thyroiditis can present with or without endocrinopathy. Hypothyroidism can follow hyperthyroidism. Initiate hormone replacement or medical management as clinically indicated. Withhold or permanently discontinue OPDIVO depending on severity [see Dosage and Administration (2.3)].
Thyroiditis
OPDIVO as a Single Agent
Thyroiditis occurred in 0.6% (12/1994) of patients receiving OPDIVO as a single agent, including Grade 2 (0.2%) adverse reactions. Thyroiditis led to permanent discontinuation of OPDIVO in no patients and withholding of OPDIVO in 0.2% of patients.
Systemic corticosteroids were required in 17% (2/12) of patients with thyroiditis. Thyroiditis resolved in 58% of the 12 patients. Of the 3 patients in whom OPDIVO was withheld for thyroiditis, 1 reinitiated OPDIVO after symptom improvement without recurrence of thyroiditis.
Hyperthyroidism
OPDIVO as a Single Agent
Hyperthyroidism occurred in 2.7% (54/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (<0.1%) and Grade 2 (1.2%) adverse reactions. Hyperthyroidism led to the permanent discontinuation of OPDIVO in no patients and withholding of OPDIVO in 0.4% of patients.
Approximately 19% of patients with hyperthyroidism received methimazole, 7% received carbimazole, and 4% received propylthiouracil. Systemic corticosteroids were required in 9% (5/54) of patients. Hyperthyroidism resolved in 76% of the 54 patients. Of the 7 patients in whom OPDIVO was withheld for hyperthyroidism, 4 reinitiated OPDIVO after symptom improvement; of these, none had recurrence of hyperthyroidism.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Hyperthyroidism occurred in 9% (42/456) of patients with melanoma or HCC who received OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 3 (0.9%) and Grade 2 (4.2%) adverse reactions. Hyperthyroidism led to the permanent discontinuation of OPDIVO with ipilimumab in no patients and withholding of OPDIVO with ipilimumab in 2.4% of patients.
Approximately 26% of patients with hyperthyroidism received methimazole and 21% received carbimazole. Systemic corticosteroids were required in 17% (7/42) of patients. Hyperthyroidism resolved in 91% of the 42 patients. Of the 11 patients in whom OPDIVO with ipilimumab was withheld for hyperthyroidism, 8 reinitiated treatment after symptom improvement; of these, 1 (13%) had recurrence of hyperthyroidism.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Hyperthyroidism occurred in 12% (80/666) of patients with RCC or CRC who received OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 3 (0.6%) and Grade 2 (4.5%) adverse reactions. Hyperthyroidism led to permanent discontinuation of OPDIVO with ipilimumab in no patients and withholding of OPDIVO with ipilimumab in 2.3% of patients with RCC or CRC.
Of the 80 patients with RCC or CRC who developed hyperthyroidism, approximately 16% received methimazole and 3% received carbimazole. Systemic corticosteroids were required in 20% (16/80) of patients with hyperthyroidism. Hyperthyroidism resolved in 85% of the 80 patients. Of the 15 patients in whom OPDIVO with ipilimumab was withheld for hyperthyroidism, 11 reinitiated treatment after symptom improvement; of these, 3 (27%) had recurrence of hyperthyroidism.
Hypothyroidism
OPDIVO as a Single Agent
Hypothyroidism occurred in 8% (163/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (0.2%) and Grade 2 (4.8%) adverse reactions. Hypothyroidism led to the permanent discontinuation of OPDIVO in no patients and withholding of OPDIVO in 0.5% of patients.
Approximately 79% of patients with hypothyroidism received levothyroxine. Systemic corticosteroids were required in 3.1% (5/163) of patients with hypothyroidism. Hypothyroidism resolved in 35% of the 163 patients. Of the 9 patients in whom OPDIVO was withheld for hypothyroidism, 3 reinitiated OPDIVO after symptom improvement; of these, 1 (33%) had recurrence of hypothyroidism.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Hypothyroidism occurred in 20% (91/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 3 (0.4%) and Grade 2 (11%) adverse reactions. Hypothyroidism led to the permanent discontinuation of OPDIVO with ipilimumab in 0.9% and withholding of OPDIVO with ipilimumab in 0.9% of patients.
Approximately 89% of patients with hypothyroidism received levothyroxine. Systemic corticosteroids were required in 2.2% (2/91) of patients with hypothyroidism. Hypothyroidism resolved in 41% of the 91 patients. Of the 4 patients in whom OPDIVO with ipilimumab was withheld for hypothyroidism, 2 reinitiated treatment after symptom improvement; of these, none had recurrence of hypothyroidism.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Hypothyroidism occurred in 18% (122/666) of patients with RCC or CRC who received OPDIVO 3 mg/kg and ipilimumab 1 mg/kg every 3 weeks, including Grade 3 (0.6%) and Grade 2 (11%) adverse reactions. Hypothyroidism led to permanent discontinuation of OPDIVO with ipilimumab in 0.2% and withholding of OPDIVO with ipilimumab in 1.4% of patients with RCC or CRC.
Of the 122 patients with RCC or CRC who developed hypothyroidism, approximately 82% received levothyroxine. Systemic corticosteroids were required in 7% (9/122) of patients with hypothyroidism. Hypothyroidism resolved in 27% of the 122 patients. Of the 9 patients in whom OPDIVO with ipilimumab was withheld for hypothyroidism, 5 reinitiated treatment after symptom improvement; of these, 1 (20%) had recurrence of hypothyroidism.
Type 1 Diabetes Mellitus, which can present with Diabetic Ketoacidosis
Monitor patients for hyperglycemia or other signs and symptoms of diabetes. Initiate treatment with insulin as clinically indicated. Withhold OPDIVO depending on severity [see Dosage and Administration (2.3)].
OPDIVO as a Single Agent
Diabetes occurred in 0.9% (17/1994) of patients receiving OPDIVO as a single agent, including Grade 3 (0.4%) and Grade 2 (0.3%) adverse reactions, and two cases of diabetic ketoacidosis. Diabetes led to the permanent discontinuation of OPDIVO in no patients and withholding of OPDIVO in 0.1% of patients.
No patients (0/17) with diabetes required systemic corticosteroids. Diabetes resolved in 29% of the 17 patients. Of the 2 patients in whom OPDIVO was withheld for diabetes, both reinitiated OPDIVO after symptom improvement; of these, neither had recurrence of diabetes.
Immune-Mediated Nephritis with Renal Dysfunction
OPDIVO can cause immune-mediated nephritis, which is defined as requiring use of steroids and no clear alternate etiology.
OPDIVO as a Single Agent
Immune-mediated nephritis and renal dysfunction occurred in 1.2% (23/1994) of patients receiving OPDIVO as a single agent, including Grade 4 (<0.1%), Grade 3 (0.5%), and Grade 2 (0.6%) adverse reactions. Immune-mediated nephritis and renal dysfunction led to permanent discontinuation of OPDIVO in 0.3% and withholding of OPDIVO in 0.4% of patients.
Systemic corticosteroids were required in 100% (23/23) of patients with nephritis and renal dysfunction. Nephritis and renal dysfunction resolved in 78% of the 23 patients. Of the 7 patients in whom OPDIVO was withheld for nephritis or renal dysfunction, 7 reinitiated OPDIVO after symptom improvement; of these, 1 (14%) had recurrence of nephritis or renal dysfunction.
Immune-Mediated Dermatologic Adverse Reactions
OPDIVO can cause immune-mediated rash or dermatitis, defined as requiring the use of steroids and no clear alternate etiology. Exfoliative dermatitis, including Stevens-Johnson Syndrome, toxic epidermal necrolysis (TEN), and DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) has occurred with PD-1/PD-L1 blocking antibodies. Topical emollients and/or topical corticosteroids may be adequate to treat mild to moderate non-exfoliative rashes. Withhold or permanently discontinue OPDIVO depending on severity [see Dosage and Administration (2.3)].
OPDIVO as a Single Agent
Immune-mediated rash occurred in 9% (171/1994) of patients, including Grade 3 (1.1%) and Grade 2 (2.2%) adverse reactions. Immune-mediated rash led to permanent discontinuation of OPDIVO in 0.3% and withholding of OPDIVO in 0.5% of patients.
Systemic corticosteroids were required in 100% (171/171) of patients with immune-mediated rash. Rash resolved in 72% of the 171 patients. Of the 10 patients in whom OPDIVO was withheld for immune-mediated rash, 9 reinitiated OPDIVO after symptom improvement; of these, 3 (33%) had recurrence of immune-mediated rash.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg: Immune-mediated rash occurred in 28% (127/456) of patients with melanoma or HCC receiving OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks, including Grade 3 (4.8%) and Grade 2 (10%) adverse reactions. Immune-mediated rash led to permanent discontinuation of OPDIVO with ipilimumab in 0.4% and withholding of OPDIVO with ipilimumab in 3.9% of patients.
Systemic corticosteroids were required in 100% (127/127) of patients with immune-mediated rash. Rash resolved in 84% of the 127 patients. Of the 18 patients in whom OPDIVO with ipilimumab was withheld for immune-mediated rash, 15 reinitiated treatment after symptom improvement; of these, 8 (53%) had recurrence of immune-mediated rash.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg: Immune-mediated rash occurred in 16% (108/666) of patients with RCC or CRC who received OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, including Grade 3 (3.5%) and Grade 2 (4.2%) adverse reactions. Immune-mediated rash led to permanent discontinuation of OPDIVO with ipilimumab in 0.5% of patients and withholding of OPDIVO with ipilimumab in 2.0% of patients with RCC or CRC.
Systemic corticosteroids were required in 100% (108/108) of patients with immune-mediated rash. Rash resolved in 75% of the 108 patients. Of the 13 patients in whom OPDIVO with ipilimumab was withheld for immune-mediated rash, 11 reinitiated treatment after symptom improvement; of these, 5 (46%) had recurrence of immune-mediated rash.
Other Immune-Mediated Adverse Reactions
The following clinically significant immune-mediated adverse reactions occurred at an incidence of <1% (unless otherwise noted) in patients who received OPDIVO or OPDIVO in combination with ipilimumab, or were reported with the use of other PD-1/PD-L1 blocking antibodies. Severe or fatal cases have been reported for some of these adverse reactions.
Cardiac/Vascular: Myocarditis, pericarditis, vasculitis
Nervous System: Meningitis, encephalitis, myelitis and demyelination, myasthenic syndrome/myasthenia gravis (including exacerbation), Guillain-Barre syndrome, nerve paresis, autoimmune neuropathy
Ocular: Uveitis, iritis, and other ocular inflammatory toxicities can occur. Some cases can be associated with retinal detachment. Various grades of visual impairment, including blindness, can occur. If uveitis occurs in combination with other immune-mediated adverse reactions, consider a Vogt-Koyanagi-Harada-like syndrome, as this may require treatment with systemic steroids to reduce the risk of permanent vision loss
Gastrointestinal: Pancreatitis to include increases in serum amylase and lipase levels, gastritis, duodenitis
Musculoskeletal and Connective Tissue: Myositis/polymyositis, rhabdomyolysis, and associated sequelae including renal failure, arthritis, polymyalgia rheumatic
Endocrine: Hypoparathyroidism
Other (Hematologic/Immune): Hemolytic anemia, aplastic anemia, hemophagocytic lymphohistiocytosis, systemic inflammatory response syndrome, histiocytic necrotizing lymphadenitis (Kikuchi lymphadenitis), sarcoidosis, immune thrombocytopenic purpura, solid organ transplant rejection, other transplant (including corneal graft) rejection
5.2 Infusion-Related Reactions
OPDIVO can cause severe infusion-related reactions, which have been reported in <1% of patients in clinical trials. Discontinue OPDIVO in patients with severe or life-threatening infusion-related reactions. Interrupt or slow the rate of infusion in patients with mild or moderate infusion-related reactions [see Dosage and Administration (2.3)].
OPDIVO as a Single Agent
In patients who received OPDIVO as a 60-minute intravenous infusion, infusion-related reactions occurred in 6.4% (127/1994) of patients.
In a trial assessing the pharmacokinetics and safety of a more rapid infusion, in which patients received OPDIVO as a 60-minute intravenous infusion or a 30-minute intravenous infusion, infusion-related reactions occurred in 2.2% (8/368) and 2.7% (10/369) of patients, respectively. Additionally, 0.5% (2/368) and 1.4% (5/369) of patients, respectively, experienced adverse reactions within 48 hours of infusion that led to dose delay, permanent discontinuation, or withholding of OPDIVO.
OPDIVO with Ipilimumab
OPDIVO 1 mg/kg with Ipilimumab 3 mg/kg
Infusion-related reactions occurred in 2.5% (10/407) of patients with melanoma and in 8% (4/49) of patients with HCC who received OPDIVO 1 mg/kg with ipilimumab 3 mg/kg every 3 weeks.
OPDIVO 3 mg/kg with Ipilimumab 1 mg/kg
Infusion-related reactions occurred in 5.1% (28/547) of patients with RCC and 4.2% (5/119) of patients with CRC who received OPDIVO 3 mg/kg with ipilimumab 1 mg/kg every 3 weeks, respectively. Infusion-related reactions occurred in 12% (37/300) of patients with malignant pleural mesothelioma who received OPDIVO 3 mg/kg every 2 weeks with ipilimumab 1 mg/kg every 6 weeks.
5.3 Complications of Allogeneic Hematopoietic Stem Cell Transplantation
Fatal and other serious complications can occur in patients who receive allogeneic hematopoietic stem cell transplantation (HSCT) before or after being treated with a PD-1 receptor blocking antibody. Transplant-related complications include hyperacute graft-versus-host-disease (GVHD), acute GVHD, chronic GVHD, hepatic veno-occlusive disease (VOD) after reduced intensity conditioning, and steroid-requiring febrile syndrome (without an identified infectious cause) [see Adverse Reactions (6.1)]. These complications may occur despite intervening therapy between PD-1 blockade and allogeneic HSCT.
Follow patients closely for evidence of transplant-related complications and intervene promptly. Consider the benefit versus risks of treatment with a PD-1 receptor blocking antibody prior to or after an allogeneic HSCT.
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action and data from animal studies, OPDIVO can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of nivolumab to cynomolgus monkeys from the onset of organogenesis through delivery resulted in increased abortion and premature infant death. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and for 5 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.5 Increased Mortality in Patients with Multiple Myeloma when OPDIVO Is Added to a Thalidomide Analogue and Dexamethasone
In randomized clinical trials in patients with multiple myeloma, the addition of a PD-1 blocking antibody, including OPDIVO, to a thalidomide analogue plus dexamethasone, a use for which no PD-1 or PD-L1 blocking antibody is indicated, resulted in increased mortality. Treatment of patients with multiple myeloma with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling.
- •
- Severe and Fatal Immune-Mediated Adverse Reactions [see Warnings and Precautions (5.1)]
- •
- Infusion-Related Reactions [see Warnings and Precautions (5.2)]
- •
- Complications of Allogeneic HSCT [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in WARNINGS AND PRECAUTIONS reflect exposure to OPDIVO as a single agent in 1994 patients enrolled in CHECKMATE-037, CHECKMATE-017, CHECKMATE-057, CHECKMATE-066, CHECKMATE-025, CHECKMATE-067, CHECKMATE-205, CHECKMATE-039 or a single-arm trial in NSCLC (n=117); OPDIVO 1 mg/kg with ipilimumab 3 mg/kg in patients enrolled in CHECKMATE-067 (n=313), CHECKMATE-040 (n=49), or another randomized trial (n=94); OPDIVO 3 mg/kg administered with ipilimumab 1 mg/kg (n=666) in patients enrolled in CHECKMATE-214 or CHECKMATE-142; OPDIVO 3 mg/kg every 2 weeks with ipilimumab 1 mg/kg every 6 weeks in patients enrolled in CHECKMATE-227 (n=576) or CHECKMATE-743 (n=300); OPDIVO 360 mg with ipilimumab 1 mg/kg and 2 cycles of platinum-doublet chemotherapy in CHECKMATE-9LA (n=361); and OPDIVO 240 mg with cabozantinib 40 mg in patients enrolled in CHECKMATE-9ER (n=320).
Unresectable or Metastatic Melanoma
Previously Treated Metastatic Melanoma
The safety of OPDIVO was evaluated in CHECKMATE-037, a randomized, open-label trial in 370 patients with unresectable or metastatic melanoma [see Clinical Studies (14.1)]. Patients had documented disease progression following treatment with ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. The trial excluded patients with autoimmune disease, prior ipilimumab-related Grade 4 adverse reactions (except for endocrinopathies) or Grade 3 ipilimumab-related adverse reactions that had not resolved or were inadequately controlled within 12 weeks of the initiating event, patients with a condition requiring chronic systemic treatment with corticosteroids (>10 mg daily prednisone equivalent) or other immunosuppressive medications, a positive test for hepatitis B or C, and a history of HIV. Patients received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (n=268) or investigator’s choice of chemotherapy (n=102): dacarbazine 1000 mg/m2 intravenously every 3 weeks or carboplatin AUC 6 mg/mL/min and paclitaxel 175 mg/m2 intravenously every 3 weeks. The median duration of exposure was 5.3 months (range: 1 day to 13.8+ months) in OPDIVO-treated patients and was 2 months (range: 1 day to 9.6+ months) in chemotherapy-treated patients. In this ongoing trial, 24% of patients received OPDIVO for >6 months and 3% of patients received OPDIVO for >1 year.
The population characteristics in the OPDIVO group and the chemotherapy group were similar: 66% male, median age 59.5 years, 98% White, baseline Eastern Cooperative Oncology Group (ECOG) performance status 0 (59%) or 1 (41%), 74% with M1c stage disease, 73% with cutaneous melanoma, 11% with mucosal melanoma, 73% received two or more prior therapies for advanced or metastatic disease, and 18% had brain metastasis. There were more patients in the OPDIVO group with elevated lactate dehydrogenase (LDH) at baseline (51% vs. 38%).
Serious adverse reactions occurred in 41% of patients receiving OPDIVO. OPDIVO was discontinued for adverse reactions in 9% of patients. Twenty-six percent of patients receiving OPDIVO had a dose interruption for an adverse reaction. Grade 3 and 4 adverse reactions occurred in 42% of patients receiving OPDIVO. The most frequent Grade 3 and 4 adverse reactions reported in 2% to <5% of patients receiving OPDIVO were abdominal pain, hyponatremia, increased aspartate aminotransferase, and increased lipase. The most common adverse reaction (reported in ≥20% of patients) was rash.
Tables 5 and 6 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-037.
| Toxicity was graded per NCI CTCAE v4. a Includes maculopapular rash, erythematous rash, pruritic rash, follicular rash, macular rash, papular rash, pustular rash, vesicular rash, and acneiform dermatitis. b Includes rhinitis, pharyngitis, and nasopharyngitis. |
||||
|
Adverse Reaction |
OPDIVO
|
Chemotherapy
|
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Skin and Subcutaneous Tissue |
||||
|
Rasha |
21 |
0.4 |
7 |
0 |
|
Pruritus |
19 |
0 |
3.9 |
0 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough |
17 |
0 |
6 |
0 |
|
Infections |
||||
|
Upper respiratory tract infectionb |
11 |
0 |
2 |
0 |
|
General |
||||
|
Peripheral edema |
10 |
0 |
5 |
0 |
Clinically important adverse reactions in <10% of patients who received OPDIVO were:
Cardiac Disorders: ventricular arrhythmia
Eye Disorders: iridocyclitis
General Disorders and Administration Site Conditions: infusion-related reactions
Investigations: increased amylase, increased lipase
Nervous System Disorders: dizziness, peripheral and sensory neuropathy
Skin and Subcutaneous Tissue Disorders: exfoliative dermatitis, erythema multiforme, vitiligo, psoriasis
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 252 to 256 patients) and chemotherapy group (range: 94 to 96 patients). | ||||
|
Laboratory Abnormality |
OPDIVO |
Chemotherapy |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Increased AST |
28 |
2.4 |
12 |
1 |
|
Hyponatremia |
25 |
5 |
18 |
1.1 |
|
Increased alkaline phosphatase |
22 |
2.4 |
13 |
1.1 |
|
Increased ALT |
16 |
1.6 |
5 |
0 |
|
Hyperkalemia |
15 |
2 |
6 |
0 |
Previously Untreated Metastatic Melanoma
CHECKMATE-066
The safety of OPDIVO was also evaluated in CHECKMATE-066, a randomized, double-blind, active-controlled trial in 411 previously untreated patients with BRAF V600 wild-type unresectable or metastatic melanoma [see Clinical Studies (14.1)]. The trial excluded patients with autoimmune disease and patients requiring chronic systemic treatment with corticosteroids (>10 mg daily prednisone equivalent) or other immunosuppressive medications. Patients received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (n=206) or dacarbazine 1000 mg/m2 intravenously every 3 weeks (n=205). The median duration of exposure was 6.5 months (range: 1 day to 16.6 months) in OPDIVO-treated patients. In this trial, 47% of patients received OPDIVO for >6 months and 12% of patients received OPDIVO for >1 year.
The trial population characteristics in the OPDIVO group and dacarbazine group: 59% male, median age 65 years, 99.5% White, 61% with M1c stage disease, 74% with cutaneous melanoma, 11% with mucosal melanoma, 4% with brain metastasis, and 37% with elevated LDH at baseline. There were more patients in the OPDIVO group with ECOG performance status 0 (71% vs. 59%).
Serious adverse reactions occurred in 36% of patients receiving OPDIVO. Adverse reactions led to permanent discontinuation of OPDIVO in 7% of patients and dose interruption in 26% of patients; no single type of adverse reaction accounted for the majority of OPDIVO discontinuations. Grade 3 and 4 adverse reactions occurred in 41% of patients receiving OPDIVO.
The most frequent Grade 3 and 4 adverse reactions reported in ≥2% of patients receiving OPDIVO were increased gamma-glutamyl transferase (3.9%) and diarrhea (3.4%). The most common adverse reactions (reported in ≥20% of patients and at a higher incidence than in the dacarbazine arm) were fatigue, musculoskeletal pain, rash, and pruritus.
Tables 7 and 8 summarize selected adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-066.
| Toxicity was graded per NCI CTCAE v4. a Includes periorbital edema, face edema, generalized edema, gravitational edema, localized edema, peripheral edema, pulmonary edema, and lymphedema. b Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, pain in jaw, and spinal pain. c Includes maculopapular rash, erythematous rash, pruritic rash, follicular rash, macular rash, papular rash, pustular rash, vesicular rash, dermatitis, allergic dermatitis, exfoliative dermatitis, acneiform dermatitis, drug eruption, and skin reaction. d Includes rhinitis, viral rhinitis, pharyngitis, and nasopharyngitis. |
||||
|
Adverse Reaction |
OPDIVO
|
Dacarbazine
|
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||
|
49 |
1.9 |
39 |
3.4 |
|
12 |
1.5 |
4.9 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
32 |
2.9 |
25 |
2.4 |
|
Skin and Subcutaneous Tissue |
||||
|
28 |
1.5 |
12 |
0 |
|
23 |
0.5 |
12 |
0 |
|
11 |
0 |
0.5 |
0 |
|
10 |
0 |
2.9 |
0 |
|
Infections |
||||
|
17 |
0 |
6 |
0 |
Clinically important adverse reactions in <10% of patients who received OPDIVO were:
Nervous System Disorders: peripheral neuropathy
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 194 to 197 patients) and dacarbazine group (range: 186 to 193 patients). | ||||
|
Laboratory Abnormality |
OPDIVO |
Dacarbazine |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Increased ALT |
25 |
3 |
19 |
0.5 |
|
Increased AST |
24 |
3.6 |
19 |
0.5 |
|
Increased alkaline phosphatase |
21 |
2.6 |
14 |
1.6 |
|
Increased bilirubin |
13 |
3.1 |
6 |
0 |
CHECKMATE-067
The safety of OPDIVO, administered with ipilimumab or as a single agent, was evaluated in CHECKMATE-067, a randomized (1:1:1), double-blind trial in 937 patients with previously untreated, unresectable or metastatic melanoma [see Clinical Studies (14.1)]. The trial excluded patients with autoimmune disease, a medical condition requiring systemic treatment with corticosteroids (more than 10 mg daily prednisone equivalent) or other immunosuppressive medication within 14 days of the start of study therapy, a positive test result for hepatitis B or C, or a history of HIV.
Patients were randomized to receive:
- •
- OPDIVO 1 mg/kg over 60 minutes with ipilimumab 3 mg/kg by intravenous infusion every 3 weeks for 4 doses followed by OPDIVO as a single agent at a dose of 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (OPDIVO and ipilimumab arm; n=313), or
- •
- OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (OPDIVO arm; n=313), or
- •
- Ipilimumab 3 mg/kg by intravenous infusion every 3 weeks for up to 4 doses (ipilimumab arm; n=311).
The median duration of exposure to OPDIVO was 2.8 months (range: 1 day to 36.4 months) for the OPDIVO and ipilimumab arm and 6.6 months (range: 1 day to 36.0 months) for the OPDIVO arm. In the OPDIVO and ipilimumab arm, 39% were exposed to OPDIVO for ≥6 months and 30% exposed for >1 year. In the OPDIVO arm, 53% were exposed for ≥6 months and 40% for >1 year.
The population characteristics were: 65% male, median age 61 years, 97% White, baseline ECOG performance status 0 (73%) or 1 (27%), 93% with American Joint Committee on Cancer (AJCC) Stage IV disease, 58% with M1c stage disease; 36% with elevated LDH at baseline, 4% with a history of brain metastasis, and 22% had received adjuvant therapy.
Serious adverse reactions (74% and 44%), adverse reactions leading to permanent discontinuation (47% and 18%) or to dosing delays (58% and 36%), and Grade 3 or 4 adverse reactions (72% and 51%) all occurred more frequently in the OPDIVO and ipilimumab arm relative to the OPDIVO arm.
The most frequent (≥10%) serious adverse reactions in the OPDIVO and ipilimumab arm and the OPDIVO arm, respectively, were diarrhea (13% and 2.2%), colitis (10% and 1.9%), and pyrexia (10% and 1%). The most frequent adverse reactions leading to discontinuation of both drugs in the OPDIVO and ipilimumab arm and of OPDIVO in the OPDIVO arm, respectively, were colitis (10% and 0.6%), diarrhea (8% and 2.2%), increased ALT (4.8% and 1%), increased AST (4.5% and 0.6%), and pneumonitis (1.9% and 0.3%).
The most common (≥20%) adverse reactions in the OPDIVO and ipilimumab arm were fatigue, diarrhea, rash, nausea, pyrexia, pruritus, musculoskeletal pain, vomiting, decreased appetite, cough, headache, dyspnea, upper respiratory tract infection, arthralgia, and increased transaminases. The most common (≥20%) adverse reactions in the OPDIVO arm were fatigue, rash, musculoskeletal pain, diarrhea, nausea, cough, pruritus, upper respiratory tract infection, decreased appetite, headache, constipation, arthralgia, and vomiting.
Tables 9 and 10 summarize the incidence of adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-067.
| Toxicity was graded per NCI CTCAE v4. a Includes asthenia and fatigue. b Includes pustular rash, dermatitis, acneiform dermatitis, allergic dermatitis, atopic dermatitis, bullous dermatitis, exfoliative dermatitis, psoriasiform dermatitis, drug eruption, exfoliative rash, erythematous rash, generalized rash, macular rash, maculopapular rash, morbilliform rash, papular rash, papulosquamous rash, and pruritic rash. c Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, and spinal pain. d Includes upper respiratory tract infection, nasopharyngitis, pharyngitis, and rhinitis. e Includes hypertension and blood pressure increased. |
||||||
|
Adverse Reaction |
OPDIVO and Ipilimumab
|
OPDIVO
|
Ipilimumab
|
|||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||||
|
62 |
7 |
59 |
1.6 |
51 |
4.2 |
|
40 |
1.6 |
16 |
0 |
18 |
0.6 |
|
Gastrointestinal |
||||||
|
54 |
11 |
36 |
5 |
47 |
7 |
|
44 |
3.8 |
30 |
0.6 |
31 |
1.9 |
|
31 |
3.8 |
20 |
1 |
17 |
1.6 |
|
Skin and Subcutaneous Tissue |
||||||
|
53 |
6 |
40 |
1.9 |
42 |
3.5 |
|
9 |
0 |
10 |
0.3 |
5 |
0 |
|
Musculoskeletal and Connective Tissue |
||||||
|
32 |
2.6 |
42 |
3.8 |
36 |
1.9 |
|
21 |
0.3 |
21 |
1 |
16 |
0.3 |
|
Metabolism and Nutrition |
||||||
|
29 |
1.9 |
22 |
0 |
24 |
1.3 |
|
Respiratory, Thoracic and Mediastinal |
||||||
|
27 |
0.3 |
28 |
0.6 |
22 |
0 |
|
24 |
2.9 |
18 |
1.3 |
17 |
0.6 |
|
Infections |
||||||
|
23 |
0 |
22 |
0.3 |
17 |
0 |
|
Endocrine |
||||||
|
19 |
0.6 |
11 |
0 |
5 |
0 |
|
11 |
1.3 |
6 |
0 |
1 |
0 |
|
Investigations |
||||||
|
12 |
0 |
7 |
0 |
7 |
0.3 |
|
Vascular |
||||||
|
7 |
2.2 |
11 |
5 |
9 |
2.3 |
Clinically important adverse reactions in <10% of patients who received OPDIVO with ipilimumab or OPDIVO as a single agent were:
Gastrointestinal Disorders: stomatitis, intestinal perforation
Skin and Subcutaneous Tissue Disorders: vitiligo
Musculoskeletal and Connective Tissue Disorders: myopathy, Sjogren’s syndrome, spondyloarthropathy, myositis (including polymyositis)
Nervous System Disorders: neuritis, peroneal nerve palsy
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab (range: 75 to 297); OPDIVO (range: 81 to 306); ipilimumab (range: 61 to 301). | ||||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab |
OPDIVO |
Ipilimumab |
|||
|
All Grades (%) |
Grade 3-4 (%) |
All Grades (%) |
Grade 3-4 (%) |
All Grades (%) |
Grade 3-4 (%) |
|
|
Chemistry |
||||||
|
Increased ALT |
55 |
16 |
25 |
3 |
29 |
2.7 |
|
Hyperglycemia |
53 |
5.3 |
46 |
7 |
26 |
0 |
|
Increased AST |
52 |
13 |
29 |
3.7 |
29 |
1.7 |
|
Hyponatremia |
45 |
10 |
22 |
3.3 |
26 |
7 |
|
Increased lipase |
43 |
22 |
32 |
12 |
24 |
7 |
|
Increased alkaline phosphatase |
41 |
6 |
27 |
2 |
23 |
2 |
|
Hypocalcemia |
31 |
1.1 |
15 |
0.7 |
20 |
0.7 |
|
Increased amylase |
27 |
10 |
19 |
2.7 |
15 |
1.6 |
|
Increased creatinine |
26 |
2.7 |
19 |
0.7 |
17 |
1.3 |
|
Hematology |
||||||
|
Anemia |
52 |
2.7 |
41 |
2.6 |
41 |
6 |
|
Lymphopenia |
39 |
5 |
41 |
4.9 |
29 |
4 |
Adjuvant Treatment of Melanoma
CHECKMATE-76K
The safety of OPDIVO as a single agent was evaluated in CHECKMATE-76K, a randomized (2:1), double-blind trial in 788 patients with completely resected Stage IIB/C melanoma who received OPDIVO 480 mg by intravenous infusion over 30 minutes every 4 weeks (n=524) or placebo by intravenous infusion over 30 minutes every 4 weeks (n=264) for up to 1 year [see Clinical Studies (14.2)]. The median duration of exposure was 11 months in patients treated with OPDIVO and 11 months in patients treated with placebo.
Serious adverse reactions occurred in 18% of patients treated with OPDIVO. A fatal adverse reaction occurred in 1 (0.2%) patient (heart failure and acute kidney injury). Permanent discontinuation of OPDIVO due to an adverse reaction occurred in 17% of patients. Adverse reactions which resulted in permanent discontinuation of OPDIVO in >1% of patients included diarrhea (1.1%), arthralgia (1.7%), and rash (1.7%).
Dosage interruptions of OPDIVO due to an adverse reaction occurred in 25% of patients. Adverse reactions which required dosage interruption in >1% of patients included COVID-19 infection, infusion related reaction, diarrhea, arthralgia, and increased ALT.
The most common adverse reactions (reported in ≥20% of patients) were fatigue, musculoskeletal pain, rash, diarrhea, and pruritus.
Tables 11 and 12 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-76K.
| Toxicity was graded per NCI CTCAE v5. a Includes asthenia. b Includes arthralgia, arthritis, back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal stiffness, myalgia, neck pain, non-cardiac chest pain, spinal pain, pain in extremity. c Includes dermatitis, dermatitis acneiform, dyshidrotic eczema, eczema, eczema asteatotic, eyelid rash, genital rash, pemphigoid, penile rash, rash erythematous, rash follicular, rash macular, rash maculo-papular, rash papular, rash pruritic, rash pustular, rash vesicular, skin exfoliation, toxic skin eruption. d Includes autoimmune colitis, colitis, diarrhea, enteritis, enterocolitis. e Includes autoimmune hypothyroidism, blood thyroid stimulating hormone increased. f Includes cluster headache, migraine. |
||||
|
Adverse Reaction |
OPDIVO |
Placebo |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||
|
Fatiguea |
36 |
0.4 |
34 |
0.4 |
|
Musculoskeletal and connective tissue |
||||
|
Musculoskeletal painb |
30 |
0.4 |
26 |
0.4 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashc |
28 |
1.1 |
15 |
0.4 |
|
Pruritus |
20 |
0.2 |
11 |
0 |
|
Gastrointestinal |
||||
|
Diarrhead |
23 |
1.3 |
16 |
0 |
|
Nausea |
14 |
0 |
11 |
0 |
|
Endocrine |
||||
|
Hypothyroidisme |
14 |
0 |
2.3 |
0 |
|
Nervous system |
||||
|
Headachef |
12 |
0.2 |
14 |
0.8 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 262 to 513 patients) and placebo group (range: 138 to 261 patients). | ||||
|
Laboratory Abnormality |
OPDIVO |
Placebo |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Anemia |
19 |
0 |
14 |
0 |
|
Lymphopenia |
17 |
1.1 |
17 |
1.7 |
|
Neutropenia |
10 |
0 |
10 |
0.4 |
|
Chemistry |
||||
|
AST increased |
25 |
2.2 |
16 |
0.4 |
|
Lipase increased |
22 |
2.9 |
21 |
2.3 |
|
ALT increased |
20 |
2.1 |
15 |
0.4 |
|
Amylase increased |
17 |
0.4 |
9 |
0 |
|
Creatinine increased |
15 |
0.4 |
13 |
0 |
|
Sodium decreased |
13 |
0.6 |
11 |
0.4 |
|
Potassium increased |
13 |
1 |
15 |
1.1 |
CHECKMATE-238
The safety of OPDIVO as a single agent was evaluated in CHECKMATE-238, a randomized (1:1), double-blind trial in 905 patients with completely resected Stage IIIB/C or Stage IV melanoma received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (n=452) or ipilimumab 10 mg/kg by intravenous infusion every 3 weeks for 4 doses then every 12 weeks beginning at Week 24 for up to 1 year (n=453) [see Clinical Studies (14.2)]. The median duration of exposure was 11.5 months in OPDIVO-treated patients and was 2.7 months in ipilimumab-treated patients. In this ongoing trial, 74% of patients received OPDIVO for >6 months.
Serious adverse reactions occurred in 18% of OPDIVO-treated patients. Study therapy was discontinued for adverse reactions in 9% of OPDIVO-treated patients and 42% of ipilimumab-treated patients. Twenty-eight percent of OPDIVO-treated patients had at least one omitted dose for an adverse reaction. Grade 3 or 4 adverse reactions occurred in 25% of OPDIVO-treated patients.
The most frequent Grade 3 and 4 adverse reactions reported in ≥2% of OPDIVO-treated patients were diarrhea and increased lipase and amylase. The most common adverse reactions (at least 20%) were fatigue, diarrhea, rash, musculoskeletal pain, pruritus, headache, nausea, upper respiratory infection, and abdominal pain. The most common immune-mediated adverse reactions were rash (16%), diarrhea/colitis (6%), and hepatitis (3%).
Tables 13 and 14 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-238.
| Toxicity was graded per NCI CTCAE v4. a Includes asthenia. b Includes abdominal discomfort, lower abdominal pain, upper abdominal pain, and abdominal tenderness. c Includes dermatitis described as acneiform, allergic, bullous, or exfoliative and rash described as generalized, erythematous, macular, papular, maculopapular, pruritic, pustular, vesicular, or butterfly, and drug eruption. d Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, spinal pain, and pain in extremity. e Includes postural dizziness and vertigo. f Includes upper respiratory tract infection including viral respiratory tract infection, lower respiratory tract infection, rhinitis, pharyngitis, and nasopharyngitis. g Includes secondary hypothyroidism and autoimmune hypothyroidism. |
||||
|
Adverse Reaction |
OPDIVO |
Ipilimumab 10 mg/kg |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||
|
Fatiguea |
57 |
0.9 |
55 |
2.4 |
|
Gastrointestinal |
||||
|
Diarrhea |
37 |
2.4 |
55 |
11 |
|
Nausea |
23 |
0.2 |
28 |
0 |
|
Abdominal painb |
21 |
0.2 |
23 |
0.9 |
|
Constipation |
10 |
0 |
9 |
0 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashc |
35 |
1.1 |
47 |
5.3 |
|
Pruritus |
28 |
0 |
37 |
1.1 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal |
32 |
0.4 |
27 |
0.4 |
|
Arthralgia |
19 |
0.4 |
13 |
0.4 |
|
Nervous System |
||||
|
Headache |
23 |
0.4 |
31 |
2.0 |
|
Dizzinesse |
11 |
0 |
8 |
0 |
|
Infections |
||||
|
Upper respiratory tract |
22 |
0 |
15 |
0.2 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough/productive |
19 |
0 |
19 |
0 |
|
Dyspnea/exertional |
10 |
0.4 |
10 |
0.2 |
|
Endocrine |
||||
|
Hypothyroidismg |
12 |
0.2 |
7.5 |
0.4 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 400 to 447 patients) and ipilimumab 10 mg/kg group (range: 392 to 443 patients). | ||||
|
Laboratory Abnormality |
OPDIVO |
Ipilimumab 10 mg/kg |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Lymphopenia |
27 |
0.4 |
12 |
0.9 |
|
Anemia |
26 |
0 |
34 |
0.5 |
|
Leukopenia |
14 |
0 |
2.7 |
0.2 |
|
Neutropenia |
13 |
0 |
6 |
0.5 |
|
Chemistry |
||||
|
Increased Lipase |
25 |
7 |
23 |
9 |
|
Increased ALT |
25 |
1.8 |
40 |
12 |
|
Increased AST |
24 |
1.3 |
33 |
9 |
|
Increased Amylase |
17 |
3.3 |
13 |
3.1 |
|
Hyponatremia |
16 |
1.1 |
22 |
3.2 |
|
Hyperkalemia |
12 |
0.2 |
9 |
0.5 |
|
Increased Creatinine |
12 |
0 |
13 |
0 |
|
Hypocalcemia |
10 |
0.7 |
16 |
0.5 |
Neoadjuvant Treatment of Resectable (Tumors ≥4 cm or Node Positive) Non-Small Cell Lung Cancer
The safety of OPDIVO in combination with platinum-doublet chemotherapy was evaluated in CHECKMATE-816, a randomized, open-label, multicenter trial in patients with resectable NSCLC [see Clinical Studies (14.3)]. Patients received either OPDIVO 360 mg administered in combination with platinum-doublet chemotherapy administered every 3 weeks for 3 cycles; or platinum-doublet chemotherapy administered every 3 weeks for 3 cycles.
The median age of patients who received OPDIVO in combination with platinum-doublet chemotherapy or platinum-doublet chemotherapy was 65 years (range: 34 – 84); 72% male; 47% White, 50% Asian, and 2% Black/African American.
Serious adverse reactions occurred in 30% of patients who were treated with OPDIVO in combination with platinum-doublet chemotherapy. Serious adverse reactions in >2% included pneumonia and vomiting. No fatal adverse reactions occurred in patients who received OPDIVO in combination with platinum-doublet chemotherapy.
Study therapy with OPDIVO in combination with platinum-doublet chemotherapy was permanently discontinued for adverse reactions in 10% of patients and 30% had at least one treatment withheld for an adverse reaction. The most common adverse reactions (≥1%) resulting in permanent discontinuation of OPDIVO in combination with platinum-doublet chemotherapy were anaphylactic reaction (1.7%), acute kidney injury (1.1%), rash (1.1%), and fatigue (1.1%).
The most common (>20%) adverse reactions were nausea, constipation, fatigue, decreased appetite, and rash. The most common Grade 3 or 4 laboratory abnormalities (≥2%) were neutropenia, hyperglycemia, leukopenia, lymphopenia, increased amylase, anemia, thrombocytopenia, and hyponatremia.
Tables 15 and 16 summarize selected adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-816.
| Toxicity was graded per NCI CTCAE v4. a Includes fatigue and asthenia. b Includes rash, dermatitis, acneiform dermatitis, atopic dermatitis, bullous dermatitis, drug eruption, maculopapular rash, and pruritic rash. c Includes peripheral neuropathy, dysesthesia, hypoesthesia, peripheral motor neuropathy, peripheral sensory neuropathy. |
||||
|
Adverse Reaction |
OPDIVO and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy |
||
|
All Grades (%) |
Grades 3 or 4 (%) |
All Grades (%) |
Grades 3 or 4 (%) |
|
|
Gastrointestinal |
||||
|
Nausea |
38 |
0.6 |
45 |
1.1 |
|
Constipation |
34 |
0 |
32 |
1.1 |
|
Vomiting |
11 |
1.1 |
13 |
0.6 |
|
General |
||||
|
Fatiguea |
26 |
2.3 |
23 |
1.1 |
|
Malaise |
15 |
0.6 |
14 |
0.6 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
20 |
1.1 |
23 |
2.3 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashb |
20 |
2.3 |
7 |
0 |
|
Alopecia |
11 |
0 |
15 |
0 |
|
Nervous System |
||||
|
Peripheral neuropathyc |
13 |
0 |
6 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and platinum-doublet chemotherapy group (range: 73 to 171 patients) and platinum-doublet chemotherapy group (range: 68 to 171 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Platinum-Doublet Chemotherapya |
Platinum-Doublet Chemotherapya |
||
|
All Grades (%) |
Grades 3 or 4 (%) |
All Grades (%) |
Grades 3 or 4 (%) |
|
|
Hematology |
||||
|
Anemia |
63 |
3.5 |
70 |
6 |
|
Neutropenia |
58 |
22 |
58 |
27 |
|
Leukopenia |
53 |
5 |
51 |
11 |
|
Lymphopenia |
38 |
4.7 |
31 |
1.8 |
|
Thrombocytopenia |
24 |
2.9 |
22 |
3 |
|
Chemistry |
||||
|
Hyperglycemia |
37 |
6 |
35 |
2.9 |
|
Hypomagnesemia |
25 |
1.2 |
29 |
1.2 |
|
Hyponatremia |
25 |
2.4 |
28 |
1.8 |
|
Increased amylase |
23 |
3.6 |
13 |
1.8 |
|
Increased ALT |
23 |
0 |
20 |
1.2 |
Neoadjuvant and Adjuvant Treatment of Resectable Non-Small Cell Lung Cancer
The safety of OPDIVO in combination with neoadjuvant platinum-doublet chemotherapy followed by surgery and continued adjuvant treatment with OPDIVO as a single agent after surgery was evaluated in CHECKMATE-77T, a randomized, double-blind, multicenter trial in patients with previously untreated resectable Stage IIA (> 4 cm) to IIIB (T3N2 or T4N2) NSCLC (per the AJCC Cancer Staging Manual 8th Edition) [see Clinical Studies (14.4)]. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible. The median duration of exposure to OPDIVO was 10.3 months (range: 1 day to 22.3 months).
The study population characteristics were: median age 66 years (range: 35 - 86); 71% male; 72% White, 25% Asian, 1.7% Black/African American, and 1.5% other race; and 6% Hispanic or Latino.
Adverse reactions occurring in patients with resectable NSCLC receiving OPDIVO in combination with platinum-doublet chemotherapy, given as neoadjuvant treatment and followed as a single agent adjuvant treatment after surgery, were generally similar to those occurring in patients in other clinical trials across tumor types receiving OPDIVO in combination with chemotherapy.
Neoadjuvant Phase of CHECKMATE-77T
A total of 228 patients received at least 1 dose of OPDIVO in combination with platinum-doublet chemotherapy as neoadjuvant treatment and 230 patients received at least 1 dose of placebo in combination with platinum-doublet chemotherapy as neoadjuvant treatment.
Serious adverse reactions occurred in 21% of patients who received OPDIVO in combination with platinum-doublet chemotherapy as neoadjuvant treatment; the most frequent (≥2%) serious adverse reactions was pneumonia. Fatal adverse reactions occurred in 2.2% of patients, due to cerebrovascular accident, COVID-19 infection, hemoptysis, pneumonia, and pneumonitis (0.4% each).
Permanent discontinuation of any study drug due to an adverse reaction occurred in 13% of patients who received OPDIVO in combination with platinum-doublet chemotherapy as neoadjuvant treatment; the most frequent (≥1%) adverse reaction that led to permanent discontinuation of any study drug was peripheral sensory neuropathy (2.2%).
Of the 228 OPDIVO-treated patients and 230 placebo-treated patients who received neoadjuvant treatment, 5.3% (n=12) and 3.5% (n=8), respectively, did not receive surgery due to adverse reactions. The adverse reactions that led to cancellation of surgery in OPDIVO-treated patients were cerebrovascular accident, pneumonia, and colitis/diarrhea (2 patients each) and acute coronary syndrome, myocarditis, hemoptysis, pneumonitis, COVID-19, and myositis (1 patient each).
Of the 178 OPDIVO-treated patients who received surgery, 4.5% (n=8) experienced delay of surgery (surgery more than 6 weeks from last neoadjuvant treatment) due to adverse reactions. Of the 178 placebo-treated patients who received surgery, 3.9% (n=7) experienced delay of surgery due to adverse reactions.
Of the 178 OPDIVO-treated patients who received surgery, 7% (n=13) did not receive adjuvant treatment due to adverse reactions. Of the 178 placebo-treated patients who received surgery, 2.8% (n=5) did not receive adjuvant treatment due to adverse reactions.
Adjuvant Phase of CHECKMATE-77T
A total of 142 patients in the OPDIVO arm and 152 patients in the placebo arm received at least 1 dose of adjuvant treatment.
Of the patients who received single agent OPDIVO as adjuvant treatment, 22% experienced serious adverse reactions; the most frequent serious adverse reaction was pneumonitis/ILD (2.8%). One fatal adverse reaction due to COVID-19 occurred. Permanent discontinuation of adjuvant OPDIVO due to an adverse reaction occurred in 14% of patients; the most frequent (≥1%) adverse reactions that led to permanent discontinuation of adjuvant OPDIVO were pneumonitis (4.2%) and diarrhea (1.4%).
Metastatic Non-Small Cell Lung Cancer
First-line Treatment of Metastatic NSCLC: In Combination with Ipilimumab
The safety of OPDIVO in combination with ipilimumab was evaluated in CHECKMATE-227, a randomized, multicenter, multi-cohort, open-label trial in patients with previously untreated metastatic or recurrent NSCLC with no EGFR or ALK genomic tumor aberrations [see Clinical Studies (14.5)]. The trial excluded patients with untreated brain metastases, carcinomatous meningitis, active autoimmune disease, or medical conditions requiring systemic immunosuppression. Patients received OPDIVO 3 mg/kg by intravenous infusion over 30 minutes every 2 weeks and ipilimumab 1 mg/kg by intravenous infusion over 30 minutes every 6 weeks or platinum-doublet chemotherapy every 3 weeks for 4 cycles. The median duration of therapy in OPDIVO and ipilimumab-treated patients was 4.2 months (range: 1 day to 25.5 months): 39% of patients received OPDIVO and ipilimumab for >6 months and 23% of patients received OPDIVO and ipilimumab for >1 year. The population characteristics were: median age 64 years (range: 26 to 87); 48% were ≥65 years of age, 76% White, and 67% male. Baseline ECOG performance status was 0 (35%) or 1 (65%), 85% were former/current smokers, 11% had brain metastases, 28% had squamous histology and 72% had non-squamous histology.
Serious adverse reactions occurred in 58% of patients. OPDIVO and ipilimumab were discontinued for adverse reactions in 24% of patients and 53% had at least one dose withheld for an adverse reaction.
The most frequent (≥2%) serious adverse reactions were pneumonia, diarrhea/colitis, pneumonitis, hepatitis, pulmonary embolism, adrenal insufficiency, and hypophysitis. Fatal adverse reactions occurred in 1.7% of patients; these included events of pneumonitis (4 patients), myocarditis, acute kidney injury, shock, hyperglycemia, multi-system organ failure, and renal failure. The most common (≥20%) adverse reactions were fatigue, rash, decreased appetite, musculoskeletal pain, diarrhea/colitis, dyspnea, cough, hepatitis, nausea, and pruritus.
Tables 17 and 18 summarize selected adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-227.
| a Includes fatigue and asthenia. b Includes eyelid edema, face edema, generalized edema, localized edema, edema, edema peripheral, and periorbital edema. c Includes autoimmune dermatitis, dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis atopic, dermatitis bullous, dermatitis contact, dermatitis exfoliative, dermatitis psoriasiform, granulomatous dermatitis, rash generalized, drug eruption, dyshidrotic eczema, eczema, exfoliative rash, nodular rash, rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic, rash pustular, toxic skin eruption. d Includes pruritus and pruritus generalized. e Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal pain, myalgia, and pain in extremity. f Includes colitis, colitis microscopic, colitis ulcerative, diarrhea, enteritis infectious, enterocolitis, enterocolitis infectious, and enterocolitis viral. g Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper, and abdominal tenderness. h Includes dyspnea and dyspnea exertional. i Includes cough and productive cough. j Includes alanine aminotransferase increased, aspartate aminotransferase increased, autoimmune hepatitis, blood bilirubin increased, hepatic enzyme increased, hepatic failure, hepatic function abnormal, hepatitis, hepatitis E, hepatocellular injury, hepatotoxicity, hyperbilirubinemia, immune-mediated hepatitis, liver function test abnormal, liver function test increased, transaminases increased. k Includes autoimmune thyroiditis, blood thyroid stimulating hormone increased, hypothyroidism, primary hypothyroidism, thyroiditis, and tri-iodothyronine free decreased. l Contains blood thyroid stimulating hormone decreased, hyperthyroidism, and tri-iodothyronine free increased. m Includes lower respiratory tract infection, lower respiratory tract infection bacterial, lung infection, pneumonia, pneumonia adenoviral, pneumonia aspiration, pneumonia bacterial, pneumonia klebsiella, pneumonia influenzal, pneumonia viral, atypical pneumonia, organizing pneumonia. |
||||
|
Adverse Reaction |
OPDIVO and Ipilimumab |
Platinum-Doublet Chemotherapy |
||
|
All Grades |
Grades 3-4 |
All Grades |
Grades 3-4 |
|
|
General |
||||
|
Fatiguea |
44 |
6 |
42 |
4.4 |
|
Pyrexia |
18 |
0.5 |
11 |
0.4 |
|
Edemab |
14 |
0.2 |
12 |
0.5 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashc |
34 |
4.7 |
10 |
0.4 |
|
Pruritusd |
21 |
0.5 |
3.3 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
31 |
2.3 |
26 |
1.4 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paine |
27 |
1.9 |
16 |
0.7 |
|
Arthralgia |
13 |
0.9 |
2.5 |
0.2 |
|
Gastrointestinal |
||||
|
Diarrhea/colitisf |
26 |
3.6 |
16 |
0.9 |
|
Nausea |
21 |
1 |
42 |
2.5 |
|
Constipation |
18 |
0.3 |
27 |
0.5 |
|
Vomiting |
13 |
1 |
18 |
2.3 |
|
Abdominal paing |
10 |
0.2 |
9 |
0.7 |
|
Respiratory, Thoracic, and Mediastinal |
||||
|
Dyspneah |
26 |
4.3 |
16 |
2.1 |
|
Coughi |
23 |
0.2 |
13 |
0 |
|
Hepatobiliary |
||||
|
Hepatitisj |
21 |
9 |
10 |
1.2 |
|
Endocrine |
||||
|
Hypothyroidismk |
16 |
0.5 |
1.2 |
0 |
|
Hyperthyroidisml |
10 |
0 |
0.5 |
0 |
|
Infections and Infestations |
||||
|
Pneumoniam |
13 |
7 |
8 |
4 |
|
Nervous System |
||||
|
Headache |
11 |
0.5 |
6 |
0 |
Other clinically important adverse reactions in CHECKMATE-227 were:
Skin and Subcutaneous Tissue: urticaria, alopecia, erythema multiforme, vitiligo
Gastrointestinal: stomatitis, pancreatitis, gastritis
Musculoskeletal and Connective Tissue: arthritis, polymyalgia rheumatica, rhabdomyolysis
Nervous System: peripheral neuropathy, autoimmune encephalitis
Blood and Lymphatic System: eosinophilia
Eye Disorders: blurred vision, uveitis
Cardiac: atrial fibrillation, myocarditis
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab group (range: 494 to 556 patients) and chemotherapy group (range: 469 to 542 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab |
Platinum-Doublet Chemotherapy |
||
|
Grades 1-4 |
Grades 3-4 |
Grades 1-4 |
Grades 3-4 |
|
|
Hematology |
||||
|
Anemia |
46 |
3.6 |
78 |
14 |
|
Lymphopenia |
46 |
5 |
60 |
15 |
|
Chemistry |
||||
|
Hyponatremia |
41 |
12 |
26 |
4.9 |
|
Increased AST |
39 |
5 |
26 |
0.4 |
|
Increased ALT |
36 |
7 |
27 |
0.7 |
|
Increased lipase |
35 |
14 |
14 |
3.4 |
|
Increased alkaline |
34 |
3.8 |
20 |
0.2 |
|
Increased amylase |
28 |
9 |
18 |
1.9 |
|
Hypocalcemia |
28 |
1.7 |
17 |
1.3 |
|
Hyperkalemia |
27 |
3.4 |
22 |
0.4 |
|
Increased creatinine |
22 |
0.9 |
17 |
0.2 |
First-line Treatment of Metastatic or Recurrent NSCLC: In Combination with Ipilimumab and Platinum-Doublet Chemotherapy
The safety of OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy was evaluated in CHECKMATE-9LA [see Clinical Studies (14.5)]. Patients received either OPDIVO 360 mg administered every 3 weeks in combination with ipilimumab 1 mg/kg administered every 6 weeks and platinum-doublet chemotherapy administered every 3 weeks for 2 cycles; or platinum-doublet chemotherapy administered every 3 weeks for 4 cycles. The median duration of therapy in OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy was 6 months (range: 1 day to 19 months): 50% of patients received OPDIVO and ipilimumab for >6 months and 13% of patients received OPDIVO and ipilimumab for >1 year.
Serious adverse reactions occurred in 57% of patients who were treated with OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy. The most frequent (>2%) serious adverse reactions were pneumonia, diarrhea, febrile neutropenia, anemia, acute kidney injury, musculoskeletal pain, dyspnea, pneumonitis, and respiratory failure. Fatal adverse reactions occurred in 7 (2%) patients, and included hepatic toxicity, acute renal failure, sepsis, pneumonitis, diarrhea with hypokalemia, and massive hemoptysis in the setting of thrombocytopenia.
Study therapy with OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy was permanently discontinued for adverse reactions in 24% of patients and 56% had at least one treatment withheld for an adverse reaction. The most common (>20%) adverse reactions were fatigue, musculoskeletal pain, nausea, diarrhea, rash, decreased appetite, constipation, and pruritus.
Tables 19 and 20 summarize selected adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-9LA.
| Toxicity was graded per NCI CTCAE v4. a Includes fatigue and asthenia. b Includes myalgia, back pain, pain in extremity, musculoskeletal pain, bone pain, flank pain, muscle spasms, musculoskeletal chest pain, musculoskeletal disorder, osteitis, musculoskeletal stiffness, non-cardiac chest pain, arthralgia, arthritis, arthropathy, joint effusion, psoriatic arthropathy, synovitis. c Includes colitis, ulcerative colitis, diarrhea, and enterocolitis. d Includes abdominal discomfort, abdominal pain, lower abdominal pain, upper abdominal pain, and gastrointestinal pain. e Includes acne, dermatitis, acneiform dermatitis, allergic dermatitis, atopic dermatitis, bullous dermatitis, generalized exfoliative dermatitis, eczema, keratoderma blennorrhagica, palmar-plantar erythrodysesthesia syndrome, rash, erythematous rash, generalized rash, macular rash, maculo-papular rash, morbilliform rash, papular rash, pruritic rash, skin exfoliation, skin reaction, skin toxicity, Stevens-Johnson syndrome, urticaria. f Includes pruritus and generalized pruritus. g Includes cough, productive cough, and upper-airway cough syndrome. h Includes dyspnea, dyspnea at rest, and exertional dyspnea. i Includes autoimmune thyroiditis, increased blood thyroid stimulating hormone, hypothyroidism, thyroiditis, and decreased free tri-iodothyronine. j Includes dizziness, vertigo and positional vertigo. |
||||
|
Adverse Reaction |
OPDIVO and Ipilimumab and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||
|
Fatiguea |
49 |
5 |
40 |
4.9 |
|
Pyrexia |
14 |
0.6 |
10 |
0.6 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal painb |
39 |
4.5 |
27 |
2 |
|
Gastrointestinal |
||||
|
Nausea |
32 |
1.7 |
41 |
0.9 |
|
Diarrheac |
31 |
6 |
18 |
1.7 |
|
Constipation |
21 |
0.6 |
23 |
0.6 |
|
Vomiting |
18 |
2 |
17 |
1.4 |
|
Abdominal paind |
12 |
0.6 |
11 |
0.9 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashe |
30 |
4.7 |
10 |
0.3 |
|
Pruritusf |
21 |
0.8 |
2.9 |
0 |
|
Alopecia |
11 |
0.8 |
10 |
0.6 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
28 |
2 |
22 |
1.7 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughg |
19 |
0.6 |
15 |
0.9 |
|
Dyspneah |
18 |
4.7 |
14 |
3.2 |
|
Endocrine |
||||
|
Hypothyroidismi |
19 |
0.3 |
3.4 |
0 |
|
Nervous System |
||||
|
Headache |
11 |
0.6 |
7 |
0 |
|
Dizzinessj |
11 |
0.6 |
6 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab and platinum-doublet chemotherapy group (range: 197 to 347 patients) and platinum-doublet chemotherapy group (range: 191 to 335 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Anemia |
70 |
9 |
74 |
16 |
|
Lymphopenia |
41 |
6 |
40 |
11 |
|
Neutropenia |
40 |
15 |
42 |
15 |
|
Leukopenia |
36 |
10 |
40 |
9 |
|
Thrombocytopenia |
23 |
4.3 |
24 |
5 |
|
Chemistry |
||||
|
Hyperglycemia |
45 |
7 |
42 |
2.6 |
|
Hyponatremia |
37 |
10 |
27 |
7 |
|
Increased ALT |
34 |
4.3 |
24 |
1.2 |
|
Increased lipase |
31 |
12 |
10 |
2.2 |
|
Increased alkaline phosphatase |
31 |
1.2 |
26 |
0.3 |
|
Increased amylase |
30 |
7 |
19 |
1.3 |
|
Increased AST |
30 |
3.5 |
22 |
0.3 |
|
Hypomagnesemia |
29 |
1.2 |
33 |
0.6 |
|
Hypocalcemia |
26 |
1.4 |
22 |
1.8 |
|
Increased creatinine |
26 |
1.2 |
23 |
0.6 |
|
Hyperkalemia |
22 |
1.7 |
21 |
2.1 |
Second-line Treatment of Metastatic NSCLC
The safety of OPDIVO was evaluated in CHECKMATE-017, a randomized open-label, multicenter trial in patients with metastatic squamous NSCLC and progression on or after one prior platinum doublet-based chemotherapy regimen and in CHECKMATE-057, a randomized, open-label, multicenter trial in patients with metastatic non-squamous NSCLC and progression on or after one prior platinum doublet-based chemotherapy regimen [see Clinical Studies (14.5)]. These trials excluded patients with active autoimmune disease, medical conditions requiring systemic immunosuppression, or with symptomatic interstitial lung disease. Patients received OPDIVO 3 mg/kg over 60 minutes by intravenous infusion every 2 weeks or docetaxel 75 mg/m2 intravenously every 3 weeks. The median duration of therapy in OPDIVO-treated patients in CHECKMATE-017 was 3.3 months (range: 1 day to 21.7+ months) and in CHECKMATE-057 was 2.6 months (range: 0 to 24.0+ months). In CHECKMATE-017, 36% of patients received OPDIVO for at least 6 months and 18% of patients received OPDIVO for at least 1 year and in CHECKMATE-057, 30% of patients received OPDIVO for >6 months and 20% of patients received OPDIVO for >1 year.
Across both trials, the median age of OPDIVO-treated patients was 61 years (range: 37 to 85); 38% were ≥65 years of age, 61% were male, and 91% were White. Ten percent of patients had brain metastases and ECOG performance status was 0 (26%) or 1 (74%).
In CHECKMATE-057, in the OPDIVO arm, seven deaths were due to infection including one case of Pneumocystis jirovecii pneumonia, four were due to pulmonary embolism, and one death was due to limbic encephalitis. Serious adverse reactions occurred in 46% of patients receiving OPDIVO. OPDIVO was discontinued in 11% of patients and was delayed in 28% of patients for an adverse reaction.
The most frequent serious adverse reactions reported in ≥2% of patients receiving OPDIVO were pneumonia, pulmonary embolism, dyspnea, pyrexia, pleural effusion, pneumonitis, and respiratory failure. Across both trials, the most common adverse reactions (≥20%) were fatigue, musculoskeletal pain, cough, dyspnea, and decreased appetite.
Tables 21 and 22 summarize selected adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-057.
| Toxicity was graded per NCI CTCAE v4. | ||||
|
Adverse Reaction |
OPDIVO
|
Docetaxel
|
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough |
31 |
0.7 |
24 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
28 |
1.4 |
23 |
1.5 |
|
Skin and Subcutaneous Tissue |
||||
|
Pruritus |
10 |
0.2 |
2 |
0 |
Other clinically important adverse reactions observed in OPDIVO-treated patients and which occurred at a similar incidence in docetaxel-treated patients and not listed elsewhere in section 6 include: fatigue/asthenia (48% all Grades, 5% Grade 3-4), musculoskeletal pain (33% all Grades), pleural effusion (4.5% all Grades), pulmonary embolism (3.3% all Grades).
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 405 to 417 patients) and docetaxel group (range: 372 to 390 patients), except for TSH: OPDIVO group n=314 and docetaxel group n=297. b Not graded per NCI CTCAE v4. |
||||
|
Laboratory Abnormality |
OPDIVO |
Docetaxel |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Hyponatremia |
35 |
7 |
34 |
4.9 |
|
Increased AST |
27 |
1.9 |
13 |
0.8 |
|
Increased alkaline phosphatase |
26 |
0.7 |
18 |
0.8 |
|
Increased ALT |
22 |
1.7 |
17 |
0.5 |
|
Increased creatinine |
18 |
0 |
12 |
0.5 |
|
Increased TSHb |
14 |
N/A |
6 |
N/A |
Malignant Pleural Mesothelioma
The safety of OPDIVO in combination with ipilimumab was evaluated in CHECKMATE-743, a randomized, open-label trial in patients with previously untreated unresectable malignant pleural mesothelioma [see Clinical Studies (14.6)]. Patients received either OPDIVO 3 mg/kg over 30 minutes by intravenous infusion every 2 weeks and ipilimumab 1 mg/kg over 30 minutes by intravenous infusion every 6 weeks for up to 2 years; or platinum-doublet chemotherapy for up to 6 cycles. The median duration of therapy in OPDIVO and ipilimumab-treated patients was 5.6 months (range: 0 to 26.2 months); 48% of patients received OPDIVO and ipilimumab for >6 months and 24% of patients received OPDIVO and ipilimumab for >1 year.
Serious adverse reactions occurred in 54% of patients who were treated with OPDIVO in combination with ipilimumab. The most frequent (≥2%) serious adverse reactions were pneumonia, pyrexia, diarrhea, pneumonitis, pleural effusion, dyspnea, acute kidney injury, infusion-related reaction, musculoskeletal pain, and pulmonary embolism. Fatal adverse reactions occurred in 4 (1.3%) patients and included pneumonitis, acute heart failure, sepsis and encephalitis.
Both OPDIVO and ipilimumab were permanently discontinued due to adverse reactions in 23% of patients and 52% had at least one dose withheld due to an adverse reaction.
The most common (≥20%) adverse reactions were fatigue, musculoskeletal pain, rash, diarrhea, dyspnea, nausea, decreased appetite, cough, and pruritus.
Tables 23 and 24 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-743.
| a Includes fatigue and asthenia. b Includes pyrexia and tumor-associated fever. c Includes edema, generalized edema, peripheral edema, and peripheral swelling. d Includes musculoskeletal pain, back pain, bone pain, flank pain, involuntary muscle contractions, muscle spasms, muscle twitching, musculoskeletal chest pain, musculoskeletal stiffness, myalgia, neck pain, non-cardiac chest pain, pain in extremity, polymyalgia rheumatica, and spinal pain. e Includes rash, acne, acneiform dermatitis, allergic dermatitis, atopic dermatitis, autoimmune dermatitis, bullous dermatitis, contact dermatitis, dermatitis, drug eruption, dyshidrotic eczema, eczema, erythematous rash, exfoliative rash, generalized exfoliative dermatitis, generalized rash, granulomatous dermatitis, keratoderma blennorrhagica, macular rash, maculopapular rash, morbilliform rash, nodular rash, papular rash, psoriasiform dermatitis, pruritic rash, pustular rash, skin exfoliation, skin reaction, skin toxicity, Stevens-Johnson syndrome, toxic skin eruption, and urticaria. f Includes pruritus, allergic pruritus, and generalized pruritus. g Includes diarrhea, colitis, enteritis, infectious enteritis, enterocolitis, infectious enterocolitis, microscopic colitis, ulcerative colitis, and viral enterocolitis. h Includes abdominal pain, abdominal discomfort, abdominal tenderness, gastrointestinal pain, lower abdominal pain, and upper abdominal pain. i Includes dyspnea, dyspnea at rest, and exertional dyspnea. j Includes cough, productive cough, and upper-airway cough syndrome. k Includes hypothyroidism, autoimmune thyroiditis, decreased free tri-iodothyronine, increased blood thyroid stimulating hormone, primary hypothyroidism, thyroiditis, and autoimmune hypothyroidism. l Includes upper respiratory tract infection, nasopharyngitis, pharyngitis, and rhinitis. m Includes pneumonia, lower respiratory tract infection, lung infection, aspiration pneumonia, and Pneumocystis jirovecii pneumonia. |
||||
|
Adverse Reaction |
OPDIVO and Ipilimumab |
Chemotherapy |
||
|
All Grades |
Grades 3-4 |
All Grades |
Grades 3-4 |
|
|
General |
||||
|
Fatiguea |
43 |
4.3 |
45 |
6 |
|
Pyrexiab |
18 |
1.3 |
4.6 |
0.7 |
|
Edemac |
17 |
0 |
8 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paind |
38 |
3.3 |
17 |
1.1 |
|
Arthralgia |
13 |
1 |
1.1 |
0 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashe |
34 |
2.7 |
11 |
0.4 |
|
Pruritusf |
21 |
1 |
1.4 |
0 |
|
Gastrointestinal |
||||
|
Diarrheag |
32 |
6 |
12 |
1.1 |
|
Nausea |
24 |
0.7 |
43 |
2.5 |
|
Constipation |
19 |
0.3 |
30 |
0.7 |
|
Abdominal painh |
15 |
1 |
10 |
0.7 |
|
Vomiting |
14 |
0 |
18 |
2.1 |
|
Respiratory, Thoracic, and Mediastinal |
||||
|
Dyspneai |
27 |
2.3 |
16 |
3.2 |
|
Coughj |
23 |
0.7 |
9 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
24 |
1 |
25 |
1.4 |
|
Endocrine |
||||
|
Hypothyroidismk |
15 |
0 |
1.4 |
0 |
|
Infections and Infestations |
||||
|
Upper respiratory tract infectionl |
12 |
0.3 |
7 |
0 |
|
Pneumoniam |
10 |
4 |
4.2 |
2.1 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab group (range: 109 to 297 patients) and chemotherapy group (range: 90 to 276 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab |
Chemotherapy |
||
|
Grades 1-4 |
Grades 3-4 |
Grades 1-4 |
Grades 3-4 |
|
|
Chemistry |
||||
|
Hyperglycemia |
53 |
3.7 |
34 |
1.1 |
|
Increased AST |
38 |
7 |
17 |
0 |
|
Increased ALT |
37 |
7 |
15 |
0.4 |
|
Increased lipase |
34 |
13 |
9 |
0.8 |
|
Hyponatremia |
32 |
8 |
21 |
2.9 |
|
Increased alkaline phosphatase |
31 |
3.1 |
12 |
0 |
|
Hyperkalemia |
30 |
4.1 |
16 |
0.7 |
|
Hypocalcemia |
28 |
0 |
16 |
0 |
|
Increased amylase |
26 |
5 |
13 |
0.9 |
|
Increased creatinine |
20 |
0.3 |
20 |
0.4 |
|
Hematology |
||||
|
Lymphopenia |
43 |
8 |
57 |
14 |
|
Anemia |
43 |
2.4 |
75 |
15 |
Advanced Renal Cell Carcinoma
First-line Renal Cell Carcinoma
CHECKMATE-214
The safety of OPDIVO with ipilimumab was evaluated in CHECKMATE-214, a randomized open-label trial in 1082 patients with previously untreated advanced RCC received OPDIVO 3 mg/kg over 60 minutes with ipilimumab 1 mg/kg intravenously every 3 weeks for 4 doses followed by OPDIVO as a single agent at a dose of 3 mg/kg by intravenous infusion every 2 weeks (n=547) or sunitinib 50 mg orally daily for the first 4 weeks of a 6-week cycle (n=535) [see Clinical Studies (14.7)]. The median duration of treatment was 7.9 months (range: 1 day to 21.4+ months) in OPDIVO and ipilimumab-treated patients and 7.8 months (range: 1 day to 20.2+ months) in sunitinib-treated patients. In this trial, 57% of patients in the OPDIVO and ipilimumab arm were exposed to treatment for >6 months and 38% of patients were exposed to treatment for >1 year.
Serious adverse reactions occurred in 59% of patients receiving OPDIVO and ipilimumab. Study therapy was discontinued for adverse reactions in 31% of OPDIVO and ipilimumab patients. Fifty-four percent (54%) of patients receiving OPDIVO and ipilimumab had a dose interruption for an adverse reaction.
The most frequent serious adverse reactions reported in ≥2% of patients treated with OPDIVO and ipilimumab were diarrhea, pyrexia, pneumonia, pneumonitis, hypophysitis, acute kidney injury, dyspnea, adrenal insufficiency, and colitis; in patients treated with sunitinib, they were pneumonia, pleural effusion, and dyspnea. The most common adverse reactions (reported in ≥20% of patients) were fatigue, rash, diarrhea, musculoskeletal pain, pruritus, nausea, cough, pyrexia, arthralgia, and decreased appetite. The most common laboratory abnormalities which have worsened compared to baseline in ≥30% of OPDIVO and ipilimumab-treated patients include increased lipase, anemia, increased creatinine, increased ALT, increased AST, hyponatremia, increased amylase, and lymphopenia.
Tables 25 and 26 summarize adverse reactions and laboratory abnormalities, respectively, that occurred in >15% of OPDIVO and ipilimumab-treated patients in CHECKMATE-214.
| Toxicity was graded per NCI CTCAE v4. a Includes asthenia. b Includes peripheral edema, peripheral swelling. c Includes dermatitis described as acneiform, bullous, and exfoliative, drug eruption, rash described as exfoliative, erythematous, follicular, generalized, macular, maculopapular, papular, pruritic, and pustular, fixed-drug eruption. d Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, spinal pain. |
||||
|
Adverse Reaction |
OPDIVO and Ipilimumab |
Sunitinib |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Adverse Reaction |
99 |
65 |
99 |
76 |
|
General |
||||
|
Fatiguea |
58 |
8 |
69 |
13 |
|
Pyrexia |
25 |
0.7 |
17 |
0.6 |
|
Edemab |
16 |
0.5 |
17 |
0.6 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashc |
39 |
3.7 |
25 |
1.1 |
|
Pruritus/generalized pruritus |
33 |
0.5 |
11 |
0 |
|
Gastrointestinal |
||||
|
Diarrhea |
38 |
4.6 |
58 |
6 |
|
Nausea |
30 |
2 |
43 |
1.5 |
|
Vomiting |
20 |
0.9 |
28 |
2.1 |
|
Abdominal pain |
19 |
1.6 |
24 |
1.9 |
|
Constipation |
17 |
0.4 |
18 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paind |
37 |
4 |
40 |
2.6 |
|
Arthralgia |
23 |
1.3 |
16 |
0 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough/productive cough |
28 |
0.2 |
25 |
0.4 |
|
Dyspnea/exertional dyspnea |
20 |
2.4 |
21 |
2.1 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
21 |
1.8 |
29 |
0.9 |
|
Nervous System |
||||
|
Headache |
19 |
0.9 |
23 |
0.9 |
|
Endocrine |
||||
|
Hypothyroidism |
18 |
0.4 |
27 |
0.2 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab group (range: 490 to 538 patients) and sunitinib group (range: 485 to 523 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab |
Sunitinib |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Increased lipase |
48 |
20 |
51 |
20 |
|
Increased creatinine |
42 |
2.1 |
46 |
1.7 |
|
Increased ALT |
41 |
7 |
44 |
2.7 |
|
Increased AST |
40 |
4.8 |
60 |
2.1 |
|
Increased amylase |
39 |
12 |
33 |
7 |
|
Hyponatremia |
39 |
10 |
36 |
7 |
|
Increased alkaline phosphatase |
29 |
2 |
32 |
1 |
|
Hyperkalemia |
29 |
2.4 |
28 |
2.9 |
|
Hypocalcemia |
21 |
0.4 |
35 |
0.6 |
|
Hypomagnesemia |
16 |
0.4 |
26 |
1.6 |
|
Hematology |
||||
|
Anemia |
43 |
3 |
64 |
9 |
|
Lymphopenia |
36 |
5 |
63 |
14 |
In addition, among patients with TSH ≤ULN at baseline, a lower proportion of patients experienced a treatment-emergent elevation of TSH > ULN in the OPDIVO and ipilimumab group compared to the sunitinib group (31% and 61%, respectively).
CHECKMATE-9ER
The safety of OPDIVO with cabozantinib was evaluated in CHECKMATE-9ER, a randomized, open-label study in patients with previously untreated advanced RCC. Patients received OPDIVO 240 mg over 30 minutes every 2 weeks with cabozantinib 40 mg orally once daily (n=320) or sunitinib 50 mg daily, administered orally for 4 weeks on treatment followed by 2 weeks off (n=320) [see Clinical Studies (14.7)]. Cabozantinib could be interrupted or reduced to 20 mg daily or 20 mg every other day. The median duration of treatment was 14 months (range: 0.2 to 27 months) in OPDIVO and cabozantinib-treated patients. In this trial, 82% of patients in the OPDIVO and cabozantinib arm were exposed to treatment for >6 months and 60% of patients were exposed to treatment for >1 year.
Serious adverse reactions occurred in 48% of patients receiving OPDIVO and cabozantinib. The most frequent (≥2%) serious adverse reactions were diarrhea, pneumonia, pneumonitis, pulmonary embolism, urinary tract infection, and hyponatremia. Fatal intestinal perforations occurred in 3 (0.9%) patients.
Adverse reactions leading to discontinuation of either OPDIVO or cabozantinib occurred in 20% of patients: 7% OPDIVO only, 8% cabozantinib only, and 6% both drugs due to same adverse reaction at the same time. Adverse reaction leading to dose interruption or reduction of either OPDIVO or cabozantinib occurred in 83% of patients: 3% OPDIVO only, 46% cabozantinib only, and 21% both drugs due to same adverse reaction at the same time, and 6% both drugs sequentially.
The most common adverse reactions reported in ≥20% of patients treated with OPDIVO and cabozantinib were diarrhea, fatigue, hepatotoxicity, palmar-plantar erythrodysesthesia syndrome, stomatitis, rash, hypertension, hypothyroidism, musculoskeletal pain, decreased appetite, nausea, dysgeusia, abdominal pain, cough, and upper respiratory tract infection.
Tables 27 and 28 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-9ER.
| Toxicity was graded per NCI CTCAE v4. a Includes abdominal discomfort, abdominal pain lower, abdominal pain upper. b Includes gastroesophageal reflux disease. c Includes asthenia. d Includes hepatotoxicity, ALT increased, AST increased, blood alkaline phosphatase increased, gamma-glutamyl transferase increased, autoimmune hepatitis, blood bilirubin increased, drug induced liver injury, hepatic enzyme increased, hepatitis, hyperbilirubinemia, liver function test increased, liver function test abnormal, transaminases increased, hepatic failure. e Includes mucosal inflammation, aphthous ulcer, mouth ulceration. f Includes dermatitis, dermatitis acneiform, dermatitis bullous, exfoliative rash, rash erythematous, rash follicular, rash macular, rash maculo-papular, rash papular, rash pruritic. g Includes blood pressure increased, blood pressure systolic increased. h Includes primary hypothyroidism. i Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, spinal pain. j Includes productive cough. k Includes nasopharyngitis, pharyngitis, rhinitis. |
||||
|
Adverse Reaction |
OPDIVO and Cabozantinib |
Sunitinib |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Gastrointestinal |
||||
|
Diarrhea |
64 |
7 |
47 |
4.4 |
|
Nausea |
27 |
0.6 |
31 |
0.3 |
|
Abdominal paina |
22 |
1.9 |
15 |
0.3 |
|
Vomiting |
17 |
1.9 |
21 |
0.3 |
|
Dyspepsiab |
15 |
0 |
22 |
0.3 |
|
General |
||||
|
Fatiguec |
51 |
8 |
50 |
8 |
|
Hepatobiliary |
||||
|
Hepatotoxicityd |
44 |
11 |
26 |
5 |
|
Skin and Subcutaneous Tissue |
||||
|
Palmar-plantar |
40 |
8 |
41 |
8 |
|
Stomatitise |
37 |
3.4 |
46 |
4.4 |
|
Rashf |
36 |
3.1 |
14 |
0 |
|
Pruritus |
19 |
0.3 |
4.4 |
0 |
|
Vascular |
||||
|
Hypertensiong |
36 |
13 |
39 |
14 |
|
Endocrine |
||||
|
Hypothyroidismh |
34 |
0.3 |
30 |
0.3 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paini |
33 |
3.8 |
29 |
3.1 |
|
Arthralgia |
18 |
0.3 |
9 |
0.3 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
28 |
1.9 |
20 |
1.3 |
|
Nervous System |
||||
|
Dysgeusia |
24 |
0 |
22 |
0 |
|
Headache |
16 |
0 |
12 |
0.6 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughj |
20 |
0.3 |
17 |
0 |
|
Dysphonia |
17 |
0.3 |
3.4 |
0 |
|
Infections and Infestations |
||||
|
Upper respiratory tract infectionk |
20 |
0.3 |
8 |
0.3 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and cabozantinib group (range: 170 to 317 patients) and sunitinib group (range: 173 to 311 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Cabozantinib |
Sunitinib |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Increased ALT |
79 |
9.8 |
39 |
3.5 |
|
Increased AST |
77 |
7.9 |
57 |
2.6 |
|
Hypophosphatemia |
69 |
28 |
48 |
10 |
|
Hypocalcemia |
54 |
1.9 |
24 |
0.6 |
|
Hypomagnesemia |
47 |
1.3 |
25 |
0.3 |
|
Hyperglycemia |
44 |
3.5 |
44 |
1.7 |
|
Hyponatremia |
43 |
11 |
36 |
12 |
|
Increased lipase |
41 |
14 |
38 |
13 |
|
Increased amylase |
41 |
10 |
28 |
6 |
|
Increased alkaline phosphatase |
41 |
2.8 |
37 |
1.6 |
|
Increased creatinine |
39 |
1.3 |
42 |
0.6 |
|
Hyperkalemia |
35 |
4.7 |
27 |
1 |
|
Hypoglycemia |
26 |
0.8 |
14 |
0.4 |
|
Hematology |
||||
|
Lymphopenia |
42 |
6.6 |
45 |
10 |
|
Thrombocytopenia |
41 |
0.3 |
70 |
9.7 |
|
Anemia |
37 |
2.5 |
61 |
4.8 |
|
Leukopenia |
37 |
0.3 |
66 |
5.1 |
|
Neutropenia |
35 |
3.2 |
67 |
12 |
Previously Treated Renal Cell Carcinoma
CHECKMATE-025
The safety of OPDIVO was evaluated in CHECKMATE-025, a randomized open-label trial in 803 patients with advanced RCC who had experienced disease progression during or after at least one anti-angiogenic treatment regimen received OPDIVO 3 mg/kg over 60 minutes by intravenous infusion every 2 weeks (n=406) or everolimus 10 mg daily (n=397) [see Clinical Studies (14.7)]. The median duration of treatment was 5.5 months (range: 1 day to 29.6+ months) in OPDIVO-treated patients and 3.7 months (range: 6 days to 25.7+ months) in everolimus-treated patients.
Rate of death on treatment or within 30 days of the last dose was 4.7% on the OPDIVO arm. Serious adverse reactions occurred in 47% of patients receiving OPDIVO. Study therapy was discontinued for adverse reactions in 16% of OPDIVO patients. Forty-four percent (44%) of patients receiving OPDIVO had a dose interruption for an adverse reaction.
The most frequent serious adverse reactions in at least 2% of patients were: acute kidney injury, pleural effusion, pneumonia, diarrhea, and hypercalcemia. The most common adverse reactions (≥20%) were fatigue, cough, nausea, rash, dyspnea, diarrhea, constipation, decreased appetite, back pain, and arthralgia. The most common laboratory abnormalities which have worsened compared to baseline in ≥30% of patients include increased creatinine, lymphopenia, anemia, increased AST, increased alkaline phosphatase, hyponatremia, increased triglycerides, and hyperkalemia. In addition, among patients with TSH < ULN at baseline, a greater proportion of patients experienced a treatment-emergent elevation of TSH >ULN in the OPDIVO group compared to the everolimus group (26% and 14%, respectively).
Tables 29 and 30 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-025.
| Toxicity was graded per NCI CTCAE v4. a Includes asthenia, decreased activity, fatigue, and malaise. b Includes nasopharyngitis, pharyngitis, rhinitis, and viral upper respiratory infection (URI). c Includes colitis, enterocolitis, and gastroenteritis. d Includes dermatitis, acneiform dermatitis, erythematous rash, generalized rash, macular rash, maculopapular rash, papular rash, pruritic rash, erythema multiforme, and erythema. |
||||
|
Adverse Reaction |
OPDIVO |
Everolimus |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Adverse Reaction |
98 |
56 |
96 |
62 |
|
General |
||||
|
Fatiguea |
56 |
6 |
57 |
7 |
|
Pyrexia |
17 |
0.7 |
20 |
0.8 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough/productive cough |
34 |
0 |
38 |
0.5 |
|
Dyspnea/exertional dyspnea |
27 |
3 |
31 |
2 |
|
Upper respiratory infectionb |
18 |
0 |
11 |
0 |
|
Gastrointestinal |
||||
|
Nausea |
28 |
0.5 |
29 |
1 |
|
Diarrheac |
25 |
2.2 |
32 |
1.8 |
|
Constipation |
23 |
0.5 |
18 |
0.5 |
|
Vomiting |
16 |
0.5 |
16 |
0.5 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashd |
28 |
1.5 |
36 |
1 |
|
Pruritus/generalized pruritus |
19 |
0 |
14 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
23 |
1.2 |
30 |
1.5 |
|
Musculoskeletal and Connective Tissue |
||||
|
Arthralgia |
20 |
1 |
14 |
0.5 |
|
Back pain |
21 |
3.4 |
16 |
2.8 |
Other clinically important adverse reactions in CHECKMATE-025 were:
General Disorders and Administration Site Conditions: peripheral edema/edema
Gastrointestinal Disorders: abdominal pain/discomfort
Musculoskeletal and Connective Tissue Disorders: extremity pain, musculoskeletal pain
Nervous System Disorders: headache/migraine, peripheral neuropathy
Investigations: weight decreased
Skin Disorders: palmar-plantar erythrodysesthesia
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 259 to 401 patients) and everolimus group (range: 257 to 376 patients). | ||||
|
Laboratory Abnormality |
OPDIVO |
Everolimus |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Lymphopenia |
42 |
6 |
53 |
11 |
|
Anemia |
39 |
8 |
69 |
16 |
|
Chemistry |
||||
|
Increased creatinine |
42 |
2 |
45 |
1.6 |
|
Increased AST |
33 |
2.8 |
39 |
1.6 |
|
Increased alkaline phosphatase |
32 |
2.3 |
32 |
0.8 |
|
Hyponatremia |
32 |
7 |
26 |
6 |
|
Hyperkalemia |
30 |
4 |
20 |
2.1 |
|
Hypocalcemia |
23 |
0.9 |
26 |
1.3 |
|
Increased ALT |
22 |
3.2 |
31 |
0.8 |
|
Hypercalcemia |
19 |
3.2 |
6 |
0.3 |
|
Lipids |
||||
|
Increased triglycerides |
32 |
1.5 |
67 |
11 |
|
Increased cholesterol |
21 |
0.3 |
55 |
1.4 |
Classical Hodgkin Lymphoma
The safety of OPDIVO was evaluated in 266 adult patients with cHL (243 patients in the CHECKMATE-205 and 23 patients in the CHECKMATE-039 trials) [see Clinical Studies (14.8)]. Patients received OPDIVO 3 mg/kg as an intravenous infusion over 60 minutes every 2 weeks until disease progression, maximal clinical benefit, or unacceptable toxicity.
The median age was 34 years (range: 18 to 72), 98% of patients had received autologous HSCT, none had received allogeneic HSCT, and 74% had received brentuximab vedotin. The median number of prior systemic regimens was 4 (range: 2 to 15). Patients received a median of 23 doses (cycles) of OPDIVO (range: 1 to 48), with a median duration of therapy of 11 months (range: 0 to 23 months).
Eleven patients died from causes other than disease progression: 3 from adverse reactions within 30 days of the last nivolumab dose, 2 from infection 8 to 9 months after completing nivolumab, and 6 from complications of allogeneic HSCT. Serious adverse reactions occurred in 26% of patients. Dose delay for an adverse reaction occurred in 34% of patients. OPDIVO was discontinued due to adverse reactions in 7% of patients.
The most frequent serious adverse reactions reported in ≥1% of patients were pneumonia, infusion-related reaction, pyrexia, colitis or diarrhea, pleural effusion, pneumonitis, and rash. The most common adverse reactions (≥20%) among all patients were upper respiratory tract infection, fatigue, cough, diarrhea, pyrexia, musculoskeletal pain, rash, nausea, and pruritus.
Tables 31 and 32 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-205 and CHECKMATE-039.
| Toxicity was graded per NCI CTCAE v4. a Includes events occurring up to 30 days after last nivolumab dose, regardless of causality. After an immune-mediated adverse reaction, reactions following nivolumab rechallenge were included if they occurred up to 30 days after completing the initial nivolumab course. b Includes nasopharyngitis, pharyngitis, rhinitis, and sinusitis. c Includes pneumonia bacterial, pneumonia mycoplasmal, pneumocystis jirovecii pneumonia. d Includes asthenia. e Includes colitis. f Includes abdominal discomfort and upper abdominal pain. g Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, and pain in extremity. h Includes dermatitis, dermatitis acneiform, dermatitis exfoliative, and rash described as macular, papular, maculopapular, pruritic, exfoliative, or acneiform. i Includes hyperesthesia, hypoesthesia, paresthesia, dysesthesia, peripheral motor neuropathy, peripheral sensory neuropathy, and polyneuropathy. These numbers are specific to treatment-emergent events. |
||
|
Adverse Reactiona |
OPDIVO (n=266) |
|
|
All Grades (%) |
Grades 3-4 (%) |
|
|
Infections |
||
|
Upper respiratory tract infectionb |
44 |
0.8 |
|
Pneumonia/bronchopneumoniac |
13 |
3.8 |
|
Nasal congestion |
11 |
0 |
|
General |
||
|
Fatigued |
39 |
1.9 |
|
Pyrexia |
29 |
<1 |
|
Respiratory, Thoracic and Mediastinal |
||
|
Cough/productive cough |
36 |
0 |
|
Dyspnea/exertional dyspnea |
15 |
1.5 |
|
Gastrointestinal |
||
|
Diarrheae |
33 |
1.5 |
|
Nausea |
20 |
0 |
|
Vomiting |
19 |
<1 |
|
Abdominal painf |
16 |
<1 |
|
Constipation |
14 |
0.4 |
|
Musculoskeletal and Connective Tissue |
||
|
Musculoskeletal paing |
26 |
1.1 |
|
Arthralgia |
16 |
<1 |
|
Skin and Subcutaneous Tissue |
||
|
Rashh |
24 |
1.5 |
|
Pruritus |
20 |
0 |
|
Nervous System |
||
|
Headache |
17 |
<1 |
|
Neuropathy peripherali |
12 |
<1 |
|
Injury, Poisoning and Procedural Complications |
||
|
Infusion-related reaction |
14 |
<1 |
|
Endocrine |
||
|
Hypothyroidism/thyroiditis |
12 |
0 |
Additional information regarding clinically important adverse reactions:
Immune-mediated pneumonitis: In CHECKMATE-205 and CHECKMATE-039, pneumonitis, including interstitial lung disease, occurred in 6.0% (16/266) of patients receiving OPDIVO. Immune-mediated pneumonitis occurred in 4.9% (13/266) of patients receiving OPDIVO (one Grade 3 and 12 Grade 2). The median time to onset was 4.5 months (range: 5 days to 12 months). All 13 patients received systemic corticosteroids, with resolution in 12. Four patients permanently discontinued OPDIVO due to pneumonitis. Eight patients continued OPDIVO (three after dose delay), of whom two had recurrence of pneumonitis.
Peripheral neuropathy: Treatment-emergent peripheral neuropathy was reported in 12% (31/266) of all patients receiving OPDIVO. Twenty-eight patients (11%) had new-onset peripheral neuropathy and 3 patients had worsening of neuropathy from baseline. The median time to onset was 50 (range: 1 to 309) days.
Complications of allogeneic HSCT after OPDIVO: Of 17 patients with cHL from the CHECKMATE-205 and CHECKMATE-039 trials who underwent allogeneic HSCT after treatment with OPDIVO, 6 patients (35%) died from transplant-related complications. Five deaths occurred in the setting of severe (Grade 3 to 4) or refractory GVHD. Hyperacute GVHD occurred in 2 patients (12%) and Grade 3 or higher GVHD was reported in 5 patients (29%). Hepatic VOD occurred in 1 patient, who received reduced-intensity conditioned allogeneic HSCT and died of GVHD and multi-organ failure.
Table 32 summarizes laboratory abnormalities in patients with cHL. The most common (≥20%) treatment-emergent laboratory abnormalities included cytopenias, liver function abnormalities, and increased lipase. Other common findings (≥10%) included increased creatinine, electrolyte abnormalities, and increased amylase.
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement: range: 203 to 266 patients. b Includes events occurring up to 30 days after last nivolumab dose. After an immune-mediated adverse reaction, reactions following nivolumab rechallenge were included if they occurred within 30 days of completing the initial nivolumab course. c In addition, in the safety population, fasting hyperglycemia (all grade 1-2) was reported in 27 of 69 (39%) evaluable patients and fasting hypoglycemia (all grade 1-2) in 11 of 69 (16%). |
||
|
Laboratory Abnormality |
OPDIVOa
|
|
|
All Grades (%)b |
Grades 3-4 (%)b |
|
|
Hematology |
||
|
Leukopenia |
38 |
4.5 |
|
Neutropenia |
37 |
5 |
|
Thrombocytopenia |
37 |
3 |
|
Lymphopenia |
32 |
11 |
|
Anemia |
26 |
2.6 |
|
Chemistryc |
||
|
Increased AST |
33 |
2.6 |
|
Increased ALT |
31 |
3.4 |
|
Increased lipase |
22 |
9 |
|
Increased alkaline phosphatase |
20 |
1.5 |
|
Hyponatremia |
20 |
1.1 |
|
Hypokalemia |
16 |
1.9 |
|
Increased creatinine |
16 |
<1 |
|
Hypocalcemia |
15 |
<1 |
|
Hyperkalemia |
15 |
1.5 |
|
Hypomagnesemia |
14 |
<1 |
|
Increased amylase |
13 |
1.5 |
|
Increased bilirubin |
11 |
1.5 |
Squamous Cell Carcinoma of the Head and Neck
The safety of OPDIVO was evaluated in CHECKMATE-141, a randomized, active-controlled, open-label, multicenter trial in patients with recurrent or metastatic SCCHN with progression during or within 6 months of receiving prior platinum-based therapy [see Clinical Studies (14.9)]. The trial excluded patients with active autoimmune disease, medical conditions requiring systemic immunosuppression, or recurrent or metastatic carcinoma of the nasopharynx, squamous cell carcinoma of unknown primary histology, salivary gland or non-squamous histologies (e.g., mucosal melanoma). Patients received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks (n=236) or investigator’s choice of either cetuximab (400 mg/m2 initial dose intravenously followed by 250 mg/m2 weekly), or methotrexate (40 to 60 mg/m2 intravenously weekly), or docetaxel (30 to 40 mg/m2 intravenously weekly). The median duration of exposure to nivolumab was 1.9 months (range: 1 day to 16.1+ months) in OPDIVO-treated patients. In this trial, 18% of patients received OPDIVO for >6 months and 2.5% of patients received OPDIVO for >1 year.
The median age of all randomized patients was 60 years (range: 28 to 83); 28% of patients in the OPDIVO group were ≥65 years of age and 37% in the comparator group were ≥65 years of age, 83% were male and 83% were White, 12% were Asian, and 4% were Black. Baseline ECOG performance status was 0 (20%) or 1 (78%), 45% of patients received only one prior line of systemic therapy, the remaining 55% of patients had two or more prior lines of therapy, and 90% had prior radiation therapy.
Serious adverse reactions occurred in 49% of patients receiving OPDIVO. OPDIVO was discontinued in 14% of patients and was delayed in 24% of patients for an adverse reaction. Adverse reactions and laboratory abnormalities occurring in patients with SCCHN were generally similar to those occurring in patients with melanoma and NSCLC.
The most frequent serious adverse reactions reported in ≥2% of patients receiving OPDIVO were pneumonia, dyspnea, respiratory failure, respiratory tract infection, and sepsis. The most common adverse reactions occurring in ≥10% of OPDIVO-treated patients and at a higher incidence than investigator’s choice were cough and dyspnea. The most common laboratory abnormalities occurring in ≥10% of OPDIVO-treated patients and at a higher incidence than investigator’s choice were increased alkaline phosphatase, increased amylase, hypercalcemia, hyperkalemia, and increased TSH.
Adjuvant Treatment of Urothelial Carcinoma (UC)
The safety of OPDIVO was evaluated in CHECKMATE-274, a randomized, double-blind, multicenter trial of adjuvant OPDIVO versus placebo in adult patients who had undergone radical resection of UC originating in the bladder or upper urinary tract (renal pelvis or ureter) and were at high risk of recurrence [see Clinical Studies (14.10)]. Patients received OPDIVO 240 mg by intravenous infusion over 30 minutes every 2 weeks (n=351) or placebo (n=348) until recurrence or unacceptable toxicity for a maximum of 1 year. The median duration of OPDIVO treatment was 8.8 months (range: 0 to 12.5).
Serious adverse reactions occurred in 30% of OPDIVO patients. The most frequent serious adverse reaction reported in ≥2% of patients was urinary tract infection. Fatal adverse reactions occurred in 1% of patients; these included events of pneumonitis (0.6%). OPDIVO was discontinued for adverse reactions in 18% of patients. OPDIVO was delayed for adverse reaction in 33% of patients.
The most common adverse reactions (reported in ≥20% of patients) were rash, fatigue, diarrhea, pruritus, musculoskeletal pain, and urinary tract infection.
Tables 33 and 34 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-274.
| Toxicity was graded per NCI CTCAE v4. a Includes acne, blister, dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis contact, eczema, eczema asteatotic, eczema nummular, erythema, erythema multiforme, lichen sclerosus, lichenoid keratosis, pemphigoid, photosensitivity reaction, pigmentation disorder, psoriasis, rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic, rosacea, skin exfoliation, skin lesion, skin reaction, toxic skin eruption, and urticaria. b Includes colitis, colitis microscopic, diarrhea, duodenitis, enteritis, immune-mediated enterocolitis. c Includes abdominal pain, abdominal discomfort, abdominal tenderness, lower and upper abdominal pain. d Includes musculoskeletal pain, back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity and spinal pain. e Includes cystitis, escherichia urinary tract infection, pyelonephritis, pyelonephritis acute, pyelonephritis chronic, urethritis, urinary tract infection, urinary tract infection bacterial, urinary tract infection staphylococcal, and urosepsis. f Includes upper respiratory tract infection, nasopharyngitis, pharyngitis and rhinitis. g Includes acute kidney injury, autoimmune nephritis, blood creatinine increased, glomerular filtration rate decreased, immune-mediated nephritis, nephritis, renal failure, and renal impairment. h Includes cough, productive cough, and upper-airway cough syndrome. i Includes dyspnea and exertional dyspnea. j Includes dizziness, postural dizziness and vertigo. k Includes aspartate aminotransferase increased, alanine aminotransferase increased, blood bilirubin increased, cholangitis, drug-induced liver injury, hepatic failure, hepatic function abnormal, hepatitis, hepatocellular injury, hyperbilirubinemia, gamma-glutamyl transferase increased, liver injury, and transaminases increased. |
||||
|
Adverse Reaction |
OPDIVO (n=351) |
Placebo (n=348) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Skin and Subcutaneous Tissue |
||||
|
Rasha |
36 |
1.7 |
19 |
0.3 |
|
Pruritus |
30 |
0 |
16 |
0 |
|
General |
||||
|
Fatigue/Asthenia |
36 |
1.1 |
32 |
0.3 |
|
Pyrexia |
10 |
0.3 |
10 |
0.3 |
|
Gastrointestinal |
||||
|
Diarrheab |
30 |
2.8 |
27 |
1.7 |
|
Nausea |
16 |
0.6 |
13 |
0 |
|
Abdominal painc |
15 |
0.9 |
15 |
0.6 |
|
Constipation |
13 |
0.3 |
15 |
0.3 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paind |
28 |
0.6 |
24 |
0.9 |
|
Arthralgia |
11 |
0.3 |
13 |
0 |
|
Infections |
||||
|
Urinary tract infectione |
22 |
6 |
23 |
9 |
|
Upper respiratory tract infectionf |
16 |
0.3 |
16 |
0.6 |
|
Endocrine |
||||
|
Hyperthyroidism |
11 |
0 |
1.1 |
0 |
|
Hypothyroidism |
11 |
0 |
2.3 |
0 |
|
Renal and Urinary Disorders |
||||
|
Renal dysfunctiong |
17 |
1.7 |
16 |
0.9 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughh |
14 |
0 |
11 |
0 |
|
Dyspneai |
11 |
0.3 |
6 |
0.3 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
13 |
0.9 |
7 |
0.3 |
|
Nervous System Disorders |
||||
|
Dizzinessj |
11 |
0.3 |
9 |
0 |
|
Hepatobiliary |
||||
|
Hepatitisk |
11 |
4 |
8 |
0.6 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 322 to 348 patients) and placebo group (range: 312 to 341 patients). | ||||
|
Laboratory Abnormality |
OPDIVO (n=351) |
Placebo (n=348) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Increased creatinine |
36 |
1.7 |
36 |
2.6 |
|
Increased amylase |
34 |
8 |
23 |
3.2 |
|
Increased lipase |
33 |
12 |
31 |
10 |
|
Hyperkalemia |
32 |
5 |
30 |
6 |
|
Increased alkaline phosphatase |
24 |
2.3 |
15 |
0.6 |
|
Increased AST |
24 |
3.5 |
16 |
0.9 |
|
Increased ALT |
23 |
2.9 |
15 |
0.6 |
|
Hyponatremia |
22 |
4.1 |
17 |
1.8 |
|
Hypocalcemia |
17 |
1.2 |
11 |
0.9 |
|
Hypomagnesemia |
16 |
0 |
9 |
0 |
|
Hypercalcemia |
12 |
0.3 |
8 |
0.3 |
|
Hematology |
||||
|
Lymphopenia |
33 |
2.9 |
27 |
1.5 |
|
Anemia |
30 |
1.4 |
28 |
0.9 |
|
Neutropenia |
11 |
0.6 |
10 |
0.3 |
First-line Treatment of Unresectable or Metastatic UC
The safety of OPDIVO was evaluated in CHECKMATE-901, a randomized, open-label trial in cisplatin-eligible patients with unresectable or metastatic UC [see Clinical Studies (14.10)]. Patients received either OPDIVO 360 mg with cisplatin and gemcitabine every 3 weeks for up to 6 cycles followed by single-agent OPDIVO 480 mg every 4 weeks up to 2 years (n=304), or cisplatin and gemcitabine chemotherapy every 3 weeks for up to 6 cycles (n=288). Patients discontinuing cisplatin alone were permitted to switch to carboplatin.
Among patients who received OPDIVO with chemotherapy, the median duration of OPDIVO exposure was 7.4 months (range: 0.03 to 47.9 months). Serious adverse reactions occurred in 48% of patients receiving OPDIVO in combination with chemotherapy. The most frequent serious adverse reactions reported in ≥2% of patients who received OPDIVO with chemotherapy were urinary tract infection (4.9%), acute kidney injury (4.3%), anemia (3%), pulmonary embolism (2.6%), sepsis (2.3%), and platelet count decreased (2.3%). The most common adverse reactions (reported in ≥20% of patients) were nausea, fatigue, musculoskeletal pain, constipation, decreased appetite, rash, vomiting, and peripheral neuropathy.
Fatal adverse reactions occurred in 3.6% of patients who received OPDIVO in combination with chemotherapy; these included sepsis (1%).
OPDIVO and/or chemotherapy were discontinued in 30% of patients and were delayed in 67% of patients for an adverse reaction.
Tables 35 and 36 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-901.
| Toxicity was graded per NCI CTCAE v4. a Includes colitis, immune-mediated enterocolitis. b Includes upper abdominal pain, lower abdominal pain, abdominal discomfort, epigastric discomfort, gastrointestinal pain, and hepatic pain. c Includes asthenia. d Includes peripheral edema, swelling, peripheral swelling, localized edema, swelling, face edema, testicular edema, gravitational edema, and edema genital. e Includes hyperthermia, body temperature increased and hyperpyrexia. f Includes back pain, arthralgia, bone pain, arthritis, musculoskeletal chest pain, non-cardiac chest pain, myalgia, neck pain, pain in extremity, and spinal pain. g Includes maculopapular rash, erythematous rash, macular rash, papular rash, pustular rash, acneiform dermatitis, dermatitis, allergic dermatitis, atopic dermatitis, exfoliative rash, eczema asteatotic, erythema multiforme, palmar-plantar erythrodysesthesia syndrome, eczema, dermatitis exfoliative generalized, and skin exfoliation. h Includes paresthesia, peripheral sensory neuropathy, hypoesthesia, dysesthesia, neuralgia, hyperesthesia, peripheral motor neuropathy, polyneuropathy. i Includes occipital neuralgia. j Includes urosepsis, cystitis, pyelonephritis, pyelonephritis acute, urinary tract infection enterococcal, escherichia urinary tract infection. k Includes blood stimulating hormone increased. l Includes acute kidney injury, renal failure, renal impairment, glomerular filtration rate decreased, anuria, azotemia. |
||||
|
Adverse Reaction |
OPDIVO and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy (n=288) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Gastrointestinal disorders |
||||
|
Nausea |
52 |
0.3 |
53 |
1 |
|
Constipation |
30 |
0 |
28 |
0.7 |
|
Vomiting |
23 |
1.3 |
19 |
2.1 |
|
Diarrheaa |
19 |
2 |
14 |
0 |
|
Abdominal painb |
14 |
0.3 |
9 |
0.3 |
|
General |
||||
|
Fatiguec |
48 |
3.9 |
43 |
4.2 |
|
Edemad |
18 |
0 |
9 |
0.3 |
|
Pyrexiae |
14 |
1 |
14 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal painf |
33 |
3 |
21 |
0.3 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
30 |
1.6 |
19 |
1 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashg |
25 |
2.3 |
7 |
0.3 |
|
Pruritus |
17 |
0.7 |
3.5 |
0 |
|
Nervous System Disorders |
||||
|
Peripheral neuropathyh |
20 |
0.7 |
14 |
0 |
|
Headachei |
11 |
0 |
5 |
0 |
|
Infections |
||||
|
Urinary tract infectionj |
19 |
8 |
18 |
8 |
|
Endocrine disorders |
||||
|
Hypothyroidismk |
17 |
0 |
0.3 |
0 |
|
Renal and Urinary Disorders |
||||
|
Renal dysfunctionl |
14 |
6 |
11 |
1.7 |
|
Hematuria |
11 |
1 |
7 |
1.4 |
|
Investigations |
||||
|
Weight decreased |
11 |
0.3 |
6 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 289-301 patients) and chemotherapy group (range: 265-281 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Platinum-Doublet |
Platinum-Doublet |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Anemia |
88 |
21 |
80 |
21 |
|
Neutropenia |
82 |
35 |
76 |
28 |
|
Lymphopenia |
71 |
17 |
56 |
13 |
|
Thrombocytopenia |
60 |
13 |
51 |
8 |
|
Chemistry |
||||
|
Increased creatinine |
53 |
2.4 |
42 |
1.1 |
|
Hypomagnesemia |
48 |
3.8 |
39 |
1.5 |
|
Hyponatremia |
43 |
13 |
39 |
8 |
|
Hyperglycemia |
41 |
3.9 |
37 |
3.2 |
|
Hypocalcemia |
36 |
2.1 |
24 |
1.1 |
|
Hyperkalemia |
33 |
3.0 |
32 |
1.1 |
|
Increased amylase |
32 |
4.2 |
23 |
3.6 |
|
Increased AST |
31 |
2.4 |
17 |
0.7 |
|
Increased ALT |
29 |
2.4 |
19 |
0.7 |
Previously Treated Advanced or Metastatic UC
The safety of OPDIVO was evaluated in CHECKMATE-275, a single arm trial in which 270 patients with locally advanced or metastatic UC had disease progression during or following platinum-containing chemotherapy or had disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy [see Clinical Studies (14.10)]. Patients received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks until disease progression or unacceptable toxicity. The median duration of treatment was 3.3 months (range: 0 to 13.4+). Forty-six percent (46%) of patients had a dose interruption for an adverse reaction.
Fourteen patients (5.2%) died from causes other than disease progression. This includes 4 patients (1.5%) who died from pneumonitis or cardiovascular failure which was attributed to treatment with OPDIVO. Serious adverse reactions occurred in 54% of patients. OPDIVO was discontinued for adverse reactions in 17% of patients.
The most frequent serious adverse reactions reported in ≥2% of patients were urinary tract infection, sepsis, diarrhea, small intestine obstruction, and general physical health deterioration. The most common adverse reactions (reported in ≥20% of patients) were fatigue, musculoskeletal pain, nausea, and decreased appetite.
Tables 37 and 38 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-275.
| Toxicity was graded per NCI CTCAE v4. a Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity and spinal pain. b Includes abdominal discomfort, lower and upper abdominal pain. c Includes dermatitis, dermatitis acneiform, dermatitis bullous, and rash described as generalized, macular, maculopapular, or pruritic. d Includes autoimmune thyroiditis, blood TSH decrease, blood TSH increase, hyperthyroidism, hypothyroidism, thyroiditis, thyroxine decreased, thyroxine free increased, thyroxine increased, tri-iodothyronine free increased, tri-iodothyronine increased. |
||
|
Adverse Reaction |
OPDIVO |
|
|
All Grades (%) |
Grades 3-4 (%) |
|
|
Adverse Reaction |
99 |
51 |
|
General |
||
|
Asthenia/fatigue/malaise |
46 |
7 |
|
Pyrexia/tumor associated fever |
17 |
0.4 |
|
Edema/peripheral edema/peripheral swelling |
13 |
0.4 |
|
Musculoskeletal and Connective Tissue |
||
|
Musculoskeletal paina |
30 |
2.6 |
|
Arthralgia |
10 |
0.7 |
|
Metabolism and Nutrition |
||
|
Decreased appetite |
22 |
2.2 |
|
Gastrointestinal |
||
|
Nausea |
22 |
0.7 |
|
Diarrhea |
17 |
2.6 |
|
Constipation |
16 |
0.4 |
|
Abdominal painb |
13 |
1.5 |
|
Vomiting |
12 |
1.9 |
|
Respiratory, Thoracic and Mediastinal |
||
|
Cough/productive cough |
18 |
0 |
|
Dyspnea/exertional dyspnea |
14 |
3.3 |
|
Infections |
||
|
Urinary tract infection/escherichia/fungal urinary tract infection |
17 |
7 |
|
Skin and Subcutaneous Tissue |
||
|
Rashc |
16 |
1.5 |
|
Pruritus |
12 |
0 |
|
Endocrine |
||
|
Thyroid disordersd |
15 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: range: 84 to 256 patients. | ||
|
Laboratory Abnormality |
OPDIVOa |
|
|
All Grades (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||
|
Hyperglycemia |
42 |
2.4 |
|
Hyponatremia |
41 |
11 |
|
Increased creatinine |
39 |
2 |
|
Increased alkaline phosphatase |
33 |
5.5 |
|
Hypocalcemia |
26 |
0.8 |
|
Increased AST |
24 |
3.5 |
|
Increased lipase |
20 |
7 |
|
Hyperkalemia |
19 |
1.2 |
|
Increased ALT |
18 |
1.2 |
|
Increased amylase |
18 |
4.4 |
|
Hypomagnesemia |
16 |
0 |
|
Hematology |
||
|
Lymphopenia |
42 |
9 |
|
Anemia |
40 |
7 |
|
Thrombocytopenia |
15 |
2.4 |
|
Leukopenia |
11 |
0 |
MSI-H or dMMR Metastatic Colorectal Cancer
Treatment of MSI-H or dMMR mCRC: In Combination with Ipilimumab
The safety of OPDIVO in combination with ipilimumab, or as a single agent, was evaluated in CHECKMATE-8HW, a randomized, open-label, three arm trial in immunotherapy naive patients with MSI-H or dMMR mCRC [see Clinical Studies (14.11)]. Patients received one of the following:
- •
- OPDIVO 240 mg every 3 weeks and ipilimumab 1 mg/kg every 3 weeks for a maximum of 4 doses, then OPDIVO 480 mg every 4 weeks.
- •
- OPDIVO 240 mg every 2 weeks for 6 doses, then OPDIVO 480 mg every 4 weeks.
- •
- Investigator’s choice chemotherapy: mFOLFOX or FOLFIRI [see Clinical Studies (14.11)].
In the OPDIVO and ipilimumab arm, the median duration of exposure to OPDIVO was 20.5 months (range: 0 to 35.9 months), 70% of patients were exposed for >6 months and 63% were exposed for >1 year. In the OPDIVO arm, the median duration of exposure to OPDIVO was 16.4 months (range: 0 to 36 months), 64% of patients were exposed for >6 months and 54% were exposed for >1 year.
Serious adverse reactions occurred in 46% of patients receiving OPDIVO in combination with ipilimumab, and 39% of patients receiving OPDIVO alone. The most frequent serious adverse reactions reported in ≥1% of patients who received OPDIVO with ipilimumab were adrenal insufficiency (2.8%), hypophysitis (2.8%), diarrhea (2.0%), abdominal pain (2.0%), small intestinal obstruction (2.0%), pneumonia (1.7%), acute kidney injury (1.4%), immune mediated enterocolitis (1.4%), pneumonitis (1.4%), colitis (1.1%), large intestinal obstruction (1.1%), and urinary tract infection (1.1%). The most frequent serious adverse reactions reported in >1% of patients who received OPDIVO, as a single agent, were intestinal obstruction (2.3%), acute kidney injury (1.7%), COVID-19 (1.7%), abdominal pain (1.4%), diarrhea (1.4%), ileus (1.4%), subileus (1.4%), pulmonary embolism (1.4%), adrenal insufficiency (1.1%) and pneumonia (1.1%).
Fatal adverse reactions occurred in 2 (0.6%) patients who received OPDIVO in combination with ipilimumab; these included myocarditis, and pneumonitis (1 each). Fatal adverse reactions occurring in 3 (0.9%) patients who received OPDIVO as a single agent; these included pneumonitis (n=2) and myasthenia gravis.
OPDIVO and/or ipilimumab were permanently discontinued in 19% of patients receiving the combination. The most frequent adverse reactions (>1%) leading to permanent discontinuation were adrenal insufficiency (1.4%), immune mediated enterocolitis (1.1%), and pneumonitis (1.1%). OPDIVO was permanently discontinued in 13% of patients receiving single agent OPDIVO. Adverse reactions leading to the delay of OPDIVO and/or ipilimumab occurred in 48% of patients receiving the combination; single agent OPDIVO was delayed in 37% of patients due to adverse reactions.
The most common adverse reactions reported in ≥20% of patients treated with OPDIVO in combination with ipilimumab were fatigue, diarrhea, pruritus, abdominal pain, musculoskeletal pain, and nausea. The most common adverse reactions reported in ≥20% of patients treated with OPDIVO as a single agent, were fatigue, diarrhea, abdominal pain, pruritus, and musculoskeletal pain.
Tables 39 and 40 summarize selected adverse reactions and selected laboratory abnormalities for OPDIVO in combination with ipilimumab and the single agent OPDIVO arms respectively, in CHECKMATE-8HW.
| Toxicity was graded per NCI CTCAE v5. a Includes colitis, diarrhea, enterocolitis, immune mediated enterocolitis |
||||
|
Adverse Reaction |
OPDIVO and ipilimumab |
OPDIVO |
||
|
All Grades (%) |
Grades 3 or 4 (%) |
All Grades (%) |
Grades 3 or4 (%) |
|
|
Gastrointestinal |
||||
|
Diarrheaa |
35 |
4.5 |
30 |
3.4 |
|
Skin and Subcutaneous Tissue |
||||
|
Pruritus |
30 |
0 |
23 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Arthralgia |
20 |
0.6 |
15 |
0.6 |
|
Endocrine |
||||
|
Hypothyroidism |
18 |
0.6 |
10 |
0 |
|
Hyperthyroidism |
12 |
0 |
5 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab group (range: 108 to 343 patients) or nivolumab group (range: 102 to 348 patients). | ||||
|
Laboratory Abnormalitya |
OPDIVO and Ipilimumab |
OPDIVO |
||
|
All Grades (%) |
Grades 3-or 4 (%) |
All Grades (%) |
Grades 3 or -4 (%) |
|
|
Hematology |
||||
|
Lymphocytes decreased |
30 |
5 |
37 |
4 |
|
Neutrophils decreased |
21 |
1.7 |
12 |
0.6 |
|
Chemistry |
||||
|
Lipase increased |
44 |
10 |
32 |
11 |
|
Amylase increased |
41 |
4.6 |
33 |
5 |
|
ALT increased |
39 |
3.5 |
32 |
1.4 |
|
AST increased |
38 |
3.2 |
29 |
1.4 |
|
Sodium decreased |
36 |
3.2 |
30 |
2.3 |
|
Creatinine increased |
32 |
2 |
25 |
1.4 |
|
Potassium increased |
29 |
1.2 |
35 |
0.9 |
|
Glucose decreased |
17 |
0 |
12 |
0 |
MSI-H or dMMR mCRC After Progression Following Treatment with a Fluoropyrimidine, Oxaliplatin, and Irinotecan
The safety of OPDIVO administered as a single agent or in combination with ipilimumab was evaluated in CHECKMATE-142, a multicenter, non-randomized, multiple parallel-cohort, open-label trial [see Clinical Studies (14.11)]. In CHECKMATE-142, 74 patients with mCRC received OPDIVO 3 mg/kg by intravenous infusion over 60 minutes every 2 weeks until disease progression or until intolerable toxicity and 119 patients with mCRC received OPDIVO 3 mg/kg and ipilimumab 1 mg/kg every 3 weeks for 4 doses, then OPDIVO 3 mg/kg every 2 weeks until disease progression or until unacceptable toxicity.
In the OPDIVO with ipilimumab cohort, serious adverse reactions occurred in 47% of patients. Treatment was discontinued in 13% of patients and delayed in 45% of patients for an adverse reaction. The most frequent serious adverse reactions reported in ≥2% of patients were colitis/diarrhea, hepatic events, abdominal pain, acute kidney injury, pyrexia, and dehydration. The most common adverse reactions (reported in ≥20% of patients) were fatigue, diarrhea, pyrexia, musculoskeletal pain, abdominal pain, pruritus, nausea, rash, decreased appetite, and vomiting.
Tables 41 and 42 summarize adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-142. Based on the design of CHECKMATE-142, the data below cannot be used to identify statistically significant differences between the two cohorts summarized below for any adverse reaction.
| Toxicity was graded per NCI CTCAE v4. a Includes asthenia. b Includes peripheral edema and peripheral swelling. c Includes upper abdominal pain, lower abdominal pain, and abdominal discomfort. d Includes back pain, pain in extremity, myalgia, neck pain, and bone pain. e Includes dermatitis, dermatitis acneiform, and rash described as maculo-papular, erythematous, and generalized. f Includes nasopharyngitis and rhinitis. |
||||
|
Adverse Reaction |
OPDIVO |
OPDIVO and Ipilimumab |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
General |
||||
|
Fatiguea |
54 |
5 |
49 |
6 |
|
Pyrexia |
24 |
0 |
36 |
0 |
|
Edemab |
12 |
0 |
7 |
0 |
|
Gastrointestinal |
||||
|
Diarrhea |
43 |
2.7 |
45 |
3.4 |
|
Abdominal painc |
34 |
2.7 |
30 |
5 |
|
Nausea |
34 |
1.4 |
26 |
0.8 |
|
Vomiting |
28 |
4.1 |
20 |
1.7 |
|
Constipation |
20 |
0 |
15 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paind |
28 |
1.4 |
36 |
3.4 |
|
Arthralgia |
19 |
0 |
14 |
0.8 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cough |
26 |
0 |
19 |
0.8 |
|
Dyspnea |
8 |
1 |
13 |
1.7 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashe |
23 |
1.4 |
25 |
4.2 |
|
Pruritus |
19 |
0 |
28 |
1.7 |
|
Dry Skin |
7 |
0 |
11 |
0 |
|
Infections |
||||
|
Upper respiratory tract infectionf |
20 |
0 |
9 |
0 |
|
Endocrine |
||||
|
Hyperglycemia |
19 |
2.7 |
6 |
1 |
|
Hypothyroidism |
5 |
0 |
14 |
0.8 |
|
Hyperthyroidism |
4 |
0 |
12 |
0 |
|
Nervous System |
||||
|
Headache |
16 |
0 |
17 |
1.7 |
|
Dizziness |
14 |
0 |
11 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
14 |
1.4 |
20 |
1.7 |
|
Psychiatric |
||||
|
Insomnia |
9 |
0 |
13 |
0.8 |
|
Investigations |
||||
|
Weight decreased |
8 |
0 |
10 |
0 |
Clinically important adverse reactions reported in <10% of patients receiving OPDIVO with ipilimumab were encephalitis (0.8%), necrotizing myositis (0.8%), and uveitis (0.8%).
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available. Number of evaluable patients ranges from 62 to 71 for the OPDIVO cohort and from 87 to 114 for the OPDIVO and ipilimumab cohort. | ||||
|
Laboratory Abnormality |
OPDIVO |
OPDIVO and Ipilimumab |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Anemia |
50 |
7 |
42 |
9 |
|
Lymphopenia |
36 |
7 |
25 |
6 |
|
Neutropenia |
20 |
4.3 |
18 |
0 |
|
Thrombocytopenia |
16 |
1.4 |
26 |
0.9 |
|
Chemistry |
||||
|
Increased alkaline |
37 |
2.8 |
28 |
5 |
|
Increased lipase |
33 |
19 |
39 |
12 |
|
Increased ALT |
32 |
2.8 |
33 |
12 |
|
Increased AST |
31 |
1.4 |
40 |
12 |
|
Hyponatremia |
27 |
4.3 |
26 |
5 |
|
Hypocalcemia |
19 |
0 |
16 |
0 |
|
Hypomagnesemia |
17 |
0 |
18 |
0 |
|
Increased amylase |
16 |
4.8 |
36 |
3.4 |
|
Increased bilirubin |
14 |
4.2 |
21 |
5 |
|
Hypokalemia |
14 |
0 |
15 |
1.8 |
|
Increased creatinine |
12 |
0 |
25 |
3.6 |
|
Hyperkalemia |
11 |
0 |
23 |
0.9 |
Hepatocellular Carcinoma
Unresectable or Metastatic Hepatocellular Carcinoma (HCC)
The safety of OPDIVO in combination with ipilimumab was evaluated in CHECKMATE-9DW, a randomized, open-label trial in adult patients with unresectable or metastatic HCC [see Clinical Studies (14.12)]. Patients received OPDIVO in combination with ipilimumab (n=332) or investigator’s choice of lenvatinib (n=275) or sorafenib (n=50) at the following dosage:
- •
- OPDIVO 1 mg/kg administered intravenously over 30 minutes in combination with ipilimumab 3 mg/kg administered intravenously over 30 minutes every 3 weeks, for a maximum of 4 doses, followed by single-agent OPDIVO at 480 mg administered intravenously over 30 minutes every 4 weeks, or
- •
- Investigator’s choice:
- o
- Lenvatinib 8 mg orally daily (if body weight <60 kg) or 12 mg orally daily (if body weight ≥60 kg), or
- o
- Sorafenib 400 mg orally twice daily
In the OPDIVO and ipilimumab arm, the median duration of exposure to OPDIVO was 4.7 months (range: <0.1 to 24.4 months), 45% were exposed for >6 months and 30% were exposed for >1 year.
Serious adverse reactions occurred in 53% of patients treated with OPDIVO in combination with ipilimumab. The most frequent non-liver-related serious adverse reactions reported in ≥2% of patients who received OPDIVO in combination with ipilimumab were diarrhea/colitis (4.5%), gastrointestinal hemorrhage (3%), and rash (2.4%).
Liver-related serious adverse reactions occurred in 17% of patients treated with OPDIVO in combination with ipilimumab, including Grade 3-4 events in 16% of patients. The most frequently reported all grade liver-related serious adverse reactions occurring in ≥1% of patients who received OPDIVO in combination with ipilimumab were immune-mediated hepatitis (3%), increased AST/ALT (3%), hepatic failure (2.4%), ascites (2.4%), and hepatotoxicity (1.2%).
Fatal adverse reactions occurred in 12 (3.6%) patients who received OPDIVO in combination with ipilimumab; these included 4 (1.2%) patients who died due to immune-mediated or autoimmune hepatitis and 4 (1.2%) patients who died of hepatic failure.
Permanent discontinuations of OPDIVO due to an adverse reaction occurred in 27% of patients. Adverse reactions leading to permanent discontinuation of OPDIVO in >1% of patients included immune-mediated hepatitis (1.8%), diarrhea/colitis (1.8%), hepatic failure (1.2%).
Dosage interruptions of OPDIVO due to an adverse reaction occurred in 62% of patients. Adverse reactions which required dosage interruption in >5% of patients included increased AST (13%), increased ALT (11%), and diarrhea/colitis (8%).
The most common (>20%) adverse reactions were rash, pruritus, fatigue, and diarrhea.
Tables 43 and 44 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-9DW.
| Toxicity was graded per NCI CTCAE v5 | ||||
| a Represents a composite of multiple related terms. | ||||
|
Adverse Reaction |
OPDIVO and Ipilimumab (n=332) |
Lenvatinib or Sorafenib (n=325) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Skin and Subcutaneous Tissue |
||||
|
Rasha |
36 |
3.6 |
15 |
1.2 |
|
Pruritus |
34 |
1.5 |
7 |
0.3 |
|
General |
||||
|
Fatiguea |
33 |
2.4 |
39 |
4 |
|
Pyrexiaa |
15 |
0.6 |
9 |
1.5 |
|
Edemaa |
13 |
1.2 |
13 |
1.5 |
|
Gastrointestinal |
||||
|
Diarrheaa |
25 |
6 |
39 |
3.4 |
|
Abdominal paina |
14 |
1.2 |
27 |
2.5 |
|
Nausea |
10 |
0.3 |
16 |
0.9 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paina |
17 |
0.6 |
23 |
0.3 |
|
Arthralgia |
12 |
0.3 |
13 |
0.6 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
16 |
1.2 |
28 |
1.8 |
|
Endocrine |
||||
|
Hypothyroidisma |
14 |
0 |
27 |
0 |
|
Hyperthyroidism |
11 |
0.6 |
1.5 |
0 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Cougha |
13 |
0 |
8 |
0 |
Clinically important adverse reactions reported in <10% of patients who received OPDIVO with ipilimumab were hyperglycemia (8%), adrenal insufficiency (4.2%), pneumonitis (2.7%), and pancreatitis (2.4%).
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and ipilimumab group (range: 168 to 331 patients) and lenvatinib or sorafenib group (range: 145 to 315 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and Ipilimumab (n=332) |
Lenvatinib or Sorafenib (n=325) |
||
|
Grades 1-4 |
Grades 3-4 |
Grades 1-4 |
Grades 3-4 |
|
|
Chemistry |
||||
|
Increased AST |
62 |
29 |
51 |
14 |
|
Increased ALT |
61 |
17 |
46 |
9 |
|
Increased lipase |
58 |
16 |
39 |
5 |
|
Decreased albumin |
48 |
0.9 |
57 |
0.6 |
|
Hyponatremia |
45 |
6 |
42 |
3.8 |
|
Hyperglycemia |
44 |
15 |
32 |
2.1 |
|
Increased bilirubin |
44 |
10 |
44 |
8 |
|
Increased amylase |
41 |
6 |
26 |
1 |
|
Increased alkaline phosphatase |
36 |
1.2 |
38 |
5 |
|
Hypocalcemia |
29 |
0.9 |
46 |
0 |
|
Increased creatinine |
26 |
2.4 |
23 |
0.6 |
|
Hypokalemia |
21 |
2.1 |
20 |
2.6 |
|
Hematology |
||||
|
Anemia |
44 |
5 |
40 |
3.8 |
|
Lymphopenia |
40 |
6.1 |
40 |
8 |
|
Thrombocytopenia |
27 |
4 |
44 |
4.8 |
|
Neutropenia |
24 |
4 |
32 |
3.5 |
Previously Treated Hepatocellular Carcinoma
The safety of OPDIVO 1 mg/kg in combination with ipilimumab 3 mg/kg was evaluated in a subgroup comprising 49 patients with HCC and Child-Pugh Class A cirrhosis enrolled in Cohort 4 of CHECKMATE-040, a multicenter, multiple-cohort, open-label trial [see Clinical Studies (14.12)] who progressed on or were intolerant to sorafenib. OPDIVO and ipilimumab were administered every 3 weeks for 4 doses, followed by single-agent OPDIVO 240 mg every 2 weeks until disease progression or unacceptable toxicity. During the OPDIVO and ipilimumab combination period, 33 of 49 (67%) patients received all 4 planned doses of OPDIVO and ipilimumab. During the entire treatment period, the median duration of exposure to OPDIVO was 5.1 months (range: 0 to 35+ months) and to ipilimumab was 2.1 months (range: 0 to 4.5 months). Forty-seven percent of patients were exposed to treatment for >6 months, and 35% of patients were exposed to treatment for >1 year. Serious adverse reactions occurred in 59% of patients. Treatment was discontinued in 29% of patients and delayed in 65% of patients for an adverse reaction.
The most frequent serious adverse reactions (reported in ≥4% of patients) were pyrexia, diarrhea, anemia, increased AST, adrenal insufficiency, ascites, esophageal varices hemorrhage, hyponatremia, increased blood bilirubin, and pneumonitis.
Tables 45 and 46 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-040.
|
Adverse Reaction |
OPDIVO and Ipilimumab (n=49) |
|
|
All Grades (%) |
Grades 3-4 (%) |
|
|
Skin and Subcutaneous Tissue |
||
|
Rash |
53 |
8 |
|
Pruritus |
53 |
4 |
|
Musculoskeletal and Connective Tissue |
||
|
Musculoskeletal pain |
41 |
2 |
|
Arthralgia |
10 |
0 |
|
Gastrointestinal |
||
|
Diarrhea |
39 |
4 |
|
Abdominal pain |
22 |
6 |
|
Nausea |
20 |
0 |
|
Ascites |
14 |
6 |
|
Constipation |
14 |
0 |
|
Dry mouth |
12 |
0 |
|
Dyspepsia |
12 |
2 |
|
Vomiting |
12 |
2 |
|
Stomatitis |
10 |
0 |
|
Respiratory, Thoracic and Mediastinal |
||
|
Cough |
37 |
0 |
|
Dyspnea |
14 |
0 |
|
Pneumonitis |
10 |
2 |
|
Metabolism and Nutrition |
||
|
Decreased appetite |
35 |
2 |
|
General |
||
|
Fatigue |
27 |
2 |
|
Pyrexia |
27 |
0 |
|
Malaise |
18 |
2 |
|
Edema |
16 |
2 |
|
Influenza-like illness |
14 |
0 |
|
Chills |
10 |
0 |
|
Nervous System |
||
|
Headache |
22 |
0 |
|
Dizziness |
20 |
0 |
|
Endocrine |
||
|
Hypothyroidism |
20 |
0 |
|
Adrenal insufficiency |
18 |
4 |
|
Investigations |
||
|
Weight decreased |
20 |
0 |
|
Psychiatric |
||
|
Insomnia |
18 |
0 |
|
Blood and Lymphatic System |
||
|
Anemia |
10 |
4 |
|
Infections |
||
|
Influenza |
10 |
2 |
|
Vascular |
||
|
Hypotension |
10 |
0 |
Clinically important adverse reactions reported in <10% of patients who received OPDIVO with ipilimumab were hyperglycemia (8%), colitis (4%), and increased blood creatine phosphokinase (2%).
|
Laboratory Abnormality |
OPDIVO and Ipilimumab (n=47) |
|
|
All Grades (%) |
Grades 3-4 (%) |
|
|
Hematology |
||
|
Lymphopenia |
53 |
13 |
|
Anemia |
43 |
4.3 |
|
Neutropenia |
43 |
9 |
|
Leukopenia |
40 |
2.1 |
|
Thrombocytopenia |
34 |
4.3 |
|
Chemistry |
||
|
Increased AST |
66 |
40 |
|
Increased ALT |
66 |
21 |
|
Increased bilirubin |
55 |
11 |
|
Increased lipase |
51 |
26 |
|
Hyponatremia |
49 |
32 |
|
Hypocalcemia |
47 |
0 |
|
Increased alkaline phosphatase |
40 |
4.3 |
|
Increased amylase |
38 |
15 |
|
Hypokalemia |
26 |
2.1 |
|
Hyperkalemia |
23 |
4.3 |
|
Increased creatinine |
21 |
0 |
|
Hypomagnesemia |
11 |
0 |
In patients who received OPDIVO with ipilimumab, virologic breakthrough occurred in 4 of 28 (14%) patients and 2 of 4 (50%) patients with active HBV or HCV at baseline, respectively. HBV virologic breakthrough was defined as at least a 1 log increase in HBV DNA for those patients with detectable HBV DNA at baseline. HCV virologic breakthrough was defined as a 1 log increase in HCV RNA from baseline.
Esophageal Cancer
Adjuvant Treatment of Resected Esophageal or Gastroesophageal Junction Cancer
The safety of OPDIVO was evaluated in CHECKMATE-577, a randomized, placebo-controlled, double-blinded, multicenter trial in 792 treated patients with completely resected (negative margins) esophageal or gastroesophageal junction cancer who had residual pathologic disease following chemoradiotherapy (CRT) [see Clinical Studies (14.13)]. The trial excluded patients who did not receive concurrent CRT prior to surgery, had stage IV resectable disease, autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily prednisone or equivalent) or other immunosuppressive medications. Patients received either OPDIVO 240 mg or placebo by intravenous infusion over 30 minutes every 2 weeks for 16 weeks followed by 480 mg or placebo by intravenous infusion over 30 minutes every 4 weeks beginning at week 17. Patients were treated until disease recurrence, unacceptable toxicity, or for up to 1-year total duration. The median duration of exposure was 10.1 months (range: <0.1 to 14 months) in OPDIVO-treated patients and 9 months (range: <0.1 to 15 months) in placebo-treated patients. Among patients who received OPDIVO, 61% were exposed for >6 months and 54% were exposed for >9 months.
Serious adverse reactions occurred in 33% of patients receiving OPDIVO. A serious adverse reaction reported in ≥2% of patients who received OPDIVO was pneumonitis. A fatal adverse reaction of myocardial infarction occurred in one patient who received OPDIVO.
OPDIVO was discontinued in 12% of patients and was delayed in 28% of patients for an adverse reaction.
Tables 47 and 48 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-577.
| a Includes upper abdominal pain, lower abdominal pain, and abdominal discomfort. b Includes gastroesophageal reflux. c Includes asthenia. d Includes productive cough. e Includes dyspnea exertional. f Includes rash pustular, dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis bullous, exfoliative rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic. g Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, myalgia intercostal, neck pain, pain in extremity, spinal pain. |
||||
|
Adverse Reaction |
OPDIVO (n=532) |
Placebo (n=260) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Adverse Reaction |
96 |
34 |
93 |
32 |
|
Gastrointestinal |
||||
|
Diarrhea |
29 |
0.9 |
29 |
0.8 |
|
Nausea |
23 |
0.8 |
21 |
0 |
|
Abdominal Paina |
17 |
0.8 |
20 |
1.5 |
|
Vomiting |
15 |
0.6 |
16 |
1.2 |
|
Dysphagia |
13 |
0.8 |
17 |
3.5 |
|
Dyspepsiab |
12 |
0.2 |
16 |
0.4 |
|
Constipation |
11 |
0 |
12 |
0 |
|
General |
||||
|
Fatiguec |
34 |
1.3 |
29 |
1.5 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughd |
20 |
0.2 |
21 |
0.4 |
|
Dyspneae |
12 |
0.8 |
12 |
0.4 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashf |
21 |
0.9 |
10 |
0.4 |
|
Pruritus |
13 |
0.4 |
6 |
0 |
|
Investigations |
||||
|
Weight decreased |
13 |
0.4 |
9 |
0 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal paing |
21 |
0.6 |
20 |
0.8 |
|
Arthralgia |
10 |
0.2 |
8 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
15 |
0.9 |
10 |
0.8 |
|
Endocrine |
||||
|
Hypothyroidism |
11 |
0 |
1.5 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (range: 163 to 526 patients) and Placebo group (range: 86 to 256 patients). b Includes alanine aminotransferase increased, aspartate aminotransferase increased. |
||||
|
Laboratory Abnormality |
OPDIVO (n=532) |
Placebo (n=260) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Increased AST |
27 |
2.1 |
22 |
0.8 |
|
Increased alkaline phosphatase |
25 |
0.8 |
18 |
0.8 |
|
Increased albumin |
21 |
0.2 |
18 |
0 |
|
Increased ALT |
20 |
1.9 |
16 |
1.2 |
|
Increased amylase |
20 |
3.9 |
13 |
1.3 |
|
Hyponatremia |
19 |
1.7 |
12 |
1.2 |
|
Hyperkalemia |
17 |
0.8 |
15 |
1.6 |
|
Hypokalemia |
12 |
1 |
11 |
1.2 |
|
Transaminases increasedb |
11 |
1.5 |
6 |
1.2 |
|
Hematology |
||||
|
Lymphopenia |
44 |
17 |
35 |
12 |
|
Anemia |
27 |
0.8 |
21 |
0.4 |
|
Neutropenia |
24 |
1.5 |
23 |
0.4 |
First-line Treatment of Unresectable Advanced or Metastatic ESCC
The safety of OPDIVO in combination with chemotherapy or in combination with ipilimumab was evaluated in CHECKMATE-648, a randomized, active-controlled, multicenter, open-label trial in patients with previously untreated unresectable advanced, recurrent or metastatic ESCC [see Clinical Studies (14.13)]. Patients received one of the following treatments:
- •
- OPDIVO 240 mg on days 1 and 15, 5-FU (fluorouracil) 800 mg/m2/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m2 intravenously on day 1 (of a 4-week cycle).
- •
- OPDIVO 3 mg/kg every 2 weeks in combination with ipilimumab 1 mg/kg every 6 weeks.
- •
- 5-FU (fluorouracil) 800 mg/m2/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m2 intravenously on day 1 (of a 4-week cycle).
Among patients who received OPDIVO with chemotherapy, the median duration of exposure was 5.7 months (range: 0.1 to 30.6 months). Among patients who received OPDIVO and ipilimumab, the median duration of exposure was 2.8 months (range: 0 to 24 months).
Serious adverse reactions occurred in 62% of patients receiving OPDIVO in combination with chemotherapy and in 69% of patients receiving OPDIVO in combination with ipilimumab. The most frequent serious adverse reactions reported in ≥2% of patients who received OPDIVO with chemotherapy were pneumonia (11%), dysphagia (7%), esophageal stenosis (2.9%), acute kidney injury (2.9%), and pyrexia (2.3%). The most frequent serious adverse reactions reported in ≥2% of patients who received OPDIVO with ipilimumab were pneumonia (10%), pyrexia (4.3%), pneumonitis (4%), aspiration pneumonia (3.7%), dysphagia (3.7%), hepatic function abnormal (2.8%), decreased appetite (2.8%), adrenal insufficiency (2.5%), and dehydration (2.5%).
Fatal adverse reactions occurred in 5 (1.6%) patients who received OPDIVO in combination with chemotherapy; these included pneumonitis, pneumatosis intestinalis, pneumonia, and acute kidney injury and in 5 (1.6%) patients who received OPDIVO in combination with ipilimumab; these included pneumonitis, interstitial lung disease, pulmonary embolism, and acute respiratory distress syndrome.
OPDIVO and/or chemotherapy were discontinued in 39% of patients and were delayed in 71% of patients for an adverse reaction. OPDIVO and/or ipilimumab were discontinued in 23% of patients and were delayed in 46% of patients for an adverse reaction.
The most common adverse reactions reported in ≥20% of patients treated with OPDIVO in combination with chemotherapy were nausea, decreased appetite, fatigue, constipation, stomatitis, diarrhea, and vomiting. The most common adverse reactions reported in ≥20% of patients treated with OPDIVO in combination with ipilimumab were rash, fatigue, pyrexia, nausea, diarrhea, and constipation.
Tables 49 and 50 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-648.
| Toxicity was graded per NCI CTCAE v4. a Includes aphthous ulcer, mouth ulceration, and mucosal inflammation. b Includes abdominal discomfort, abdominal pain lower, and abdominal pain upper. c Includes asthenia and malaise. d Includes tumor associated fever. e Includes swelling, generalized edema, edema peripheral, and peripheral swelling. f Includes hyperesthesia, hypoesthesia, peripheral motor neuropathy, peripheral sensorimotor neuropathy, and peripheral sensory neuropathy. g Includes dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis bullous, drug eruption, exfoliative rash, rash erythematous, rash follicular, rash macular, rash maculo-papular, rash papular, and rash pruritic. h Includes productive cough. i Includes organizing pneumonia, pneumonia bacterial, and pneumonia pseudomonal. j Includes back pain, bone pain, musculoskeletal chest pain, myalgia, neck pain, pain in extremity, and spinal pain. |
||||||
|
Adverse Reaction |
OPDIVO with Cisplatin and 5‑FU (n=310) |
OPDIVO and Ipilimumab (n=322) |
Cisplatin and 5-FU |
|||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Gastrointestinal |
||||||
|
Nausea |
65 |
4.2 |
22 |
0.6 |
56 |
2.6 |
|
Constipation |
44 |
1.0 |
20 |
0.3 |
43 |
1 |
|
Stomatitisa |
44 |
9 |
11 |
0.6 |
35 |
3 |
|
Diarrhea |
29 |
2.9 |
22 |
1.9 |
20 |
2 |
|
Vomiting |
23 |
2.3 |
15 |
1.6 |
19 |
3 |
|
Dysphagia |
14 |
7 |
12 |
5 |
12 |
4.9 |
|
Abdominal painb |
13 |
1.9 |
10 |
0.9 |
11 |
0.7 |
|
Metabolism and Nutrition |
||||||
|
Decreased |
51 |
7 |
17 |
4 |
50 |
6 |
|
General |
||||||
|
Fatiguec |
47 |
3.5 |
28 |
2.5 |
41 |
4.9 |
|
Pyrexiad |
19 |
0.3 |
23 |
0.9 |
12 |
0.3 |
|
Edemae |
16 |
0 |
7 |
0 |
13 |
0 |
|
Nervous System |
||||||
|
Peripheral |
18 |
1.3 |
2.8 |
0 |
13 |
1 |
|
Psychiatric |
||||||
|
Insomnia |
16 |
0 |
8 |
0 |
10 |
0.3 |
|
Skin and Subcutaneous Tissue |
||||||
|
Rashg |
16 |
0.6 |
31 |
3.1 |
7 |
0 |
|
Pruritus |
11 |
0 |
17 |
0.9 |
3.6 |
0 |
|
Alopecia |
10 |
0 |
11 |
0 |
||
|
Respiratory, Thoracic and Mediastinal |
||||||
|
Coughh |
16 |
0.3 |
13 |
0.3 |
13 |
0.3 |
|
Infections and Infestations |
||||||
|
Pneumoniai |
13 |
5 |
14 |
8 |
10 |
2.6 |
|
Endocrine |
||||||
|
Hypothyroidism |
7 |
0 |
14 |
0 |
0.3 |
0 |
|
Investigations |
||||||
|
Weight decreased |
12 |
0.6 |
12 |
1.9 |
11 |
1 |
|
Musculoskeletal and Connective Tissue |
||||||
|
Musculoskeletal |
11 |
0.3 |
14 |
0.6 |
8 |
0.3 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO with cisplatin and 5-FU group (range: 60 to 305 patients), OPDIVO and ipilimumab group (range: 59 to 307 patients) or cisplatin and 5-FU group (range: 56 to 283 patients). | ||||||
|
Laboratory Abnormality |
OPDIVO with Cisplatin and 5-FU |
OPDIVO and Ipilimumab |
Cisplatin and 5-FU |
|||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||||
|
Anemia |
81 |
21 |
52 |
7 |
66 |
14 |
|
Lymphopenia |
67 |
23 |
50 |
13 |
44 |
8 |
|
Neutropenia |
61 |
18 |
13 |
1.3 |
48 |
13 |
|
Leukopenia |
53 |
11 |
39 |
5 |
||
|
Thrombocytopenia |
43 |
3.3 |
12 |
1 |
29 |
2.8 |
|
Chemistry |
||||||
|
Hyponatremia |
52 |
15 |
45 |
11 |
40 |
8 |
|
Hypocalcemia |
43 |
3 |
32 |
0 |
23 |
0.7 |
|
Increased creatinine |
41 |
2.3 |
15 |
0.7 |
31 |
0.7 |
|
Hypomagnesemia |
35 |
1.7 |
15 |
0 |
25 |
1.8 |
|
Hyperglycemia |
34 |
0 |
43 |
4.3 |
36 |
0.8 |
|
Hyperkalemia |
33 |
2.3 |
23 |
1.6 |
24 |
0.7 |
|
Hypokalemia |
29 |
9 |
19 |
5 |
17 |
6 |
|
Increased alkaline phosphatase |
26 |
1.3 |
31 |
3.3 |
15 |
0 |
|
Increased AST |
23 |
3.3 |
39 |
6 |
11 |
1.4 |
|
Increased ALT |
23 |
2.3 |
33 |
6 |
8 |
0.7 |
|
Hypoglycemia |
18 |
0.4 |
15 |
1.2 |
7 |
0 |
|
Hypercalcemia |
11 |
2.6 |
15 |
2 |
8 |
0 |
Previously-Treated Unresectable Advanced, Recurrent or Metastatic Esophageal Squamous Cell Carcinoma (ESCC)
The safety of OPDIVO was evaluated in ATTRACTION-3, a randomized, active-controlled, open-label, multicenter trial in 209 patients with unresectable advanced, recurrent or metastatic ESCC refractory or intolerant to at least one fluoropyrimidine- and platinum-based chemotherapy [see Clinical Studies (14.13)]. The trial excluded patients who were refractory or intolerant to taxane therapy, had brain metastases that were symptomatic or required treatment, had autoimmune disease, used systemic corticosteroids or immunosuppressants, had apparent tumor invasion of organs adjacent to the esophageal tumor or had stents in the esophagus or respiratory tract. Patients received OPDIVO 240 mg by intravenous infusion over 30 minutes every 2 weeks (n=209) or investigator’s choice: docetaxel 75 mg/m2 intravenously every 3 weeks (n=65) or paclitaxel 100 mg/m2 intravenously once a week for 6 weeks followed by 1 week off (n=143). Patients were treated until disease progression or unacceptable toxicity. The median duration of exposure was 2.6 months (range: 0 to 29.2 months) in OPDIVO-treated patients and 2.6 months (range: 0 to 21.4 months) in docetaxel- or paclitaxel-treated patients. Among patients who received OPDIVO, 26% were exposed for >6 months and 10% were exposed for >1 year.
Serious adverse reactions occurred in 38% of patients receiving OPDIVO. Serious adverse reactions reported in ≥2% of patients who received OPDIVO were pneumonia, esophageal fistula, interstitial lung disease and pyrexia. The following fatal adverse reactions occurred in patients who received OPDIVO: interstitial lung disease or pneumonitis (1.4%), pneumonia (1.0%), septic shock (0.5%), esophageal fistula (0.5%), gastrointestinal hemorrhage (0.5%), pulmonary embolism (0.5%), and sudden death (0.5%).
OPDIVO was discontinued in 13% of patients and was delayed in 27% of patients for an adverse reaction.
Tables 51 and 52 summarize the adverse reactions and laboratory abnormalities, respectively, in ATTRACTION-3.
| Toxicity was graded per NCI CTCAE v4. a Includes urticaria, drug eruption, eczema, eczema asteatotic, eczema nummular, palmar-plantar erythrodysesthesia syndrome, erythema, erythema multiforme, blister, skin exfoliation, Stevens-Johnson syndrome, dermatitis, dermatitis described as acneiform, bullous, or contact, and rash described as maculo-papular, generalized, or pustular. b Includes hypophagia, and food aversion. c Includes colitis. d Includes spondylolisthesis, periarthritis, musculoskeletal chest pain, neck pain, arthralgia, back pain, myalgia, pain in extremity, arthritis, bone pain, and periarthritis calcarea. e Includes influenza, influenza like illness, pharyngitis, nasopharyngitis, tracheitis, and bronchitis and upper respiratory infection with bronchitis. f Includes pneumonia aspiration, pneumonia bacterial, and lung infection. Two patients (1.0%) died of pneumonia in the OPDIVO treatment arm. Two patients (1.0%) died of pneumonia in the chemotherapy treatment arm; these deaths occurred with paclitaxel only. g Includes productive cough. h Includes tumor-associated fever. i Includes asthenia. j Includes hemoglobin decreased, and iron deficiency anemia. k Includes blood thyroid stimulating hormone increased. |
||||
|
Adverse Reaction |
OPDIVO (n=209) |
Docetaxel or Paclitaxel (n=208) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Skin and Subcutaneous Tissue |
||||
|
Rasha |
22 |
1.9 |
28 |
1 |
|
Pruritus |
12 |
0 |
7 |
0 |
|
Metabolism and Nutrition |
||||
|
Decreased appetiteb |
21 |
1.9 |
35 |
5 |
|
Gastrointestinal |
||||
|
Diarrheac |
18 |
1.9 |
17 |
1.4 |
|
Constipation |
17 |
0 |
19 |
0 |
|
Nausea |
11 |
0 |
20 |
0.5 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal |
17 |
0 |
26 |
1.4 |
|
Infections |
||||
|
Upper respiratory |
17 |
1 |
14 |
0 |
|
Pneumoniaf |
13 |
5 |
19 |
9 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughg |
16 |
0 |
14 |
0.5 |
|
General |
||||
|
Pyrexiah |
16 |
0.5 |
19 |
0.5 |
|
Fatiguei |
12 |
1.4 |
27 |
4.8 |
|
Blood and Lymphatic System |
||||
|
Anemiaj |
13 |
8 |
30 |
13 |
|
Endocrine |
||||
|
Hypothyroidismk |
11 |
0 |
1.4 |
0 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO group (209 patients) and Docetaxel or Paclitaxel group (range: 207 to 208 patients). | ||||
|
Laboratory Abnormality |
OPDIVO (n=209) |
Docetaxel or Paclitaxel (n=208) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Chemistry |
||||
|
Increased creatinine |
78 |
0.5 |
68 |
0.5 |
|
Hyperglycemia |
52 |
5 |
62 |
5 |
|
Hyponatremia |
42 |
11 |
50 |
12 |
|
Increased AST |
40 |
6 |
30 |
1 |
|
Increased alkaline phosphatase |
33 |
4.8 |
24 |
1.0 |
|
Increased ALT |
31 |
5 |
22 |
1.9 |
|
Hypercalcemia |
22 |
6 |
14 |
2.9 |
|
Hyperkalemia |
22 |
0.5 |
31 |
1 |
|
Hypoglycemia |
14 |
1.4 |
14 |
0.5 |
|
Hypokalemia |
11 |
2.9 |
13 |
3.4 |
|
Hematology |
||||
|
Lymphopenia |
46 |
19 |
72 |
43 |
|
Anemia |
42 |
9 |
71 |
17 |
|
Leukopenia |
11 |
0.5 |
79 |
45 |
Gastric Cancer, Gastroesophageal Junction Cancer, and Esophageal Adenocarcinoma
The safety of OPDIVO in combination with chemotherapy was evaluated in CHECKMATE-649, a randomized, multicenter, open-label trial in patients with previously untreated advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma [see Clinical Studies (14.14)]. The trial excluded patients who were known human epidermal growth factor receptor 2 (HER2) positive or had untreated central nervous system (CNS) metastases. Patients were randomized to receive OPDIVO in combination with chemotherapy or chemotherapy. Patients received one of the following treatments:
- •
- OPDIVO 240 mg in combination with mFOLFOX6 (fluorouracil, leucovorin and oxaliplatin) every 2 weeks or mFOLFOX6 every 2 weeks.
- •
- OPDIVO 360 mg in combination with CapeOX (capecitabine and oxaliplatin) every 3 weeks or CapeOX every 3 weeks.
Patients were treated with OPDIVO in combination with chemotherapy or chemotherapy until disease progression, unacceptable toxicity, or up to 2 years. The median duration of exposure was 6.8 months (range: 0 to 33.5 months) in OPDIVO and chemotherapy-treated patients. Among patients who received OPDIVO and chemotherapy, 54% were exposed for >6 months and 28% were exposed for >1 year.
Fatal adverse reactions occurred in 16 (2.0%) patients who were treated with OPDIVO in combination with chemotherapy; these included pneumonitis (4 patients), febrile neutropenia (2 patients), stroke (2 patients), gastrointestinal toxicity, intestinal mucositis, septic shock, pneumonia, infection, gastrointestinal bleeding, mesenteric vessel thrombosis, and disseminated intravascular coagulation. Serious adverse reactions occurred in 52% of patients treated with OPDIVO in combination with chemotherapy. OPDIVO and/or chemotherapy were discontinued in 44% of patients and at least one dose was withheld in 76% of patients due to an adverse reaction.
The most frequent serious adverse reactions reported in ≥2% of patients treated with OPDIVO in combination with chemotherapy were vomiting (3.7%), pneumonia (3.6%), anemia (3.6%), pyrexia (2.8%), diarrhea (2.7%), febrile neutropenia (2.6%), and pneumonitis (2.4%). The most common adverse reactions reported in ≥20% of patients treated with OPDIVO in combination with chemotherapy were peripheral neuropathy, nausea, fatigue, diarrhea, vomiting, decreased appetite, abdominal pain, constipation, and musculoskeletal pain.
Tables 53 and 54 summarize the adverse reactions and laboratory abnormalities, respectively, in CHECKMATE-649.
| Toxicity was graded per NCI CTCAE v4. a Includes dysesthesia, hypoesthesia, peripheral motor neuropathy, peripheral sensorimotor neuropathy, and peripheral sensory neuropathy. b Includes abdominal discomfort, abdominal pain lower, and abdominal pain upper. c Includes aphthous ulcer, mouth ulceration, and mucosal inflammation. d Includes asthenia. e Includes tumor associated fever. f Includes swelling, generalized edema, edema peripheral, and peripheral swelling. g Includes blood albumin decreased. h Includes back pain, bone pain, musculoskeletal chest pain, musculoskeletal discomfort, myalgia, neck pain, pain in extremity, and spinal pain. i Includes dermatitis, dermatitis acneiform, dermatitis allergic, dermatitis bullous, drug eruption, exfoliative rash, nodular rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic, and rash vesicular. j Includes productive cough. k Includes nasopharyngitis, pharyngitis, and rhinitis. |
||||
|
Adverse Reaction |
OPDIVO and mFOLFOX6 or CapeOX (n=782) |
mFOLFOX6 or CapeOX (n=767) |
||
|
All Grades (%) |
Grades 3-4 (%) |
All Grades (%) |
Grades 3-4 (%) |
|
|
Adverse Reaction |
99 |
69 |
98 |
59 |
|
Nervous System |
||||
|
Peripheral neuropathya |
53 |
7 |
46 |
4.8 |
|
Headache |
11 |
0.8 |
6 |
0.3 |
|
Gastrointestinal |
||||
|
Nausea |
48 |
3.2 |
44 |
3.7 |
|
Diarrhea |
39 |
5 |
34 |
3.7 |
|
Vomiting |
31 |
4.2 |
29 |
4.2 |
|
Abdominal painb |
27 |
2.8 |
24 |
2.6 |
|
Constipation |
25 |
0.6 |
21 |
0.4 |
|
Stomatitisc |
17 |
1.8 |
13 |
0.8 |
|
General |
||||
|
Fatigued |
44 |
7 |
40 |
5 |
|
Pyrexiae |
19 |
1 |
11 |
0.4 |
|
Edemaf |
12 |
0.5 |
8 |
0.1 |
|
Metabolism and Nutrition |
||||
|
Decreased appetite |
29 |
3.6 |
26 |
2.5 |
|
Hypoalbuminemiag |
14 |
0.3 |
9 |
0.3 |
|
Investigations |
||||
|
Weight decreased |
17 |
1.3 |
15 |
0.7 |
|
Increased lipase |
14 |
7 |
8 |
3.7 |
|
Increased amylase |
12 |
3.1 |
5 |
0.4 |
|
Musculoskeletal and Connective Tissue |
||||
|
Musculoskeletal painh |
20 |
1.3 |
14 |
2 |
|
Skin and Subcutaneous Tissue |
||||
|
Rashi |
18 |
1.7 |
4.4 |
0.1 |
|
Palmar-plantar erythrodysesthesia syndrome |
13 |
1.5 |
12 |
0.8 |
|
Respiratory, Thoracic and Mediastinal |
||||
|
Coughj |
13 |
0.1 |
9 |
0 |
|
Infections and Infestations |
||||
|
Upper respiratory tract infectionk |
10 |
0.1 |
7 |
0.1 |
| a Each test incidence is based on the number of patients who had both baseline and at least one on-study laboratory measurement available: OPDIVO and mFOLFOX6 or CapeOX group (407 to 767 patients) or mFOLFOX6 or CapeOX group (range: 405 to 735 patients). | ||||
|
Laboratory Abnormality |
OPDIVO and mFOLFOX6 or CapeOX |
mFOLFOX6 or CapeOX |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Hematology |
||||
|
Neutropenia |
73 |
29 |
62 |
23 |
|
Leukopenia |
69 |
12 |
59 |
9 |
|
Thrombocytopenia |
68 |
7 |
63 |
4.4 |
|
Anemia |
59 |
14 |
60 |
10 |
|
Lymphopenia |
59 |
12 |
49 |
9 |
|
Chemistry |
||||
|
Increased AST |
52 |
4.6 |
47 |
1.9 |
|
Hypocalcemia |
42 |
1.6 |
37 |
1 |
|
Hyperglycemia |
41 |
3.9 |
38 |
2.7 |
|
Increased ALT |
37 |
3.4 |
30 |
1.9 |
|
Hyponatremia |
34 |
6 |
24 |
5 |
|
Hypokalemia |
27 |
7 |
24 |
4.8 |
|
Hyperbilirubinemia |
24 |
2.8 |
21 |
2 |
|
Increased creatinine |
15 |
1 |
9 |
0.5 |
|
Hyperkalemia |
14 |
1.4 |
11 |
0.7 |
|
Hypoglycemia |
12 |
0.7 |
9 |
0.2 |
|
Hypernatremia |
11 |
0.5 |
7.1 |
0 |
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of OPDIVO. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Eye: Vogt-Koyanagi-Harada (VKH) syndrome
Complications of OPDIVO Treatment After Allogeneic HSCT: Treatment refractory, severe acute and chronic GVHD
Blood and lymphatic system disorders: Hemophagocytic lymphohistiocytosis (HLH) (including fatal cases), autoimmune hemolytic anemia (including fatal cases)
Metabolism and nutrition disorders: tumor lysis syndrome
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on data from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], OPDIVO can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, administration of nivolumab to cynomolgus monkeys from the onset of organogenesis through delivery resulted in increased abortion and premature infant death (see Data). Human IgG4 is known to cross the placental barrier and nivolumab is an immunoglobulin G4 (IgG4); therefore, nivolumab has the potential to be transmitted from the mother to the developing fetus. The effects of OPDIVO are likely to be greater during the second and third trimesters of pregnancy. There are no available data on OPDIVO use in pregnant women to evaluate a drug-associated risk. Advise pregnant women of the potential risk to a fetus.
The background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
A central function of the PD-1/PD-L1 pathway is to preserve pregnancy by maintaining maternal immune tolerance to the fetus. Blockade of PD-L1 signaling has been shown in murine models of pregnancy to disrupt tolerance to the fetus and to increase fetal loss. The effects of nivolumab on prenatal and postnatal development were evaluated in monkeys that received nivolumab twice weekly from the onset of organogenesis through delivery, at exposure levels of between 9 and 42 times higher than those observed at the clinical dose of 3 mg/kg (based on AUC). Nivolumab administration resulted in a non-dose-related increase in spontaneous abortion and increased neonatal death. Based on its mechanism of action, fetal exposure to nivolumab may increase the risk of developing immune-mediated disorders or altering the normal immune response and immune-mediated disorders have been reported in PD-1 knockout mice. In surviving infants (18 of 32 compared to 11 of 16 vehicle-exposed infants) of cynomolgus monkeys treated with nivolumab, there were no apparent malformations and no effects on neurobehavioral, immunological, or clinical pathology parameters throughout the 6-month postnatal period.
8.2 Lactation
Risk Summary
There are no data on the presence of nivolumab in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment and for 5 months after the last dose of OPDIVO.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating OPDIVO [see Use in Specific Populations (8.1)].
Contraception
OPDIVO can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and for 5 months following the last dose.
8.4 Pediatric Use
The safety and effectiveness of OPDIVO have been established in pediatric patients aged 12 years and older for the following indications: as a single agent and in combination with ipilimumab for the treatment of unresectable or metastatic melanoma, as a single agent for the adjuvant treatment of completely resected Stage IIB, Stage IIC, Stage III, or Stage IV melanoma in combination with ipilimumab for the treatment of MSI-H or dMMR unresectable and metastatic CRC, and as a single agent for the treatment of MSI-H or dMMR mCRC that has progressed following treatment with a fluoropyrimidine, oxaliplatin, and irinotecan. Use of OPDIVO for these indications is supported by evidence from adequate and well-controlled studies in adults with melanoma or MSI-H or dMMR mCRC and additional pharmacokinetic data in pediatric patients. Nivolumab exposure in pediatric patients 12 years and older is comparable to that of adults and the courses of melanoma and MSI-H or dMMR mCRC are similar in pediatric patients aged 12 years and older to that of adults to allow extrapolation of safety and efficacy [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.1, 14.11)].
The safety and effectiveness of OPDIVO have not been established for pediatric patients younger than 12 years old with melanoma or MSI-H or dMMR mCRC.
The safety and effectiveness of OPDIVO have not been established in pediatric patients with non-small cell lung cancer, malignant pleural mesothelioma, advanced renal cell carcinoma, classical Hodgkin lymphoma, squamous cell carcinoma of the head and neck, urothelial carcinoma, hepatocellular carcinoma, esophageal cancer, gastric cancer, gastroesophageal cancer and esophageal adenocarcinoma.
8.5 Geriatric Use
Single Agent
Of 3569 patients with melanoma, NSCLC, renal cell carcinoma, urothelial carcinoma, ESCC, and esophageal or gastroesophageal junction cancer who were randomized to single agent OPDIVO in clinical studies, 41% were 65 years and over and 10% were 75 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients [see Clinical Studies (14.1, 14.2, 14.5, 14.7, 14.10, 14.13, 14.14)].
In patients with cHL, recurrent head and neck SCC, or dMMR or MSI-H metastatic CRC (mCRC) who were treated with single agent OPDIVO in clinical studies did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients [see Clinical Studies (14.8, 14.9, 14.11)].
OPDIVO in Combination with Ipilimumab
Of the 314 patients with melanoma who were randomized to OPDIVO in combination with ipilimumab, 41% were 65 years or older and 11% were 75 years or older. No overall differences in safety or effectiveness were reported between elderly patients and younger patients [see Clinical Studies (14.1)].
Of the 576 patients with NSCLC who were randomized to OPDIVO in combination with ipilimumab, 48% were 65 years or older and 10% were 75 years or older. No overall difference in safety was reported between older patients and younger patients; however, there was a higher discontinuation rate due to adverse reactions in patients aged 75 years or older (29%) relative to all patients who received OPDIVO with ipilimumab (18%). Of the 396 patients in the primary efficacy population (PD-L1 ≥1%) randomized to OPDIVO in combination with ipilimumab, the hazard ratio for overall survival was 0.70 (95% CI: 0.55, 0.89) in the 199 patients younger than 65 years compared to 0.91 (95% CI: 0.72, 1.15) in the 197 patients 65 years or older [see Clinical Studies (14.3)].
Of the 303 patients with malignant pleural mesothelioma who were randomized to OPDIVO in combination with ipilimumab, 77% were 65 years old or older and 26% were 75 years or older. No overall difference in safety was reported between older patients and younger patients; however, there were higher rates of serious adverse reactions and discontinuation due to adverse reactions in patients aged 75 years or older (68% and 35%, respectively) relative to all patients who received OPDIVO with ipilimumab (54% and 28%, respectively). For patients aged 75 years or older who received chemotherapy, the rate of serious adverse reactions was 34% and the discontinuation rate due to adverse reactions was 26% relative to 28% and 19% respectively for all patients. The hazard ratio for overall survival was 0.76 (95% CI: 0.52, 1.11) in the 71 patients younger than 65 years compared to 0.74 (95% CI: 0.59, 0.93) in the 232 patients 65 years or older randomized to OPDIVO in combination with ipilimumab [see Clinical Studies (14.6)].
Of the 550 patients with renal cell carcinoma who were randomized to OPDIVO in combination with ipilimumab, 38% were 65 years or older and 8% were 75 years or older. No overall difference in safety was reported between elderly patients and younger patients. In elderly patients with intermediate or poor risk, no overall difference in effectiveness was reported [see Clinical Studies (14.7)].
Of the 354 patients with dMMR or MSI-H metastatic CRC (mCRC) who were randomized to OPDIVO in combination with ipilimumab, 44% were 65 years or older and 14% were 75 years or older. Of the 353 patients randomized to OPDIVO, as a single agent, 45% were 65 years or older and 13% were 75 years or older. There was a higher incidence of any Grade 3 or 4 adverse reactions (55%) in patients aged 65 years or older receiving OPDIVO in combination with ipilimumab compared to those younger than 65 receiving the combination (42%). There was also a higher incidence of any Grade 3 or 4 adverse reactions (55%) in patients aged 65 years or older receiving OPDIVO in combination with ipilimumab relative to patients aged 65 years or older receiving single-agent OPDIVO (41%). There was a similar incidence of any Grade 3 or 4 adverse reactions in patients receiving single-agent OPDIVO aged 65 years or older (41%) relative to patients younger than 65 years (45%). Patients 65 years or older who received OPDIVO with ipilimumab discontinued treatment due to adverse reaction at a higher rate (23%) relative to patients 65 years or older receiving nivolumab (15%). No overall differences in effectiveness were reported between elderly patients and younger patients receiving either OPDIVO in combination with ipilimumab or single-agent OPDIVO [see Clinical Studies (14.11)].
Of the 335 patients with unresectable hepatocellular carcinoma who were randomized to OPDIVO in combination with ipilimumab, 52% were 65 years or older and 14% were 75 years or older. No overall difference in safety was reported between elderly patients and younger patients, however, there were higher rates of serious adverse reactions and discontinuation due to adverse reactions in patients aged 75 years or older (67% and 35%, respectively) relative to all patients who received OPDIVO with ipilimumab (53% and 27%, respectively).
Of the 49 patients with hepatocellular carcinoma who were treated with OPDIVO in combination with ipilimumab, 29% were between 65 years and 74 years of age and 8% were 75 years or older. Clinical studies of OPDIVO in combination with ipilimumab did not include sufficient numbers of patients with hepatocellular carcinoma aged 65 and over to determine whether they respond differently from younger patients [see Clinical Studies (14.12)].
Of the 325 patients with ESCC who were randomized to OPDIVO in combination with ipilimumab, 43% were 65 years old or older and 7% were 75 years or older. No overall difference in safety was reported between older patients and younger patients; however, there was a higher discontinuation rate due to adverse reactions in patients aged 75 years or older (38%) relative to all patients who received OPDIVO with ipilimumab (23%). For patients aged 75 years or older who received chemotherapy, the discontinuation rate due to adverse reactions was 33% relative to 23% for all patients [see Clinical Studies (14.13)].
OPDIVO in Combination with Platinum-Containing Chemotherapy
Of the 179 patients with NSCLC who were randomized to OPDIVO in combination with platinum-doublet chemotherapy, 48% were 65 years old or older and 6% were 75 years old or older. No overall differences in safety or effectiveness were reported between patients older and younger than 65 years [see Clinical Studies (14.3)].
Of the 229 patients with NSCLC who were randomized to OPDIVO 360 mg in combination with platinum-doublet chemotherapy every 3 weeks for up to 4 cycles, followed by OPDIVO 480 mg every 4 weeks, 56% were 65 years old or older and 7% were 75 years old or older. No overall differences in safety or effectiveness were reported between patients older and younger than 65 years.
Of the 1,110 patients with ESCC, GC, GEJC, or EAC who were randomized to OPDIVO in combination with fluoropyrimidine- and platinum-containing chemotherapy, 42% were 65 years or older and 10% were 75 years or older. No overall difference in safety was reported between elderly patients and younger patients [see Clinical Studies (14.13, 14.14)].
Of the 304 patients with UC who were treated with OPDIVO in combination with gemcitabine and platinum-doublet chemotherapy, 40% were 65 years or older and 11% were 75 years or older. No overall differences in safety or effectiveness were observed between patients 65 years of age and over and younger patients. Clinical studies of OPDIVO with platinum-doublet chemotherapy did not include sufficient numbers of patients aged 75 years and over to determine whether safety and effectiveness differs compared to younger patients. [see Clinical Studies (14.10)].
OPDIVO in Combination with Ipilimumab and Platinum-Doublet Chemotherapy
Of the 361 patients with NSCLC who were randomized to OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy, 51% were 65 years or older and 10% were 75 years or older. No overall difference in safety was reported between older patients and younger patients; however, there was a higher discontinuation rate due to adverse reactions in patients aged 75 years or older (43%) relative to all patients who received OPDIVO with ipilimumab and chemotherapy (24%). For patients aged 75 years or older who received chemotherapy only, the discontinuation rate due to adverse reactions was 16% relative to all patients who had a discontinuation rate of 13%. Based on an updated analysis for overall survival, of the 361 patients randomized to OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy, the hazard ratio for overall survival was 0.61 (95% CI: 0.47, 0.80) in the 176 patients younger than 65 years compared to 0.73 (95% CI: 0.56, 0.95) in the 185 patients 65 years or older [see Clinical Studies (14.5)].
OPDIVO in Combination with Cabozantinib
Of the 320 patients with renal cell carcinoma who were treated with OPDIVO in combination with cabozantinib, 41% were 65 years or older and 9% were 75 years or older. No overall difference in safety was reported between elderly patients and younger patients [see Clinical Studies (14.7)].
11. Opdivo Description
Nivolumab is a programmed death receptor-1 (PD-1) blocking antibody. Nivolumab is an IgG4 kappa immunoglobulin that has a calculated molecular mass of 146 kDa. It is expressed in a recombinant Chinese Hamster Ovary (CHO) cell line.
OPDIVO is a sterile, preservative-free, non-pyrogenic, clear to opalescent, colorless to pale-yellow liquid that may contain light (few) particles.
OPDIVO (nivolumab) injection for intravenous use is supplied in single-dose vials. Each mL of OPDIVO solution contains nivolumab 10 mg, mannitol (30 mg), pentetic acid (0.008 mg), polysorbate 80 (0.2 mg), sodium chloride (2.92 mg), sodium citrate dihydrate (5.88 mg), and Water for Injection, USP. May contain hydrochloric acid and/or sodium hydroxide to adjust pH to 6.
12. Opdivo - Clinical Pharmacology
12.1 Mechanism of Action
Binding of the PD-1 ligands, PD-L1 and PD-L2, to the PD-1 receptor found on T cells, inhibits T-cell proliferation and cytokine production. Upregulation of PD-1 ligands occurs in some tumors and signaling through this pathway can contribute to inhibition of active T-cell immune surveillance of tumors. Nivolumab is a human immunoglobulin G4 (IgG4) monoclonal antibody that binds to the PD-1 receptor and blocks its interaction with PD-L1 and PD-L2, releasing PD-1 pathway-mediated inhibition of the immune response, including the anti-tumor immune response. In syngeneic mouse tumor models, blocking PD-1 activity resulted in decreased tumor growth.
Combined nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) mediated inhibition results in enhanced T-cell function that is greater than the effects of either antibody alone, and results in improved anti-tumor responses in metastatic melanoma and advanced RCC. In murine syngeneic tumor models, dual blockade of PD-1 and CTLA-4 resulted in increased anti-tumor activity.
12.2 Pharmacodynamics
There are no clinically significant exposure-response relationships for efficacy or safety for nivolumab monotherapy across the approved dosing regimens, regardless of cancer type.
12.3 Pharmacokinetics
Nivolumab pharmacokinetics (PK) was assessed using a population PK approach for both single agent OPDIVO and OPDIVO with ipilimumab. The PK of nivolumab was studied in patients over a dose range of 0.1 mg/kg to 20 mg/kg administered as a single dose or as multiple doses of OPDIVO as a 60-minute intravenous infusion every 2 or 3 weeks. The exposure to nivolumab increases dose proportionally over the dose range of 0.1 to 10 mg/kg administered every 2 weeks. The predicted exposure of nivolumab after a 30-minute infusion is comparable to that observed with a 60-minute infusion. Steady-state concentrations of nivolumab were reached by 12 weeks when administered at 3 mg/kg every 2 weeks, and systemic accumulation was 3.7-fold.
Distribution
The geometric mean volume of distribution at steady state (Vss) and coefficient of variation (CV%) is 6.8 L (27.3%).
Elimination
Nivolumab clearance (CL) decreases over time, with a mean maximal reduction from baseline values (CV%) of 24.5% (47.6%) resulting in a geometric mean steady-state clearance (CLss) (CV%) of 8.2 mL/h (53.9%) in patients with metastatic tumors; the decrease in CLss is not considered clinically relevant. Nivolumab clearance does not decrease over time in patients with completely resected melanoma, as the geometric mean population clearance is 24% lower in this patient population compared with patients with metastatic melanoma at steady state.
The geometric mean elimination half-life (t1/2) is 25 days (77.5%).
Specific Populations
The following factors had no clinically important effect on the clearance of nivolumab: age (29 to 87 years), weight (35 to 160 kg), sex, race, baseline LDH, PD-L1 expression, solid tumor type, tumor size, renal impairment (eGFR ≥ 15 mL/min/1.73 m2), and mild (total bilirubin [TB] less than or equal to the ULN and AST greater than ULN or TB greater than 1 to 1.5 times ULN and any AST) or moderate hepatic impairment (TB greater than 1.5 to 3 times ULN and any AST). Nivolumab has not been studied in patients with severe hepatic impairment (TB greater than 3 times ULN and any AST).
Pediatric Patients
The exposures of nivolumab, as monotherapy or in combination with ipilimumab, in pediatric patients 12 years of age or older are within range of those in adults at the recommended dosage [see Dosage and Administration (2.2)].
Drug Interaction Studies
When OPDIVO 3 mg/kg every 3 weeks was administered in combination with ipilimumab 1 mg/kg every 3 weeks, the CL of nivolumab and ipilimumab were unchanged compared to nivolumab or ipilimumab administered alone.
When OPDIVO 1 mg/kg every 3 weeks was administered in combination with ipilimumab 3 mg/kg every 3 weeks, the CL of nivolumab was increased by 29% compared to OPDIVO administered alone and the CL of ipilimumab was unchanged compared to ipilimumab administered alone.
When OPDIVO 3 mg/kg every 2 weeks was administered in combination with ipilimumab 1 mg/kg every 6 weeks, the CL of nivolumab was unchanged compared to OPDIVO administered alone and the CL of ipilimumab was increased by 30% compared to ipilimumab administered alone.
When OPDIVO 360 mg every 3 weeks was administered in combination with ipilimumab 1 mg/kg every 6 weeks and chemotherapy, the CL of nivolumab was unchanged compared to OPDIVO administered alone and the CL of ipilimumab increased by 22% compared to ipilimumab administered alone.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of OPDIVO or of other nivolumab products.
Anti-drug antibody and neutralizing antibody responses were monitored throughout the treatment period where the benefit to risk ratio was assessed. Incidence of anti-drug antibodies and neutralizing antibodies are presented in Table 55.
| a Details of each treatment regimen are described in Section 14 [see Clinical Studies (14)]. b NAb incidence is reported among the subset of patients positive for ADA. c Includes unresectable or metastatic melanoma, metastatic NSCLC, advanced RCC, cHL, recurrent or metastatic SCCHN, and UC indications. ADA = treatment-emergent anti-nivolumab antibodies, NAb = neutralizing antibodies, HCC = hepatocellular carcinoma, RCC = renal cell carcinoma, CRC = colorectal cancer, NSCLC = non-small cell lung cancer. |
|||
|
Treatment Regimena |
Indication(s) |
ADA |
NAbb |
|
OPDIVO as a single agent |
Multiplec |
11% |
7% |
|
OPDIVO with ipilimumab for 4 doses followed by OPDIVO as a single agent |
Melanoma |
38% |
12% |
|
HCC |
56% |
41% |
|
|
RCC and CRC |
26% |
3% |
|
|
OPDIVO with ipilimumab |
Malignant Pleural Mesothelioma |
26% |
2.9% |
|
NSCLC |
37% |
3.9% |
|
|
OPDIVO with ipilimumab and 2 cycles of platinum‑doublet chemotherapy |
NSCLC |
34% |
8% |
Effects of Anti-Drug Antibodies
Presence of treatment-emergent anti-nivolumab antibodies increased nivolumab clearance by up to 20% after administration of nivolumab as monotherapy or in combination with ipilimumab. These anti-drug antibody-associated pharmacokinetic changes were not considered to be clinically significant. There was no identified clinically significant effect of anti-drug antibodies on incidence of infusion-related reactions. The effects of anti-drug antibodies on effectiveness have not been fully characterized.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been performed to assess the potential of nivolumab for carcinogenicity or genotoxicity. Fertility studies have not been performed with nivolumab. In 1-month and 3-month repeat-dose toxicology studies in monkeys, there were no notable effects in the male and female reproductive organs; however, most animals in these studies were not sexually mature.
13.2 Animal Toxicology and/or Pharmacology
In animal models, inhibition of PD-1 signaling increased the severity of some infections and enhanced inflammatory responses. M. tuberculosis–infected PD-1 knockout mice exhibit markedly decreased survival compared with wild-type controls, which correlated with increased bacterial proliferation and inflammatory responses in these animals. PD-1 knockout mice have also shown decreased survival following infection with lymphocytic choriomeningitis virus.
14. Clinical Studies
14.1 Unresectable or Metastatic Melanoma
Previously Treated Metastatic Melanoma
CHECKMATE-037 (NCT01721746) was a multicenter, open-label trial that randomized (2:1) patients with unresectable or metastatic melanoma to receive OPDIVO 3 mg/kg intravenously every 2 weeks or investigator’s choice of chemotherapy, either single-agent dacarbazine 1000 mg/m2 every 3 weeks or the combination of carboplatin AUC 6 intravenously every 3 weeks and paclitaxel 175 mg/m2 intravenously every 3 weeks. Patients were required to have progression of disease on or following ipilimumab treatment and, if BRAF V600 mutation positive, a BRAF inhibitor. The trial excluded patients with autoimmune disease, medical conditions requiring systemic immunosuppression, ocular melanoma, active brain metastasis, or a history of Grade 4 ipilimumab-related adverse reactions (except for endocrinopathies) or Grade 3 ipilimumab-related adverse reactions that had not resolved or were inadequately controlled within 12 weeks of the initiating event. Tumor assessments were conducted 9 weeks after randomization then every 6 weeks for the first year, and every 12 weeks thereafter.
Efficacy was evaluated in a single-arm, non-comparative, planned interim analysis of the first 120 patients who received OPDIVO in CHECKMATE-037 and in whom the minimum duration of follow-up was 6 months. The major efficacy outcome measures in this population were confirmed overall response rate (ORR) as measured by blinded independent central review using Response Evaluation Criteria in Solid Tumors (RECIST 1.1) and duration of response.
Among the 120 patients treated with OPDIVO, the median age was 58 years (range: 25 to 88), 65% of patients were male, 98% were White, and the ECOG performance score was 0 (58%) or 1 (42%). Disease characteristics were M1c disease (76%), BRAF V600 mutation positive (22%), elevated LDH (56%), history of brain metastases (18%), and two or more prior systemic therapies for metastatic disease (68%).
The ORR was 32% (95% confidence interval [CI]: 23, 41), consisting of 4 complete responses and 34 partial responses in OPDIVO-treated patients. Of 38 patients with responses, 87% had ongoing responses with durations ranging from 2.6+ to 10+ months, which included 13 patients with ongoing responses of 6 months or longer.
There were responses in patients with and without BRAF V600 mutation-positive melanoma. A total of 405 patients were randomized and the median duration of OS was 15.7 months (95% CI: 12.9, 19.9) in OPDIVO-treated patients compared to 14.4 months (95% CI: 11.7, 18.2) (HR 0.95; 95.54% CI: 0.73, 1.24) in patients assigned to investigator’s choice of treatment. Figure 1 summarizes the OS results.
Figure 1: Overall Survival - CHECKMATE-037*
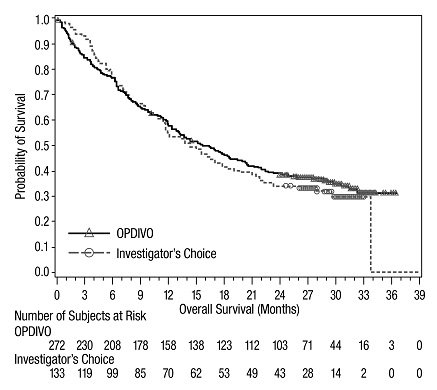
* The primary OS analysis was not adjusted to account for subsequent therapies, with 54 (40.6%) patients in the chemotherapy arm subsequently receiving an anti-PD1 treatment. OS may be confounded by dropout, imbalance of subsequent therapies, and differences in baseline factors.
Previously Untreated Metastatic Melanoma
CHECKMATE-066
CHECKMATE-066 (NCT01721772) was a multicenter, double-blind, randomized (1:1) trial in 418 patients with BRAF V600 wild-type unresectable or metastatic melanoma. Patients were randomized to receive either OPDIVO 3 mg/kg by intravenous infusion every 2 weeks or dacarbazine 1000 mg/m2 intravenously every 3 weeks until disease progression or unacceptable toxicity. Randomization was stratified by PD-L1 status (≥5% of tumor cell membrane staining by immunohistochemistry vs. <5% or indeterminate result) and M stage (M0/M1a/M1b versus M1c). Key eligibility criteria included histologically confirmed, unresectable or metastatic, cutaneous, mucosal, or acral melanoma; no prior therapy for metastatic disease; completion of prior adjuvant or neoadjuvant therapy at least 6 weeks prior to randomization; ECOG performance status 0 or 1; absence of autoimmune disease; and absence of active brain or leptomeningeal metastases. The trial excluded patients with ocular melanoma. Tumor assessments were conducted 9 weeks after randomization then every 6 weeks for the first year and then every 12 weeks thereafter. The major efficacy outcome measure was overall survival (OS). Additional outcome measures included investigator-assessed progression-free survival (PFS) and ORR per RECIST v1.1.
The trial population characteristics were: median age was 65 years (range: 18 to 87), 59% were male, and 99.5% were White. Disease characteristics were M1c stage disease (61%), cutaneous melanoma (74%), mucosal melanoma (11%), elevated LDH level (37%), PD-L1 ≥5% tumor cell membrane expression (35%), and history of brain metastasis (4%). More patients in the OPDIVO arm had an ECOG performance status of 0 (71% vs. 58%).
CHECKMATE-066 demonstrated a statistically significant improvement in OS for the OPDIVO arm compared with the dacarbazine arm in an interim analysis based on 47% of the total planned events for OS. At the time of analysis, 88% (63/72) of OPDIVO-treated patients had ongoing responses, which included 43 patients with ongoing response of 6 months or longer. Efficacy results are shown in Table 56 and Figure 2.
| a Not Reached. b Based on a stratified proportional hazards model. c Based on stratified log-rank test. d p-value is compared with the allocated alpha of 0.0021 for this interim analysis. |
||
|
OPDIVO
|
Dacarbazine
|
|
|
Overall Survival |
||
|
Deaths (%) |
50 (24) |
96 (46) |
|
Median (months) (95% CI) |
NRa |
10.8 (9.3, 12.1) |
|
Hazard ratio (95% CI)b |
0.42 (0.30, 0.60) |
|
|
p-valuec,d |
<0.0001 |
|
|
Progression-free Survival |
||
|
Disease progression or death (%) |
108 (51) |
163 (78) |
|
Median (months) (95% CI) |
5.1 (3.5, 10.8) |
2.2 (2.1, 2.4) |
|
Hazard ratio (95% CI)b |
0.43 (0.34, 0.56) |
|
|
p-valuec,d |
<0.0001 |
|
|
Overall Response Rate |
34% |
9% |
|
(95% CI) |
(28, 41) |
(5, 13) |
|
Complete response rate |
4% |
1% |
|
Partial response rate |
30% |
8% |
Figure 2: Overall Survival - CHECKMATE-066
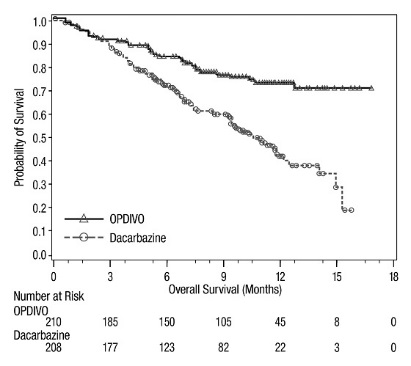
CHECKMATE-067
CHECKMATE-067 (NCT01844505) was a multicenter, randomized (1:1:1), double-blind trial in 945 patients with previously untreated, unresectable or metastatic melanoma to one of the following arms: OPDIVO and ipilimumab, OPDIVO, or ipilimumab. Patients were required to have completed adjuvant or neoadjuvant treatment at least 6 weeks prior to randomization and have no prior treatment with anti-CTLA-4 antibody and no evidence of active brain metastasis, ocular melanoma, autoimmune disease, or medical conditions requiring systemic immunosuppression.
Patients were randomized to receive:
- •
- OPDIVO 1 mg/kg with ipilimumab 3 mg/kg intravenously every 3 weeks for 4 doses, followed by OPDIVO as a single agent at a dose of 3 mg/kg by intravenous infusion every 2 weeks (OPDIVO and ipilimumab arm),
- •
- OPDIVO 3 mg/kg by intravenous infusion every 2 weeks (OPDIVO arm), or
- •
- Ipilimumab 3 mg/kg intravenously every 3 weeks for 4 doses, followed by placebo every 2 weeks (ipilimumab arm).
Randomization was stratified by PD-L1 expression (≥5% vs. <5% tumor cell membrane expression) as determined by a clinical trial assay, BRAF V600 mutation status, and M stage per the AJCC staging system (M0, M1a, M1b vs. M1c). Tumor assessments were conducted 12 weeks after randomization then every 6 weeks for the first year, and every 12 weeks thereafter. The major efficacy outcome measures were investigator-assessed PFS per RECIST v1.1 and OS. Additional efficacy outcome measures were confirmed ORR and duration of response.
The trial population characteristics were: median age 61 years (range: 18 to 90); 65% male; 97% White; ECOG performance score 0 (73%) or 1 (27%). Disease characteristics were: AJCC Stage IV disease (93%); M1c disease (58%); elevated LDH (36%); history of brain metastases (4%); BRAF V600 mutation-positive melanoma (32%); PD-L1 ≥5% tumor cell membrane expression as determined by the clinical trials assay (46%); and prior adjuvant therapy (22%).
CHECKMATE-067 demonstrated statistically significant improvements in OS and PFS for patients randomized to either OPDIVO-containing arm as compared with the ipilimumab arm. The trial was not designed to assess whether adding ipilimumab to OPDIVO improves PFS or OS compared to OPDIVO as a single agent. Efficacy results are shown in Table 57 and Figure 3.
| a OS results are based on final OS analysis with 28 months of minimum follow-up; PFS (co-primary endpoint) and ORR (secondary endpoint) results were based on primary analysis with 9 months of minimum follow-up. b Based on a stratified proportional hazards model. c Based on stratified log-rank test. d If the maximum of the two OS p-values is less than 0.04 (a significance level assigned by the Hochberg procedure), then both p-values are considered significant. e p-value is compared with .005 of the allocated alpha for final PFS treatment comparisons. f Based on the stratified Cochran-Mantel-Haenszel test. + Censored observation |
|||
|
OPDIVO and Ipilimumab
|
OPDIVO
|
Ipilimumab
|
|
|
Overall Survivala |
|||
|
Deaths (%) |
128 (41) |
142 (45) |
197 (63) |
|
Hazard ratiob (vs. ipilimumab) |
0.55 |
0.63 | |
|
p-valuec, d |
<0.0001 |
<0.0001 | |
|
Progression-free Survivala |
|||
|
Disease progression or death |
151 (48%) |
174 (55%) |
234 (74%) |
|
Median (months) |
11.5 |
6.9 |
2.9 |
|
Hazard ratiob (vs. ipilimumab) |
0.42 |
0.57 | |
|
p-valuec, e |
<0.0001 |
<0.0001 | |
|
Confirmed Overall Response Ratea |
50% |
40% |
14% |
|
(95% CI) |
(44, 55) |
(34, 46) |
(10, 18) |
|
p-valuef |
<0.0001 |
<0.0001 | |
|
Complete response |
8.9% |
8.5% |
1.9% |
|
Partial response |
41% |
31% |
12% |
|
Duration of Response |
|||
|
Proportion ≥6 months in duration |
76% |
74% |
63% |
|
Range (months) |
1.2+ to 15.8+ |
1.3+ to 14.6+ |
1.0+ to 13.8+ |
Figure 3: Overall Survival - CHECKMATE-067
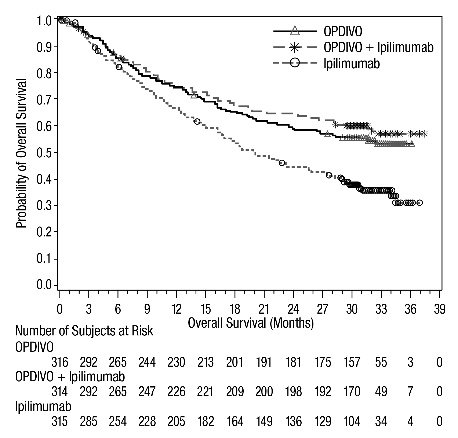
Based on a minimum follow-up of 48 months, the median OS was not reached (95% CI: 38.2, NR) in the OPDIVO and ipilimumab arm. The median OS was 36.9 months (95% CI: 28.3, NR) in the OPDIVO arm and 19.9 months (95% CI: 16.9, 24.6) in the ipilimumab arm.
Based on a minimum follow-up of 28 months, the median PFS was 11.7 months (95% CI: 8.9, 21.9) in the OPDIVO and ipilimumab arm, 6.9 months (95% CI: 4.3, 9.5) in the OPDIVO arm, and 2.9 months (95% CI: 2.8, 3.2) in the ipilimumab arm. Based on a minimum follow-up of 28 months, the proportion of responses lasting ≥24 months was 55% in the OPDIVO and ipilimumab arm, 56% in the OPDIVO arm, and 39% in the ipilimumab arm.
14.2 Adjuvant Treatment of Melanoma
CHECKMATE-76K
CHECKMATE-76K (NCT04099251) was a randomized, double-blind trial in 790 patients with completely resected Stage IIB/C melanoma. Patients were randomized (2:1) to receive OPDIVO 480 mg or placebo by intravenous infusion every 4 weeks for up to 1 year or until disease recurrence or unacceptable toxicity. Enrollment required complete resection of the primary melanoma with negative margins and a negative sentinel lymph node within 12 weeks prior to randomization, and ECOG performance status of 0 or 1. The trial excluded patients with ocular/uveal or mucosal melanoma, autoimmune disease, any condition requiring systemic treatment with either corticosteroids (≥10 mg daily prednisone or equivalent) or other immunosuppressive medications, as well as patients with prior therapy for melanoma except surgery. Randomization was stratified by AJCC 8th staging system edition (T3b vs. T4a vs. T4b). The major efficacy outcome measure was recurrence-free survival (RFS) defined as the time between the date of randomization and the date of first recurrence (local, regional, or distant metastasis), new primary melanoma, or death, from any cause, whichever occurred first and as assessed by the investigator. Tumor assessments were conducted every 26 weeks during years 1-3 and every 52 weeks thereafter until year 5.
The trial population characteristics were: median age 62 years (range: 19 to 92), 61% were male, 98% were White, 0.4% Black or African American, 0.1% Asian, and 1.1% race unknown, 2.2% Hispanic or Latino, 58% Not Hispanic or Latino, 40% ethnicity unknown, and 94% had an ECOG performance status of 0. Sixty one percent had stage IIB and 39% had stage IIC melanoma.
CHECKMATE-76K demonstrated a statistically significant improvement in RFS for patients randomized to the OPDIVO arm compared with the placebo arm. Efficacy results are shown in Table 58 and Figure 4.
| a Not reached. b Based on Kaplan-Meier estimates. c Hazard Ratio is OPDIVO over placebo based on a stratified Cox proportional hazard model. d Based on a 2-sided stratified log-rank test. Boundary for statistical significance: p‑value <0.033. |
||
|
OPDIVO |
Placebo |
|
|
Recurrence-free Survival |
||
|
Number of events, n (%) |
66 (13%) |
69 (26%) |
|
Median (months)b
|
NRa
|
NRa
|
|
Hazard ratioc
|
0.42 |
|
Figure 4: Recurrence-free Survival - CHECKMATE-76K
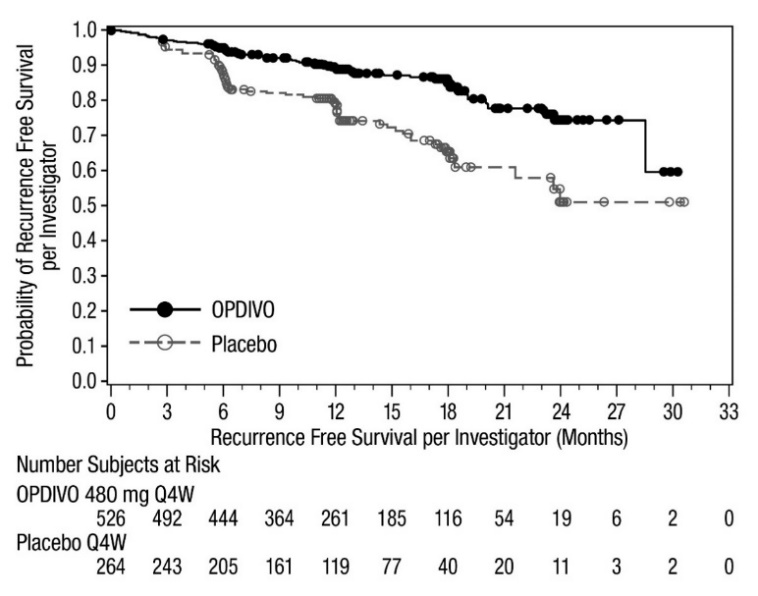
CHECKMATE-238
CHECKMATE-238 (NCT02388906) was a randomized, double-blind trial in 906 patients with completely resected Stage IIIB/C or Stage IV melanoma. Patients were randomized (1:1) to receive OPDIVO 3 mg/kg by intravenous infusion every 2 weeks or ipilimumab 10 mg/kg intravenously every 3 weeks for 4 doses then every 12 weeks beginning at Week 24 for up to 1 year. Enrollment required complete resection of melanoma with margins negative for disease within 12 weeks prior to randomization. The trial excluded patients with a history of ocular/uveal melanoma, autoimmune disease, and any condition requiring systemic treatment with either corticosteroids (≥10 mg daily prednisone or equivalent) or other immunosuppressive medications, as well as patients with prior therapy for melanoma except surgery, adjuvant radiotherapy after neurosurgical resection for lesions of the central nervous system, and prior adjuvant interferon completed ≥6 months prior to randomization. Randomization was stratified by PD-L1 status (positive [based on 5% level] vs. negative/indeterminate) and AJCC stage (Stage IIIB/C vs. Stage IV M1a-M1b vs. Stage IV M1c). The major efficacy outcome measure was recurrence-free survival (RFS) defined as the time between the date of randomization and the date of first recurrence (local, regional, or distant metastasis), new primary melanoma, or death, from any cause, whichever occurs first and as assessed by the investigator. Patients underwent imaging for tumor recurrence every 12 weeks for the first 2 years then every 6 months thereafter.
The trial population characteristics were: median age was 55 years (range: 18 to 86), 58% were male, 95% were White, and 90% had an ECOG performance status of 0. Disease characteristics were AJCC Stage IIIB (34%), Stage IIIC (47%), Stage IV (19%), M1a-b (14%), BRAF V600 mutation positive (42%), BRAF wild-type (45%), elevated LDH (8%), PD-L1 ≥5% tumor cell membrane expression determined by clinical trial assay (34%), macroscopic lymph nodes (48%), and tumor ulceration (32%).
CHECKMATE-238 demonstrated a statistically significant improvement in RFS for patients randomized to the OPDIVO arm compared with the ipilimumab 10 mg/kg arm. Efficacy results are shown in Table 59 and Figure 5.
| a Not reached. b Based on a stratified proportional hazards model. c Based on a stratified log-rank test. d p-value is compared with 0.0244 of the allocated alpha for this analysis. e At the time of the final OS analysis, fewer overall survival events were observed than originally anticipated (approximately 302). |
||
|
OPDIVO |
Ipilimumab 10 mg/kg |
|
|
Recurrence-free Survival |
||
|
Number of events, n (%) |
154 (34%) |
206 (45%) |
|
Median (months) |
NRa
|
NRa
|
|
Hazard ratiob
|
0.65 |
|
|
Overall Survival |
||
|
Number of events, n (%)e |
100 (22%) |
111 (25%) |
|
Median (months) |
NRa |
NRa |
|
Hazard ratiob
|
0.87 |
|
Figure 5: Recurrence-free Survival - CHECKMATE-238
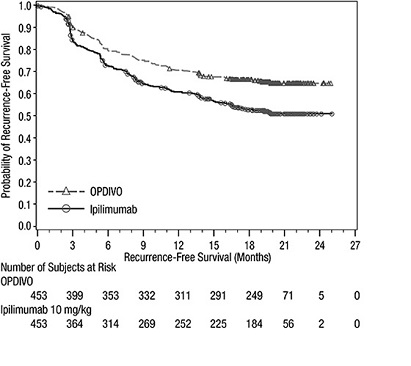
14.3 Neoadjuvant Treatment of Resectable (Tumors ≥4 cm or Node Positive) Non-Small Cell Lung Cancer
CHECKMATE-816 (NCT02998528) was a randomized, open label trial in patients with resectable NSCLC. The trial included patients with resectable, histologically confirmed Stage IB (≥4 cm), II, or IIIA NSCLC (per the 7th edition American Joint Committee on Cancer/Union for International Cancer Control (AJCC/UICC) staging criteria), ECOG performance status 0 or 1, and measurable disease (per RECIST version 1.1). Patients with unresectable or metastatic NSCLC, known EGFR mutations or ALK translocations, Grade 2 or greater peripheral neuropathy, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study.
Patients were randomized to receive either:
- •
- OPDIVO 360 mg administered intravenously over 30 minutes and platinum-doublet chemotherapy administered intravenously every 3 weeks for up to 3 cycles, or
- •
- platinum-doublet chemotherapy administered every 3 weeks for up to 3 cycles.
Platinum-doublet chemotherapy consisted of paclitaxel 175 mg/m2 or 200 mg/m2 and carboplatin AUC 5 or AUC 6 (any histology); pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 (non-squamous histology); or gemcitabine 1000 mg/m2 or 1250 mg/m2 and cisplatin 75 mg/m2 (squamous histology). In the platinum-doublet chemotherapy arm, two additional treatment regimen options included vinorelbine 25 mg/m2 or 30 mg/m2 and cisplatin 75 mg/m2; or docetaxel 60 mg/m2 or 75 mg/m2 and cisplatin 75 mg/m2 (any histology).
Stratification factors for randomization were tumor PD-L1 expression level (≥1% versus <1% or non-quantifiable), disease stage (IB/II versus IIIA), and sex (male versus female). Tumor assessments were performed at baseline, within 14 days of surgery, every 12 weeks after surgery for 2 years, then every 6 months for 3 years, and every year for 5 years until disease recurrence or progression. The major efficacy outcome measures were event-free survival (EFS) based on blinded independent central review (BICR) assessment and pathologic complete response (pCR) as evaluated by blinded independent pathology review (BIPR). Additional efficacy outcome measures included OS.
A total of 358 patients were randomized to receive either OPDIVO in combination with platinum-doublet chemotherapy (n=179) or platinum-doublet chemotherapy (n=179). The median age was 65 years (range: 34 to 84) with 51% of patients ≥65 years and 7% of patients ≥75 years, 50% were Asian, 47% were White, 2% were Black, and 71% were male. Baseline ECOG performance status was 0 (67%) or 1 (33%); 50% had tumors with PD-L1 expression ≥1%; 35% had stage IB/II and 64% had stage IIIA disease; 51% had tumors with squamous histology and 49% had tumors with non-squamous histology; and 89% were former/current smokers.
Eighty-three percent of patients in the OPDIVO in combination with platinum-doublet chemotherapy arm had definitive surgery compared to 75% of patients in the platinum-doublet chemotherapy arm.
The study demonstrated statistically significant improvements in EFS and pCR. Efficacy results are presented in Table 60 and Figure 6.
| Minimum follow-up for EFS was 21 months. a Kaplan-Meier estimate. b Based on a stratified Cox proportional hazard model. c Based on a stratified log-rank test. Boundary for statistical significance: p-value <0.0262. d Based on Clopper and Pearson method. e Strata-adjusted difference based on Cochran-Mantel-Haenszel method of weighting. f From stratified CMH test. |
||
|
OPDIVO and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy |
|
|
Event-free Survival (EFS) per BICR |
||
|
Events (%) |
64 (35.8) |
87 (48.6) |
|
Median (months)a (95% CI) |
31.6 (30.2, NR) |
20.8 (14.0, 26.7) |
|
Hazard Ratiob (95% CI) |
0.63 (0.45, 0.87) |
|
|
Stratified log-rank p-valuec |
0.0052 |
|
|
Pathologic Complete Response (pCR) per BIPR |
||
|
Number of patients with pCR |
43 |
4 |
|
pCR Rate (%), (95% CI)d |
24.0 (18.0, 31.0) |
2.2 (0.6, 5.6) |
|
Estimated treatment difference (95% CI)e |
21.6 (15.1, 28.2) |
|
|
p-valuef |
<0.0001 |
|
Figure 6: Event-Free Survival - CHECKMATE-816
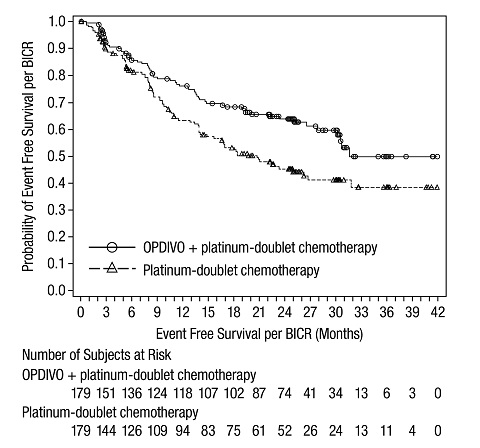
At the time of the EFS analysis, 26% of the patients had died. A prespecified interim analysis for OS resulted in a HR of 0.57 (95% CI: 0.38, 0.87), which did not cross the boundary for statistical significance.
14.4 Neoadjuvant and Adjuvant Treatment of Resectable Non-Small Cell Lung Cancer
The efficacy of OPDIVO, in combination with platinum-doublet chemotherapy, followed by surgery, and continued adjuvant treatment with OPDIVO as a single agent, was investigated in CHECKMATE-77T (NCT04025879), a randomized, double-blind trial in 461 patients with resectable NSCLC. The trial included patients with resectable, suspected or histologically confirmed Stage IIA (>4 cm) to IIIB (T3-T4 N2) NSCLC (per the 8th edition American Joint Committee on Cancer (AJCC) Staging Manual), and ECOG performance status 0 or 1. Patients with unresectable or metastatic NSCLC, EGFR mutations or known ALK translocations, brain metastasis, Grade 2 or greater peripheral neuropathy, interstitial lung disease or active, non-infectious pneumonitis (symptomatic and/or requiring treatment), active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Randomization was stratified by tumor PD-L1 expression level (≥1% versus <1% versus indeterminate/not evaluable), disease stage (Stage II versus Stage III), and tumor histology (squamous versus nonsquamous).
Patients were randomized (1:1) to receive either:
- •
- Neoadjuvant OPDIVO 360 mg administered intravenously over 30 minutes in combination with one of the following platinum-doublet chemotherapy regimens every 3 weeks for four cycles:
- o
- Paclitaxel 175 mg/m2 or 200 mg/m2 and carboplatin AUC 5 or AUC 6 (any histology)
- o
- Pemetrexed 500 mg/m2, and cisplatin 75 mg/m2 or carboplatin AUC 5 or AUC 6 (nonsquamous histology)
- o
- Cisplatin 75 mg/m2 and docetaxel 75 mg/m2 (squamous histology).
- Within 90 days after the surgery, OPDIVO 480 mg was administered intravenously over 30 minutes every 4 weeks.
or
- •
- Neoadjuvant placebo administered intravenously over 30 minutes in combination with platinum-doublet chemotherapy (see above) every 3 weeks for four cycles. Within 90 days after the surgery, placebo was administered intravenously over 30 minutes every 4 weeks.
All study medications were administered via intravenous infusion. Treatment continued until disease progression, recurrence, or unacceptable toxicity for up to 13 cycles (1 year). Tumor assessments were performed every 12 weeks for 2 years, then every 24 weeks for up to 5 years or until disease recurrence or progression was confirmed by BICR.
The trial was not designed to isolate the effect of OPDIVO in each phase (neoadjuvant or adjuvant) of treatment.
The major efficacy outcome measure was event-free survival (EFS) based on BICR assessment. Additional efficacy outcome measures included overall survival (OS), pathologic complete response (pCR), and major pathologic response (MPR).
The median age was 66 years (range: 35 to 86); 71% were male; 72% were White, 25% were Asian, 1.7% were Black, and 1.5% were mixed race/ race unknown/ not reported; and 6% were Hispanic or Latino. Baseline ECOG performance status was 0 (62%) or 1 (38%); 56% had tumors with PD-L1 expression ≥1% and 40% had tumors with PD-L1 expression <1%; 35% had stage II and 64% had stage III disease; 23% had N1 disease and 39% had N2 disease; 51% had tumors with squamous histology and 49% had tumors with nonsquamous histology; and 90% were former/current smokers.
Seventy-eight percent of patients in the neoadjuvant OPDIVO in combination with platinum-doublet chemotherapy followed by adjuvant OPDIVO arm had definitive surgery compared to 77% of patients in the neoadjuvant placebo and platinum-doublet chemotherapy followed by placebo arm.
The study demonstrated a statistically significant improvement in EFS for patients treated with neoadjuvant OPDIVO in combination with platinum-doublet chemotherapy followed by single agent OPDIVO compared with patients randomized to placebo in combination with platinum-doublet chemotherapy followed by placebo. Efficacy results are presented in Table 61 and Figure 7.
| a Kaplan-Meier estimate. b Based on a stratified Cox proportional hazard model. c Based on a stratified log-rank test. Boundary for statistical significance: p-value <0.0264. |
||
|
Neoadjuvant OPDIVO and Platinum-Doublet Chemotherapy/Adjuvant OPDIVO (n=229) |
Neoadjuvant Placebo and Platinum-Doublet Chemotherapy/Adjuvant Placebo (n=232) |
|
|
Event-free Survival (EFS) per BICR |
||
|
Events (%) |
76 (33%) |
113 (49%) |
|
Median (months)a (95% CI) |
NR (28.9, NR) |
18.4 (13.6, 28.1) |
|
Hazard Ratiob (95% CI) |
0.58 (0.43, 0.78) |
|
|
Stratified log-rank p-valuec |
0.00025 |
|
Figure 7: Event-Free Survival - CHECKMATE-77T
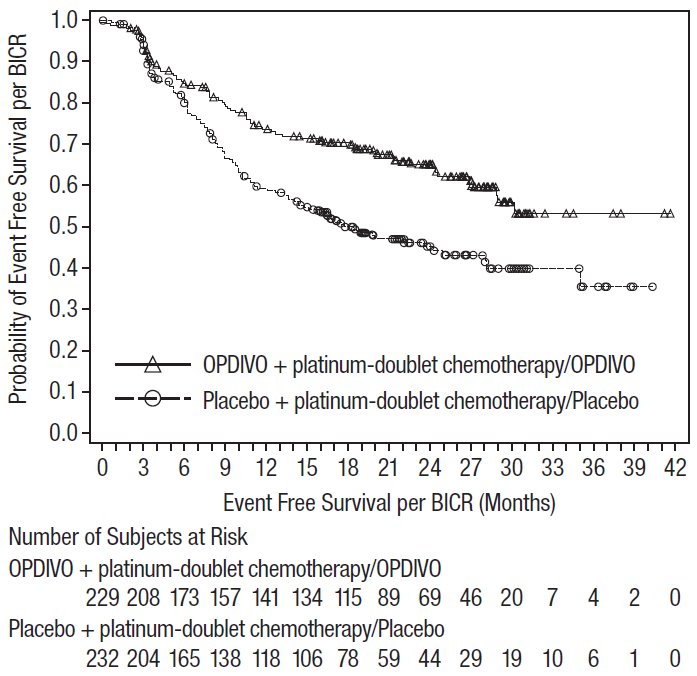
In a pre-specified descriptive analysis, the pCR rate was 25% (95% CI: 20, 31) in the OPDIVO arm and 4.7% (95% CI: 2.4, 8) in the placebo arm.
At the time of the EFS analysis, OS data were immature.
14.5 Metastatic Non-Small Cell Lung Cancer
First-line Treatment of Metastatic Non-Small Cell Lung Cancer (NSCLC) Expressing PD-L1 (≥1%): In Combination with Ipilimumab
CHECKMATE-227 (NCT02477826) was a randomized, open-label, multi-part trial in patients with metastatic or recurrent NSCLC. The study included patients (18 years of age or older) with histologically confirmed Stage IV or recurrent NSCLC (per the 7th International Association for the Study of Lung Cancer [IASLC] classification), ECOG performance status 0 or 1, and no prior anticancer therapy. Patients were enrolled regardless of their tumor PD-L1 status. Patients with known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, untreated brain metastases, carcinomatous meningitis, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Patients with treated brain metastases were eligible if neurologically returned to baseline at least 2 weeks prior to enrolment, and either off corticosteroids, or on a stable or decreasing dose of <10 mg daily prednisone equivalents.
Primary efficacy results were based on Part 1a of the study, which was limited to patients with PD-L1 tumor expression ≥1%. Tumor specimens were evaluated prospectively using the PD-L1 IHC 28-8 pharmDx assay at a central laboratory. Randomization was stratified by tumor histology (non-squamous versus squamous). The evaluation of efficacy relied on the comparison between:
- •
- OPDIVO 3 mg/kg administered intravenously over 30 minutes every 2 weeks in combination with ipilimumab 1 mg/kg administered intravenously over 30 minutes every 6 weeks; or
- •
- Platinum-doublet chemotherapy
Chemotherapy regimens consisted of pemetrexed (500 mg/m2) and cisplatin (75 mg/m2) or pemetrexed (500 mg/m2) and carboplatin (AUC 5 or 6) for non-squamous NSCLC or gemcitabine (1000 or 1250 mg/m2) and cisplatin (75 mg/m2) or gemcitabine (1000 mg/m2) and carboplatin (AUC 5) (gemcitabine was administered on Days 1 and 8 of each cycle) for squamous NSCLC.
Study treatment continued until disease progression, unacceptable toxicity, or for up to 24 months. Treatment continued beyond disease progression if a patient was clinically stable and was considered to be deriving clinical benefit by the investigator. Patients who discontinued combination therapy because of an adverse event attributed to ipilimumab were permitted to continue OPDIVO as a single agent. Tumor assessments were performed every 6 weeks from the first dose of study treatment for the first 12 months, then every 12 weeks until disease progression or study treatment was discontinued. The primary efficacy outcome measure was OS. Additional efficacy outcome measures included PFS, ORR, and duration of response as assessed by BICR.
In Part 1a, a total of 793 patients were randomized to receive either OPDIVO in combination with ipilimumab (n=396) or platinum-doublet chemotherapy (n=397). The median age was 64 years (range: 26 to 87) with 49% of patients ≥65 years and 10% of patients ≥75 years, 76% White, and 65% male. Baseline ECOG performance status was 0 (34%) or 1 (65%), 50% with PD-L1 ≥50%, 29% with squamous and 71% with non-squamous histology, 10% had brain metastases, and 85% were former/current smokers.
The study demonstrated a statistically significant improvement in OS for PD-L1 ≥1% patients randomized to the OPDIVO and ipilimumab arm compared with the platinum-doublet chemotherapy arm. The OS results are presented in Table 62 and Figure 8.
| a Kaplan-Meier estimate. b Based on a stratified Cox proportional hazard model. |
||
|
OPDIVO and Ipilimumab |
Platinum-Doublet Chemotherapy |
|
|
Overall Survival |
||
|
Events (%) |
258 (65%) |
298 (75%) |
|
Median (months)a (95% CI) |
17.1 (15, 20.1) |
14.9 (12.7, 16.7) |
|
Hazard ratio (95% CI)b |
0.79 (0.67, 0.94) |
|
|
Stratified log-rank p-value |
0.0066 |
|
Figure 8: Overall Survival (PD-L1 ≥1%) - CHECKMATE-227
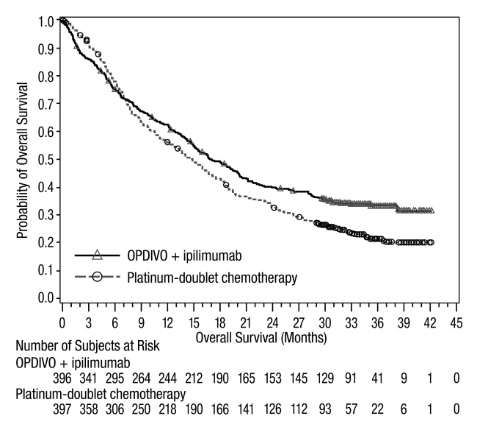
BICR-assessed PFS showed a HR of 0.82 (95% CI: 0.69, 0.97), with a median PFS of 5.1 months (95% CI: 4.1, 6.3) in the OPDIVO and ipilimumab arm and 5.6 months (95% CI: 4.6, 5.8) in the platinum-doublet chemotherapy arm. The BICR-assessed confirmed ORR was 36% (95% CI: 31, 41) in the OPDIVO and ipilimumab arm and 30% (95% CI: 26, 35) in the platinum-doublet chemotherapy arm. Median duration of response observed in the OPDIVO and ipilimumab arm was 23.2 months and 6.2 months in the platinum-doublet chemotherapy arm.
First-line Treatment of Metastatic or Recurrent NSCLC: In Combination with Ipilimumab and Platinum-Doublet Chemotherapy
CHECKMATE-9LA (NCT03215706) was a randomized, open-label trial in patients with metastatic or recurrent NSCLC. The trial included patients (18 years of age or older) with histologically confirmed Stage IV or recurrent NSCLC (per the 7th International Association for the Study of Lung Cancer classification [IASLC]), ECOG performance status 0 or 1, and no prior anticancer therapy (including EGFR and ALK inhibitors) for metastatic disease. Patients were enrolled regardless of their tumor PD-L1 status. Patients with known EGFR mutations or ALK translocations sensitive to available targeted inhibitor therapy, untreated brain metastases, carcinomatous meningitis, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Patients with stable brain metastases were eligible for enrollment.
Patients were randomized 1:1 to receive either:
- •
- OPDIVO 360 mg administered intravenously over 30 minutes every 3 weeks, ipilimumab 1 mg/kg administered intravenously over 30 minutes every 6 weeks, and platinum-doublet chemotherapy administered intravenously every 3 weeks for 2 cycles, or
- •
- platinum-doublet chemotherapy administered every 3 weeks for 4 cycles.
Platinum-doublet chemotherapy consisted of either carboplatin (AUC 5 or 6) and pemetrexed 500 mg/m2, or cisplatin 75 mg/m2 and pemetrexed 500 mg/m2 for non-squamous NSCLC; or carboplatin (AUC 6) and paclitaxel 200 mg/m2 for squamous NSCLC. Patients with non-squamous NSCLC in the control arm could receive optional pemetrexed maintenance therapy. Stratification factors for randomization were tumor PD-L1 expression level (≥1% versus <1% or non-quantifiable), histology (squamous versus non-squamous), and sex (male versus female). Study treatment continued until disease progression, unacceptable toxicity, or for up to 2 years. Treatment could continue beyond disease progression if a patient was clinically stable and was considered to be deriving clinical benefit by the investigator. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue OPDIVO as a single agent as part of the study. Tumor assessments were performed every 6 weeks from the first dose of study treatment for the first 12 months, then every 12 weeks until disease progression or study treatment was discontinued. The primary efficacy outcome measure was OS. Additional efficacy outcome measures included PFS, ORR, and duration of response as assessed by BICR.
A total of 719 patients were randomized to receive either OPDIVO in combination with ipilimumab and platinum-doublet chemotherapy (n=361) or platinum-doublet chemotherapy (n=358). The median age was 65 years (range: 26 to 86) with 51% of patients ≥65 years and 10% of patients ≥75 years. The majority of patients were White (89%) and male (70%). Baseline ECOG performance status was 0 (31%) or 1 (68%), 57% had tumors with PD-L1 expression ≥1% and 37% had tumors with PD-L1 expression that was <1%, 32% had tumors with squamous histology and 68% had tumors with non-squamous histology, 17% had CNS metastases, and 86% were former or current smokers.
The study demonstrated a statistically significant benefit in OS, PFS, and ORR. Efficacy results from the prespecified interim analysis when 351 events were observed (87% of the planned number of events for final analysis) are presented in Table 63.
| a Based on a stratified Cox proportional hazard model. b p-value is compared with the allocated alpha of 0.033 for this interim analysis. c p-value is compared with the allocated alpha of 0.0252 for this interim analysis. d Kaplan-Meier estimate. e Confidence interval based on the Clopper and Pearson Method. f p-value is compared with the allocated alpha of 0.025 for this interim analysis. |
||
|
OPDIVO and Ipilimumab and Platinum-Doublet Chemotherapy |
Platinum-Doublet Chemotherapy |
|
|
Overall Survival |
||
|
Events (%) |
156 (43.2) |
195 (54.5) |
|
Median (months) (95% CI) |
14.1 |
10.7 |
|
Hazard ratio (96.71% CI)a |
0.69 (0.55, 0.87) |
|
|
Stratified log-rank p-valueb |
0.0006 |
|
|
Progression-free Survival per BICR |
||
|
Events (%) |
232 (64.3) |
249 (69.6) |
|
Hazard ratio (97.48% CI)a |
0.70 (0.57, 0.86) |
|
|
Stratified log-rank p-valuec |
0.0001 |
|
|
Median (months)d (95% CI) |
6.8 |
5.0 |
|
Overall Response Rate per BICR (%) |
38 |
25 |
|
(95% CI)e |
(33, 43) |
(21, 30) |
|
Stratified CMH test p-valuef |
0.0003 |
|
|
Duration of Response per BICR |
||
|
Median (months) (95% CI)d |
10.0 |
5.1 |
With an additional 4.6 months of follow-up, the hazard ratio for overall survival was 0.66 (95% CI: 0.55, 0.80) and median survival was 15.6 months (95% CI: 13.9, 20.0) and 10.9 months (95% CI: 9.5, 12.5) for patients receiving OPDIVO and ipilimumab and platinum-doublet chemotherapy or platinum-doublet chemotherapy, respectively (Figure 9).
Figure 9: Overall Survival - CHECKMATE-9LA
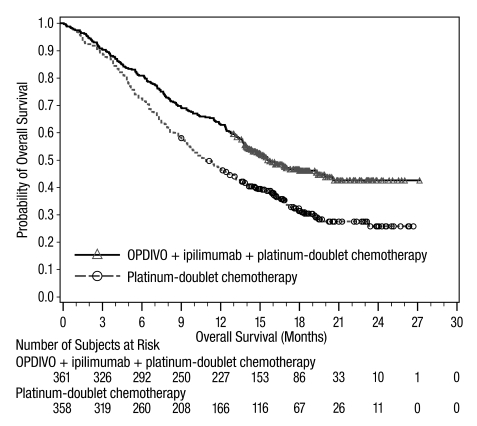
Second-line Treatment of Metastatic Squamous NSCLC
CHECKMATE-017 (NCT01642004) was a randomized (1:1), open-label trial in 272 patients with metastatic squamous NSCLC who had experienced disease progression during or after one prior platinum doublet-based chemotherapy regimen. Patients received OPDIVO 3 mg/kg by intravenous infusion every 2 weeks (n=135) or docetaxel 75 mg/m2 intravenously every 3 weeks (n=137). Randomization was stratified by prior paclitaxel vs. other prior treatment and region (US/Canada vs. Europe vs. Rest of World). This trial included patients regardless of their PD-L1 status. The trial excluded patients with autoimmune disease, medical conditions requiring systemic immunosuppression, symptomatic interstitial lung disease, or untreated brain metastasis. Patients with treated brain metastases were eligible if neurologically returned to baseline at least 2 weeks prior to enrollment, and either off corticosteroids, or on a stable or decreasing dose of <10 mg daily prednisone equivalents. The first tumor assessments were conducted 9 weeks after randomization and continued every 6 weeks thereafter. The major efficacy outcome measure was OS. Additional efficacy outcome measures were investigator-assessed ORR and PFS.
The trial population characteristics were: median age was 63 years (range: 39 to 85) with 44% ≥65 years of age and 11% ≥75 years of age. The majority of patients were White (93%) and male (76%); the majority of patients were enrolled in Europe (57%) with the remainder in US/Canada (32%) and the rest of the world (11%). Baseline ECOG performance status was 0 (24%) or 1 (76%) and 92% were former/current smokers. Baseline disease characteristics of the population as reported by investigators were Stage IIIb (19%), Stage IV (80%), and brain metastases (6%). All patients received prior therapy with a platinum-doublet regimen and 99% of patients had tumors of squamous-cell histology.
The trial demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO as compared with docetaxel at the prespecified interim analysis when 199 events were observed (86% of the planned number of events for final analysis). Efficacy results are shown in Table 64 and Figure 10.
| a Based on a stratified proportional hazards model. b Based on stratified log-rank test. c p-value is compared with 0.0315 of the allocated alpha for this interim analysis. d Based on the stratified Cochran-Mantel-Haenszel test. e Not Reached. |
||
|
OPDIVO
|
Docetaxel
|
|
|
Overall Survival |
||
|
Deaths (%) |
86 (64%) |
113 (82%) |
|
Median (months) |
9.2 |
6.0 |
|
Hazard ratio (95% CI)a |
0.59 (0.44, 0.79) |
|
|
p-valueb,c |
0.0002 |
|
|
Overall Response Rate |
27 (20%) |
12 (9%) |
|
(95% CI) |
(14, 28) |
(5, 15) |
|
p-valued |
0.0083 |
|
|
Complete response |
1 (0.7%) |
0 |
|
Median duration of response (months) |
NRe
|
8.4 |
|
Progression-free Survival |
||
|
Disease progression or death (%) |
105 (78%) |
122 (89%) |
|
Median (months) |
3.5 |
2.8 |
|
Hazard ratio (95% CI)a |
0.62 (0.47, 0.81) |
|
|
p-valueb |
0.0004 |
|
Figure 10: Overall Survival - CHECKMATE-017
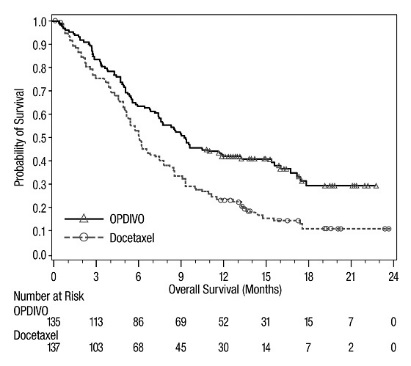
Archival tumor specimens were retrospectively evaluated for PD-L1 expression. Across the trial population, 17% of 272 patients had non-quantifiable results. Among the 225 patients with quantifiable results, 47% had PD-L1 negative squamous NSCLC, defined as <1% of tumor cells expressing PD-L1 and 53% had PD-L1 positive squamous NSCLC defined as ≥1% of tumor cells expressing PD-L1. In pre-specified exploratory subgroup analyses, the hazard ratios for survival were 0.58 (95% CI: 0.37, 0.92) in the PD-L1 negative subgroup and 0.69 (95% CI: 0.45, 1.05) in the PD-L1 positive subgroup.
Second-line Treatment of Metastatic Non-Squamous NSCLC
CHECKMATE-057 (NCT01673867) was a randomized (1:1), open-label trial in 582 patients with metastatic non-squamous NSCLC who had experienced disease progression during or after one prior platinum doublet-based chemotherapy regimen. Appropriate prior targeted therapy in patients with known sensitizing EGFR mutation or ALK translocation was allowed. Patients received OPDIVO 3 mg/kg by intravenous infusion every 2 weeks (n=292) or docetaxel 75 mg/m2 intravenously every 3 weeks (n=290). Randomization was stratified by prior maintenance therapy (yes vs. no) and number of prior therapies (1 vs. 2). The trial excluded patients with autoimmune disease, medical conditions requiring systemic immunosuppression, symptomatic interstitial lung disease, or untreated brain metastasis. Patients with treated brain metastases were eligible if neurologically stable. The first tumor assessments were conducted 9 weeks after randomization and continued every 6 weeks thereafter. The major efficacy outcome measure was OS. Additional efficacy outcome measures were investigator-assessed ORR and PFS. In addition, prespecified analyses were conducted in subgroups defined by PD-L1 expression.
The trial population characteristics: median age was 62 years (range: 21 to 85) with 42% of patients ≥65 years and 7% of patients ≥75 years. The majority of patients were White (92%) and male (55%); the majority of patients were enrolled in Europe (46%) followed by the US/Canada (37%) and the rest of the world (17%). Baseline ECOG performance status was 0 (31%) or 1 (69%), 79% were former/current smokers, 3.6% had NSCLC with ALK rearrangement, 14% had NSCLC with EGFR mutation, and 12% had previously treated brain metastases. Prior therapy included platinum-doublet regimen (100%) and 40% received maintenance therapy as part of the first-line regimen. Histologic subtypes included adenocarcinoma (93%), large cell (2.4%), and bronchoalveolar (0.9%).
CHECKMATE-057 demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO as compared with docetaxel at the prespecified interim analysis when 413 events were observed (93% of the planned number of events for final analysis). Efficacy results are shown in Table 65 and Figure 11.
| a Based on a stratified proportional hazards model. b Based on stratified log-rank test. c p-value is compared with .0408 of the allocated alpha for this interim analysis. d Based on the stratified Cochran-Mantel-Haenszel test. e Not Reached. |
||
|
OPDIVO
|
Docetaxel
|
|
|
Overall Survival |
||
|
Deaths (%) |
190 (65%) |
223 (77%) |
|
Median (months) |
12.2 |
9.4 |
|
Hazard ratio (95% CI)a |
0.73 (0.60, 0.89) |
|
|
p-valueb,c |
0.0015 |
|
|
Overall Response Rate |
56 (19%) |
36 (12%) |
|
(95% CI) |
(15, 24) |
(9, 17) |
|
p-valued |
0.02 |
|
|
Complete response |
4 (1.4%) |
1 (0.3%) |
|
Median duration of response (months) |
17 |
6 |
|
Progression-free Survival |
||
|
Disease progression or death (%) |
234 (80%) |
245 (84%) |
|
Median (months) |
2.3 |
4.2 |
|
Hazard ratio (95% CI)a |
0.92 (0.77, 1.11) |
|
|
p-valueb |
0.39 |
|
Figure 11: Overall Survival - CHECKMATE-057
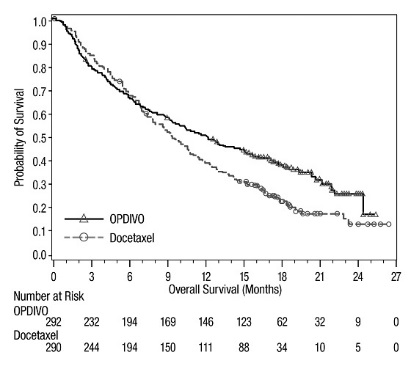
Archival tumor specimens were evaluated for PD-L1 expression following completion of the trial. Across the trial population, 22% of 582 patients had non-quantifiable results. Of the remaining 455 patients, the proportion of patients in retrospectively determined subgroups based on PD-L1 testing using the PD-L1 IHC 28-8 pharmDx assay were: 46% PD-L1 negative, defined as <1% of tumor cells expressing PD-L1 and 54% had PD-L1 expression, defined as ≥1% of tumor cells expressing PD-L1. Among the 246 patients with tumors expressing PD-L1, 26% had ≥1% but <5% tumor cells with positive staining, 7% had ≥5% but <10% tumor cells with positive staining, and 67% had ≥10% tumor cells with positive staining. Figures 12 and 13 summarize the results of prespecified analyses of OS and PFS in subgroups determined by percentage of tumor cells expressing PD-L1.
Figure 12: Forest Plot: OS Based on PD-L1 Expression - CHECKMATE-057
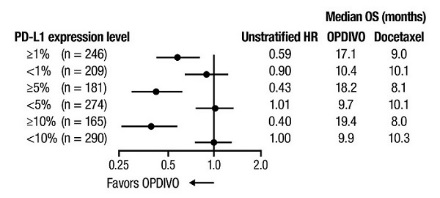
Figure 13: Forest Plot: PFS Based on PD-L1 Expression - CHECKMATE-057
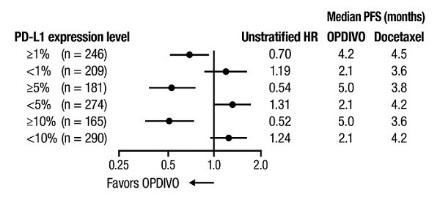
14.6 Malignant Pleural Mesothelioma
CHECKMATE-743 (NCT02899299) was a randomized, open-label trial in patients with unresectable malignant pleural mesothelioma. The trial included patients with histologically confirmed and previously untreated malignant pleural mesothelioma with no palliative radiotherapy within 14 days of initiation of therapy. Patients with interstitial lung disease, active autoimmune disease, medical conditions requiring systemic immunosuppression, or active brain metastasis were excluded from the trial.
Patients were randomized 1:1 to receive either:
- •
- OPDIVO 3 mg/kg over 30 minutes by intravenous infusion every 2 weeks and ipilimumab 1 mg/kg over 30 minutes by intravenous infusion every 6 weeks for up to 2 years, or
- •
- cisplatin 75 mg/m2 and pemetrexed 500 mg/m2, or carboplatin 5 AUC and pemetrexed 500 mg/m2 administered every 3 weeks for 6 cycles.
Stratification factors for randomization were tumor histology (epithelioid vs. sarcomatoid or mixed histology subtypes) and sex (male vs. female). Study treatment continued for up to 2 years, or until disease progression or unacceptable toxicity. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue OPDIVO as a single agent. Treatment could continue beyond disease progression if a patient was clinically stable and was considered to be deriving clinical benefit by the investigator. Tumor assessments were performed every 6 weeks from the first dose of study treatment for the first 12 months, then every 12 weeks until disease progression or study treatment was discontinued. The primary efficacy outcome measure was OS. Additional efficacy outcome measures included PFS, ORR, and duration of response as assessed by BICR utilizing modified RECIST criteria.
A total of 605 patients were randomized to receive either OPDIVO in combination with ipilimumab (n=303) or chemotherapy (n=302). The median age was 69 years (range: 25 to 89), with 72% of patients ≥65 years and 26% ≥75 years; 85% were White, 11% were Asian, and 77% were male. Baseline ECOG performance status was 0 (40%) or 1 (60%), 35% had Stage III and 51% had Stage IV disease, 75% had epithelioid and 25% had non-epithelioid histology, 75% had tumors with PD-L1 expression ≥1%, and 22% had tumors with PD-L1 expression <1%.
The trial demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO in combination with ipilimumab compared to chemotherapy. Efficacy results from the prespecified interim analysis are presented in Table 66 and Figure 14.
| a At the time of the interim analysis, 419 deaths (89% of the deaths needed for the final analysis) had occurred. b Kaplan-Meier estimate. c Stratified Cox proportional hazard model. d p-value is compared with the allocated alpha of 0.0345 for this interim analysis. e Based on confirmed response by BICR. |
||
|
OPDIVO and Ipilimumab |
Chemotherapy |
|
|
Overall Survivala |
||
|
Events (%) |
200 (66) |
219 (73) |
|
Median (months)b (95% CI) |
18.1 |
14.1 |
|
Hazard ratio (95% CI)c |
0.74 (0.61, 0.89) |
|
|
Stratified log-rank p-valued |
0.002 |
|
|
Progression-free Survival |
||
|
Events (%) |
218 (72) |
209 (69) |
|
Hazard ratio (95% CI)c |
1.0 (0.82, 1.21) |
|
|
Median (months)b (95% CI) |
6.8 |
7.2 |
|
Overall Response Ratee |
40% |
43% |
|
(95% CI) |
(34, 45) |
(37, 49) |
|
Duration of Response |
||
|
Median (months)b
|
11.0 |
6.7 |
Figure 14: Overall Survival - CHECKMATE-743
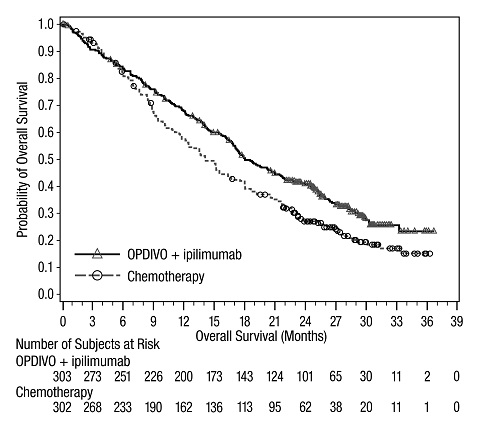
In a prespecified exploratory analysis based on histology, in the subgroup of patients with epithelioid histology, the hazard ratio (HR) for OS was 0.85 (95% CI: 0.68, 1.06), with median OS of 18.7 months in the OPDIVO and ipilimumab arm and 16.2 months in the chemotherapy arm. In the subgroup of patients with non-epithelioid histology, the HR for OS was 0.46 (95% CI: 0.31, 0.70), with median OS of 16.9 months in the OPDIVO and ipilimumab arm and 8.8 months in the chemotherapy arm.
14.7 Advanced Renal Cell Carcinoma
First-line Renal Cell Carcinoma
CHECKMATE-214
CHECKMATE-214 (NCT02231749) was a randomized (1:1), open-label trial in patients with previously untreated advanced RCC. Patients were included regardless of their PD-L1 status. CHECKMATE-214 excluded patients with any history of or concurrent brain metastases, active autoimmune disease, or medical conditions requiring systemic immunosuppression. Patients were stratified by International Metastatic RCC Database Consortium (IMDC) prognostic score and region.
Efficacy was evaluated in intermediate/poor risk patients with at least 1 or more of 6 prognostic risk factors as per the IMDC criteria (less than one year from time of initial renal cell carcinoma diagnosis to randomization, Karnofsky performance status <80%, hemoglobin less than the lower limit of normal, corrected calcium of >10 mg/dL, platelet count greater than the upper limit of normal, and absolute neutrophil count greater than the upper limit of normal).
Patients were randomized to OPDIVO 3 mg/kg and ipilimumab 1 mg/kg intravenously every 3 weeks for 4 doses followed by OPDIVO 3 mg/kg intravenously every two weeks (n=425), or sunitinib 50 mg orally daily for the first 4 weeks of a 6-week cycle (n=422). Treatment continued until disease progression or unacceptable toxicity.
The trial population characteristics were: median age was 61 years (range: 21 to 85) with 38% ≥65 years of age and 8% ≥75 years of age. The majority of patients were male (73%) and White (87%) and 26% and 74% of patients had a baseline KPS of 70% to 80% and 90% to 100%, respectively.
The major efficacy outcome measures were OS, PFS (independent radiographic review committee [IRRC]-assessed) and confirmed ORR (IRRC-assessed) in intermediate/poor risk patients. In this population, the trial demonstrated statistically significant improvement in OS and ORR for patients randomized to OPDIVO and ipilimumab as compared with sunitinib (Table 67 and Figure 15). OS benefit was observed regardless of PD-L1 expression level. The trial did not demonstrate a statistically significant improvement in PFS. Efficacy results are shown in Table 67 and Figure 15.
| a Not Reached. b Based on a stratified proportional hazards model. c Based on a stratified log-rank test. d p-value is compared to alpha 0.002 in order to achieve statistical significance. e Based on the stratified DerSimonian-Laird test. f p-value is compared to alpha 0.001 in order to achieve statistical significance. g Not Significant at alpha level of 0.009. |
||
|
Intermediate/Poor-Risk |
||
|
OPDIVO and Ipilimumab |
Sunitinib |
|
|
Overall Survival |
||
|
Deaths (%) |
140 (32.9) |
188 (44.5) |
|
Median survival (months) |
NRa |
25.9 |
|
Hazard ratio (99.8% CI)b |
0.63 (0.44, 0.89) |
|
|
p-valuec,d |
<0.0001 |
|
|
Confirmed Overall Response Rate (95% CI) |
41.6% (36.9, 46.5) |
26.5% (22.4, 31.0) |
|
p-valuee,f |
<0.0001 |
|
|
Complete response (CR) |
40 (9.4) |
5 (1.2) |
|
Partial response (PR) |
137 (32.2) |
107 (25.4) |
|
Median duration of response (months) (95% CI) |
NRa (21.8, NRa) |
18.2 (14.8, NRa) |
|
Progression-free Survival | ||
|
Disease progression or death (%) |
228 (53.6) |
228 (54.0) |
|
Median (months) |
11.6 |
8.4 |
|
Hazard ratio (99.1% CI)a |
0.82 (0.64, 1.05) |
|
|
p-valuec |
NSg |
|
Figure 15: Overall Survival (Intermediate/Poor Risk Population) - CHECKMATE-214
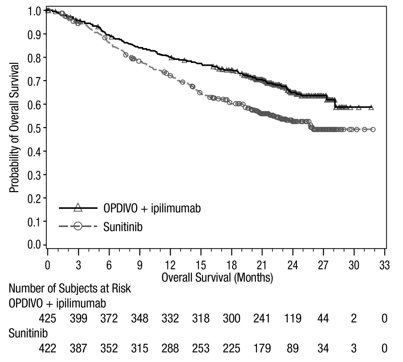
CHECKMATE-214 also randomized 249 favorable risk patients as per IMDC criteria to OPDIVO and ipilimumab (n=125) or to sunitinib (n=124). These patients were not evaluated as part of the efficacy analysis population. OS in favorable risk patients receiving OPDIVO and ipilimumab compared to sunitinib has a hazard ratio of 1.45 (95% CI: 0.75, 2.81). The efficacy of OPDIVO and ipilimumab in previously untreated renal cell carcinoma with favorable-risk disease has not been established.
CHECKMATE-9ER
CHECKMATE-9ER (NCT03141177) was a randomized, open-label study of OPDIVO combined with cabozantinib versus sunitinib in patients with previously untreated advanced RCC. CHECKMATE-9ER excluded patients with autoimmune disease or other medical conditions requiring systemic immunosuppression. Patients were stratified by IMDC prognostic score (favorable vs. intermediate vs. poor), PD-L1 tumor expression (≥1% vs. <1% or indeterminate), and region (US/Canada/Western Europe/Northern Europe vs. Rest of World).
Patients were randomized to OPDIVO 240 mg intravenously every 2 weeks and cabozantinib 40 mg orally daily (n=323), or sunitinib 50 mg orally daily for the first 4 weeks of a 6-week cycle (4 weeks on treatment followed by 2 weeks off) (n=328). Treatment continued until disease progression per RECIST v1.1 or unacceptable toxicity. Treatment beyond RECIST-defined disease progression was permitted if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Tumor assessments were performed at baseline, after randomization at Week 12, then every 6 weeks until Week 60, and then every 12 weeks thereafter.
The trial population characteristics were: median age 61 years (range: 28 to 90) with 38% ≥65 years of age and 10% ≥75 years of age. The majority of patients were male (74%) and White (82%) and 23% and 77% of patients had a baseline KPS of 70% to 80% and 90% to 100%, respectively. Patient distribution by IMDC risk categories was 22% favorable, 58% intermediate, and 20% poor.
The major efficacy outcome measure was PFS (BICR assessed). Additional efficacy outcome measures were OS and ORR (BICR assessed). The trial demonstrated a statistically significant improvement in PFS, OS, and ORR for patients randomized to OPDIVO and cabozantinib compared with sunitinib. Consistent results for PFS were observed across pre-specified subgroups of IMDC risk categories and PD-L1 tumor expression status. An updated OS analysis was conducted when 271 deaths were observed based on the pre-specified number of deaths for the pre-planned final analysis of OS. Efficacy results are shown in Table 68 and Figures 16 and 17.
| a Based on Kaplan-Meier estimates. b Stratified Cox proportional hazards model. c Based on stratified log-rank test. d 2-sided p-values from stratified log-rank test. e Not Reached. f p-value is compared with the allocated alpha of 0.0111 for this interim analysis. g CI based on the Clopper-Pearson method. h 2-sided p-value from Cochran-Mantel-Haenszel test. |
||
|
OPDIVO and Cabozantinib |
Sunitinib |
|
|
Progression-free Survival |
||
|
Disease progression or death (%) |
144 (45) |
191 (58) |
|
Median PFS (months)a (95% CI) |
16.6 (12.5, 24.9) |
8.3 (7.0, 9.7) |
|
Hazard ratio (95% CI)b |
0.51 (0.41, 0.64) |
|
|
p-valuec,d |
<0.0001 |
|
|
Overall Survival |
||
|
Deaths (%) |
67 (21) |
99 (30) |
|
Median OS (months)a (95% CI) |
NRe |
NR (22.6, NRe) |
|
Hazard ratio (98.89% CI)b |
0.60 (0.40, 0.89) |
|
|
p-valuec,d,f |
0.0010 |
|
|
Updated Overall Survival |
||
|
Deaths (%) |
121 (37) |
150 (46) |
|
Median OS (months)a (95% CI) |
37.7 (35.5, NR) |
34.3 (29.0, NR) |
|
Hazard ratio (95% CI)b |
0.70 (0.55, 0.90) |
|
|
Confirmed Objective Response Rate (95% CI)g |
55.7% (50.1, 61.2) |
27.1% (22.4, 32.3) |
|
p-valueh |
<0.0001 |
|
|
Complete Response |
26 (8%) |
15 (4.6%) |
|
Partial Response |
154 (48%) |
74 (23%) |
|
Median duration of response in months (95% CI)a |
20.2 (17.3, NRe) |
11.5 (8.3, 18.4) |
Figure 16: Progression-free Survival - CHECKMATE-9ER
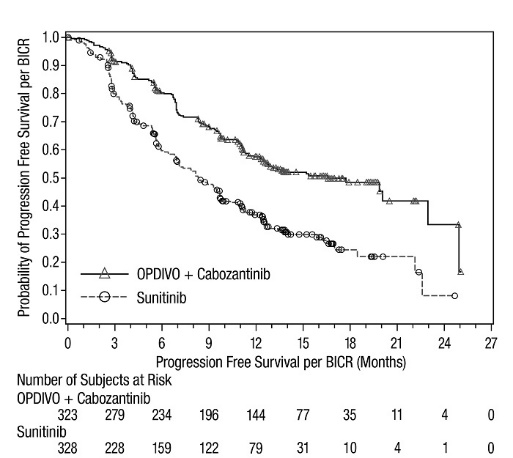
Figure 17: Updated Overall Survival - CHECKMATE-9ER
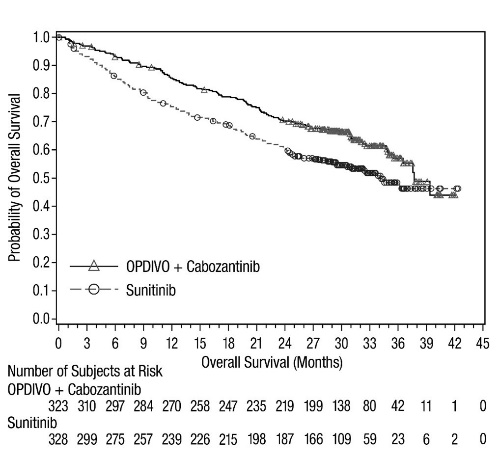
In an exploratory analysis, the updated analysis of OS in patients with IMDC favorable, intermediate, intermediate/poor, and poor risk demonstrated a HR (95% CI) of 1.03 (0.55, 1.92), 0.74 (0.54, 1.01), 0.65 (0.50, 0.85), and 0.49 (0.31, 0.79), respectively.
Previously Treated Renal Cell Carcinoma
CHECKMATE-025
CHECKMATE-025 (NCT01668784) was a randomized (1:1), open-label trial in patients with advanced RCC who had experienced disease progression during or after one or two prior anti-angiogenic therapy regimens. Patients had to have a Karnofsky Performance Score (KPS) ≥70% and patients were included regardless of their PD-L1 status. The trial excluded patients with any history of or concurrent brain metastases, prior treatment with an mTOR inhibitor, active autoimmune disease, or medical conditions requiring systemic immunosuppression. Patients were stratified by region, Memorial Sloan Kettering Cancer Center (MSKCC) Risk Group and the number of prior anti-angiogenic therapies. Patients were randomized OPDIVO 3 mg/kg by intravenous infusion every 2 weeks (n=410) or everolimus 10 mg orally daily (n=411). The first tumor assessments were conducted 8 weeks after randomization and continued every 8 weeks thereafter for the first year and then every 12 weeks until progression or treatment discontinuation, whichever occurred later. The major efficacy outcome measure was overall survival (OS).
The trial population characteristics were: median age was 62 years (range: 18 to 88) with 40% ≥65 years of age and 9% ≥75 years of age. The majority of patients were male (75%) and White (88%) and 34% and 66% of patients had a baseline KPS of 70% to 80% and 90% to 100%, respectively. The majority of patients (77%) were treated with one prior anti-angiogenic therapy. Patient distribution by MSKCC risk groups was 34% favorable, 47% intermediate, and 19% poor.
The trial demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO as compared with everolimus at the prespecified interim analysis when 398 events were observed (70% of the planned number of events for final analysis). OS benefit was observed regardless of PD-L1 expression level. Efficacy results are shown in Table 69 and Figure 18.
| a Not Reached. b Based on a stratified proportional hazards model. c Based on a stratified log-rank test. d p-value is compared with 0.0148 of the allocated alpha for this interim analysis. |
||
|
OPDIVO
|
Everolimus
|
|
|
Overall Survival | ||
|
Deaths (%) |
183 (45) |
215 (52) |
|
Median survival (months) (95% CI) |
25.0 (21.7, NRa) |
19.6 (17.6, 23.1) |
|
Hazard ratio (95% CI)b |
0.73 (0.60, 0.89) |
|
|
p-valuec,d |
0.0018 |
|
|
Confirmed Overall Response Rate (95% CI) |
21.5% (17.6, 25.8) |
3.9% (2.2, 6.2) |
|
Median duration of response (months) (95% CI) |
23.0 (12.0, NRa) |
13.7 (8.3, 21.9) |
|
Median time to onset of confirmed response (months) (min, max) |
3.0 (1.4, 13.0) |
3.7 (1.5, 11.2) |
Figure 18: Overall Survival - CHECKMATE-025
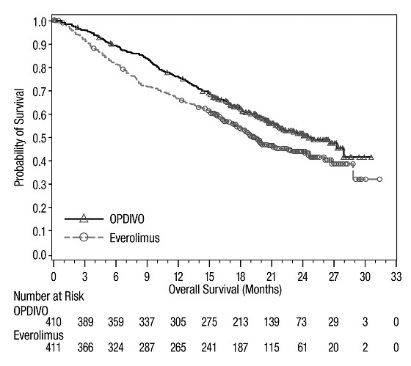
14.8 Classical Hodgkin Lymphoma
Two studies evaluated the efficacy of OPDIVO as a single agent in adult patients with cHL after failure of autologous HSCT.
CHECKMATE-205 (NCT02181738) was a single-arm, open-label, multicenter, multicohort trial in cHL. CHECKMATE-039 (NCT01592370) was an open-label, multicenter, dose escalation trial that included cHL. Both studies included patients regardless of their tumor PD-L1 status and excluded patients with ECOG performance status of 2 or greater, autoimmune disease, symptomatic interstitial lung disease, hepatic transaminases more than 3 times ULN, creatinine clearance <40 mL/min, prior allogeneic HSCT, or chest irradiation within 24 weeks. In addition, both studies required an adjusted diffusion capacity of the lungs for carbon monoxide (DLCO) of over 60% in patients with prior pulmonary toxicity.
Patients received OPDIVO 3 mg/kg by intravenous infusion every 2 weeks until disease progression, maximal clinical benefit, or unacceptable toxicity. A cycle consisted of one dose. Dose reduction was not permitted.
Efficacy was evaluated by ORR as determined by an IRRC. Additional outcome measures included duration of response (DOR).
Efficacy was evaluated in 95 patients in CHECKMATE-205 and CHECKMATE-039 combined who had failure of autologous HSCT and post-transplantation brentuximab vedotin. The median age was 37 years (range: 18 to 72). The majority were male (64%) and White (87%). Patients had received a median of 5 prior systemic regimens (range: 2 to 15). They received a median of 27 doses of OPDIVO (range: 3 to 48), with a median duration of therapy of 14 months (range: 1 to 23 months). Efficacy results are shown in Table 70.
| a Per 2007 revised International Working Group criteria. b Kaplan-Meier estimate. Among responders, the median follow-up for DOR, measured from the date of first response, was 9.9 months. c A + sign indicates a censored value. d Not Reached. |
|
|
CHECKMATE-205 and CHECKMATE-039 |
|
|
Overall Response Rate, n (%)a
|
63 (66%) |
|
Complete remission rate |
6 (6%) |
|
Partial remission rate |
57 (60%) |
|
Duration of Response (months)
|
|
|
Time to Response (months)
|
|
Efficacy was also evaluated in 258 patients in CHECKMATE-205 and CHECKMATE-039 combined who had relapsed or progressive cHL after autologous HSCT. The analysis included the group described above. The median age was 34 years (range: 18 to 72). The majority were male (59%) and White (86%). Patients had a median of 4 prior systemic regimens (range: 2 to 15), with 85% having 3 or more prior systemic regimens and 76% having prior brentuximab vedotin. Of the 195 patients having prior brentuximab vedotin, 17% received it only before autologous HSCT, 78% received it only after HSCT, and 5% received it both before and after HSCT. Patients received a median of 21 doses of OPDIVO (range: 1 to 48), with a median duration of therapy of 10 months (range: 0 to 23 months). Efficacy results are shown in Table 71.
| a Kaplan-Meier estimate. Among responders, the median follow-up for DOR, measured from the date of first response, was 6.7 months. b The estimated median duration of PR was 13.1 months (95% CI, 9.5, NE). The median duration of CR was not reached. c Not Reached. |
|
|
CHECKMATE-205 and CHECKMATE-039 |
|
|
Overall Response Rate, n (%)
|
179 (69%) |
|
Complete remission rate |
37 (14%) |
|
Partial remission rate |
142 (55%) |
|
Duration of Response (months)
|
|
|
Time to Response (months)
|
|
14.9 Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck
CHECKMATE-141 (NCT02105636) was a randomized (2:1), active-controlled, open-label trial enrolling patients with metastatic or recurrent SCCHN who had experienced disease progression during or within 6 months of receiving platinum-based therapy administered in either the adjuvant, neo-adjuvant, primary (unresectable locally advanced) or metastatic setting. The trial excluded patients with autoimmune disease, medical conditions requiring immunosuppression, recurrent or metastatic carcinoma of the nasopharynx, squamous cell carcinoma of unknown primary histology, salivary gland or non-squamous histologies (e.g., mucosal melanoma), or untreated brain metastasis. Patients with treated brain metastases were eligible if neurologically stable. Patients were randomized to receive OPDIVO 3 mg/kg by intravenous infusion every 2 weeks or investigator’s choice of cetuximab (400 mg/m2 initial dose intravenously followed by 250 mg/m2 weekly), or methotrexate (40 to 60 mg/m2 intravenously weekly), or docetaxel (30 to 40 mg/m2 intravenously weekly).
Randomization was stratified by prior cetuximab treatment (yes/no). The first tumor assessments were conducted 9 weeks after randomization and continued every 6 weeks thereafter. The major efficacy outcome measure was OS. Additional efficacy outcome measures were PFS and ORR.
A total of 361 patients were randomized; 240 patients to the OPDIVO arm and 121 patients to the investigator’s choice arm (docetaxel: 45%; methotrexate: 43%; and cetuximab: 12%). The trial population characteristics were: median age was 60 years (range: 28 to 83) with 31% ≥65 years of age, 83% were White, 12% Asian, and 4% were Black, and 83% male. Baseline ECOG performance status was 0 (20%) or 1 (78%), 76% were former/current smokers, 90% had Stage IV disease, 45% of patients received only one prior line of systemic therapy, the remaining 55% received two or more prior lines of systemic therapy, and 25% had HPVp16-positive tumors, 24% had HPV p16-negative tumors, and 51% had unknown status.
The trial demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO as compared with investigator’s choice at a pre-specified interim analysis (78% of the planned number of events for final analysis). There were no statistically significant differences between the two arms for PFS (HR=0.89; 95% CI: 0.70, 1.13) or ORR (13.3% [95% CI: 9.3, 18.3] vs. 5.8% [95% CI: 2.4, 11.6] for nivolumab and investigator’s choice, respectively). Efficacy results are shown in Table 72 and Figure 19.
| a Based on stratified proportional hazards model. b Based on stratified log-rank test. c p-value is compared with 0.0227 of the allocated alpha for this interim analysis. |
||
|
OPDIVO |
Cetuximab, Methotrexate or Docetaxel |
|
|
Overall Survival |
||
|
Deaths (%) |
133 (55%) |
85 (70%) |
|
Median (months) |
7.5 |
5.1 |
|
Hazard ratio (95% CI)a |
0.70 (0.53, 0.92) |
|
|
p-valueb,c |
0.0101 |
|
Figure 19: Overall Survival - CHECKMATE-141
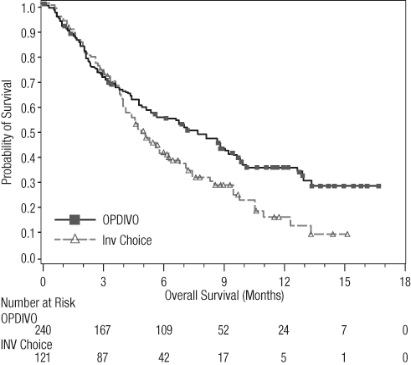
Archival tumor specimens were retrospectively evaluated for PD-L1 expression using the PD-L1 IHC 28-8 pharmDx assay. Across the trial population, 28% (101/361) of patients had non-quantifiable results. Among the 260 patients with quantifiable results, 43% (111/260) had PD-L1 negative SCCHN, defined as <1% of tumor cells expressing PD-L1, and 57% (149/260) had PD-L1 positive SCCHN, defined as ≥1% of tumor cells expressing PD-L1. In pre-specified exploratory subgroup analyses, the hazard ratio for survival was 0.89 (95% CI: 0.54, 1.45) with median survivals of 5.7 and 5.8 months for the nivolumab and chemotherapy arms, respectively, in the PD-L1 negative subgroup. The HR for survival was 0.55 (95% CI: 0.36, 0.83) with median survivals of 8.7 and 4.6 months for the nivolumab and chemotherapy arms, respectively, in the PD-L1 positive SCCHN subgroup.
14.10 Urothelial Carcinoma
Adjuvant Treatment of UC at High Risk of Recurrence
CHECKMATE-274 (NCT02632409) was a randomized, double-blind, placebo-controlled study of adjuvant OPDIVO in patients who were within 120 days of radical resection (R0) of UC of the bladder or upper urinary tract (renal pelvis or ureter) at high risk of recurrence. High risk of recurrence was defined as either 1) ypT2-ypT4a or ypN+ for patients who received neoadjuvant cisplatin or 2) pT3-pT4a or pN+ for patients who did not receive neoadjuvant cisplatin and who also either were ineligible for or refused adjuvant cisplatin. Patients were randomized 1:1 to receive OPDIVO 240 mg or placebo by intravenous infusion every 2 weeks until recurrence or until unacceptable toxicity for a maximum treatment duration of 1 year. Patients were stratified by pathologic nodal status (N+ vs. N0/x with <10 nodes removed vs. N0 with ≥10 nodes removed), tumor cells expressing PD-L1 (≥1% vs. <1%/indeterminate as determined by the central lab using the PD-L1 IHC 28-8 pharmDx assay), and use of neoadjuvant cisplatin (yes vs. no).
The trial population characteristics were: median age of 67 years (range: 30 to 92); 76% male; 76% White, 22% Asian, 0.7% Black, and 0.1% American Indian or Alaska Native. Of the 335 (47%) of patients with node-positive UC, 44 (6%) had non–muscle-invasive (<pT2) primary tumors. ECOG performance status was 0 (63%), 1 (35%), or 2 (2%). Prior neoadjuvant cisplatin had been given to 43% of patients; of the 57% who did not receive prior neoadjuvant cisplatin, reasons listed were ineligibility (22%), patient preference (33%), and other/not reported (2%). Tumor PD-L1 expression was ≥1% in 40% of patients, and 21% of patients had upper tract UC.
The major efficacy outcome measures were investigator-assessed DFS in all randomized patients and in patients with tumors expressing PD-L1 ≥1%. DFS was defined as time to first recurrence (local urothelial tract, local non-urothelial tract, or distant metastasis), or death. Additional efficacy outcome measures included OS.
At the pre-specified interim analysis, CHECKMATE-274 demonstrated a statistically significant improvement in DFS for patients randomized to OPDIVO vs. placebo in the all randomized patient population, as well as in the subpopulation of patients with PD-L1 ≥1%, as shown in Table 73 and Figure 20.
In exploratory subgroup analyses in patients with upper tract UC (n=149), no improvement in DFS was observed in the nivolumab arm compared to the placebo arm. The unstratified DFS hazard ratio estimate was 1.15 (95% CI: 0.74, 1.80).
In an exploratory subgroup analysis in patients with PD-L1 expression of <1% (n=414), the unstratified DFS hazard ratio estimate was 0.83 (95% CI: 0.64, 1.08).
OS data is immature with 33% of deaths in the overall randomized population. In the UTUC subpopulation, 37 deaths occurred (20 in the nivolumab arm, 17 in the placebo arm).
| N.R. Not Reached, N.E. Not Estimable a Includes disease at baseline events (protocol deviations): n=1 in OPDIVO arm and n=3 in placebo arm. b Based on Kaplan-Meier estimates. c Stratified Cox proportional hazard model. Hazard ratio is OPDIVO over placebo. d Log-rank test stratified by prior neoadjuvant cisplatin, pathological nodal status, PD-L1 status (≥1% vs <1%/indeterminate). Boundary for statistical significance in all randomized patients: p-value <0.01784. e Log-rank test stratified by prior neoadjuvant cisplatin, pathological nodal status. Boundary for statistical significance in all randomized patients with PD-L1 ≥1%: p-value <0.01282. |
||||
|
All Randomized |
PD-L1 ≥1% |
|||
|
OPDIVO |
Placebo |
OPDIVO |
Placebo |
|
|
Disease-free Survival |
||||
|
Eventsa, n (%) |
170 (48) |
204 (57) |
55 (39) |
81 (57) |
|
Median DFS (months)b
|
20.8 |
10.8 |
N.R. |
8.4 |
|
Hazard ratioc
|
0.70 |
0.55 |
||
|
p-value |
0.0008d |
0.0005e |
||
Figure 20: Disease-free Survival in All Randomized Patients - CHECKMATE-274
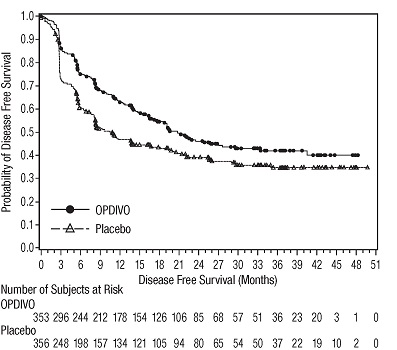
First-line Treatment of Unresectable or Metastatic UC
CHECKMATE-901 (NCT 03036098) was a randomized, open-label study in patients with previously untreated unresectable or metastatic UC. Prior neoadjuvant or adjuvant chemotherapy were permitted as long as the disease recurrence took place ≥12 months from completion of therapy. Patients who were ineligible for cisplatin and those with active CNS metastases were excluded. Stratification factors for randomization were PD-L1 status (≥1% vs. <1% or indeterminate) and liver metastasis. Patients were randomized 1:1 to receive either:
- •
- OPDIVO 360 mg and cisplatin 70 mg/m2 on Day 1 and gemcitabine 1000 mg/m2 on Days 1 and 8 of a 21-day cycle of a 21-day cycle for up to 6 cycles followed by single-agent OPDIVO 480 mg every 4 weeks until disease progression or unacceptable toxicity. In the absence of disease progression or unacceptable toxicity, OPDIVO was continued for up to 2 years from first dose.
- •
- Cisplatin 70 mg/m2 on Day 1 and gemcitabine 1000 mg/m2 on Days 1 and 8 of a 21-day cycle for up to 6 cycles, until disease progression or unacceptable toxicity.
The major efficacy outcome measures were OS and PFS as assessed by BICR using RECIST v1.1. Additional efficacy outcome measures included ORR as assessed by BICR.
The median age was 65 years of age (range: 32 to 86) with 51% of patients ≥65 years of age and 12% of patients ≥75 years of age, 23% were Asian, 72% were White, 0.3% were Black, 0.3% were American Indian or Alaska Native, 4.9% were Other, 12% were Hispanic or Latino, and 77% were male. Baseline ECOG performance status was 0 (53%) or 1 (46%). At baseline, 87% of patients had metastatic UC, including 20% with liver metastases, 11% had locally advanced UC, and 51% had UC histologic variants. Forty-nine (16%) in the OPDIVO in combination with cisplatin-based chemotherapy arm and 43 (14%) in the cisplatin-based chemotherapy arm switched from cisplatin to carboplatin after at least one cycle of cisplatin.
Efficacy results are presented in Table 74 and Figures 21 and 22.
| a Based on Kaplan-Meier Estimates. b Stratified Cox proportional hazard model. c 2 sided p values from stratified log-rank test. d Assessed by BICR. |
||
|
OPDIVO and Cisplatin and Gemcitabine |
Cisplatin and Gemcitabine (n=304) |
|
|
Overall Survival (OS) |
||
|
Events, n (%) |
172 (56.6) |
193 (63.5) |
|
Median (months) |
21.7 |
18.9 |
|
Hazard ratio (95% CI)b |
0.78 |
|
|
p-valuec |
0.0171 |
|
|
Progression-free Survival (PFS)d |
||
|
Events, n (%) |
211 (69.4) |
191 (62.8) |
|
Median (months) |
7.9 |
7.6 |
|
Hazard ratio (95% CI)b |
0.72 |
|
|
p-valuec |
0.0012 |
|
|
Objective Response Rate (ORR)d |
||
|
Response rate, n (%) (95% CI) |
175 (57.6%) (51.8, 63.2) |
131 (43.1%) (37.5, 48.9) |
|
Complete response rate, n (%) |
66 (22%) |
36 (12%) |
|
Partial response rate, n (%) |
109 (36%) |
95 (31%) |
|
Duration of Response (DoR) |
||
|
Median (months) |
9.5 |
7.3 |
Figure 21: Overall Survival - CHECKMATE-901
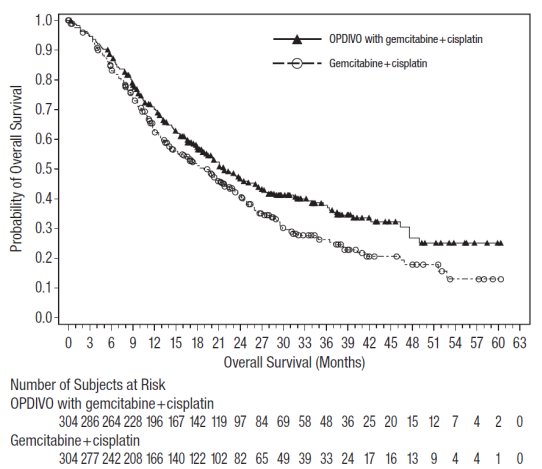
Figure 22: Progression-free Survival - CHECKMATE-901
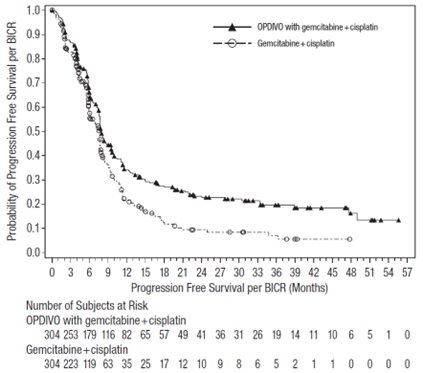
Previously Treated Advanced or Metastatic UC
CHECKMATE-275 (NCT02387996) was a single-arm trial in 270 patients with locally advanced or metastatic UC who had disease progression during or following platinum-containing chemotherapy or who had disease progression within 12 months of treatment with a platinum-containing neoadjuvant or adjuvant chemotherapy regimen. Patients were excluded for active brain or leptomeningeal metastases, active autoimmune disease, medical conditions requiring systemic immunosuppression, and ECOG performance status >1. Patients received OPDIVO 3 mg/kg by intravenous infusion every 2 weeks until unacceptable toxicity or either radiographic or clinical progression. Tumor response assessments were conducted every 8 weeks for the first 48 weeks and every 12 weeks thereafter. Major efficacy outcome measures included confirmed ORR as assessed by IRRC using RECIST v1.1 and DOR.
The median age was 66 years (range: 38 to 90), 78% were male, 86% were White. Twenty-seven percent had non-bladder urothelial carcinoma and 84% had visceral metastases. Thirty-four percent of patients had disease progression following prior platinum-containing neoadjuvant or adjuvant therapy. Twenty-nine percent of patients had received ≥2 prior systemic regimens in the metastatic setting. Thirty-six percent of patients received prior cisplatin only, 23% received prior carboplatin only, and 7% were treated with both cisplatin and carboplatin in the metastatic setting. Forty-six percent of patients had an ECOG performance status of 1. Eighteen percent of patients had a hemoglobin <10 g/dL, and twenty-eight percent of patients had liver metastases at baseline. Patients were included regardless of their PD-L1 status.
Tumor specimens were evaluated prospectively using the PD-L1 IHC 28-8 pharmDx assay at a central laboratory and the results were used to define subgroups for pre-specified analyses. Of the 270 patients, 46% were defined as having PD-L1 expression of ≥1% (defined as ≥1% of tumor cells expressing PD-L1). The remaining 54% of patients were classified as having PD-L1 expression of <1% (defined as <1% of tumor cells expressing PD-L1). Confirmed ORR in all patients and the two PD-L1 subgroups are shown in Table 75. Median time to response was 1.9 months (range: 1.6-7.2). In 77 patients who received prior systemic therapy only in the neoadjuvant or adjuvant setting, the ORR was 23.4% (95% CI: 14.5%, 34.4%).
| a Estimated from the Kaplan-Meier Curve. b Not Reached. |
|||
|
All Patients |
PD-L1 <1% |
PD-L1 ≥1% |
|
|
Confirmed Overall Response Rate, n (%)
|
53 (19.6%) |
22 (15.1%) |
31 (25.0%) |
|
Complete response rate |
7 (2.6%) |
1 (0.7%) |
6 (4.8%) |
|
Partial response rate |
46 (17.0%) |
21 (14.4%) |
25 (20.2%) |
|
Median Duration of Responsea (months) (range) |
10.3 (1.9+, 12.0+) |
7.6 (3.7, 12.0+) |
NRb (1.9+, 12.0+) |
14.11 Microsatellite Instability-High or Mismatch Repair Deficient Metastatic Colorectal Cancer
Treatment of MSI-H or dMMR mCRC In Combination with Ipilimumab
CHECKMATE-8HW (NCT03143153) was a randomized, 3-arm, open-label trial in immunotherapy-naive patients across all lines of therapy with unresectable or metastatic CRC with known tumor MSI-H or dMMR (MSI-H/dMMR) status as determined in accordance with local standard of practice using PCR, NGS, or IHC assays. Central assessment of MSI-H status using PCR (Idylla MSI) test and dMMR status using IHC (Omnis MMR) test was conducted retrospectively on patient tumor specimens used for local MSI-H/dMMR status determination. Patients with confirmed MSI-H/dMMR status by either central test comprised the primary study population.
The trial excluded patients with brain metastasis that were symptomatic, had active autoimmune disease, used systemic corticosteroids or immunosuppressants, or had been treated with checkpoint inhibitors.
Patients were randomized to receive one of the following treatments:
- •
- OPDIVO 240 mg every 3 weeks and ipilimumab 1 mg/kg every 3 weeks for a maximum of 4 doses, then OPDIVO 480 mg every 4 weeks.
- •
- OPDIVO 240 mg every 2 weeks for 6 doses, then OPDIVO 480 mg every 4 weeks.
- •
- Investigator’s choice chemotherapy
- o
- mFOLFOX6 (oxaliplatin, leucovorin, and FU) with or without either bevacizumab or cetuximab: Oxaliplatin 85 mg/m2, leucovorin 400 mg/m2, and FU 400 mg/m2 bolus followed by FU 2400 mg/m2 over 46 hours every 2 weeks. Bevacizumab 5 mg/kg or cetuximab 500 mg/m2 administered prior to mFOLFOX6 every 2 weeks.
- o
- FOLFIRI (irinotecan, leucovorin, and FU) with or without either bevacizumab or cetuximab: Irinotecan 180 mg/m2, leucovorin 400 mg/m2, and FU 400 mg/m2 bolus and FU 2400 mg/m2 over 46 hours every 2 weeks. Bevacizumab 5 mg/kg on or cetuximab 500 mg/m2 administered prior to FOLFIRI every 2 weeks.
Randomization was stratified by tumor location (right vs left) and by prior lines of therapy (0, 1, 2L+). Patients randomized to the chemotherapy arm could receive OPDIVO in combination with ipilimumab upon progression assessed by BICR.
Study treatment was administered until disease progression, unacceptable toxicity, or for up to 2 years for patients who received OPDIVO plus ipilimumab or nivolumab monotherapy. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue OPDIVO as a single agent. OPDIVO with or without ipilimumab could be administered beyond RECIST 1.1-assessed progressive disease if there was a clinical benefit as determined by investigator and therapy was tolerated. Tumor assessments per RECIST v1.1 were conducted every 6 weeks for the first 24 weeks, then every 8 weeks thereafter up until week 96, then every 16 weeks thereafter up until week 144, and then every 24 weeks.
The evaluation of efficacy relied on the comparison of patients with centrally confirmed MSI-H/dMMR mCRC randomized to OPDIVO in combination with ipilimumab versus chemotherapy in the first-line (1L) setting and the comparison of patients with centrally confirmed MSI-H/dMMR mCRC randomized to OPDIVO plus ipilimumab vs nivolumab in all lines setting.
The major efficacy outcome measure was BICR-assessed PFS per RECIST 1.1. Additional efficacy outcome measures included ORR and duration of response assessed by BICR and OS.
The baseline characteristics of the total of 839 patients randomized were: the median age was 63 years (range: 20 to 87), with 46% ≥65 years of age and 14% ≥75 years of age; 50% were male and 87% were White, 9.3% were Asian, 1.5% Black or African American, and 2.3% other race; 9.2% were Hispanic or Latino, 50% Not Hispanic or Latino, 41% ethnicity unknown. Baseline ECOG performance status was 0 (52%) and 1 (48%); number of prior lines of therapy was 0 (56%), 1 (24%), and ≥2 (19%); and tumor location was right-sided or left-sided for 69% and 31% of patients. The baseline characteristics in patients with centrally confirmed MSI-H/dMMR is consistent with that of all randomized patients.
First Line OPDIVO in combination with ipilimumab
Among 303 patients in the first-line setting who were randomly assigned to OPDIVO in combination with ipilimumab (202) and to chemotherapy (101), 171 and 84 patients had centrally confirmed MSI-H/dMMR status in OPDIVO in combination with ipilimumab arm and chemotherapy arm, respectively.
In the 1L setting 200 of 202 patients assigned to receive OPDIVO combined with ipilimumab and 88 of 101 patients assigned to receive chemotherapy received at least 1 dose of study treatment. Among the 88 patients who received chemotherapy, 58% and 42% of patients received oxaliplatin-containing regimens and irinotecan-containing regimens, respectively, and 66 (75%) patients received a targeted agent, either bevacizumab (64%) or cetuximab (11%).
The BICR-assessed PFS efficacy results for patients with centrally confirmed MSI-H/dMMR randomized to the OPDIVO and ipilimumab arm compared with chemotherapy in the 1L setting are presented in Table 76 and Figure 23. The comparative results of ORR and OS between arms were not available at the time of the PFS analysis due to statistical testing strategy.
| NR: Not Reached; NE: Not Estimable. | ||
| Minimum follow-up was 6.1 months at data cutoff date 12Oct2023. | ||
| a Based on log-rank test stratified by the same factors as used in the Cox proportional hazards model. The p-value threshold for statistical significance was 0.0209. | ||
| b Based on Kaplan-Meier estimates. | ||
| c HR from a Cox proportional hazards model stratified by tumor sidedness (left vs right) per IRT. | ||
|
OPDIVO and Ipilimumab |
Chemotherapy |
|
|
Progression-free Survival |
||
|
Disease progression or death (%) |
48 (28) |
52 (62) |
|
Median in monthsb (95% CI) |
NR (38.4, NE) |
5.8 (4.4, 7.8) |
|
Hazard ratioc (95% CI) |
0.21 (0.14, 0.32) |
|
|
p-valuea |
<0.0001 |
|
Figure 23: Progression-free Survival (First Line OPDIVO + Ipilimumab vs Chemotherapy) - CHECKMATE-8HW
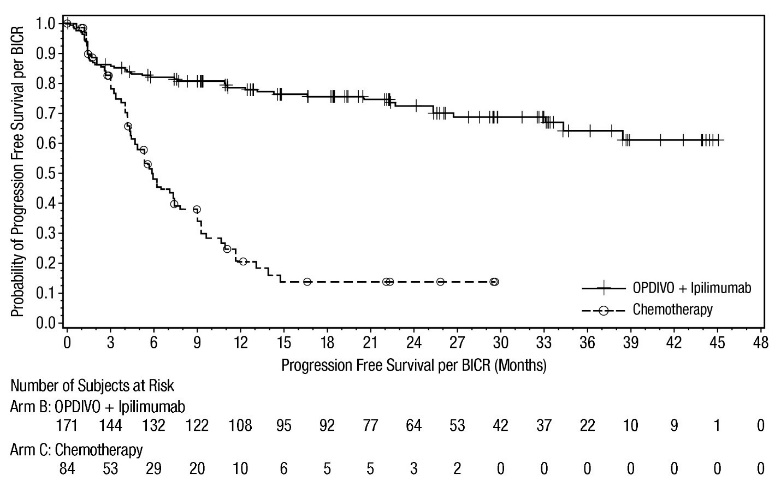
All Lines OPDIVO in combination with ipilimumab
Among 707 patients across all treatment lines who were randomly assigned to OPDIVO in combination with ipilimumab (354) and to OPDIVO (353) single agent, 296 and 286 patients had centrally confirmed MSI-H/dMMR status in the OPDIVO in combination with ipilimumab arm and in the OPDIVO arm, respectively. Patients receiving at least 1 dose of study treatment included 352 of 354 patients randomized to OPDIVO in combination with ipilimumab, and 351 of 353 patients randomized to single agent OPDIVO.
The BICR-assessed PFS and ORR efficacy results for patients with centrally confirmed MSI-H/dMMR randomized to the OPDIVO in combination with ipilimumab compared with nivolumab single agent across all treatment lines setting are presented in Table 77 and Figure 24. The comparative results of OS between arms were not available at the time of the PFS analysis due to statistical testing strategy.
| OPDIVO and Ipilimumab (n=296) | OPDIVO (n=286) |
|
|---|---|---|
| NR: Not Reached; NE: Not Estimable. | ||
| Minimum follow-up was 16.7 months at data cutoff date 28Aug2024. | ||
| a Based on log-rank test stratified by the same factors as used in the Cox proportional hazards model. The p-value threshold for statistical significance was 0.0095. | ||
| b Based on Kaplan-Meier estimates. | ||
| c HR from a Cox proportional hazards model stratified by tumor sidedness (left vs right) and prior lines of therapy (0, 1, ≥2) per IRT. | ||
| d Based on Cochran-Mantel-Haenszel test stratified by the same factors as used in the Cox proportional hazards model. The p-value threshold for statistical significance was 0.006. | ||
|
Progression-free Survival |
||
|
Disease progression or death n (%) |
101 (34) |
136 (48) |
|
Median (months)b (95% CI) |
NR (53.8, NE) |
39.3 (22.1, NE) |
|
Hazard ratioc (95% CI) |
0.62 (0.48, 0.81) |
|
|
p-valuea |
0.0003 |
|
|
Objective Response Rate (ORR) |
||
|
Response Rate, n (%) (95% CI) |
209 (71%) (65, 76) |
165 (58%) (52, 63) |
|
Complete Response Rate, n (%) |
90 (30%) |
80 (28%) |
|
Partial Response Rate, n (%) |
119 (40%) |
85 (30%) |
|
p-valued |
0.0011 |
|
Figure 24: Progression-free Survival (All lines OPDIVO + Ipilimumab vs OPDIVO) - CHECKMATE-8HW
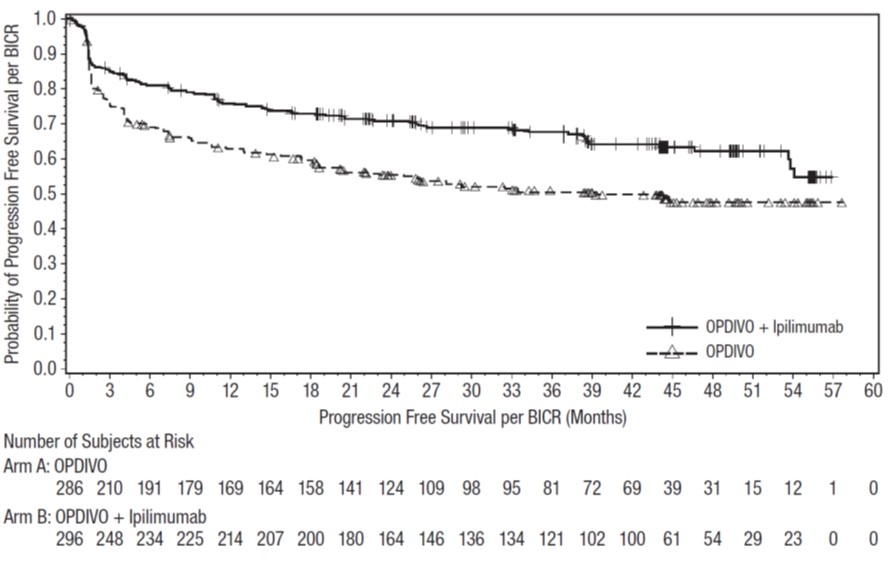
Treatment of MSI-H or dMMR mCRC after Progression Following Treatment with a Fluoropyrimidine, Oxaliplatin, and Irinotecan
CHECKMATE-142 (NCT02060188) was a multicenter, non-randomized, multiple parallel-cohort, open-label trial conducted in patients with locally determined dMMR or MSI-H metastatic CRC (mCRC) who had disease progression during or after prior treatment with fluoropyrimidine- , oxaliplatin- , or irinotecan-based chemotherapy. Key eligibility criteria were at least one prior line of treatment for metastatic disease, ECOG performance status 0 or 1, and absence of the following: active brain metastases, active autoimmune disease, or medical conditions requiring systemic immunosuppression.
Patients enrolled in the single agent OPDIVO MSI-H mCRC cohort received OPDIVO 3 mg/kg by intravenous infusion (IV) every 2 weeks. Patients enrolled in the OPDIVO and ipilimumab MSI-H mCRC cohort received OPDIVO 3 mg/kg and ipilimumab 1 mg/kg intravenously every 3 weeks for 4 doses, followed by OPDIVO as a single agent at a dose of 3 mg/kg as intravenous infusion every 2 weeks. Treatment in both cohorts continued until unacceptable toxicity or radiographic progression.
Tumor assessments were conducted every 6 weeks for the first 24 weeks and every 12 weeks thereafter. Efficacy outcome measures included ORR and DOR as assessed by BICR using RECIST v1.1.
A total of 74 patients were enrolled in the single-agent MSI-H mCRC OPDIVO cohort. The median age was 53 years (range: 26 to 79) with 23% ≥65 years of age and 5% ≥75 years of age, 59% were male and 88% were White. Baseline ECOG performance status was 0 (43%), 1 (55%), or 3 (1.4%) and 36% were reported to have Lynch Syndrome. Across the 74 patients, 72% received prior treatment with a fluoropyrimidine, oxaliplatin, and irinotecan; 7%, 30%, 28%, 19%, and 16% received 0, 1, 2, 3, or ≥4 prior lines of therapy for metastatic disease, respectively, and 42% of patients had received an anti-EGFR antibody.
A total of 119 patients were enrolled in the OPDIVO and ipilimumab MSI-H mCRC cohort. The median age was 58 years (range: 21 to 88), with 32% ≥65 years of age and 9% ≥75 years of age; 59% were male and 92% were White. Baseline ECOG performance status was 0 (45%) and 1 (55%), and 29% were reported to have Lynch Syndrome. Across the 119 patients, 69% had received prior treatment with a fluoropyrimidine, oxaliplatin, and irinotecan; 10%, 40%, 24%, and 15% received 1, 2, 3, or ≥4 prior lines of therapy for metastatic disease, respectively, and 29% had received an anti-EGFR antibody.
Efficacy results for each of these single-arm cohorts are shown in Table 78.
| a Minimum follow-up 33.7 months for all patients treated with OPDIVO (n=74). b Minimum follow-up 27.5 months for all patients treated with OPDIVO and ipilimumab (n=119). c Estimated using the Clopper-Pearson method. |
||||
|
OPDIVOa
|
OPDIVO and Ipilimumabb
|
|||
|
All Patients |
Prior Treatment |
All Patients |
Prior Treatment |
|
|
Overall Response Rate per BICR; n (%) |
28 (38%) |
17 (32%) |
71 (60%) |
46 (56%) |
|
(95% CI)c |
(27, 50) |
(20, 46) |
(50, 69) |
(45, 67) |
|
Complete Response (%) |
8 (11%) |
5 (9%) |
17 (14%) |
11 (13%) |
|
Partial Response (%) |
20 (27%) |
12 (23%) |
54 (45%) |
35 (43%) |
|
Duration of Response |
||||
|
Proportion of responders with ≥6 months response duration |
86% |
94% |
89% |
87% |
|
Proportion of responders with ≥12 months response duration |
82% |
88% |
77% |
74% |
14.12 Hepatocellular Carcinoma
Treatment of Unresectable or Metastatic Hepatocellular Carcinoma (HCC)
CHECKMATE-9DW (NCT04039607) was a randomized (1:1), open-label trial in adults (18 years of age or older) with unresectable or metastatic HCC. Patients had histologically confirmed HCC, Child Pugh Class A, ECOG performance status 0 or 1, and no prior systemic therapy for advanced disease. Esophagogastroduodenoscopy was not mandated prior to enrollment. The trial excluded patients with active autoimmune disease, brain or leptomeningeal metastases, a history of hepatic encephalopathy (within 12 months of randomization), a platelet count <60,000, clinically significant ascites, medical conditions requiring systemic immunosuppression, infection with HIV, or active co-infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) or HBV and hepatitis D virus (HDV).
Patients were randomized to receive either:
- •
- OPDIVO 1 mg/kg administered intravenously over 30 minutes in combination with ipilimumab 3 mg/kg administered intravenously over 30 minutes every 3 weeks, for a maximum of 4 doses, followed by single agent OPDIVO at 480 mg administered intravenously over 30 minutes every 4 weeks, or
- •
- Investigator’s choice:
- o
- Lenvatinib 8 mg orally daily (if body weight <60 kg) or 12 mg orally daily (if body weight ≥60 kg), or
- o
- Sorafenib 400 mg orally twice daily
Randomization was stratified by etiology (HBV vs. HCV vs. non-viral), macrovascular invasion and/or extrahepatic spread (present or absent), and alpha-fetoprotein levels (≥400 or <400 ng/mL). Study treatment for OPDIVO in combination with ipilimumab continued until disease progression, unacceptable toxicity, or up to 2 years. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue OPDIVO as a single agent. Treatment beyond RECIST 1.1 defined disease progression was permitted if the patient was clinically stable and considered to be deriving clinical benefit by the investigator. Tumor assessments were performed at baseline, after randomization at week 9 and week 16, then every 8 weeks up to 48 weeks, and then every 12 weeks thereafter until disease progression, treatment discontinuation, or initiation of subsequent therapy. The primary efficacy outcome measure was OS in all randomized patients. Additional efficacy measures included BICR-assessed ORR and DOR based on RECIST 1.1 criteria.
A total of 668 patients were randomized to receive OPDIVO in combination with ipilimumab (n=335) or investigator’s choice (n=333) of lenvatinib or sorafenib. In the investigator arm, 85% and 15% of treated patients received lenvatinib or sorafenib, respectively. The trial population characteristics were median age 66 years (range: 20 to 89), with 53% ≥65 years old; 82% male; 53% White, 44% Asian, 2.2% Black; 12% Hispanic or Latino, 48% Not Hispanic or Latino, 40% not reported. Baseline ECOG performance status was 0 (71%) or 1 (29%). Thirty-four percent (34%) of patients had HBV infection, 28% had HCV infection, and 36% had no evidence of HBV or HCV infection.
Nineteen percent (19%) of patients had alcoholic liver disease and 11% had non-alcoholic fatty liver disease. The majority of patients had BCLC stage C (73%) disease at baseline, 19% had stage B, and 6% had stage A. Patients with Child-Pugh scores of 5, 6, and 7 were 77%, 20%, and 3%, respectively; 1 patient with Child Pugh 8 was enrolled. A total of 54% of patients had extrahepatic spread; 25% had macrovascular invasion; and 33% had AFP levels ≥400 µg/L.
CHECKMATE-9DW demonstrated a statistically significant improvement in OS and ORR. The minimum follow-up was 26.8 months. Efficacy results are shown in Table 79 and Figure 25.
| OPDIVO and Ipilimumab (n=335) | Lenvatinib or Sorafenib (n=333) |
|
|---|---|---|
| a Based on stratified Cox proportional hazard model. | ||
| b Based on a 2-sided stratified log-rank test. Boundary for statistical significance: p-value ≤0.0257. | ||
| c Assessed by BICR using RECIST 1.1. | ||
| d Based on a 2-sided stratified Cochran-Mantel-Haenszel test. Boundary for statistical significance: p-value ≤0.025. | ||
| e NR: Not Reached. | ||
| + Censored observation. | ||
|
Overall Survival |
||
|
Deaths (%) |
194 (58%) |
228 (68%) |
|
Median (months) (95% CI) |
23.7 (18.8, 29.4) |
20.6 (17.5, 22.5) |
|
Hazard ratio (95% CI)a |
0.79 (0.65, 0.96) |
|
|
p-valueb |
0.0180 |
|
|
Overall Response Rate, n (%)c |
121 (36.1) |
44 (13.2) |
|
(95% CI) |
(31.0, 41.5) |
(9.8, 17.3) |
|
p-valued |
<0.0001 |
|
|
Complete response (%) |
23 (6.9) |
6 (1.8) |
|
Partial response (%) |
98 (29.3) |
38 (11.4) |
|
Duration of Response (months)c |
||
|
Median (95% CI) |
30.4 (21.2, NRe) |
12.9 (10.2, 31.2) |
|
Range |
1.5+, 36.9+ |
2.1+, 32.5+ |
Figure 25: Overall Survival - CHECKMATE-9DW
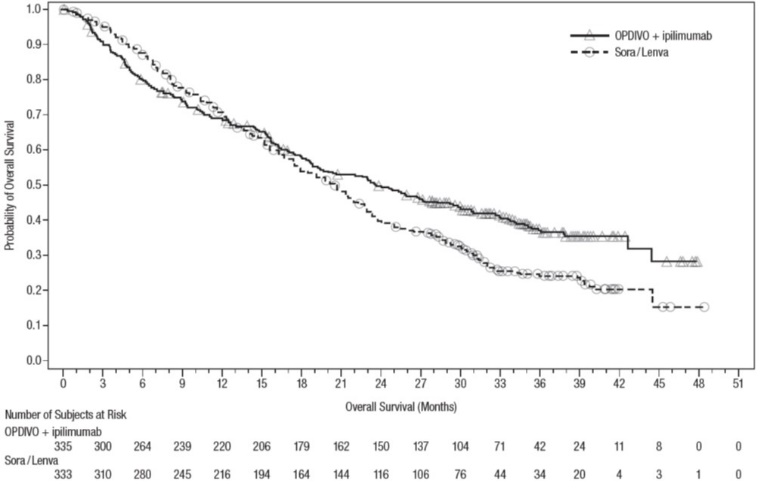
Previously Treated Hepatocellular Carcinoma
CHECKMATE-040 (NCT01658878) was a multicenter, multiple cohort, open-label trial that evaluated the efficacy of OPDIVO as a single agent and in combination with ipilimumab in patients with hepatocellular carcinoma (HCC) who progressed on or were intolerant to sorafenib. Additional eligibility criteria included histologic confirmation of HCC and Child-Pugh Class A cirrhosis. The trial excluded patients with active autoimmune disease, brain metastasis, a history of hepatic encephalopathy, clinically significant ascites, infection with HIV, or active co-infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) or HBV and hepatitis D virus (HDV); however, patients with only active HBV or HCV were eligible.
Tumor assessments were conducted every 6 weeks for 48 weeks and then every 12 weeks thereafter. The major efficacy outcome measure was confirmed overall response rate as assessed by BICR using RECIST v1.1 and modified RECIST (mRECIST) for HCC. Duration of response was also assessed.
The efficacy of OPDIVO in combination with ipilimumab was evaluated in 49 patients (Cohort 4) who received OPDIVO 1 mg/kg and ipilimumab 3 mg/kg administered every 3 weeks for 4 doses, followed by single-agent OPDIVO at 240 mg every 2 weeks until disease progression or unacceptable toxicity. The median age was 60 years (range: 18 to 80), 88% were male, 74% were Asian, and 25% were White. Baseline ECOG performance status was 0 (61%) or 1 (39%). Fifty-seven (57%) percent of patients had active HBV infection, 8% had active HCV infection, and 35% had no evidence of active HBV or HCV. The etiology for HCC was alcoholic liver disease in 16% and non-alcoholic fatty liver disease in 6% of patients. Child-Pugh class and score was A5 for 82% and A6 for 18%; 80% of patients had extrahepatic spread; 35% had vascular invasion; and 51% had AFP levels ≥400 µg/L. Prior cancer treatment history included surgery (74%), radiotherapy (29%), or local treatment (59%). All patients had received prior sorafenib, of whom 10% were unable to tolerate sorafenib; 29% of patients had received 2 or more prior systemic therapies.
Efficacy results are shown in Table 80. The results for OPDIVO in combination with ipilimumab in Cohort 4 are based on a minimum follow-up of 28 months.
| a Confirmed by BICR. b Confidence interval is based on the Clopper and Pearson method. |
|
|
OPDIVO and Ipilimumab |
|
|
Overall Response Rate per BICR,a n (%), RECIST v1.1 |
16 (33%) |
|
(95% CI)b |
(20, 48) |
|
Complete response |
4 (8%) |
|
Partial response |
12 (24%) |
|
Duration of Response per BICR,a RECIST v1.1 |
n=16 |
|
Range (months) |
4.6, 30.5+ |
|
Percent with duration ≥6 months |
88% |
|
Percent with duration ≥12 months |
56% |
|
Percent with duration ≥24 months |
31% |
|
Overall Response Rate per BICR,a n (%), mRECIST |
17 (35%) |
|
(95% CI)b |
(22, 50) |
|
Complete response |
6 (12%) |
|
Partial response |
11 (22%) |
14.13 Esophageal Cancer
Adjuvant Treatment of Resected Esophageal or Gastroesophageal Junction Cancer
CHECKMATE-577 (NCT02743494) was a randomized, multicenter, double-blind trial in 794 patients with completely resected (negative margins) esophageal or gastroesophageal junction cancer who had residual pathologic disease following concurrent chemoradiotherapy (CRT). Patients were randomized (2:1) to receive either OPDIVO 240 mg or placebo by intravenous infusion over 30 minutes every 2 weeks for 16 weeks followed by 480 mg or placebo by intravenous infusion over 30 minutes every 4 weeks beginning at week 17. Treatment was until disease recurrence, unacceptable toxicity, or for up to 1 year in total duration. Enrollment required complete resection within 4 to 16 weeks prior to randomization. The trial excluded patients who did not receive CRT prior to surgery, had stage IV resectable disease, autoimmune disease, or any condition requiring systemic treatment with either corticosteroids (>10 mg daily prednisone or equivalent) or other immunosuppressive medications. Randomization was stratified by tumor PD-L1 status (≥1% vs. <1% or indeterminate or non-evaluable), pathologic lymph node status (positive ≥ypN1 vs. negative ypN0), and histology (squamous vs. adenocarcinoma). The major efficacy outcome measure was disease-free survival (DFS) defined as the time between the date of randomization and the date of first recurrence (local, regional, or distant from the primary resected site) or death, from any cause, whichever occurred first as assessed by the investigator prior to subsequent anti-cancer therapy. Patients on treatment underwent imaging for tumor recurrence every 12 weeks for 2 years, and a minimum of one scan every 6 to 12 months for years 3 to 5.
The trial population characteristics were: median age 62 years (range: 26 to 86), 36% were ≥65 years of age, 85% were male, 15% were Asian, 82% were White, and 1.1% were Black. Disease characteristics were AJCC Stage II (35%) or Stage III (65%) at initial diagnosis carcinoma, EC (60%) or GEJC (40%) at initial diagnosis, with pathologic positive lymph node status (58%) at study entry and histological confirmation of predominant adenocarcinoma (71%) or squamous cell carcinoma (29%). The baseline Tumor PD-L1 status ≥1% was positive for 16% of patients and negative for 72% of patients. Baseline ECOG performance status was 0 (58%) or 1 (42%).
CHECKMATE-577 demonstrated a statistically significant improvement in DFS for patients randomized to the OPDIVO arm as compared with the placebo arm. DFS benefit was observed regardless of tumor PD-L1 expression and histology.
Efficacy results are shown in Table 81 and Figure 26.
| a Based on a stratified proportional hazards model. b Based on a stratified log-rank test. |
||
|
OPDIVO |
Placebo |
|
|
Disease-free Survival |
||
|
Number of events, n (%) |
241 (45%) |
155 (59%) |
|
Median (months) |
22.4 |
11.0 |
|
Hazard ratioa (95% CI) |
0.69 (0.56, 0.85) |
|
|
p-valueb |
0.0003 |
|
Figure 26: Disease-free Survival - CHECKMATE-577
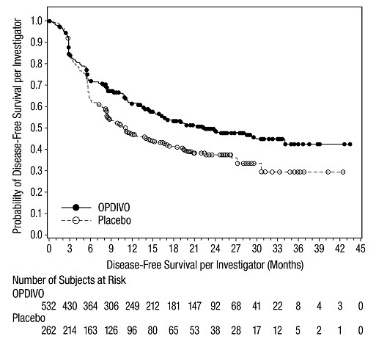
First-line Treatment of Unresectable Advanced or Metastatic ESCC Whose Tumors Express PD-L1 (≥1)
CHECKMATE-648 (NCT03143153) was a randomized, active-controlled, open-label trial in patients with previously untreated unresectable advanced, recurrent or metastatic ESCC (squamous or adenosquamous histology). The trial enrolled patients whose tumor was evaluable for tumor cell (TC) PD-L1 expression [also called PD-L1 tumor proportion score (TPS)], which was evaluated using the PD-L1 IHC 28-8 pharmDx assay at a central laboratory. A retrospective scoring of a patient’s tumor PD-L1 status using Combined Positive Score (CPS), was also conducted using the PD-L1-stained tumor specimens used for randomization. Patients were not amenable to chemoradiation or surgery with curative intent. Prior treatment with curative intent was allowed if completed more than six months prior to trial enrollment. The trial excluded patients with brain metastasis that were symptomatic, had active autoimmune disease, used systemic corticosteroids or immunosuppressants, or patients at high risk of bleeding or fistula due to apparent invasion of tumor to organs adjacent to the esophageal tumor. Patients were randomized to receive one of the following treatments:
- •
- OPDIVO 240 mg on days 1 and 15, fluorouracil 800 mg/m2/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m2 intravenously on day 1 (of a 4-week cycle).
- •
- OPDIVO 3 mg/kg every 2 weeks in combination with ipilimumab 1 mg/kg every 6 weeks.
- •
- Fluorouracil 800 mg/m2/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m2 intravenously on day 1 (of a 4-week cycle).
Patients received OPDIVO until disease progression, unacceptable toxicity, or up to 2 years. In patients who received OPDIVO in combination with chemotherapy and in whom either fluorouracil and/or cisplatin were discontinued, other components of the treatment regimen were allowed to be continued. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue OPDIVO as a single agent.
Randomization was stratified by TC PD-L1 expression (≥1% vs. <1% or indeterminate), region (East Asia vs. Rest of Asia vs. Rest of World), ECOG performance status (0 vs. 1), and number of organs with metastases (≤1 vs. ≥2). The major efficacy outcome measures were OS and BICR-assessed PFS in patients with TC PD-L1 expression ≥ 1%. Additional efficacy measures included OS in all randomized patients, BICR-assessed PFS in all randomized patients, and ORR assessed by BICR in TC PD-L1 expression ≥ 1% and in all randomized patients. The tumor assessments per RECIST v1.1 were conducted every 6 weeks up to and including week 48, then every 12 weeks thereafter.
A total of 970 patients were randomized in the CHECKMATE-648 study, among whom 965 and 906 patients had quantifiable TC PD-L1 expression and CPS at baseline, respectively; 85% (824/970) had tumors with PD-L1 CPS ≥1. The trial population characteristics in patients with PD-L1 CPS ≥1 were: median age 63 years (range: 26 to 90), 46% were ≥65 years of age, 82% were male, 71% were Asian, 25% were White, and 1.2% were Black or African American. Patients had histological confirmation of squamous cell carcinoma (99%) or adenosquamous cell carcinoma (1.7%) in the esophagus. Baseline ECOG performance status was 0 (44.0%) or 1 (54%).
A statistically significant improvement in OS was demonstrated in patients randomized to OPDIVO in combination with chemotherapy and patients randomized to OPDIVO in combination with ipilimumab compared with chemotherapy. An exploratory analysis of OS in patients with PD-L1 CPS <1 showed a HR of 0.98 (95% CI 0.50, 1.95) for the comparison of OPDIVO in combination with chemotherapy, and the exploratory analysis OS in patients with PD-L1 CPS <1 showed a HR of 1.0 (95% CI 0.52, 1.94) for the comparison of OPDIVO in combination with ipilimumab; these results indicate that the improvement in the ITT population was primarily attributed to the results observed in the subgroup of patients with PD-L1 CPS ≥1. Efficacy results are shown in Table 82 and Figures 27 and 28.
| a Assessed by BICR. b Based on stratified Cox proportional hazard model. Hazard ratios are reported for each OPDIVO containing arm compared to chemotherapy within each analysis population. c Based on a stratified 2-sided log-rank test. S1, S2, S3 Significant p-value compared to stopping boundary of 0.005, 0.014, and 0.015 respectively. NS: Not Statistically significant, NT: Not evaluated for statistical significance as per pre-specified hierarchical testing procedure. |
||||||
|
OPDIVO with Cisplatin and Fluorouracil |
OPDIVO and Ipilimumab |
Cisplatin and Fluorouracil |
OPDIVO with Cisplatin and Fluorouracil |
OPDIVO and Ipilimumab |
Cisplatin and Fluorouracil |
|
|
TC PD-L1 expression ≥1% |
PD-L1 CPS ≥1 |
|||||
|
Overall Survival |
||||||
|
Deaths (%) |
98 (62) |
106 (67) |
121 (77) |
177 (64) |
179 (67) |
205 (73) |
|
Median (months) (95% CI) |
15.4 (11.9, 19.5) |
13.7 (11.2, 17.0) |
9.1 (7.7, 10) |
13.8 (12.0, 16.1) |
12.7 (10.9, 15.5) |
9.8 (8.8, 11.6) |
|
Hazard ratio (95% CI)b |
0.54 (0.41, 0.71) |
0.64 (0.49, 0.84) |
- |
0.69 (0.57, 0.85) |
0.76 (0.62, 0.93) |
- |
|
p-valuec |
<0.0001S1 |
0.0010S2 |
- |
- |
- |
- |
|
Progression-free Survivala |
||||||
|
Disease progression or death (%) |
117 (74) |
123 (78) |
100 (64) |
201 (72) |
206 (77) |
184 (66) |
|
Median (months) (95% CI) |
6.9 (5.7, 8.3) |
4.0 (2.4, 4.9) |
4.4 (2.9, 5.8) |
5.8 (5.5, 7.0) |
2.8 (2.6, 4.2) |
5.6 (4.2, 5.9) |
|
Hazard ratio (95% CI)b |
0.65 (0.49, 0.86) |
1.02 (0.78, 1.34) |
- |
0.8 (0.7, 1.0) |
1.2 (1.0, 1.5) |
- |
|
p-valuec |
0.0023S3 |
NS |
- |
- |
- |
- |
|
Overall Response Rate, n (%)a, NT |
84 (53.2) |
56 (35.4) |
31 (19.7) |
135 (49) |
74 (28) |
76 (27) |
|
(95% CI) |
(45.1, 61.1) |
(28.0, 43.4) |
(13.8, 26.8) |
(42.5, 54.6) |
(22.5, 33.6) |
(22.0, 32.8) |
|
Complete response (%) |
26 (16.5) |
28 (17.7) |
8 (5.1) |
39 (14) |
32 (12.0) |
18 (6.4) |
|
Partial response (%) |
58 (36.7) |
28 (17.7) |
23 (14.6) |
96 (35) |
42 (15.8) |
58 (20.7) |
|
Duration of Response (months)a |
||||||
|
Median (95% CI) |
8.4 (6.9, 12.4) |
11.8 (7.1, 27.4) |
5.7 (4.4, 8.7) |
8.2 (6.7, 11.1) |
11.8 (7.1, 23.6) |
6.9 (5.7, 8.2) |
|
Range |
1.4+, 34.6 |
1.4+, 34.5+ |
1.4+, 31.8+ |
1.4+, 35.9+ |
1.4+, 34.5+ |
1.4+, 31.8+ |
Figure 27: Overall Survival – CHECKMATE-648
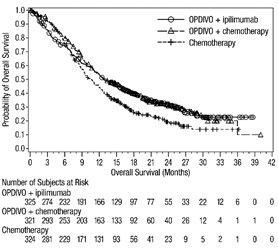
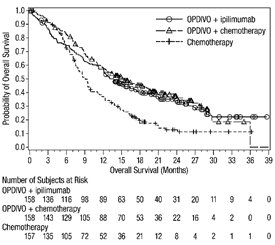
Figure 28: Progression-free Survival – CHECKMATE-648
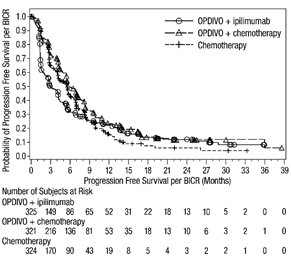
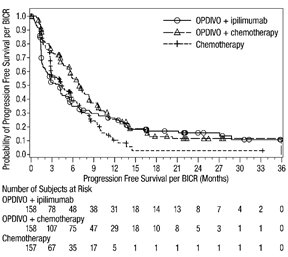
Previously Treated Unresectable Advanced, Recurrent or Metastatic Esophageal Squamous Cell Carcinoma (ESCC)
ATTRACTION-3 (NCT02569242) was a multicenter, randomized (1:1), active-controlled, open-label trial in patients with unresectable advanced, recurrent, or metastatic ESCC, who were refractory or intolerant to at least one fluoropyrimidine- and platinum-based regimen. The trial enrolled patients regardless of PD-L1 status, but tumor specimens were evaluated prospectively using the PD-L1 IHC 28-8 pharmDx assay at a central laboratory. The trial excluded patients who were refractory or intolerant to taxane therapy, had brain metastases that were symptomatic or required treatment, had autoimmune disease, used systemic corticosteroids or immunosuppressants, or had apparent tumor invasion of organs adjacent to the esophageal tumor or had stents in the esophagus or respiratory tract. Patients were randomized to receive OPDIVO 240 mg by intravenous infusion over 30 minutes every 2 weeks or investigator’s choice of taxane chemotherapy consisting of docetaxel (75 mg/m2 intravenously every 3 weeks) or paclitaxel (100 mg/m2 intravenously once a week for 6 weeks followed by 1 week off).
Randomization was stratified by region (Japan vs. Rest of World), number of organs with metastases (≤1 vs. ≥2), and PD-L1 status (≥1% vs. <1% or indeterminate). Patients were treated until disease progression, assessed by the investigator per RECIST v1.1, or unacceptable toxicity. The tumor assessments were conducted every 6 weeks for 1 year, and every 12 weeks thereafter. The major efficacy outcome measure was OS. Additional efficacy outcome measures were ORR and PFS as assessed by the investigator using RECIST v1.1 and DOR.
A total of 419 patients were randomized; 210 to the OPDIVO arm and 209 to the investigator’s choice arm (docetaxel: 31%, paclitaxel: 69%). The trial population characteristics were: median age 65 years (range: 33 to 87), 53% were ≥65 years of age, 87% were male, 96% were Asian and 4% were White. Sixty-seven percent of patients had received one prior systemic therapy regimen and 26% had received two prior systemic therapy regimens prior to enrolling in ATTRACTION-3. Baseline ECOG performance status was 0 (50%) or 1 (50%).
ATTRACTION-3 demonstrated a statistically significant improvement in OS for patients randomized to OPDIVO as compared with investigator’s choice of taxane chemotherapy. OS benefit was observed regardless of PD-L1 expression level. OS results by PD-L1 CPS level (<1 and ≥1) were not studied. The minimum follow-up was 17.6 months. Efficacy results are shown in Table 83 and Figure 29.
| a Based on ITT analysis b Based on a stratified proportional hazards model. c Based on a stratified log-rank test. d Based on Response Evaluable Set (RES) analysis, n=171 in OPDIVO group and n=158 in investigator’s choice group. e Based on stratified Cochran-Mantel-Haenszel test; p-value not significant. f PFS not tested due to pre-specified hierarchical testing strategy. |
||
|
OPDIVO |
Docetaxel or Paclitaxel |
|
|
Overall Survivala |
||
|
Deaths (%) |
160 (76%) |
173 (83%) |
|
Median (months) |
10.9 |
8.4 |
|
Hazard ratio (95% CI)b |
0.77 (0.62, 0.96) |
|
|
p-valuec |
0.0189 |
|
|
Overall Response Rated |
33 (19.3) |
34 (21.5) |
|
(95% CI) |
(13.7, 26.0) |
(15.4, 28.8) |
|
Complete response (%) |
1 (0.6) |
2 (1.3) |
|
Partial response (%) |
32 (18.7) |
32 (20.3) |
|
Median duration of response (months) |
6.9 |
3.9 |
|
p-valuee |
0.6323 |
|
|
Progression-free Survivala, f |
||
|
Disease progression or death (%) |
187 (89) |
176 (84) |
|
Median (months) |
1.7 |
3.4 |
|
Hazard ratio (95% CI)b |
1.1 (0.9, 1.3) |
|
Figure 29: Overall Survival - ATTRACTION-3
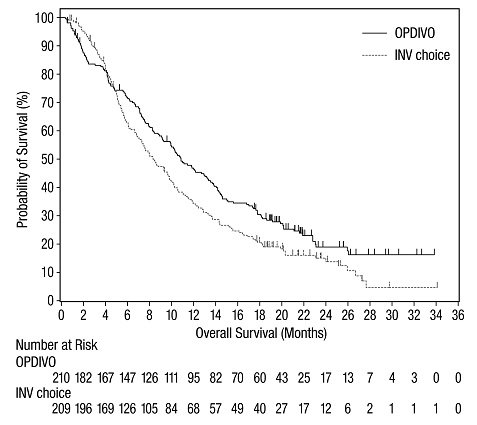
Of the 419 patients, 48% had PD-L1 positive ESCC, defined as ≥1% of tumor cells expressing PD-L1. The remaining 52% had PD-L1 negative ESCC defined as <1% of tumor cells expressing PD-L1.
In a pre-specified exploratory analysis by PD-L1 status, the hazard ratio (HR) for OS was 0.69 (95% CI: 0.51, 0.94) with median survivals of 10.9 and 8.1 months for the OPDIVO and investigator’s choice arms, respectively, in the PD-L1 positive subgroup. In the PD-L1 negative subgroup, the HR for OS was 0.84 (95% CI: 0.62, 1.14) with median survivals of 10.9 and 9.3 months for the OPDIVO and investigator’s choice arms, respectively.
14.14 Gastric Cancer, Gastroesophageal Junction Cancer, and Esophageal Adenocarcinoma Whose Tumors Express PD-L1 (≥1)
CHECKMATE-649 (NCT02872116) was a randomized, multicenter, open-label trial in patients (n=1581) with previously untreated advanced or metastatic gastric cancer, gastroesophageal junction cancer, and esophageal adenocarcinoma. The trial enrolled patients regardless of PD-L1 status, and tumor specimens were evaluated using the PD-L1 IHC 28-8 pharmDx assay at a central laboratory (tumor cell [TC] and Combined Positive Score [CPS]). The trial excluded patients who were known human epidermal growth factor receptor 2 (HER2) positive, or had untreated CNS metastases. Patients were randomized to receive OPDIVO in combination with chemotherapy (n=789) or chemotherapy (n=792). Patients received one of the following treatments:
- •
- OPDIVO 240 mg in combination with mFOLFOX6 (fluorouracil, leucovorin and oxaliplatin) every 2 weeks or mFOLFOX6 every 2 weeks.
- •
- OPDIVO 360 mg in combination with CapeOX (capecitabine and oxaliplatin) every 3 weeks or CapeOX every 3 weeks.
Patients were treated until disease progression, unacceptable toxicity, or up to 2 years. In patients who received OPDIVO in combination with chemotherapy and in whom chemotherapy was discontinued, OPDIVO monotherapy was allowed to be given at 240 mg every 2 weeks, 360 mg every 3 weeks, or 480 mg every 4 weeks up to 2 years after treatment initiation.
Randomization was stratified by tumor cell PD-L1 status (≥1% vs. <1% or indeterminate), region (Asia vs. US vs. Rest of World), ECOG performance status (0 vs. 1), and chemotherapy regimen (mFOLFOX6 vs. CapeOX). The major efficacy outcome measures, assessed in patients with PD-L1 CPS ≥5, were PFS assessed by BICR and OS. Additional efficacy outcome measures included OS and PFS in patients with PD-L1 CPS ≥1 and in all randomized patients, and ORR and DOR as assessed by BICR in patients with PD-L1 CPS ≥1 and ≥5, and in all randomized patients. Tumor assessments were conducted per RECIST v1.1 every 6 weeks up to and including week 48, then every 12 weeks thereafter.
A total of 1581 patients were randomized in the CHECKMATE-649 study, among whom 1296 and 955 had baseline PD-L1 CPS ≥1 and CPS ≥5 respectively. The trial population characteristics in patients with PD-L1 CPS ≥1 were: median age 62 years (range: 18 to 90), 40% were ≥65 years of age, 72% were male, 23% were Asian, and 69% were White, and 1% were Black or African American. Baseline ECOG performance status was 0 (42%) or 1 (58%). Seventy percent of patients had adenocarcinoma tumors in the stomach, 17% in the gastroesophageal junction, and 13% in the esophagus.
CHECKMATE-649 demonstrated a statistically significant improvement in OS and PFS for patients with PD-L1 CPS ≥5. Statistically significant improvement in OS was also demonstrated for all randomized patients and patients with PD-L1 CPS ≥1. Exploratory analysis of OS in the CPS <1 population showed a hazard ratio of 0.85 (95% CI: 0.63, 1.15), indicating that the improvement in the ITT population was primarily attributed to the results observed in the subgroup of patients with PD-L1 CPS ≥1. The minimum follow-up was 12.1 months. Efficacy results are shown in Table 84 and Figures 30, and 31.
| a Based on stratified Cox proportional hazard model. b Based on stratified log-rank test. c Assessed by BICR. d Based on confirmed response. e Not evaluated for statistical significance. |
||||
|
OPDIVO and mFOLFOX6 |
mFOLFOX6 |
OPDIVO and mFOLFOX6 |
mFOLFOX6 |
|
|
PD-L1 CPS ≥1 |
PD-L1 CPS ≥5 |
|||
|
Overall Survival |
||||
|
Deaths (%) |
434 (68) |
492 (75) |
309 (65) |
362 (75) |
|
Median |
14.0 |
11.3 |
14.4 |
11.1 |
|
Hazard ratio |
0.77 (0.68, 0.88) |
0.71 (0.61, 0.83) |
||
|
p-valueb |
<0.0001 |
<0.0001 |
||
|
Progression-free Survivalc |
||||
|
Disease |
454 (70.8) |
472 (72.1) |
328 (69.3) |
350 (72.6) |
|
Median |
7.5 |
6.9 |
7.7 |
6.0 |
|
Hazard ratio (95% CI)a |
0.74 (0.65, 0.85) |
0.68 (0.58, 0.79) |
||
|
p-valueb |
e |
<0.0001 |
||
|
Overall Response Rate, n (%)c,d |
314 (49) |
249 (38) |
237 (50) |
184 (38) |
|
(95% CI) |
(45, 53) |
(34, 42) |
(46, 55) |
(34, 43) |
|
Complete |
65 (10) |
42 (6) |
55 (12) |
34 (7) |
|
Partial |
249 (39) |
207 (32) |
182 (38) |
150 (31) |
|
Duration of Response (months)c,d |
||||
|
Median |
8.5 |
6.9 |
9.5 |
6.9 |
Figure 30: Overall Survival (PD-L1 CPS ≥1) - CHECKMATE-649
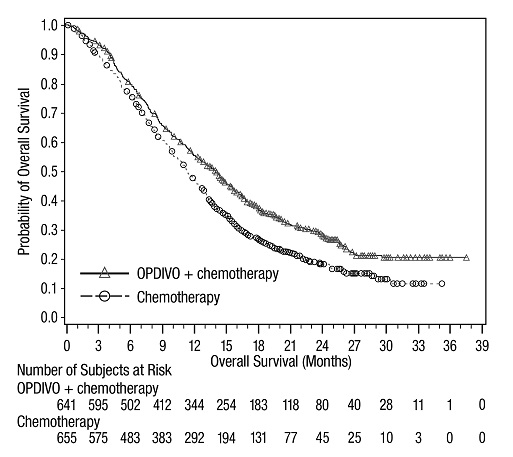
Figure 31: Overall Survival (PD-L1 CPS ≥5) - CHECKMATE-649
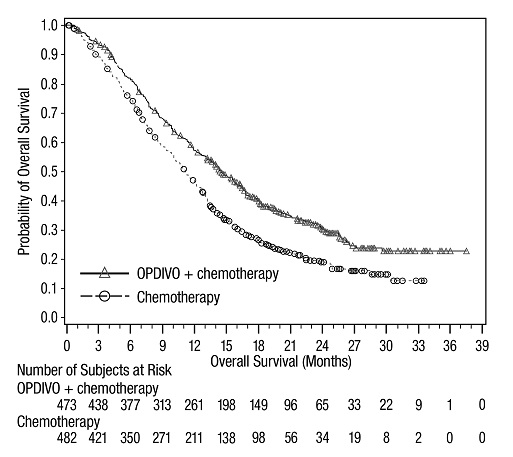
An exploratory analysis of OS in the 44 patients with MSI-H tumors showed a HR of 0.37 (95% CI: 0.16, 0.87).
16. How is Opdivo supplied
OPDIVO® (nivolumab) Injection is a clear to opalescent, colorless to pale-yellow solution in a single-dose vial available as follows:
|
Carton Contents |
NDC |
|
40 mg/4 mL (10 mg/mL) single-dose vial |
0003-3772-11 |
|
100 mg/10 mL (10 mg/mL) single-dose vial |
0003-3774-12 |
|
120 mg/12 mL (10 mg/mL) single-dose vial |
0003-3756-14 |
|
240 mg/24 mL (10 mg/mL) single-dose vial |
0003-3734-13 |
Store under refrigeration at 2°C to 8°C (36°F to 46°F). Protect from light by storing in the original package until time of use. Do not freeze or shake.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Immune-Mediated Adverse Reactions
Inform patients of the risk of immune-mediated adverse reactions that may require corticosteroid treatment and withholding or discontinuation of OPDIVO, including:
- •
- Pneumonitis: Advise patients to contact their healthcare provider immediately for any new or worsening cough, chest pain, or shortness of breath [see Warnings and Precautions (5.1)].
- •
- Colitis: Advise patients to contact their healthcare provider immediately for diarrhea or severe abdominal pain [see Warnings and Precautions (5.1)].
- •
- Hepatitis: Advise patients to contact their healthcare provider immediately for jaundice, severe nausea or vomiting, pain on the right side of abdomen, lethargy, or easy bruising or bleeding [see Warnings and Precautions (5.1)].
- •
- Endocrinopathies: Advise patients to contact their healthcare provider immediately for signs or symptoms of hypophysitis, adrenal insufficiency, hypothyroidism, hyperthyroidism, and diabetes mellitus [see Warnings and Precautions (5.1)].
- •
- Nephritis and Renal Dysfunction: Advise patients to contact their healthcare provider immediately for signs or symptoms of nephritis including decreased urine output, blood in urine, swelling in ankles, loss of appetite, and any other symptoms of renal dysfunction [see Warnings and Precautions (5.1)].
- •
- Skin Adverse Reactions: Advise patients to contact their healthcare provider immediately for rash [see Warnings and Precautions (5.1)].
Infusion-Related Reactions
- •
- Advise patients of the potential risk of infusion-related reactions [see Warnings and Precautions (5.2)].
Complications of Allogeneic HSCT
- •
- Advise patients of potential risk of post-transplant complications [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- •
- Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Populations (8.1)].
- •
- Advise females of reproductive potential to use effective contraception during treatment with OPDIVO and for at least 5 months following the last dose [see Use in Specific Populations (8.3)].
Lactation
- •
- Advise women not to breastfeed during treatment with OPDIVO and for 5 months after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
U.S. License No. 1713
Medication Guide
|
MEDICATION GUIDE |
|||
|
Read this Medication Guide before you start receiving OPDIVO and before each infusion. There may be new information. If your healthcare provider prescribes OPDIVO in combination with ipilimumab, also read the Medication Guide that comes with ipilimumab. If your healthcare provider prescribes OPDIVO in combination with cabozantinib, also read the Patient Information that comes with cabozantinib. This Medication Guide does not take the place of talking with your healthcare provider about your medical condition or your treatment. |
|||
|
What is the most important information I should know about OPDIVO? OPDIVO is a medicine that may treat certain cancers by working with your immune system. OPDIVO can cause your immune system to attack normal organs and tissues in any area of your body and can affect the way they work. These problems can sometimes become severe or life-threatening and can lead to death. These problems may happen anytime during treatment or even after your treatment has ended. You may have more than one of these problems at the same time. Some of these problems may happen more often when OPDIVO is used in combination with another therapy. Call or see your healthcare provider right away if you develop any new or worsening signs or symptoms, including: Lung problems. |
|||
|
|
|
|
|
Intestinal problems.
Liver problems. |
|||
|
|
||
|
Hormone gland problems. |
|||
|
|
||
|
Kidney problems. |
|||
|
|
||
|
Skin problems. |
|||
|
|
||
|
Problems can also happen in other organs and tissues. These are not all of the signs and symptoms of immune system problems that can happen with OPDIVO. Call or see your healthcare provider right away for any new or worsening signs or symptoms, which may include: |
|||
Rejection of a transplanted organ or tissue. Your healthcare provider should tell you what signs and symptoms you should report and monitor you depending on the type of organ transplant that you have had. Getting medical treatment right away may help keep these problems from becoming more serious. Your healthcare provider will check you for these problems during treatment with OPDIVO. Your healthcare provider may treat you with corticosteroid or hormone replacement medicines. Your healthcare provider may also need to delay or completely stop treatment with OPDIVO, if you have severe side effects. |
|||
|
What is OPDIVO? OPDIVO is a prescription medicine used to treat:
It is not known if OPDIVO is safe and effective in children younger than 12 years of age with melanoma or MSI-H or dMMR metastatic colorectal cancer. It is not known if OPDIVO is safe and effective in children for the treatment of any other cancers. |
|||
|
Before receiving OPDIVO, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
|||
|
How will I receive OPDIVO?
|
|||
|
What are the possible side effects of OPDIVO? OPDIVO can cause serious side effects, including:
|
|||
|
|
||
The most common side effects of OPDIVO when used alone include: |
|||
|
|
||
|
The most common side effects of OPDIVO when used in combination with ipilimumab include: |
|||
|
|
||
|
The most common side effects of OPDIVO when used in combination with a platinum-containing chemotherapy and another chemotherapy medicine include: |
|||
|
|
||
|
The most common side effects of OPDIVO when used in combination with ipilimumab, a platinum-containing chemotherapy, and another chemotherapy medicine include: |
|||
|
|
||
|
The most common side effects of OPDIVO when used in combination with cabozantinib include: |
|||
|
|
||
|
The most common side effects of OPDIVO when used in combination with fluoropyrimidine and platinum-containing chemotherapy include: |
|||
|
|
||
|
These are not all the possible side effects of OPDIVO. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
|
General information about the safe and effective use of OPDIVO. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about OPDIVO that is written for health professionals. |
|||
|
What are the ingredients in OPDIVO? Active ingredient: nivolumab Inactive ingredients: mannitol, pentetic acid, polysorbate 80, sodium chloride, sodium citrate dihydrate, and Water for Injection. May contain hydrochloric acid and/or sodium hydroxide. Manufactured by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA U.S. License No. 1713 OPDIVO® is a trademark of Bristol-Myers Squibb Company. Other brands listed are the trademarks of their respective owners. For more information, call 1-855-673-4861 or go to www.OPDIVO.com. |
|||
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: May 2025
OPDIVO 40 mg/4 mL Representative Packaging
See How Supplied section for a complete list of available packages of OPDIVO.
NDC 0003-3772-11
Rx only
Opdivo®
(nivolumab)
injection
40 mg/4 mL
(10 mg/mL)
For intravenous Infusion Only
Single-dose vial; Discard unused portion.
Please provide enclosed Medication Guide to the patient.
Bristol Myers Squibb
OPDIVO 100 mg/10 mL Representative Packaging
NDC 0003-3774-12
Rx only
Opdivo®
(nivolumab)
injection
100 mg/10 mL
(10 mg/mL)
For Intravenous Infusion Only
Single-dose vial; Discard unused portion.
Please provide enclosed Medication Guide to the patient.
Bristol Myers Squibb
| OPDIVO
nivolumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| OPDIVO
nivolumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| OPDIVO
nivolumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| OPDIVO
nivolumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - E.R. Squibb & Sons, L.L.C. (011550092) |
Biological Products Related to Opdivo
Find detailed information on biosimilars for this medication.
Frequently asked questions
- What are Monoclonal Antibodies and how do they work?
- How long does it prolong life and what’s the success rate?
- How long does Opdivo take to work and how do you know if it's working?
- Pembrolizumab vs. nivolumab: How do they compare?
- What is the difference between Opdivo and Keytruda?
- How long does Opdivo stay in your system?
- What happens when you stop taking Opdivo for melanoma?
- How effective are Opdivo and Yervoy when taken together?
- Opdivo vs Opdivo Qvantig: What is the Difference?
More about Opdivo (nivolumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (97)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- FDA approval history
- Drug class: anti-PD-1 and PD-L1 monoclonal antibodies (immune checkpoint inhibitors)
- Breastfeeding
- En español