Ocrevus: Package Insert / Prescribing Info
Package insert / product label
Generic name: ocrelizumab
Dosage form: injection
Drug class: CD20 monoclonal antibodies
J Code (medical billing code): J2350 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on Aug 31, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
OCREVUS® (ocrelizumab) injection, for intravenous use
Initial U.S. Approval: 2017
Indications and Usage for Ocrevus
Ocrevus Dosage and Administration
- Before initiating OCREVUS, screen for Hepatitis B virus and obtain serum quantitative immunoglobulins, aminotransferases, alkaline phosphatase, and bilirubin (2.1)
- Pre-medicate with methylprednisolone (or an equivalent corticosteroid) and an antihistamine (e.g., diphenhydramine) prior to each infusion (2.2)
- Administer OCREVUS by intravenous infusion
- Must be diluted prior to administration (2.3, 2.6)
- Monitor patients closely during and for at least one hour after infusion (2.3, 2.5)
Dosage Forms and Strengths
- Injection: 300 mg/10 mL (30 mg/mL) in a single-dose vial (3)
Contraindications
Warnings and Precautions
- Infusion Reactions: Management recommendations for infusion reactions depend on the type and severity of the reaction. Permanently discontinue OCREVUS if a life-threatening or disabling infusion reaction occurs (2.3, 5.1)
- Infections: Serious, including life-threatening and fatal infections, have occurred. Delay OCREVUS administration in patients with an active infection until the infection is resolved. Vaccination with live-attenuated or live vaccines is not recommended during treatment with OCREVUS and after discontinuation, until B-cell repletion (5.2)
- Progressive Multifocal Leukoencephalopathy (PML): Withhold OCREVUS at the first sign or symptom suggestive of PML (5.3)
- Reduction in Immunoglobulins: Monitor the level of immunoglobulins at the beginning of treatment. Monitor during and after discontinuation of treatment with OCREVUS, until B-cell repletion, and especially when recurrent serious infections are suspected. Consider discontinuing OCREVUS in patients with serious opportunistic or recurrent serious infections, and if prolonged hypogammaglobulinemia requires treatment with intravenous immunoglobulins (2.1, 5.4)
- Malignancies: An increased risk of malignancy, including breast cancer, may exist with OCREVUS (5.5)
- Immune-Mediated Colitis: Immune-mediated colitis has been reported in the postmarketing setting. Monitor patients for new or persistent diarrhea or other gastrointestinal symptoms, and evaluate promptly if colitis is suspected (5.6)
- Liver Injury: Clinically significant liver injury has occurred. Obtain serum aminotransferases, alkaline phosphatase, and bilirubin levels before initiating OCREVUS, and during treatment as clinically indicated. Discontinue OCREVUS in patients with evidence of liver injury in the absence of an alternative etiology (5.7).
Adverse Reactions/Side Effects
The most common adverse reactions were:
- RMS (incidence ≥10% and > REBIF®): upper respiratory tract infections and infusion reactions (6.1)
- PPMS (incidence ≥10% and > placebo): upper respiratory tract infections, infusion reactions, skin infections, and lower respiratory tract infections (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 8/2025
Full Prescribing Information
1. Indications and Usage for Ocrevus
OCREVUS is indicated for the treatment of:
- Relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults
- Primary progressive MS, in adults
2. Ocrevus Dosage and Administration
2.1 Assessments Prior to First Dose of OCREVUS
Hepatitis B Virus Screening
Prior to initiating OCREVUS, perform Hepatitis B virus (HBV) screening. OCREVUS is contraindicated in patients with active HBV confirmed by positive results for HBsAg and anti-HBV tests. For patients who are negative for surface antigen [HBsAg] and positive for HB core antibody [HBcAb+] or are carriers of HBV [HBsAg+], consult liver disease experts before starting and during treatment [see Warnings and Precautions (5.2)].
Serum Immunoglobulins
Prior to initiating OCREVUS, perform testing for quantitative serum immunoglobulins [see Warnings and Precautions (5.4)]. For patients with low serum immunoglobulins, consult immunology experts before initiating treatment with OCREVUS.
Vaccinations
Because vaccination with live-attenuated or live vaccines is not recommended during treatment and after discontinuation until B-cell repletion, administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of OCREVUS for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to initiation of OCREVUS for non-live vaccines [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)].
Liver Function Tests
Prior to initiating OCREVUS, obtain serum aminotransferases (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]), alkaline phosphatase, and bilirubin levels [see Warnings and Precautions (5.7)].
2.2 Preparation Before Every Infusion
Infection Assessment
Prior to every infusion of OCREVUS, determine whether there is an active infection. In case of active infection, delay infusion of OCREVUS until the infection resolves [see Warnings and Precautions (5.2)].
Recommended Premedication
Pre-medicate with 100 mg of methylprednisolone (or an equivalent corticosteroid) administered intravenously approximately 30 minutes prior to each OCREVUS infusion to reduce the frequency and severity of infusion reactions [see Warnings and Precautions (5.1)]. Pre-medicate with an antihistamine (e.g., diphenhydramine) approximately 30-60 minutes prior to each OCREVUS infusion to further reduce the frequency and severity of infusion reactions.
The addition of an antipyretic (e.g., acetaminophen) may also be considered.
2.3 Recommended Dosage and Dose Administration
Administer OCREVUS under the close supervision of an experienced healthcare professional with access to appropriate medical support to manage severe reactions such as serious infusion reactions.
- Initial dose: 300 mg intravenous infusion, followed two weeks later by a second 300 mg intravenous infusion.
- Subsequent doses: single 600 mg intravenous infusion every 6 months.
- Observe the patient for at least one hour after the completion of the infusion [see Warnings and Precautions (5.1)].
| Amount and Volume* | Infusion Rate and Duration† | ||
|---|---|---|---|
|
|||
| Initial Dose
(two infusions) | Infusion 1 | 300 mg in 250 mL |
|
| Infusion 2 (2 weeks later) | 300 mg in 250 mL | ||
| Subsequent Doses
(one infusion) every 6 months)‡ | Option 1 Infusion of approximately 3.5 hours duration† | 600 mg in 500 mL |
|
| OR | |||
| Option 2 (If no prior serious infusion reaction with any previous OCREVUS infusion)§ Infusion of approximately 2 hours duration† | 600 mg in 500 mL |
|
|
2.4 Delayed or Missed Doses
If a planned infusion of OCREVUS is missed, administer OCREVUS as soon as possible; do not wait until the next scheduled dose. Reset the dose schedule to administer the next sequential dose 6 months after the missed dose is administered. Doses of OCREVUS must be separated by at least 5 months [see Dosage and Administration (2.3)].
2.5 Dose Modifications Because of Infusion Reactions
Dose modifications in response to infusion reactions depends on the severity.
Life-threatening Infusion Reactions
Immediately stop and permanently discontinue OCREVUS if there are signs of a life-threatening or disabling infusion reaction [see Warnings and Precautions (5.1)]. Provide appropriate supportive treatment.
Severe Infusion Reactions
Immediately interrupt the infusion and administer appropriate supportive treatment, as necessary [see Warnings and Precautions (5.1)]. Restart the infusion only after all symptoms have resolved. When restarting, begin at half of the infusion rate at the time of onset of the infusion reaction. If this rate is tolerated, increase the rate as described in Table 1 [see Dosage and Administration (2.3)]. This change in rate will increase the total duration of the infusion but not the total dose.
Mild to Moderate Infusion Reactions
Reduce the infusion rate to half the rate at the onset of the infusion reaction and maintain the reduced rate for at least 30 minutes [see Warnings and Precautions (5.1)]. If this rate is tolerated, increase the rate as described in Table 1 [see Dosage and Administration (2.3)]. This change in rate will increase the total duration of the infusion but not the total dose.
2.6 Preparation and Storage of the Dilute Solution for Infusion
Preparation
OCREVUS must be prepared by a healthcare professional using aseptic technique. A sterile needle and syringe should be used to prepare the diluted infusion solution.
Visually inspect for particulate matter and discoloration prior to administration. Do not use the solution if discolored or if the solution contains discrete foreign particulate matter. Do not shake.
Withdraw intended dose and further dilute into an infusion bag containing 0.9% Sodium Chloride Injection, to a final drug concentration of approximately 1.2 mg/mL.
- Withdraw 10 mL (300 mg) of OCREVUS and inject into 250 mL
- Withdraw 20 mL (600 mg) of OCREVUS and inject into 500 mL
Do not use other diluents to dilute OCREVUS since their use has not been tested. The product contains no preservative and is intended for single use only.
Storage of Infusion Solution
Prior to the start of the intravenous infusion, the content of the infusion bag should be at room temperature.
Use the prepared infusion solution immediately. If not used immediately, store up to 24 hours in the refrigerator at 2°C to 8°C (36°F to 46°F) and 8 hours at room temperature up to 25°C (77°F), which includes infusion time. In the event an intravenous infusion cannot be completed the same day, discard the remaining solution.
No incompatibilities between OCREVUS and polyvinyl chloride (PVC) or polyolefin (PO) bags and intravenous (IV) administration sets have been observed.
3. Dosage Forms and Strengths
Injection: 300 mg/10 mL (30 mg/mL) clear or slightly opalescent, and colorless to pale brown solution in a single-dose vial.
4. Contraindications
OCREVUS is contraindicated in patients with:
- Active HBV infection [see Dosage and Administration (2.1) and Warnings and Precautions (5.2)]
- A history of life-threatening infusion reaction to OCREVUS [see Warnings and Precautions (5.1)]
5. Warnings and Precautions
5.1 Infusion Reactions
OCREVUS can cause infusion reactions, which can include pruritus, rash, urticaria, erythema, bronchospasm, throat irritation, oropharyngeal pain, dyspnea, pharyngeal or laryngeal edema, flushing, hypotension, pyrexia, fatigue, headache, dizziness, nausea, tachycardia, and anaphylaxis. In multiple sclerosis (MS) clinical trials, the incidence of infusion reactions in OCREVUS-treated patients [who received methylprednisolone (or an equivalent steroid) and possibly other pre-medication to reduce the risk of infusion reactions prior to each infusion] was 34 to 40%, with the highest incidence with the first infusion. There were no fatal infusion reactions, but 0.3% of OCREVUS-treated MS patients experienced infusion reactions that were serious, some requiring hospitalization.
Observe patients treated with OCREVUS for infusion reactions during the infusion and for at least one hour after completion of the infusion. Inform patients that infusion reactions can occur up to 24 hours after the infusion.
Reducing the Risk of Infusion Reactions and Managing Infusion Reactions
Administer pre-medication (e.g., methylprednisolone or an equivalent corticosteroid, and an antihistamine) to reduce the frequency and severity of infusion reactions. The addition of an antipyretic (e.g., acetaminophen) may also be considered [see Dosage and Administration (2.2)].
Management recommendations for infusion reactions depend on the type and severity of the reaction [see Dosage and Administration (2.5)]. For life-threatening infusion reactions, immediately and permanently stop OCREVUS and administer appropriate supportive treatment. For less severe infusion reactions, management may involve temporarily stopping the infusion, reducing the infusion rate, and/or administering symptomatic treatment.
5.2 Infections
Serious, including life-threatening or fatal, bacterial, viral, parasitic and fungal infections have been reported in patients receiving OCREVUS. An increased risk of infections (including serious and fatal bacterial, fungal, and new or reactivated viral infections) has been observed in patients during and following completion of treatment with anti-CD20 B-cell depleting therapies.
A higher proportion of OCREVUS-treated patients experienced infections compared to patients taking REBIF or placebo. In RMS trials, 58% of OCREVUS-treated patients experienced one or more infections compared to 52% of REBIF-treated patients. In the PPMS trial, 70% of OCREVUS-treated patients experienced one or more infections compared to 68% of patients on placebo. OCREVUS increased the risk for upper respiratory tract infections, lower respiratory tract infections, skin infections, and herpes-related infections [see Adverse Reactions (6.1)].
OCREVUS was not associated with an increased risk of serious infections in MS patients in controlled trials.
Delay OCREVUS administration in patients with an active infection until the infection is resolved.
Respiratory Tract Infections
A higher proportion of OCREVUS-treated patients experienced respiratory tract infections compared to patients taking REBIF or placebo. In RMS trials, 40% of OCREVUS-treated patients experienced upper respiratory tract infections compared to 33% of REBIF-treated patients, and 8% of OCREVUS-treated patients experienced lower respiratory tract infections compared to 5% of REBIF-treated patients. In the PPMS trial, 49% of OCREVUS-treated patients experienced upper respiratory tract infections compared to 43% of patients on placebo and 10% of OCREVUS-treated patients experienced lower respiratory tract infections compared to 9% of patients on placebo. The infections were predominantly mild to moderate and consisted mostly of upper respiratory tract infections and bronchitis.
Herpes
In active-controlled (RMS) clinical trials, herpes infections were reported more frequently in OCREVUS-treated patients than in REBIF-treated patients, including herpes zoster (2.1% vs. 1.0%), herpes simplex (0.7% vs. 0.1%), oral herpes (3.0% vs. 2.2%), genital herpes (0.1% vs. 0%), and herpes virus infection (0.1% vs. 0%). Infections were predominantly mild to moderate in severity.
In the placebo-controlled (PPMS) clinical trial, oral herpes was reported more frequently in the OCREVUS-treated patients than in the patients on placebo (2.7% vs 0.8%).
Serious cases of infections caused by herpes simplex virus and varicella zoster virus, including central nervous system infections (encephalitis and meningitis), intraocular infections, and disseminated skin and soft tissue infections, have been reported in the postmarketing setting in multiple sclerosis patients receiving OCREVUS. Serious herpes virus infections may occur at any time during treatment with OCREVUS. Some cases were life-threatening.
If serious herpes infections occur, OCREVUS should be discontinued or withheld until the infection has resolved, and appropriate treatment should be administered [see Patient Counseling Information (17)].
Hepatitis B Virus (HBV) Reactivation
Hepatitis B reactivation has been reported in MS patients treated with OCREVUS in the postmarketing setting. Fulminant hepatitis, hepatic failure, and death caused by HBV reactivation have occurred in patients treated with anti-CD20 antibodies. Perform HBV screening in all patients before initiation of treatment with OCREVUS. Do not administer OCREVUS to patients with active HBV confirmed by positive results for HBsAg and anti-HB tests. For patients who are negative for surface antigen [HBsAg] and positive for HB core antibody [HBcAb+] or are carriers of HBV [HBsAg+], consult liver disease experts before starting and during treatment.
Possible Increased Risk of Immunosuppressant Effects with Other Immunosuppressants
When initiating OCREVUS after an immunosuppressive therapy or initiating an immunosuppressive therapy after OCREVUS, consider the potential for increased immunosuppressive effects [see Drug Interactions (7.1) and Clinical Pharmacology (12.1, 12.2)]. OCREVUS has not been studied in combination with other MS therapies.
Vaccinations
Administer all immunizations according to immunization guidelines at least 4 weeks prior to initiation of OCREVUS for live or live-attenuated vaccines and, whenever possible, at least 2 weeks prior to initiation of OCREVUS for non-live vaccines.
OCREVUS may interfere with the effectiveness of non-live vaccines [see Drug Interactions (7.2)].
The safety of immunization with live or live-attenuated vaccines following OCREVUS therapy has not been studied, and vaccination with live-attenuated or live vaccines is not recommended during treatment and until B-cell repletion [see Clinical Pharmacology (12.2)].
Vaccination of Infants Born to Mothers Treated with OCREVUS During Pregnancy
In infants of mothers exposed to OCREVUS during pregnancy, do not administer live or live-attenuated vaccines before confirming the recovery of B-cell counts as measured by CD19+ B-cells. Depletion of B-cells in these infants may increase the risks from live or live-attenuated vaccines.
You may administer non-live vaccines, as indicated, prior to recovery from B-cell depletion, but should consider assessing vaccine immune responses, including consultation with a qualified specialist, to assess whether a protective immune response was mounted [see Use in Specific Populations (8.1)].
5.3 Progressive Multifocal Leukoencephalopathy (PML)
Cases of progressive multifocal leukoencephalopathy (PML) have been reported in patients with MS treated with OCREVUS in the postmarketing setting. PML is an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability. PML has occurred in OCREVUS-treated patients who had not been treated previously with natalizumab (which has a known association with PML), were not taking any immunosuppressive or immunomodulatory medications associated with the risk of PML prior to or concomitantly with OCREVUS, and did not have any known ongoing systemic medical conditions resulting in compromised immune system function.
JCV infection resulting in PML has also been observed in patients treated with other anti-CD20 antibodies and other MS therapies.
At the first sign or symptom suggestive of PML, withhold OCREVUS and perform an appropriate diagnostic evaluation. Typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes.
MRI findings may be apparent before clinical signs or symptoms. Cases of PML, diagnosed based on MRI findings and the detection of JCV DNA in the cerebrospinal fluid in the absence of clinical signs or symptoms specific to PML, have been reported in patients treated with other MS medications associated with PML. Many of these patients subsequently became symptomatic with PML. Therefore, monitoring with MRI for signs that may be consistent with PML may be useful, and any suspicious findings should lead to further investigation to allow for an early diagnosis of PML, if present. Following discontinuation of another MS medication associated with PML, lower PML-related mortality and morbidity have been reported in patients who were initially asymptomatic at diagnosis compared to patients who had characteristic clinical signs and symptoms at diagnosis.
It is not known whether these differences are due to early detection and discontinuation of MS treatment or due to differences in disease in these patients.
If PML is confirmed, treatment with OCREVUS should be discontinued.
5.4 Reduction in Immunoglobulins
As expected with any B-cell depleting therapy, decreased immunoglobulin levels are observed with OCREVUS treatment. The pooled data of OCREVUS clinical studies (RMS and PPMS) and their open-label extensions (up to approximately 7 years of exposure) have shown an association between decreased levels of immunoglobulin G (IgG<LLN) and increased rates of serious infections. Monitor the levels of quantitative serum immunoglobulins during OCREVUS treatment and after discontinuation of treatment, until B-cell repletion, and especially in the setting of recurrent serious infections. Consider discontinuing OCREVUS therapy in patients with serious opportunistic or recurrent serious infections, and if prolonged hypogammaglobulinemia requires treatment with intravenous immunoglobulins [see Adverse Reactions (6.1)].
5.5 Malignancies
An increased risk of malignancy with OCREVUS may exist. In controlled trials, malignancies, including breast cancer, occurred more frequently in OCREVUS-treated patients. Breast cancer occurred in 6 of 781 females treated with OCREVUS and none of 668 females treated with REBIF or placebo. Patients should follow standard breast cancer screening guidelines.
5.6 Immune-Mediated Colitis
Immune-mediated colitis, which can present as a severe and acute-onset form of colitis, has been reported in patients receiving OCREVUS in the postmarketing setting. Some cases of colitis were serious, requiring hospitalization, with a few patients requiring surgical intervention. Systemic corticosteroids were required in many of these patients. The time from treatment initiation to onset of symptoms in these cases ranged from a few weeks to years. Monitor patients for immune-mediated colitis during OCREVUS treatment, and evaluate promptly if signs and symptoms that may indicate immune-mediated colitis, such as new or persistent diarrhea or other gastrointestinal signs and symptoms, occur.
5.7 Liver Injury
Clinically significant liver injury, without findings of viral hepatitis, has been reported in the postmarketing setting in patients treated with anti-CD20 B-cell depleting therapies approved for the treatment of MS, including OCREVUS. Signs of liver injury, including markedly elevated serum hepatic enzymes with elevated total bilirubin, have occurred from weeks to months after administration.
Patients treated with OCREVUS found to have an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than 3× the upper limit of normal (ULN) with serum total bilirubin greater than 2× ULN are potentially at risk for severe drug-induced liver injury.
Obtain liver function tests prior to initiating treatment with OCREVUS [see Dosage and Administration (2.1)], and monitor for signs and symptoms of any hepatic injury during treatment. Measure serum aminotransferases, alkaline phosphatase, and bilirubin levels promptly in patients who report symptoms that may indicate liver injury, including new or worsening fatigue, anorexia, nausea, vomiting, right upper abdominal discomfort, dark urine, or jaundice. If liver injury is present and an alternative etiology is not identified, discontinue OCREVUS.
6. Adverse Reactions/Side Effects
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Infusion Reactions [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Progressive Multifocal Leukoencephalopathy [see Warnings and Precautions (5.3)]
- Reduction in Immunoglobulins [see Warnings and Precautions (5.4)]
- Malignancies [see Warnings and Precautions (5.5)]
- Immune-Mediated Colitis [see Warnings and Precautions (5.6)]
- Liver Injury [see Warnings and Precautions (5.7)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reactions rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety of OCREVUS has been evaluated in 1311 patients across MS clinical studies, which included 825 patients in active-controlled clinical trials in patients with relapsing forms of MS (RMS) and 486 patients in a placebo-controlled study in patients with primary progressive MS (PPMS).
Adverse Reactions in Patients with Relapsing Forms of MS
In active-controlled clinical trials (Study 1 and Study 2), 825 patients with RMS received OCREVUS 600 mg intravenously every 24 weeks (initial treatment was given as two separate 300 mg infusions at Weeks 0 and 2) [see Clinical Studies (14.1)]. The overall exposure in the 96-week controlled treatment periods was 1448 patient-years.
The most common adverse reactions in RMS trials (incidence ≥ 10%) were upper respiratory tract infections and infusion reactions. Table 2 summarizes the adverse reactions that occurred in RMS trials (Study 1 and Study 2).
| Adverse Reactions | Studies 1 and 2 | |
|---|---|---|
| OCREVUS 600 mg IV Every 24 Weeks* (n=825) % | REBIF 44 mcg SQ 3 Times per Week (n=826) % |
|
|
||
| Upper respiratory tract infections | 40 | 33 |
| Infusion reactions | 34 | 10 |
| Depression | 8 | 7 |
| Lower respiratory tract infections | 8 | 5 |
| Back pain | 6 | 5 |
| Herpes virus- associated infections | 6 | 4 |
| Pain in extremity | 5 | 4 |
Adverse Reactions in Patients with Primary Progressive MS
In a placebo-controlled clinical trial (Study 3), a total of 486 patients with PPMS received one course of OCREVUS (600 mg of OCREVUS administered as two 300 mg infusions two weeks apart) given intravenously every 24 weeks and 239 patients received placebo intravenously [see Clinical Studies (14.2)]. The overall exposure in the controlled treatment period was 1416 patient-years, with median treatment duration of 3 years.
The most common adverse reactions in the PPMS trial (incidence ≥ 10%) were upper respiratory tract infections, infusion reactions, skin infections, and lower respiratory tract infections. Table 3 summarizes the adverse reactions that occurred in the PPMS trial (Study 3).
| Adverse Reactions | Study 3 | |
|---|---|---|
| OCREVUS 600 mg IV Every 24 Weeks* | Placebo | |
| (n=486) % | (n=239) % |
|
|
||
| Upper respiratory tract infections | 49 | 43 |
| Infusion reactions | 40 | 26 |
| Skin infections | 14 | 11 |
| Lower respiratory tract infections | 10 | 9 |
| Cough | 7 | 3 |
| Diarrhea | 6 | 5 |
| Edema peripheral | 6 | 5 |
| Herpes virus associated infections | 5 | 4 |
Adverse Reactions in Patients who Received 2-hour Infusions
Study 4 was designed to characterize the safety profile of OCREVUS infusions administered over 2 hours in patients with Relapsing-Remitting Multiple Sclerosis who did not experience a serious infusion reaction with any previous OCREVUS infusion. In this study, the incidence, intensity, and types of symptoms of infusion reactions were consistent with those of infusions administered over 3.5 hours [see Clinical Studies (14.3)].
Laboratory Abnormalities
Decreased Immunoglobulins
OCREVUS decreased total immunoglobulins with the greatest decline seen in IgM levels; however, a decrease in IgG levels was associated with an increased rate of serious infections.
In the active-controlled (RMS) trials (Study 1 and Study 2), the proportion of patients at baseline reporting IgG, IgA, and IgM below the lower limit of normal (LLN) in OCREVUS-treated patients was 0.5%, 1.5%, and 0.1%, respectively. Following treatment, the proportion of OCREVUS-treated patients reporting IgG, IgA, and IgM below the LLN at 96 weeks was 1.5%, 2.4%, and 16.5%, respectively.
In the placebo-controlled (PPMS) trial (Study 3), the proportion of patients at baseline reporting IgG, IgA, and IgM below the LLN in OCREVUS-treated patients was 0.0%, 0.2%, and 0.2%, respectively. Following treatment, the proportion of OCREVUS-treated patients reporting IgG, IgA, and IgM below the LLN at 120 weeks was 1.1%, 0.5%, and 15.5%, respectively.
The pooled data of OCREVUS clinical studies (RMS and PPMS) and their open-label extensions (up to approximately 7 years of exposure) have shown an association between decreased levels of IgG and increased rates of serious infections. The type, severity, latency, duration, and outcome of serious infections observed during episodes of immunoglobulins below LLN were consistent with the overall serious infections observed in patients treated with OCREVUS.
Decreased Neutrophil Levels
In the PPMS clinical trial (Study 3), decreased neutrophil counts occurred in 13% of OCREVUS-treated patients compared to 10% in placebo patients. The majority of the decreased neutrophil counts were only observed once for a given patient treated with OCREVUS and were between LLN - 1.5 × 109/L and 1.0 × 109/L. Overall, 1% of the patients in the OCREVUS group had neutrophil counts less than 1.0 × 109/L and these were not associated with an infection.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. Immunogenicity data are highly dependent on the sensitivity and specificity of the test methods used. Additionally, the observed incidence of a positive result in a test method may be influenced by several factors, including sample handling, timing of sample collection, drug interference, concomitant medication, and the underlying disease. Therefore, comparison of the incidence of antibodies to OCREVUS with the incidence of antibodies to other products may be misleading.
Patients in MS trials (Study 1, Study 2, and Study 3) were tested at multiple time points (baseline and every 6 months post-treatment for the duration of the trial) for anti-drug antibodies (ADAs). Out of 1311 patients treated with OCREVUS, 12 (~1%) tested positive for ADAs, of which 2 patients tested positive for neutralizing antibodies. These data are not adequate to assess the impact of ADAs on the safety and efficacy of OCREVUS.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of OCREVUS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal Disorders: Immune-mediated colitis [see Warnings and Precautions (5.6)]
Hepatobiliary Disorders: Liver injury [see Warnings and Precautions (5.7)]
Infections and Infestations: Serious herpes infections [see Warnings and Precautions (5.2)], progressive multifocal leukoencephalopathy [see Warnings and Precautions (5.3)], and babesiosis
Skin: Pyoderma gangrenosum
Related/similar drugs
7. Drug Interactions
7.1 Immunosuppressive or Immune-Modulating Therapies
The concomitant use of OCREVUS and other immune-modulating or immunosuppressive therapies, including immunosuppressant doses of corticosteroids, is expected to increase the risk of immunosuppression. Consider the risk of additive immune system effects when coadministering immunosuppressive therapies with OCREVUS. When switching from drugs with prolonged immune effects, such as daclizumab, fingolimod, natalizumab, teriflunomide, or mitoxantrone, consider the duration and mode of action of these drugs because of additive immunosuppressive effects when initiating OCREVUS [see Warnings and Precautions (5.2)].
7.2 Vaccinations
A Phase 3b randomized, open-label study examined the concomitant use of OCREVUS and several non-live vaccines in adults 18-55 years of age with relapsing forms of MS (68 subjects undergoing treatment with OCREVUS at the time of vaccination and 34 subjects not undergoing treatment with OCREVUS at the time of vaccination). Concomitant exposure to OCREVUS attenuated antibody responses to tetanus toxoid-containing vaccine, pneumococcal polysaccharide, pneumococcal conjugate vaccines, and seasonal inactivated influenza vaccines. The impact of the observed attenuation on vaccine effectiveness in this patient population is unknown. The safety and effectiveness of live or live-attenuated vaccines administered concomitantly with OCREVUS have not been assessed [see Warnings and Precautions (5.2)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
OCREVUS is a humanized monoclonal antibody of an immunoglobulin G1 subtype and immunoglobulins are known to cross the placental barrier. There are no adequate data on the developmental risk associated with use of OCREVUS in pregnant women. However, transient peripheral B-cell depletion and lymphocytopenia have been reported in infants born to mothers exposed to other anti-CD20 antibodies during pregnancy. B-cell levels in infants following maternal exposure to OCREVUS have not been studied in clinical trials. The potential duration of B-cell depletion in such infants, and the impact of B-cell depletion on vaccine safety and effectiveness, is unknown [see Warnings and Precautions (5.2)].
Following administration of ocrelizumab to pregnant monkeys at doses similar to or greater than those used clinically, increased perinatal mortality, depletion of B-cell populations, renal, bone marrow, and testicular toxicity were observed in the offspring in the absence of maternal toxicity [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Following intravenous administration of OCREVUS to monkeys during organogenesis (loading doses of 15 or 75 mg/kg on gestation days 20, 21, and 22, followed by weekly doses of 20 or 100 mg/kg), depletion of B-lymphocytes in lymphoid tissue (spleen and lymph nodes) was observed in fetuses at both doses.
Intravenous administration of OCREVUS (three daily loading doses of 15 or 75 mg/kg, followed by weekly doses of 20 or 100 mg/kg) to pregnant monkeys throughout the period of organogenesis and continuing through the neonatal period resulted in perinatal deaths (some associated with bacterial infections), renal toxicity (glomerulopathy and inflammation), lymphoid follicle formation in the bone marrow, and severe decreases in circulating B-lymphocytes in neonates. The cause of the neonatal deaths is uncertain; however, both affected neonates were found to have bacterial infections. Reduced testicular weight was observed in neonates at the high dose.
A no-effect dose for adverse developmental effects was not identified; the doses tested in monkey are 2 and 10 times the recommended human dose of 600 mg, on a mg/kg basis.
8.2 Lactation
Risk Summary
There are no data on the presence of ocrelizumab in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Ocrelizumab was excreted in the milk of ocrelizumab-treated monkeys. Human IgG is excreted in human milk, and the potential for absorption of ocrelizumab to lead to B-cell depletion in the infant is unknown. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for OCREVUS and any potential adverse effects on the breastfed infant from OCREVUS or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Women of childbearing potential should use effective contraception while receiving OCREVUS and for 6 months after the last infusion of OCREVUS [see Clinical Pharmacology (12.3)].
11. Ocrevus Description
Ocrelizumab is a recombinant humanized monoclonal antibody directed against CD20-expressing B-cells. Ocrelizumab is a glycosylated immunoglobulin G1 (IgG1) with a molecular mass of approximately 145 kDa.
OCREVUS (ocrelizumab) injection for intravenous infusion is a preservative-free, sterile, clear or slightly opalescent, and colorless to pale brown solution supplied in single-dose vials. Each mL of solution contains 30 mg ocrelizumab, glacial acetic acid (0.25 mg), polysorbate 20 (0.2 mg), sodium acetate trihydrate (2.14 mg), and trehalose dihydrate (40 mg) at pH 5.3.
12. Ocrevus - Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism by which ocrelizumab exerts its therapeutic effects in multiple sclerosis is unknown, but is presumed to involve binding to CD20, a cell surface antigen present on pre-B and mature B lymphocytes. Following cell surface binding to B lymphocytes, ocrelizumab results in antibody-dependent cellular cytolysis and complement-mediated lysis.
12.2 Pharmacodynamics
For B-cell counts, assays for CD19+ B-cells are used because the presence of OCREVUS interferes with the CD20 assay. Treatment with OCREVUS reduces CD19+ B-cell counts in blood by 14 days after infusion. In clinical studies, B-cell counts rose to above the lower limit of normal (LLN) or above baseline counts between infusions of OCREVUS at least one time in 0.3% to 4.1% of patients. In a clinical study of 51 patients, the median time for B-cell counts to return to either baseline or LLN was 72 weeks (range 27-175 weeks) after the last OCREVUS infusion. Within 2.5 years after the last infusion, B-cell counts rose to either baseline or LLN in 90% of patients.
12.3 Pharmacokinetics
Pharmacokinetics (PK) of OCREVUS in MS clinical studies fit a two compartment model with time-dependent clearance. The overall exposure at the steady-state (AUC over the 24 week dosing intervals) of OCREVUS was 3,510 mcg/mL per day. In clinical studies in MS patients, maintenance doses of ocrelizumab were either 600 mg every 6 months (RMS patients) or two 300 mg infusions separated by 14 days every 6 months (PPMS patients). The mean maximum concentration was 212 mcg/mL in patients with RMS (600 mg infusion over 3.5 hours) and 141 mcg/mL in patients with PPMS (two 300 mg infusions over 2.5 hours administered within two weeks). The mean maximum peak concentrations (Cmax) of ocrelizumab in patients with relapsing-remitting multiple sclerosis (RRMS) observed after the 3.5-hour infusion and 2-hour infusion were 202 ± 42 (mean ± SD) and 200 ± 46 mcg/mL, respectively, compared to the previously reported Cmax of 212 mcg/mL. The pharmacokinetics of ocrelizumab was essentially linear and dose proportional between 400 mg and 2000 mg.
Distribution
The population PK estimate of the central volume of distribution was 2.78 L. Peripheral volume and inter-compartment clearance were estimated at 2.68 L and 0.29 L/day, respectively.
Elimination
Constant clearance was estimated at 0.17 L/day, and initial time-dependent clearance at 0.05 L/day, which declined with a half-life of 33 weeks. The terminal elimination half-life was 26 days.
Metabolism
The metabolism of OCREVUS has not been directly studied because antibodies are cleared principally by catabolism.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity studies have been performed to assess the carcinogenic potential of OCREVUS.
No studies have been performed to assess the mutagenic potential of OCREVUS. As an antibody, OCREVUS is not expected to interact directly with DNA.
No effects on reproductive organs were observed in male monkeys administered ocrelizumab by intravenous injection (three loading doses of 15 or 75 mg/kg, followed by weekly doses of 20 or 100 mg/kg) for 8 weeks. There were also no effects on estrus cycle in female monkeys administered ocrelizumab over three menstrual cycles using the same dosing regimen. The doses tested in monkey are 2 and 10 times the recommended human dose of 600 mg, on a mg/kg basis.
14. Clinical Studies
14.1 Relapsing Forms of Multiple Sclerosis (RMS)
The efficacy of OCREVUS was demonstrated in two randomized, double-blind, double-dummy, active comparator-controlled clinical trials of identical design, in patients with RMS treated for 96 weeks (Study 1 and Study 2). The dose of OCREVUS was 600 mg every 24 weeks (initial treatment was given as two 300 mg IV infusions administered 2 weeks apart, and subsequent doses were administered as a single 600 mg IV infusion) and placebo subcutaneous injections were given 3 times per week. The dose of REBIF, the active comparator, was 44 mcg given as subcutaneous injections 3 times per week and placebo IV infusions were given every 24 weeks. Both studies included patients who had experienced at least one relapse within the prior year, or two relapses within the prior two years, and had an Expanded Disability Status Scale (EDSS) score from 0 to 5.5. Patients with primary progressive forms of multiple sclerosis (MS) were excluded. Neurological evaluations were performed every 12 weeks and at the time of a suspected relapse. Brain MRIs were performed at baseline and at Weeks 24, 48, and 96.
The primary outcome of both Study 1 and Study 2 was the annualized relapse rate (ARR). Additional outcome measures included the proportion of patients with confirmed disability progression, the mean number of MRI T1 gadolinium (Gd)-enhancing lesions at Weeks 24, 48, and 96, and new or enlarging MRI T2 hyperintense lesions. Progression of disability was defined as an increase of 1 point or more from the baseline EDSS score attributable to MS when the baseline EDSS score was 5.5 or less, or 0.5 points or more when the baseline EDSS score was above 5.5. Disability progression was considered confirmed when the increase in the EDSS was confirmed at a regularly scheduled visit 12 weeks after the initial documentation of neurological worsening. The primary population for analysis of confirmed disability progression was the pooled population from Studies 1 and 2.
In Study 1, 410 patients were randomized to OCREVUS and 411 to REBIF; 11% of OCREVUS-treated and 17% of REBIF-treated patients did not complete the 96-week double-blind treatment period. The baseline demographic and disease characteristics were balanced between the two treatment groups. At baseline, the mean age of patients was 37 years; 66% were female. The mean time from MS diagnosis to randomization was 3.8 years, the mean number of relapses in the previous year was 1.3, and the mean EDSS score was 2.8; 74% of patients had not been treated with a non-steroid therapy for MS in the 2 years prior to the study. At baseline, 40% of patients had one or more T1 Gd-enhancing lesions (mean 1.8).
In Study 2, 417 patients were randomized to OCREVUS and 418 to REBIF; 14% of OCREVUS-treated and 23% of REBIF-treated patients did not complete the 96-week double-blind treatment period. The baseline demographic and disease characteristics were balanced between the two treatment groups. At baseline, the mean age of patients was 37 years; 66% were female. The mean time from MS diagnosis to randomization was 4.1 years, the mean number of relapses in the previous year was 1.3, and the mean EDSS score was 2.8; 74% of patients had not been treated with a non-steroid therapy for MS in the 2 years prior to the study. At baseline, 40% of OCREVUS-treated patients had one or more T1 Gd-enhancing lesions (mean 1.9).
In Study 1 and Study 2, OCREVUS significantly lowered the annualized relapse rate and the proportion of patients with disability progression confirmed at 12 weeks after onset compared to REBIF. Results for Study 1 and Study 2 are presented in Table 4 and Figure 1.
| Endpoints | Study 1 | Study 2 | ||
|---|---|---|---|---|
| OCREVUS 600 mg every 24 weeks | REBIF 44 mcg three times a week | OCREVUS 600 mg every 24 weeks | REBIF 44 mcg three times a week | |
| N=410 | N=411 | N=417 | N=418 | |
|
||||
| Clinical Endpoints | ||||
| Annualized Relapse Rate (Primary Endpoint) | 0.156 | 0.292 | 0.155 | 0.290 |
| Relative Reduction | 46% (p<0.0001) | 47% (p<0.0001) | ||
| Proportion Relapse-free | 83% | 71% | 82% | 72% |
| Proportion of Patients with 12-week Confirmed Disability Progression* | 9.8% OCREVUS vs 15.2% REBIF | |||
| Risk Reduction (Pooled Analysis†) | 40%; p=0.0006 | |||
| MRI Endpoints | ||||
| Mean number of T1 Gd-enhancing lesions per MRI scan | 0.016 | 0.286 | 0.021 | 0.416 |
| Relative Reduction | 94% (p<0.0001) | 95% (p<0.0001) | ||
| Mean number of new and/or enlarging T2 hyperintense lesions per MRI | 0.323 | 1.413 | 0.325 | 1.904 |
| Relative Reduction | 77% (p<0.0001) | 83% (p<0.0001) | ||
|
| Figure 1 Kaplan-Meier Plot* of Time to Onset of Confirmed Disability Progression Sustained for at Least 12 Weeks with the Initial Event of Neurological Worsening Occurring During the Double-blind Treatment Period in Pooled Studies 1 and 2 in Patients with RMS (Pooled ITT Population) |
|
|
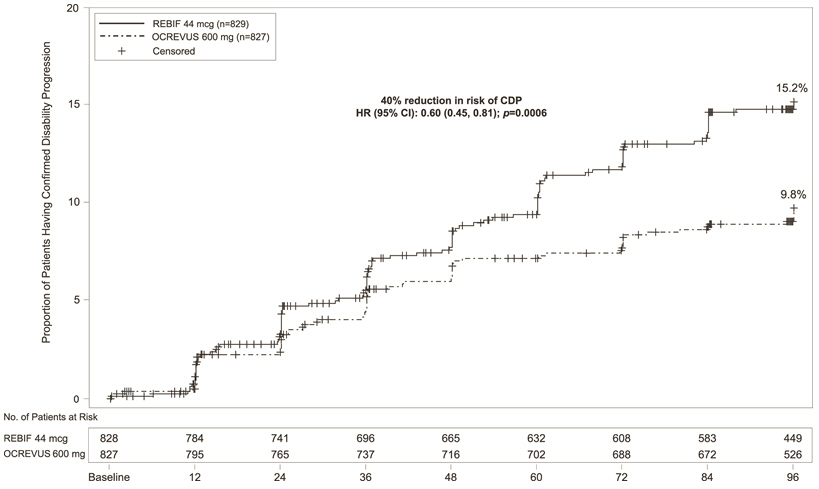
In exploratory subgroup analyses of Study 1 and Study 2, the effect of OCREVUS on annualized relapse rate and disability progression was similar in male and female patients.
14.2 Primary Progressive Multiple Sclerosis (PPMS)
Study 3 was a randomized, double-blind, placebo-controlled clinical trial in patients with PPMS. Patients were randomized 2:1 to receive either OCREVUS 600 mg or placebo as two 300 mg intravenous infusions 2 weeks apart every 24 weeks for at least 120 weeks. Selection criteria required a baseline EDSS of 3 to 6.5 and a score of 2 or greater for the EDSS pyramidal functional system due to lower extremity findings. Neurological assessments were conducted every 12 weeks. An MRI scan was obtained at baseline and at Weeks 24, 48, and 120.
In Study 3, the primary outcome was the time to onset of disability progression attributable to MS confirmed to be present at the next neurological assessment at least 12 weeks later. Disability progression occurred when the EDSS score increased by 1 point or more from the baseline EDSS if the baseline EDSS was 5.5 points or less, or by 0.5 points or more if the baseline EDSS was more than 5.5 points. In Study 3, confirmed disability progression also was deemed to have occurred if patients who had onset of disability progression discontinued participation in the study before the next assessment. Additional outcome measures included timed 25-foot walk, and percentage change in T2 hyperintense lesion volume.
Study 3 randomized 488 patients to OCREVUS and 244 to placebo; 21% of OCREVUS-treated patients and 34% of placebo-treated patients did not complete the trial. The baseline demographic and disease characteristics were balanced between the two treatment groups. At baseline, the mean age of patients was 45; 49% were female. The mean time since symptom onset was 6.7 years, the mean EDSS score was 4.7, and 26% had one or more T1 Gd-enhancing lesions at baseline; 88% of patients had not been treated previously with a non-steroid treatment for MS. The time to onset of disability progression confirmed at 12 weeks after onset was significantly longer for OCREVUS-treated patients than for placebo-treated patients (see Figure 2). Results for Study 3 are presented in Table 5 and Figure 2.
| Endpoints | Study 3 | |
|---|---|---|
| OCREVUS 600 mg (two 300 mg infusions two weeks apart every 24 weeks) | Placebo | |
| N=488 | N=244 | |
|
||
| Clinical Outcomes | ||
| Proportion of patients with 12-week Confirmed Disability Progression* | 32.9% | 39.3% |
| Risk reduction | 24%; p=0.0321 | |
| MRI Endpoints | ||
| Mean change in volume of T2 lesions, from baseline to Week 120 (cm3) | -0.39 | 0.79 |
| p<0.0001 | ||
|
| Figure 2 Kaplan-Meier Plot of Time to Onset of Confirmed Disability Progression Sustained for at Least 12 Weeks with the Initial Event of Neurological Worsening Occurring During the Double-blind Treatment Period in Study 3* |
|
|
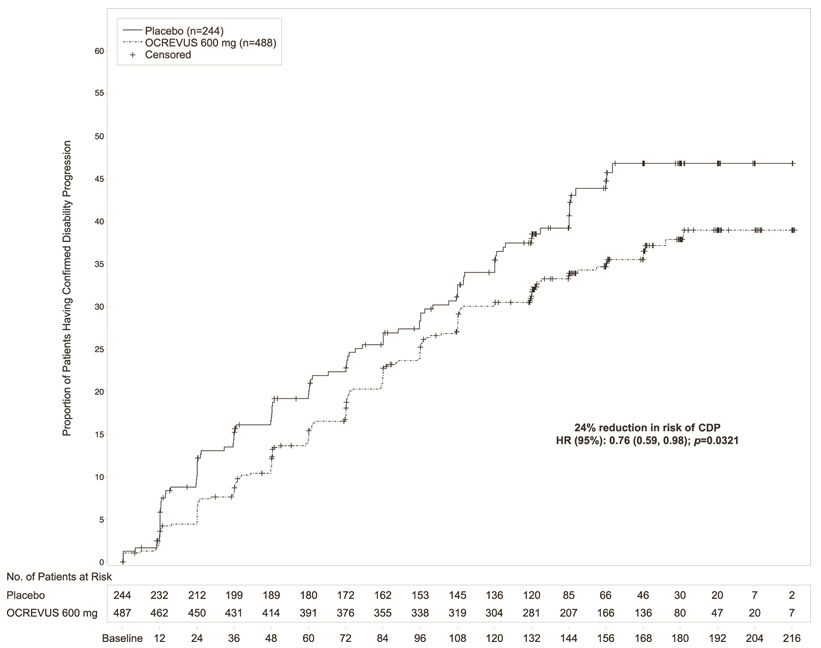
In the overall population in Study 3, the proportion of patients with 20 percent worsening of the timed 25-foot walk confirmed at 12 weeks was 49% in OCREVUS-treated patients compared to 59% in placebo-treated patients (25% risk reduction).
In exploratory subgroup analyses of Study 3, the proportion of female patients with disability progression confirmed at 12 weeks after onset was similar in OCREVUS-treated patients and placebo-treated patients (approximately 36% in each group). In male patients, the proportion of patients with disability progression confirmed at 12 weeks after onset was approximately 30% in OCREVUS-treated patients and 43% in placebo-treated patients. Clinical and MRI endpoints that generally favored OCREVUS numerically in the overall population, and that showed similar trends in both male and female patients, included annualized relapse rate, change in T2 lesion volume, and number of new or enlarging T2 lesions.
14.3 Safety Study of 2-Hour Infusions
The safety of the 2-hour OCREVUS infusion was evaluated in Study 4 (NCT03085810), a prospective, multicenter, randomized, double-blind, controlled, parallel arm substudy in patients with Relapsing-Remitting Multiple Sclerosis who were naïve to other non-steroid therapies for MS and did not experience a serious infusion reaction with any previous OCREVUS infusion. The first dose of OCREVUS was administered as two 300 mg infusions (600 mg total) separated by 14 days. After enrollment in the substudy, patients were randomized in a 1:1 ratio to receive infusions over approximately 3.5-hours or 2-hours, after appropriate premedication [see Dosage and Administration (2.2)], every 24 weeks. The randomization was stratified by region and the dose at which patients were first randomized.
The primary endpoint of the substudy was the proportion of patients with infusion reactions occurring during or within 24 hours following the first randomized infusion of OCREVUS. The primary analysis was performed when 580 patients were randomized, at which time 469/579 (81%) of the treated patients had received only a single randomized infusion of OCREVUS. The proportions of patients with infusion reactions occurring during or within 24 hours following the first randomized infusion in this substudy were similar between the 2-hour and 3.5-hour infusion groups (24.4% versus 23.3%, respectively). Overall, in all randomized doses, 27.1% of the patients in the 2-hour infusion group and 25.0% of the patients in the 3.5-hour infusion group reported mild or moderate infusion reactions; two infusion reactions were severe in intensity, with one severe infusion reaction (0.3%) reported in one patient in each group in this substudy [see Warnings and Precautions (5.1)]. There were no life-threatening, fatal, or serious infusion reactions in this substudy.
16. How is Ocrevus supplied
OCREVUS (ocrelizumab) injection is a preservative-free, sterile, clear or slightly opalescent, and colorless to pale brown solution supplied as a carton containing one 300 mg/10 mL (30 mg/mL) single-dose vial (NDC 50242-150-01).
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Infusion Reactions
Inform patients about the signs and symptoms of infusion reactions, and that infusion reactions can occur up to 24 hours after infusion. Advise patients to contact their healthcare provider immediately for signs or symptoms of infusion reactions [see Warnings and Precautions (5.1)].
Infection
Advise patients to contact their healthcare provider for any signs of infection during treatment or after the last dose [see Clinical Pharmacology (12.2)]. Signs include fever, chills, constant cough, dysuria, or signs of herpes such as cold sores, shingles, or genital sores [see Warnings and Precautions (5.2)].
Advise patients that OCREVUS may cause reactivation of hepatitis B infection and that monitoring will be required if they are at risk [see Warnings and Precautions (5.2)].
Advise patients that herpes infections, including serious herpes infections affecting the central nervous system, skin, and eyes, have occurred during treatment with OCREVUS. Advise patients to promptly contact their healthcare provider if they experience any signs or symptoms of herpes infections including oral or genital symptoms, fever, skin rash, pain, itching, decreased visual acuity, eye redness, eye pain, headache, neck stiffness, or change in mental status [see Warnings and Precautions (5.2)].
Vaccination
Advise patients to complete any required live or live-attenuated vaccinations at least 4 weeks and, whenever possible, non-live vaccinations at least 2 weeks prior to initiation of OCREVUS. Administration of live-attenuated or live vaccines is not recommended during OCREVUS treatment and until B-cell recovery [see Warnings and Precautions (5.2)].
Progressive Multifocal Leukoencephalopathy
Inform patients that PML has occurred in patients who received OCREVUS. Inform the patient that PML is characterized by a progression of deficits and usually leads to death or severe disability over weeks or months. Instruct the patient of the importance of contacting their healthcare provider if they develop any symptoms suggestive of PML. Inform the patient that typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes [see Warnings and Precautions (5.3)].
Malignancies
Advise patients that an increased risk of malignancy, including breast cancer, may exist with OCREVUS. Advise patients that they should follow standard breast cancer screening guidelines [see Warnings and Precautions (5.5)].
Immune-Mediated Colitis
Advise patients to promptly contact their healthcare provider if they experience any signs and symptoms of colitis, including diarrhea, abdominal pain, and blood in stool [see Warnings and Precautions (5.6)].
Liver Injury
Inform patients that liver injury has been reported with anti-CD20 B-cell depleting therapies, including OCREVUS. Instruct patients treated with OCREVUS to promptly report any symptoms that may indicate liver injury, including fatigue, anorexia, nausea, vomiting, right upper abdominal discomfort, dark urine, or jaundice. A blood test should be obtained before patients start therapy, and during treatment as clinically indicated [see Warnings and Precautions (5.7)].
Contraception
Females of childbearing potential should use effective contraception while receiving OCREVUS and for 6 months after the last infusion of OCREVUS [see Clinical Pharmacology (12.3)]. Instruct patients that if they are pregnant or plan to become pregnant while taking OCREVUS, they should inform their healthcare provider [see Use in Specific Populations (8.1)].
OCREVUS® [ocrelizumab]
Manufactured by:
Genentech, Inc.
A Member of the Roche Group
1 DNA Way
South San Francisco, CA 94080-4990
OCREVUS is a registered trademark of Genentech, Inc.
©2025 Genentech, Inc.
U.S. License No. 1048
| This Medication Guide has been approved by the U.S. Food and Drug Administration | Revised: 8/2025 | ||||||
| MEDICATION GUIDE | |||||||
| OCREVUS® (oak-rev-us) (ocrelizumab) injection, for intravenous use |
|||||||
|
What is the most important information I should know about OCREVUS? |
|||||||
OCREVUS can cause serious side effects, including:
|
|||||||
|
|
|
|
||||
| These infusion reactions can happen for up to 24 hours after your infusion. It is important that you call your healthcare provider right away if you get any of the signs or symptoms listed above after each infusion. If you get infusion reactions, your healthcare provider may need to stop or slow down the rate of your infusion. |
|||||||
|
|||||||
|
|
|
|||||
|
|||||||
|
|
|
|||||
|
|||||||
|
|||||||
|
|||||||
|
|
||||||
|
|||||||
| What is OCREVUS? | |||||||
OCREVUS is a prescription medicine used to treat:
|
|||||||
| It is not known if OCREVUS is safe and effective in children. | |||||||
Who should not receive OCREVUS?
|
|||||||
Before receiving OCREVUS, tell your healthcare provider about all of your medical conditions, including if you:
|
|||||||
| Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. | |||||||
How will I receive OCREVUS?
|
|||||||
OCREVUS may cause serious side effects, including:
|
|||||||
| These are not all the possible side effects of OCREVUS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||||||
| General information about the safe and effective use of OCREVUS. | |||||||
| Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your pharmacist or healthcare provider for information about OCREVUS that is written for health professionals. | |||||||
| Active ingredient: ocrelizumab. | |||||||
| Inactive ingredients: glacial acetic acid, polysorbate 20, sodium acetate trihydrate, trehalose dihydrate. | |||||||
| Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990 | |||||||
| U.S. License No. 1048 | |||||||
| For more information, go to www.OCREVUS.com or call 1-844-627-3887. | |||||||
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
PRINCIPAL DISPLAY PANEL - 300 mg/10 mL Vial Carton
NDC 50242-150-01
Ocrevus®
(ocrelizumab)
Injection
300 mg/10 mL
(30 mg/mL)
For Intravenous Infusion.
Must Be Diluted.
Single-Dose Vial.
Discard Unused Portion.
Attention Pharmacist: Dispense the
accompanying Medication Guide to each
patient.
Rx only
1 vial
Genentech
10233065
| OCREVUS
ocrelizumab injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Genentech, Inc. (080129000) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| F. Hoffmann-La Roche Ltd | 485244961 | MANUFACTURE(50242-150) , ANALYSIS(50242-150) , LABEL(50242-150) , PACK(50242-150) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Roche Singapore Technical Operations Pte. Ltd. | 937189173 | ANALYSIS(50242-150) , API MANUFACTURE(50242-150) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Roche Diagnostics GmbH | 315028860 | ANALYSIS(50242-150) , MANUFACTURE(50242-150) , PACK(50242-150) , LABEL(50242-150) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Roche Diagnostics GmbH | 323105205 | ANALYSIS(50242-150) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genentech, Inc. | 080129000 | ANALYSIS(50242-150) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genentech, Inc. | 146373191 | ANALYSIS(50242-150) | |
Biological Products Related to Ocrevus
Find detailed information on biosimilars for this medication.
Frequently asked questions
- What are Monoclonal Antibodies and how do they work?
- Is Kesimpta better than Ocrevus?
- How much does Ocrevus cost?
- Briumvi vs Ocrevus: How do they compare?
- Ocrevus side effects: 5 key side effects
- How long does it take Ocrevus to work?
- How long does an Ocrevus infusion take?
- Is Ocrevus a form of chemotherapy?
- How does Ocrevus work for MS?
More about Ocrevus (ocrelizumab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (181)
- Drug images
- Side effects
- Dosage information
- Patient tips
- During pregnancy
- Support group
- FDA approval history
- Drug class: CD20 monoclonal antibodies
- Breastfeeding
- En español