Letairis: Package Insert / Prescribing Info
Package insert / product label
Generic name: ambrisentan
Dosage form: tablet, film coated
Drug class: Agents for pulmonary hypertension
Medically reviewed by Drugs.com. Last updated on Aug 31, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
Letairis (ambrisentan) tablets, for oral use
Initial U.S. Approval: 2007
WARNING: EMBRYO-FETAL TOXICITY
See full prescribing information for complete boxed warning.
- Based on animal data Letairis may cause fetal harm if used during pregnancy (4.1, 5.1, 8.1).
- Females of reproductive potential: Exclude pregnancy before the start of treatment. Use effective contraception prior to initiation of treatment, during treatment, and for one month after treatment with Letairis (2.2, 4.1, 5.1, 8.1, 8.3)
- When pregnancy is detected, discontinue Letairis as soon as possible (5.1).
Recent Major Changes
| 04/2025 04/2025 04/2025 04/2025 (removed) 04/2025 |
Indications and Usage for Letairis
Letairis is an endothelin receptor antagonist indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) in adult patients:
- To improve exercise ability and delay clinical worsening.
- In combination with tadalafil to reduce the risks of disease progression and hospitalization for worsening PAH, and to improve exercise ability.
Studies establishing effectiveness included trials predominantly in patients with WHO Functional Class II–III symptoms and etiologies of idiopathic or heritable PAH (60%) or PAH associated with connective tissue diseases (34%) (1).
Letairis Dosage and Administration
Dosage Forms and Strengths
Tablet: 5 mg and 10 mg (3)
Warnings and Precautions
- Fluid retention may require intervention (5.2).
- If patients develop acute pulmonary edema during initiation of therapy with Letairis, consider underlying pulmonary veno-occlusive disease and discontinue treatment if necessary (5.3).
- Decreases in sperm count have been observed in patients taking endothelin receptor antagonists (5.4).
- Decreases in hemoglobin have been observed within the first few weeks; measure hemoglobin at initiation, at 1 month, and periodically thereafter (5.5).
Adverse Reactions/Side Effects
- Most common adverse reactions (>3% compared to placebo) are peripheral edema, nasal congestion, sinusitis, and flushing (6.1).
- When used in combination with tadalafil, most common adverse reactions (>5% compared with either monotherapy) are peripheral edema, headache, nasal congestion, cough, anemia, dyspepsia, and bronchitis (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Gilead Sciences, Inc. at (1-800-445-3235, Option 3) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Cyclosporine increases ambrisentan exposure; limit ambrisentan dose to 5 mg once daily (7).
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2025
Full Prescribing Information
WARNING: EMBRYO-FETAL TOXICITY
Letairis is contraindicated for use during pregnancy because it may cause major birth defects if used by pregnant patients, based on studies in animals [see Contraindications (4.1), Warnings and Precautions (5.1), and Use in Specific Populations (8.1)].
Therefore, for females of reproductive potential, exclude pregnancy before the initiation of treatment with Letairis. Advise use of effective contraception before initiation, during treatment, and for one month after treatment with Letairis [see Dosage and Administration (2.2) Contraindications (4.1), Warnings and Precautions (5.1), and Use in Specific Populations (8.1, 8.3)]. When pregnancy is detected, discontinue Letairis as soon as possible (5.1).
1. Indications and Usage for Letairis
Letairis is indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) in adult patients:
- To improve exercise ability and delay clinical worsening.
- In combination with tadalafil to reduce the risks of disease progression and hospitalization for worsening PAH, and to improve exercise ability [see Clinical Studies (14.2)].
Studies establishing effectiveness included predominantly patients with WHO Functional Class II–III symptoms and etiologies of idiopathic or heritable PAH (60%) or PAH associated with connective tissue diseases (34%).
2. Letairis Dosage and Administration
2.1 Adult Dosage
Initiate treatment at 5 mg once daily, with or without tadalafil 20 mg once daily. At 4-week intervals, either the dose of Letairis or tadalafil can be increased, as needed and tolerated, to Letairis 10 mg or tadalafil 40 mg.
Do not split, crush, or chew tablets.
2.2 Pregnancy Testing in Females of Reproductive Potential
Exclude pregnancy before initiating treatment with Letairis in females of reproductive potential [see Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
3. Dosage Forms and Strengths
5 mg and 10 mg film-coated tablets for oral administration
- Each 5 mg tablet is square convex, pale pink, with “5” on one side and “GSI” on the other side.
- Each 10 mg tablet is oval convex, deep pink, with “10” on one side and “GSI” on the other side.
4. Contraindications
4.1 Pregnancy
Letairis may cause fetal harm when administered to a pregnant female. Letairis is contraindicated in females who are pregnant. Letairis was consistently shown to have teratogenic effects when administered to animals. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to a fetus [see Dosage and Administration (2.2), Warnings and Precautions (5.1), and Use in Specific Populations (8.1)].
4.2 Idiopathic Pulmonary Fibrosis
Letairis is contraindicated in patients with Idiopathic Pulmonary Fibrosis (IPF), including IPF patients with pulmonary hypertension (WHO Group 3) [see Clinical Studies (14.4)].
5. Warnings and Precautions
5.1 Embryo-fetal Toxicity
Based on data from animal reproduction studies, Letairis may cause fetal harm when administered during pregnancy and is contraindicated during pregnancy. The available human data for endothelin receptor antagonists do not establish the presence or absence of major birth defects related to the use of Letairis. Advise patients who can become pregnant of the potential risk to a fetus. Obtain a pregnancy test prior to initiation of treatment with Letairis. Advise patients who can become pregnant to use effective contraception prior to initiation of treatment, during treatment, and for one month after discontinuation of treatment with Letairis. When pregnancy is detected, discontinue use as soon as possible [see Dosage and Administration (2.2), and Use in Specific Populations (8.1, 8.3)].
5.2 Fluid Retention
Peripheral edema is a known class effect of endothelin receptor antagonists, and is also a clinical consequence of PAH and worsening PAH. In the placebo-controlled studies, there was an increased incidence of peripheral edema in patients treated with doses of 5 or 10 mg Letairis compared to placebo [see Adverse Reactions (6.1)]. Most edema was mild to moderate in severity.
In addition, there have been postmarketing reports of fluid retention in patients with pulmonary hypertension, occurring within weeks after starting Letairis. Patients required intervention with a diuretic, fluid management, or, in some cases, hospitalization for decompensating heart failure.
If clinically significant fluid retention develops, with or without associated weight gain, further evaluation should be undertaken to determine the cause, such as Letairis or underlying heart failure, and the possible need for specific treatment or discontinuation of Letairis therapy.
Peripheral edema/fluid retention is more common with Letairis plus tadalafil than with Letairis or tadalafil alone.
5.3 Pulmonary Edema with Pulmonary Veno-occlusive Disease (PVOD)
If patients develop acute pulmonary edema during initiation of therapy with vasodilating agents such as Letairis, the possibility of PVOD should be considered, and if confirmed Letairis should be discontinued.
5.4 Decreased Sperm Counts
Decreased sperm counts have been observed in human and animal studies with another endothelin receptor antagonist and in animal fertility studies with ambrisentan. Letairis may have an adverse effect on spermatogenesis. Counsel patients about potential effects on fertility [see Use in Specific Populations (8.6) and Nonclinical Toxicology (13.1)].
5.5 Hematological Changes
Decreases in hemoglobin concentration and hematocrit have followed administration of other endothelin receptor antagonists and were observed in clinical studies with Letairis. These decreases were observed within the first few weeks of treatment with Letairis, and stabilized thereafter. The mean decrease in hemoglobin from baseline to end of treatment for those patients receiving Letairis in the 12-week placebo-controlled studies was 0.8 g/dL.
Marked decreases in hemoglobin (>15% decrease from baseline resulting in a value below the lower limit of normal) were observed in 7% of all patients receiving Letairis (and 10% of patients receiving 10 mg) compared to 4% of patients receiving placebo. The cause of the decrease in hemoglobin is unknown, but it does not appear to result from hemorrhage or hemolysis.
In the long-term open-label extension of the two pivotal clinical studies, mean decreases from baseline (ranging from 0.9 to 1.2 g/dL) in hemoglobin concentrations persisted for up to 4 years of treatment.
There have been postmarketing reports of decreases in hemoglobin concentration and hematocrit that have resulted in anemia requiring transfusion.
Measure hemoglobin prior to initiation of Letairis, at one month, and periodically thereafter. Initiation of Letairis therapy is not recommended for patients with clinically significant anemia. If a clinically significant decrease in hemoglobin is observed and other causes have been excluded, consider discontinuing Letairis.
6. Adverse Reactions/Side Effects
Clinically significant adverse reactions that appear in other sections of the labeling include:
- Embryo-fetal Toxicity [see Warnings and Precautions (5.1), Use in Specific Populations (8.1)]
- Fluid Retention [see Warnings and Precautions (5.2)]
- Pulmonary Edema with PVOD [see Warnings and Precautions (5.3)]
- Decreased Sperm Count [see Warnings and Precautions (5.4)]
- Hematologic Changes [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Safety data for Letairis are presented from two 12-week, placebo-controlled studies (ARIES-1 and ARIES-2) in patients with pulmonary arterial hypertension (PAH), and one randomized, double-blind, active-controlled trial in 605 patients with PAH (AMBITION) comparing Letairis plus tadalafil to Letairis or tadalafil alone. The exposure to Letairis in these studies ranged from 1 day to 4 years (N=357 for at least 6 months and N=279 for at least 1 year).
In ARIES-1 and ARIES-2, a total of 261 patients received Letairis at doses of 2.5, 5, or 10 mg once daily and 132 patients received placebo. The adverse reactions that occurred in >3% more patients receiving Letairis than receiving placebo are shown in Table 1.
| Placebo (N=132) | Letairis (N=261) |
||
|---|---|---|---|
| Adverse Reaction | n (%) | n (%) | Placebo-adjusted (%) |
| Peripheral edema | 14 (11) | 45 (17) | 6 |
| Nasal congestion | 2 (2) | 15 (6) | 4 |
| Sinusitis | 0 (0) | 8 (3) | 3 |
| Flushing | 1 (1) | 10 (4) | 3 |
Most adverse drug reactions were mild to moderate and only nasal congestion was dose-dependent.
Few notable differences in the incidence of adverse reactions were observed for patients by age or sex. Peripheral edema was similar in younger patients (<65 years) receiving Letairis (14%; 29/205) or placebo (13%; 13/104), and was greater in elderly patients (≥65 years) receiving Letairis (29%; 16/56) compared to placebo (4%; 1/28). The results of such subgroup analyses must be interpreted cautiously.
The incidence of treatment discontinuations due to adverse events other than those related to PAH during the clinical trials in patients with PAH was similar for Letairis (2%; 5/261 patients) and placebo (2%; 3/132 patients). The incidence of patients with serious adverse events other than those related to PAH during the clinical trials in patients with PAH was similar for placebo (7%; 9/132 patients) and for Letairis (5%; 13/261 patients).
During 12-week controlled clinical trials, the incidence of aminotransferase elevations >3 × upper limit of normal (ULN) were 0% on Letairis and 2.3% on placebo. In practice, cases of hepatic injury should be carefully evaluated for cause.
Combination Use with Tadalafil
The mean exposure to Letairis + tadalafil in the AMBITION study was 78.7 weeks. The adverse reactions that occurred in >5% more patients receiving Letairis + tadalafil than receiving Letairis or tadalafil monotherapy in AMBITION are shown in Table 2.
| Adverse Reactions | Letairis + Tadalafil Combination Therapy (N=302) n (%) | Letairis Monotherapy (N=152) n (%) | Tadalafil Monotherapy (N=151) n (%) |
|---|---|---|---|
| Peripheral edema | 135 (45%) | 58 (38%) | 43 (28%) |
| Headache | 125 (41%) | 51 (34%) | 53 (35%) |
| Nasal congestion | 58 (19%) | 25 (16%) | 17 (11%) |
| Cough | 53 (18%) | 20 (13%) | 24 (16%) |
| Anemia | 44 (15%) | 11 (7%) | 17 (11%) |
| Dyspepsia | 32 (11%) | 5 (3%) | 18 (12%) |
| Bronchitis | 31 (10%) | 6 (4%) | 13 (9%) |
Peripheral edema was more frequent on combination therapy; however, there was no notable difference observed in the incidence of peripheral edema in elderly patients (≥65 years) versus younger patients (<65 years) on combination therapy (44% vs. 45%) or Letairis monotherapy (37% vs. 39%) in AMBITION.
Treatment discontinuations due to adverse events while on randomized treatment were similar across treatment groups: 16% for Letairis + tadalafil, 14% for Letairis alone, and 13% for tadalafil alone.
Use in Patients with Prior Endothelin Receptor Antagonist (ERA) Related Serum Liver Enzyme Abnormalities
In an uncontrolled, open-label study, 36 patients who had previously discontinued endothelin receptor antagonists (ERAs: bosentan, an investigational drug, or both) due to aminotransferase elevations >3 × ULN were treated with Letairis. Prior elevations were predominantly moderate, with 64% of the ALT elevations <5 × ULN, but 9 patients had elevations >8 × ULN. Eight patients had been re-challenged with bosentan and/or the investigational ERA and all eight had a recurrence of aminotransferase abnormalities that required discontinuation of ERA therapy. All patients had to have normal aminotransferase levels on entry to this study. Twenty-five of the 36 patients were also receiving prostanoid and/or phosphodiesterase type 5 (PDE5) inhibitor therapy. Two patients discontinued early (including one of the patients with a prior 8 × ULN elevation). Of the remaining 34 patients, one patient experienced a mild aminotransferase elevation at 12 weeks on Letairis 5 mg that resolved with decreasing the dosage to 2.5 mg, and that did not recur with later escalations to 10 mg. With a median follow-up of 13 months and with 50% of patients increasing the dose of Letairis to 10 mg, no patients were discontinued for aminotransferase elevations. While the uncontrolled study design does not provide information about what would have occurred with re-administration of previously used ERAs or show that Letairis led to fewer aminotransferase elevations than would have been seen with those drugs, the study indicates that Letairis may be tried in patients who have experienced asymptomatic aminotransferase elevations on other ERAs after aminotransferase levels have returned to normal.
6.2 Postmarketing Experience
The following adverse reactions were identified during post-approval use of Letairis. Because these reactions were reported voluntarily from a population of uncertain size, it is not possible to estimate reliably the frequency or to establish a causal relationship to drug exposure: anemia requiring transfusion [see Warnings and Precautions (5.5)] heart failure (associated with fluid retention), symptomatic hypotension, and hypersensitivity (e.g., angioedema, rash).
Elevations of liver aminotransferases (ALT, AST) have been reported with Letairis use; in most cases alternative causes of the liver injury could be identified (heart failure, hepatic congestion, hepatitis, alcohol use, hepatotoxic medications). Other endothelin receptor antagonists have been associated with elevations of aminotransferases, hepatotoxicity, and cases of liver failure [see Adverse Reactions (6.1)].
Related/similar drugs
7. Drug Interactions
Multiple dose coadministration of ambrisentan and cyclosporine resulted in an approximately 2-fold increase in ambrisentan exposure in healthy volunteers; therefore, limit the dose of ambrisentan to 5 mg once daily when coadministered with cyclosporine [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on data from animal reproduction studies, Letairis may cause fetal harm, including birth defects and fetal death, when administered to a pregnant woman and is contraindicated during pregnancy. There are limited data on Letairis use in pregnant women. Available data from post-marketing reports and published literature over decades of use with endothelin receptor antagonists in the same class as Letairis have not identified an increased risk of major birth defects; however, these data are limited. Methodological limitations of these postmarketing reports and published literature include lack of a control group; limited information regarding dose, duration, and timing of drug exposure; and missing data. These limitations preclude establishing a reliable estimate of the risk of adverse fetal and neonatal outcomes with maternal endothelin receptor antagonist use. In animal reproduction studies, Letairis was teratogenic in rats and rabbits at doses which resulted in exposures of 3.5 and 1.7 times, respectively, the human dose of 10 mg per day [see Animal Data]. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, advise the patient of the potential hazard to a fetus [see Contraindications (4.1), Warnings and Precautions (5.1)].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Data
Animal Data
Letairis was teratogenic at oral dosages of ≥15 mg/kg/day (AUC 51.7 h•µg/mL) in rats and ≥7 mg/kg/day (24.7 h•µg/mL) in rabbits; it was not studied at lower dosages. These dosages are of 3.5 and 1.7 times, respectively, the human dose of 10 mg per day (14.8 h•µg/mL) based on AUC. In both species, there were abnormalities of the lower jaw and hard and soft palate, malformation of the heart and great vessels, and failure of formation of the thymus and thyroid.
A preclinical study in rats has shown decreased survival of newborn pups (mid and high dosages) and effects on testicle size and fertility of pups (high dosage) following maternal treatment with ambrisentan from late gestation through weaning. The mid and high dosages were 51 ×, and 170 × (on a mg/m2 body surface area basis) the maximum oral human dose of 10 mg and an average adult body weight of 70 kg. These effects were absent at a maternal dosage 17 × the human dose based on mg/m2.
8.2 Lactation
Risk Summary
It is not known whether ambrisentan is present in human milk. Because many drugs are present in human milk and because of the potential for serious adverse reactions in breastfed infants from Letairis, a decision should be made whether to discontinue breastfeeding or discontinue Letairis, taking into account the importance of the drug to the mother.
8.3 Females and Males of Reproductive Potential
Based on data from animal reproductive toxicity studies, ambrisentan may cause fetal harm, including birth defects and fetal death, when administered to a pregnant patient and is contraindicated during pregnancy [see Contraindications (4.1), Use in Specific Populations (8.1)].
Pregnancy Testing
Verify that patients who can become pregnant are not pregnant prior to initiating ambrisentan. The patient should contact their physician immediately for pregnancy testing if onset of menses is delayed or pregnancy is suspected. If the pregnancy test is positive, the physician and patient should discuss the risks to the pregnancy and the fetus.
Contraception
Patients who can become pregnant who are using ambrisentan should use effective contraception prior to initiation of treatment, during treatment, and for one month after discontinuation of treatment with ambrisentan to prevent pregnancy [see Warnings and Precautions (5.1)].
Infertility
Males
In a 6-month study of another endothelin receptor antagonist, bosentan, 25 male patients with WHO functional class III and IV PAH and normal baseline sperm count were evaluated for effects on testicular function. There was a decline in sperm count of at least 50% in 25% of the patients after 3 or 6 months of treatment with bosentan. One patient developed marked oligospermia at 3 months, and the sperm count remained low with 2 follow-up measurements over the subsequent 6 weeks.
Bosentan was discontinued and after 2 months the sperm count had returned to baseline levels. In 22 patients who completed 6 months of treatment, sperm count remained within the normal range and no changes in sperm morphology, sperm motility, or hormone levels were observed. Based on these findings and preclinical data [see Nonclinical Toxicology (13.1)] from endothelin receptor antagonists, it cannot be excluded that endothelin receptor antagonists such as Letairis have an adverse effect on spermatogenesis. Counsel patients about the potential effects on fertility [see Warnings and Precautions (5.5)].
8.4 Pediatric Use
Safety and effectiveness of Letairis in pediatric patients have not been established.
Juvenile Animal Data
In juvenile rats administered ambrisentan orally once daily during postnatal day 7 to 26, 36, or 62, a decrease in brain weight (−3% to −8%) with no morphologic or neurobehavioral changes occurred after breathing sounds, apnea, and hypoxia were observed, at exposures approximately 1.8 to 7.0 times human pediatric exposures at 10 mg, based on AUC.
8.5 Geriatric Use
In the two placebo-controlled clinical studies of Letairis, 21% of patients were ≥65 years old and 5% were ≥75 years old. The elderly (age ≥65 years) showed less improvement in walk distances with Letairis than younger patients did, but the results of such subgroup analyses must be interpreted cautiously. Peripheral edema was more common in the elderly than in younger patients.
8.6 Renal Impairment
The impact of renal impairment on the pharmacokinetics of ambrisentan has been examined using a population pharmacokinetic approach in PAH patients with creatinine clearances ranging between 20 and 150 mL/min. There was no significant impact of mild or moderate renal impairment on exposure to ambrisentan [see Clinical Pharmacology (12.3)]. Dose adjustment of Letairis in patients with mild or moderate renal impairment is therefore not required. There is no information on the exposure to ambrisentan in patients with severe renal impairment.
The impact of hemodialysis on the disposition of ambrisentan has not been investigated.
8.7 Hepatic Impairment
Pre-existing Hepatic Impairment
The influence of pre-existing hepatic impairment on the pharmacokinetics of ambrisentan has not been evaluated. Because there is in vitro and in vivo evidence of significant metabolic and biliary contribution to the elimination of ambrisentan, hepatic impairment might be expected to have significant effects on the pharmacokinetics of ambrisentan [see Clinical Pharmacology (12.3)]. Letairis is not recommended in patients with moderate or severe hepatic impairment. There is no information on the use of Letairis in patients with mild pre-existing impaired liver function; however, exposure to ambrisentan may be increased in these patients.
Elevation of Liver Transaminases
Other endothelin receptor antagonists (ERAs) have been associated with aminotransferase (AST, ALT) elevations, hepatotoxicity, and cases of liver failure [see Adverse Reactions (6.1, 6.2)]. In patients who develop hepatic impairment after Letairis initiation, the cause of liver injury should be fully investigated. Discontinue Letairis if elevations of liver aminotransferases are >5 × ULN or if elevations are accompanied by bilirubin >2 × ULN, or by signs or symptoms of liver dysfunction and other causes are excluded.
10. Overdosage
There is no experience with overdosage of Letairis. The highest single dose of Letairis administered to healthy volunteers was 100 mg, and the highest daily dose administered to patients with PAH was 10 mg once daily. In healthy volunteers, single doses of 50 mg and 100 mg (5 to 10 times the maximum recommended dose) were associated with headache, flushing, dizziness, nausea, and nasal congestion. Massive overdosage could potentially result in hypotension that may require intervention.
11. Letairis Description
Letairis contains ambrisentan, an endothelin receptor antagonist. The chemical name of ambrisentan is (+)-(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy-3,3-diphenylpropanoic acid. It has a molecular formula of C22H22N2O4 and a molecular weight of 378.42 and has the following structural formula:
Figure 1 Ambrisentan Structural Formula
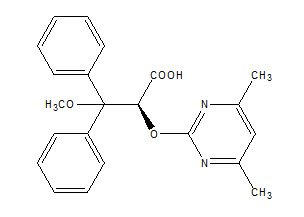
Ambrisentan is a white to off-white, crystalline solid. It is a carboxylic acid with a pKa of 4.0. Ambrisentan is practically insoluble in water and in aqueous solutions at low pH. Solubility increases in aqueous solutions at higher pH. In the solid state ambrisentan is very stable, is not hygroscopic, and is not light sensitive.
Letairis is available as 5 mg and 10 mg film-coated tablets for once daily oral administration. The tablets include the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate and microcrystalline cellulose. The tablets are film-coated with a coating material containing FD&C Red #40 aluminum lake, lecithin, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. Each square, pale pink Letairis tablet contains 5 mg of ambrisentan. Each oval, deep pink Letairis tablet contains 10 mg of ambrisentan. Letairis tablets are unscored.
12. Letairis - Clinical Pharmacology
12.1 Mechanism of Action
Endothelin-1 (ET-1) is a potent autocrine and paracrine peptide. Two receptor subtypes, ETA and ETB, mediate the effects of ET-1 in the vascular smooth muscle and endothelium. The primary actions of ETA are vasoconstriction and cell proliferation, while the predominant actions of ETB are vasodilation, antiproliferation, and ET-1 clearance.
In patients with PAH, plasma ET-1 concentrations are increased as much as 10-fold and correlate with increased mean right atrial pressure and disease severity. ET-1 and ET-1 mRNA concentrations are increased as much as 9-fold in the lung tissue of patients with PAH, primarily in the endothelium of pulmonary arteries. These findings suggest that ET-1 may play a critical role in the pathogenesis and progression of PAH.
Ambrisentan is a high-affinity (Ki=0.011 nM) ETA receptor antagonist with a high selectivity for the ETA versus ETB receptor (>4000-fold). The clinical impact of high selectivity for ETA is not known.
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a randomized, positive- and placebo-controlled, parallel-group study, healthy subjects received either Letairis 10 mg daily followed by a single dose of 40 mg, placebo followed by a single dose of moxifloxacin 400 mg, or placebo alone. Letairis 10 mg daily had no significant effect on the QTc interval. The 40 mg dose of Letairis increased mean QTc at tmax by 5 ms with an upper 95% confidence limit of 9 ms. For patients receiving Letairis 5–10 mg daily and not taking metabolic inhibitors, no significant QT prolongation is expected.
N-terminal pro-B-type natriuretic peptide (NT-proBNP)
In AMBITION [see Clinical Studies (14.2)], the decrease in NT-proBNP in patients on Letairis plus tadalafil was observed early (Week 4) and was sustained, with a reduction of 63% on Letairis plus tadalafil, 50% on Letairis alone, and 41% on tadalafil alone at Week 24.
12.3 Pharmacokinetics
The pharmacokinetics of ambrisentan (S-ambrisentan) in healthy subjects is dose proportional. The absolute bioavailability of ambrisentan is not known. Ambrisentan is absorbed with peak concentrations occurring approximately 2 hours after oral administration in healthy subjects and PAH patients. Food does not affect its bioavailability. In vitro studies indicate that ambrisentan is a substrate of P-gp. Ambrisentan is highly bound to plasma proteins (99%). The elimination of ambrisentan is predominantly by non-renal pathways, but the relative contributions of metabolism and biliary elimination have not been well characterized. In plasma, the AUC of 4-hydroxymethyl ambrisentan accounts for approximately 4% relative to parent ambrisentan AUC. The in vivo inversion of S-ambrisentan to R-ambrisentan is negligible. The mean oral clearance of ambrisentan is 38 mL/min and 19 mL/min in healthy subjects and in PAH patients, respectively. Although ambrisentan has a 15-hour terminal half-life, the mean trough concentration of ambrisentan at steady-state is about 15% of the mean peak concentration and the accumulation factor is about 1.2 after long-term daily dosing, indicating that the effective half-life of ambrisentan is about 9 hours.
Drug Interactions
In Vitro Studies
Studies with human liver tissue indicate that ambrisentan is metabolized by CYP3A, CYP2C19, and uridine 5'-diphosphate glucuronosyltransferases (UGTs) 1A9S, 2B7S, and 1A3S. In vitro studies suggest that ambrisentan is a substrate of the Organic Anion Transporting Polypeptides OATP1B1 and OATP1B3, and P-glycoprotein (P-gp). Drug interactions might be expected because of these factors; however, a clinically relevant interaction has been demonstrated only with cyclosporine [see Drug Interactions (7)]. In vitro studies found ambrisentan to have little to no inhibition of human hepatic transporters. Ambrisentan demonstrated weak dose-dependent inhibition of OATP1B1, OATP1B3, and NTCP (IC50 of 47 µM, 45 µM, and approximately 100 µM, respectively) and no transporter-specific inhibition of BSEP, BRCP, P-gp, or MRP2. Ambrisentan does not inhibit or induce drug metabolizing enzymes at clinically relevant concentrations.
In Vivo Studies
The effects of other drugs on ambrisentan pharmacokinetics and the effects of ambrisentan on the exposure to other drugs are shown in Figure 2 and Figure 3, respectively.
Figure 2 Effects of Other Drugs on Ambrisentan Pharmacokinetics
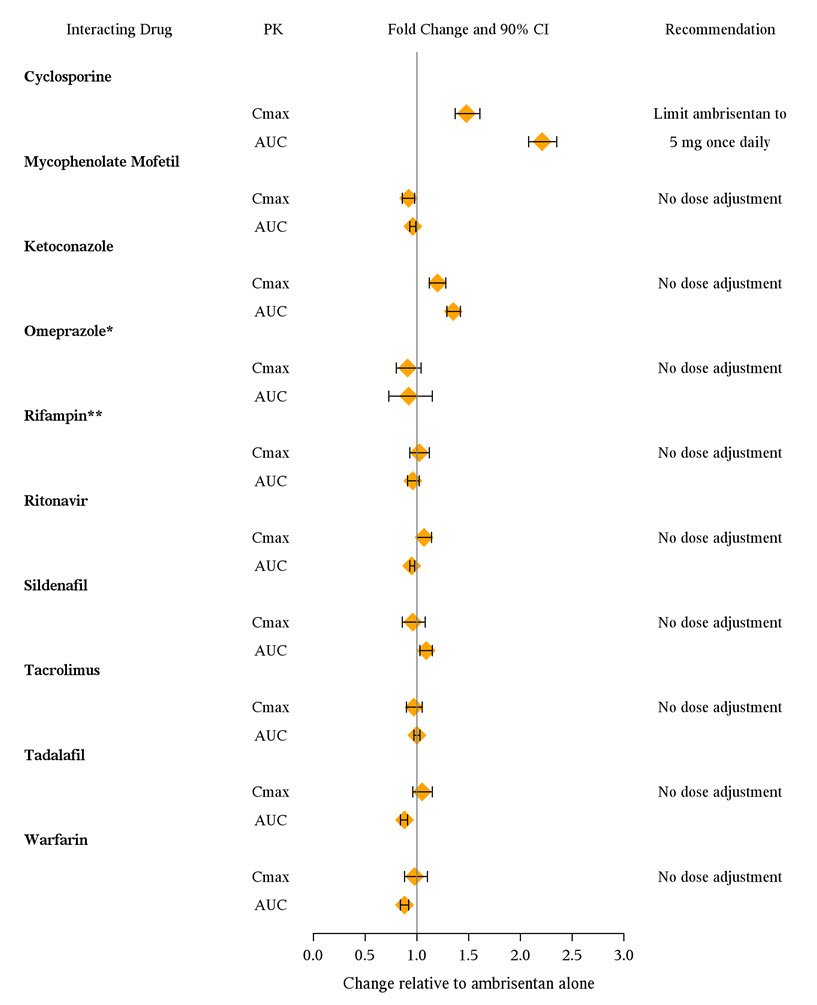
* Omeprazole: based on population pharmacokinetic analysis in PAH patients
** Rifampin: AUC and Cmax were measured at steady-state. On Day 3 of coadministration a transient 2-fold increase in AUC was noted that was no longer evident by Day 7. Day 7 results are presented.
Figure 3 Effects of Ambrisentan on Other Drugs

* Active metabolite of mycophenolate mofetil
** GMR (95% CI) for INR
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Oral carcinogenicity studies of up to two years duration were conducted at starting doses of 10, 30, and 60 mg/kg/day in rats (8 to 48 times the maximum recommended human dose [MRHD] on a mg/m2 basis) and at 50, 150, and 250 mg/kg/day in mice (28 to 140 times the MRHD). In the rat study, the high- and mid-dose male and female groups had their doses lowered to 40 and 20 mg/kg/day, respectively, in week 51 because of effects on survival. The high-dose males and females were taken off drug completely in weeks 69 and 93, respectively. The only evidence of ambrisentan-related carcinogenicity was a positive trend in male rats, for the combined incidence of benign basal cell tumor and basal cell carcinoma of skin/subcutis in the mid-dose group (high-dose group excluded from analysis), and the occurrence of mammary fibroadenomas in males in the high-dose group. In the mouse study, high-dose male and female groups had their doses lowered to 150 mg/kg/day in week 39 and were taken off drug completely in week 96 (males) or week 76 (females). In mice, ambrisentan was not associated with excess tumors in any dosed group.
Positive findings of clastogenicity were detected, at drug concentrations producing moderate to high toxicity, in the chromosome aberration assay in cultured human lymphocytes. There was no evidence for genetic toxicity of ambrisentan when tested in vitro in bacteria (Ames test) or in vivo in rats (micronucleus assay, unscheduled DNA synthesis assay).
The development of testicular tubular atrophy and impaired fertility has been linked to the chronic administration of endothelin receptor antagonists in rodents. Testicular tubular degeneration was observed in rats treated with ambrisentan for two years at doses ≥10 mg/kg/day (8-fold MRHD). Increased incidences of testicular findings were also observed in mice treated for two years at doses ≥50 mg/kg/day (28-fold MRHD). Effects on sperm count, sperm morphology, mating performance, and fertility were observed in fertility studies in which male rats were treated with ambrisentan at oral doses of 300 mg/kg/day (236-fold MRHD). At doses of ≥10 mg/kg/day, observations of testicular histopathology in the absence of fertility and sperm effects were also present.
14. Clinical Studies
14.1 Pulmonary Arterial Hypertension (PAH)
Two 12-week, randomized, double-blind, placebo-controlled, multicenter studies were conducted in 393 patients with PAH (WHO Group 1). The two studies were identical in design except for the doses of Letairis and the geographic region of the investigational sites. ARIES-1 compared once-daily doses of 5 mg and 10 mg Letairis to placebo, while ARIES-2 compared once-daily doses of 2.5 mg and 5 mg Letairis to placebo. In both studies, Letairis or placebo was added to current therapy, which could have included a combination of anticoagulants, diuretics, calcium channel blockers, or digoxin, but not epoprostenol, treprostinil, iloprost, bosentan, or sildenafil. The primary study endpoint was 6-minute walk distance. In addition, clinical worsening, WHO functional class, dyspnea, and SF-36® Health Survey were assessed.
Patients had idiopathic or heritable PAH (64%) or PAH associated with connective tissue diseases (32%), HIV infection (3%), or anorexigen use (1%). There were no patients with PAH associated with congenital heart disease.
Patients had WHO functional class I (2%), II (38%), III (55%), or IV (5%) symptoms at baseline. The mean age of patients was 50 years, 79% of patients were female, and 77% were Caucasian.
Submaximal Exercise Ability
Results of the 6-minute walk distance at 12 weeks for the ARIES-1 and ARIES-2 studies are shown in Table 3 and Figure 4.
| ARIES-1 | ARIES-2 | |||||
|---|---|---|---|---|---|---|
| Placebo
(N=67) | 5 mg
(N=67) | 10 mg
(N=67) | Placebo
(N=65) | 2.5 mg
(N=64) | 5 mg
(N=63) |
|
| Mean ± standard deviation | ||||||
|
||||||
| Baseline | 342 ± 73 | 340 ± 77 | 342 ± 78 | 343 ± 86 | 347 ± 84 | 355 ± 84 |
| Mean change from baseline | -8 ± 79 | 23 ± 83 | 44 ± 63 | -10 ± 94 | 22 ± 83 | 49 ± 75 |
| Placebo-adjusted mean change from baseline | _ | 31 | 51 | _ | 32 | 59 |
| Placebo-adjusted median change from baseline | _ | 27 | 39 | _ | 30 | 45 |
| p-value* | _ | 0.008 | <0.001 | _ | 0.022 | <0.001 |
Figure 4 Mean Change in 6-Minute Walk Distance (ARIES-1 and ARIES-2)
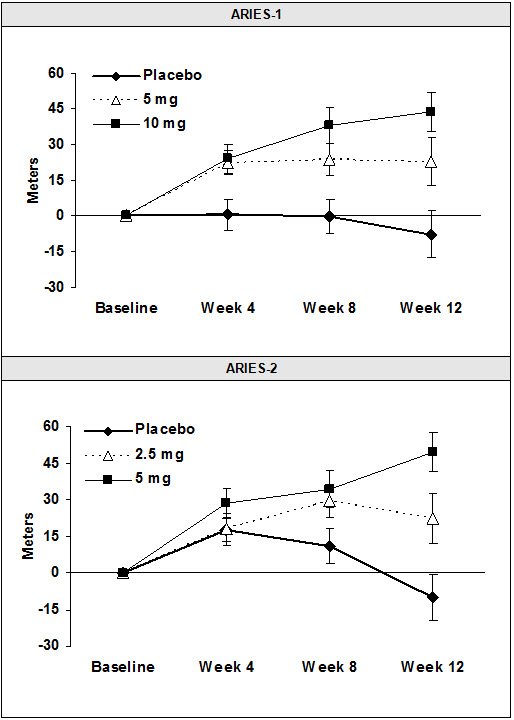
Mean change from baseline in 6-minute walk distance in the placebo and Letairis groups.
Values are expressed as mean ± standard error of the mean.
In both studies, treatment with Letairis resulted in a significant improvement in 6-minute walk distance for each dose of Letairis and the improvements increased with dose. An increase in 6-minute walk distance was observed after 4 weeks of treatment with Letairis, with a dose-response observed after 12 weeks of treatment. Improvements in walk distance with Letairis were smaller for elderly patients (age ≥65) than younger patients and for patients with secondary PAH than for patients with idiopathic or heritable PAH. The results of such subgroup analyses must be interpreted cautiously.
Clinical Worsening
Time to clinical worsening of PAH was defined as the first occurrence of death, lung transplantation, hospitalization for PAH, atrial septostomy, study withdrawal due to the addition of other PAH therapeutic agents, or study withdrawal due to early escape. Early escape was defined as meeting two or more of the following criteria: a 20% decrease in the 6-minute walk distance; an increase in WHO functional class; worsening right ventricular failure; rapidly progressing cardiogenic, hepatic, or renal failure; or refractory systolic hypotension. The clinical worsening events during the 12-week treatment period of the Letairis clinical trials are shown in Table 4 and Figure 5.
| ARIES-1 | ARIES-2 | |||
|---|---|---|---|---|
| Placebo
(N=67) | Letairis
(N=134) | Placebo
(N=65) | Letairis
(N=127) |
|
| Intention-to-treat population.
Note: Patients may have had more than one reason for clinical worsening. Nominal p-values |
||||
| Clinical worsening, no. (%) | 7 (10%) | 4 (3%) | 13 (22%) | 8 (6%) |
| Hazard ratio | _ | 0.28 | _ | 0.30 |
| p-value, Log-rank test | _ | 0.030 | _ | 0.005 |
There was a significant delay in the time to clinical worsening for patients receiving Letairis compared to placebo. Results in subgroups such as the elderly were also favorable.
Figure 5 Time to Clinical Worsening (ARIES-1 and ARIES-2)
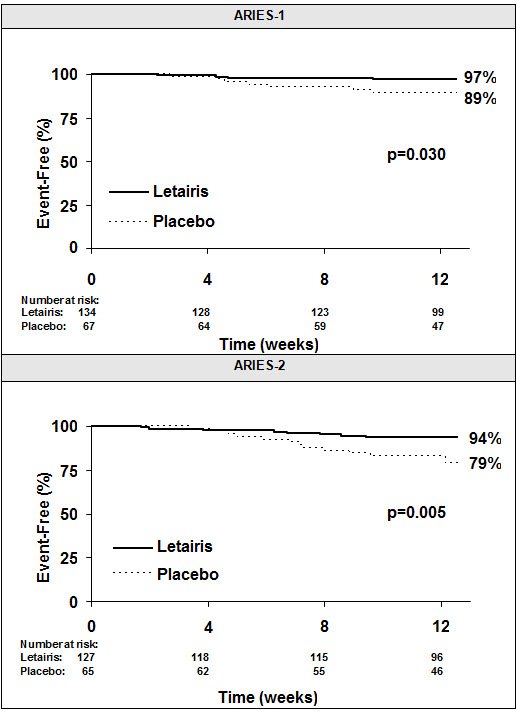
Time from randomization to clinical worsening with Kaplan-Meier estimates of the proportions of patients without events in ARIES-1 and ARIES-2.
p-values shown are the log-rank comparisons of Letairis to placebo stratified by idiopathic or heritable PAH and non-idiopathic, non-heritable PAH patients.
14.2 Combination Treatment of PAH
In a randomized, double-blind, active-controlled trial (AMBITION), 605 patients with WHO Functional Class II or III PAH were randomized 2:1:1 to once daily Letairis plus tadalafil or to Letairis or tadalafil alone. Treatment was initiated with Letairis 5 mg and tadalafil 20 mg. If tolerated, tadalafil was increased to 40 mg at 4 weeks and Letairis was increased to 10 mg at 8 weeks.
The primary endpoint was time to first occurrence of (a) death, (b) hospitalization for worsening PAH, (c) >15% decrease from baseline in 6MWD combined with WHO Functional Class III or IV symptoms sustained over 14 days (short term clinical worsening), or (d) reduction in 6MWD sustained over 14 days combined with WHO Functional Class III or IV symptoms sustained over 6 months (inadequate long term clinical response).
Patients had idiopathic PAH (55%), heritable PAH (3%), or PAH associated with connective tissue diseases, congenital heart disease, stable HIV infection, or drugs or toxins (APAH, 43%). Median time from diagnosis to first study drug administration was 25 days. Approximately 32% and 68% of patients were in WHO Functional Class II and III, respectively. The mean patient age was 55.7 years (34% were ≥65 years old). Most patients were white (90%) and female (76%); 45% were North American.
Principal results are shown in Figures 6 and 7.
Figure 6 Time to Primary Endpoint Event (AMBITION)
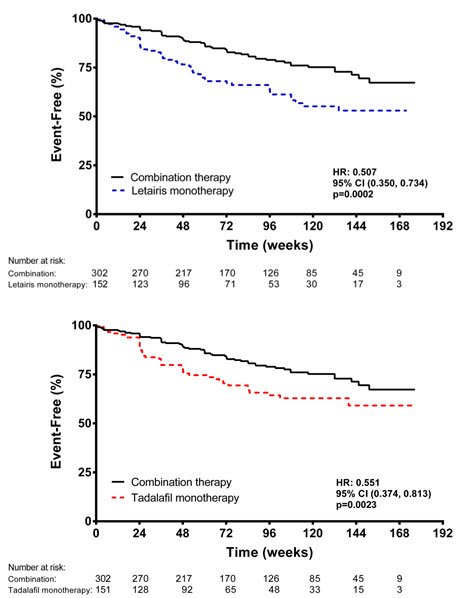
Figure 7 Primary Endpoint Events and First Occurrences of Each Component at Any Time (AMBITION)
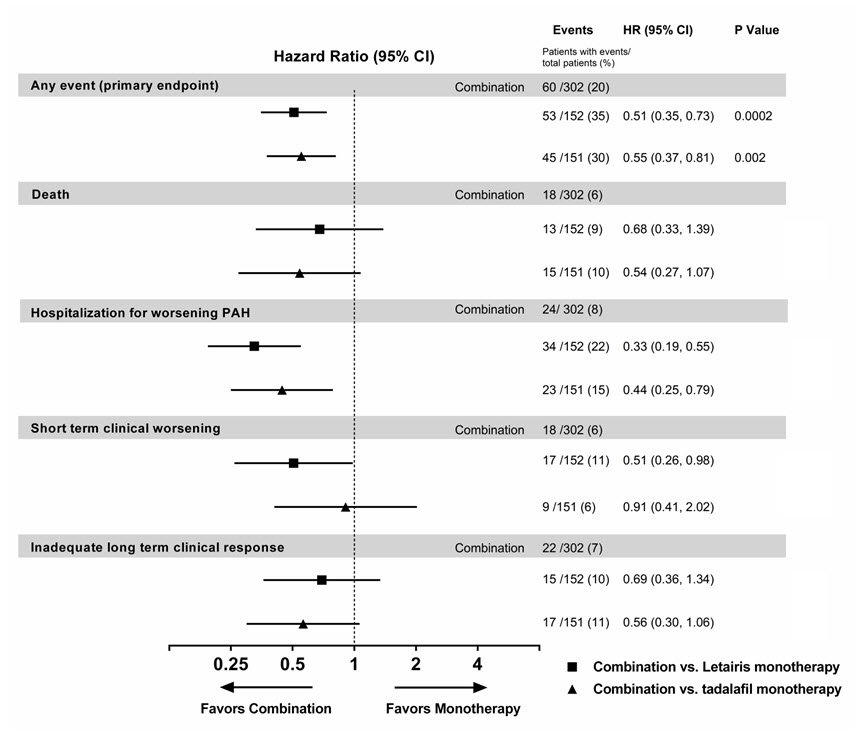
The treatment effect of Letairis plus tadalafil compared with individual monotherapy on time to first primary endpoint event was consistent across subgroups. (Figure 8).
Figure 8 Primary Endpoint by Subgroups (AMBITION)
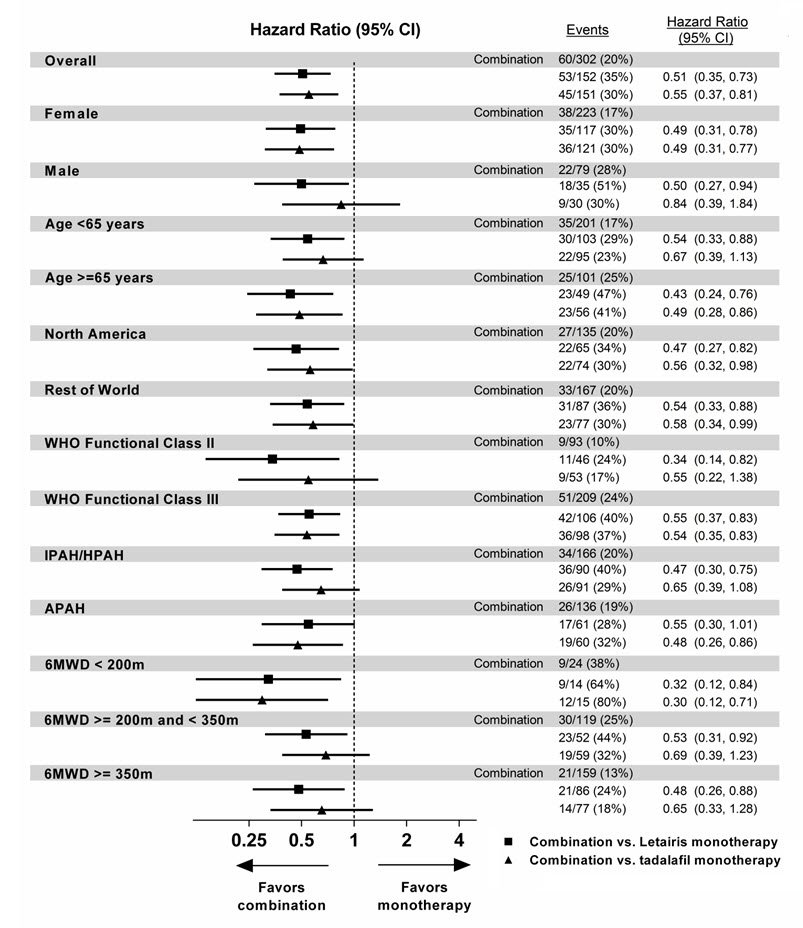
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and all of which were pre-specified, if not the groupings. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over interpreted.
Exercise Ability
Results of the 6MWD at 24 weeks for the AMBITION study are shown in Table 5 and Figure 9.
| Letairis + Tadalafil
(N=302) | Letairis Monotherapy
(N=152) | Tadalafil Monotherapy
(N=151) |
|
|---|---|---|---|
|
|||
| Baseline (median) | 356 | 366 | 352 |
| Change from baseline (median) | 43 | 23 | 22 |
| Median difference from Letairis + Tadalafil
(95% CI) | 24
(11, 37) | 20
(8, 32) |
|
| P-Value | 0.0004 | 0.0016 | |
Figure 9 Median Change in 6-Minute Walk Distance (meters) in AMBITION
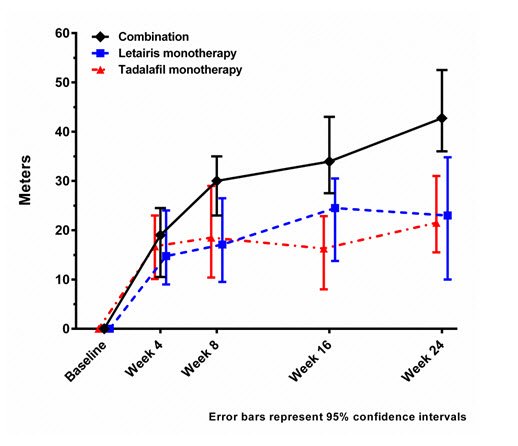
14.3 Long-term Treatment of PAH
In long-term follow-up of patients who were treated with Letairis (2.5 mg, 5 mg, or 10 mg once daily) in the two pivotal studies and their open-label extension (N=383), Kaplan-Meier estimates of survival at 1, 2, and 3 years were 93%, 85%, and 79%, respectively. Of the patients who remained on Letairis for up to 3 years, the majority received no other treatment for PAH. These uncontrolled observations do not allow comparison with a group not given Letairis and cannot be used to determine the long-term effect of Letairis on mortality.
14.4 Adverse Effects in Idiopathic Pulmonary Fibrosis (IPF)
A randomized controlled study in patients with IPF, with or without pulmonary hypertension (WHO Group 3), compared Letairis (N=329) to placebo (N=163). The study was terminated after 34 weeks for lack of efficacy, and was found to demonstrate a greater risk of disease progression or death on Letairis. More patients taking Letairis died (8% vs. 4%), had a respiratory hospitalization (13% vs. 6%), and had a decrease in FVC/DLCO (17% vs. 12%) [see Contraindications (4.2)].
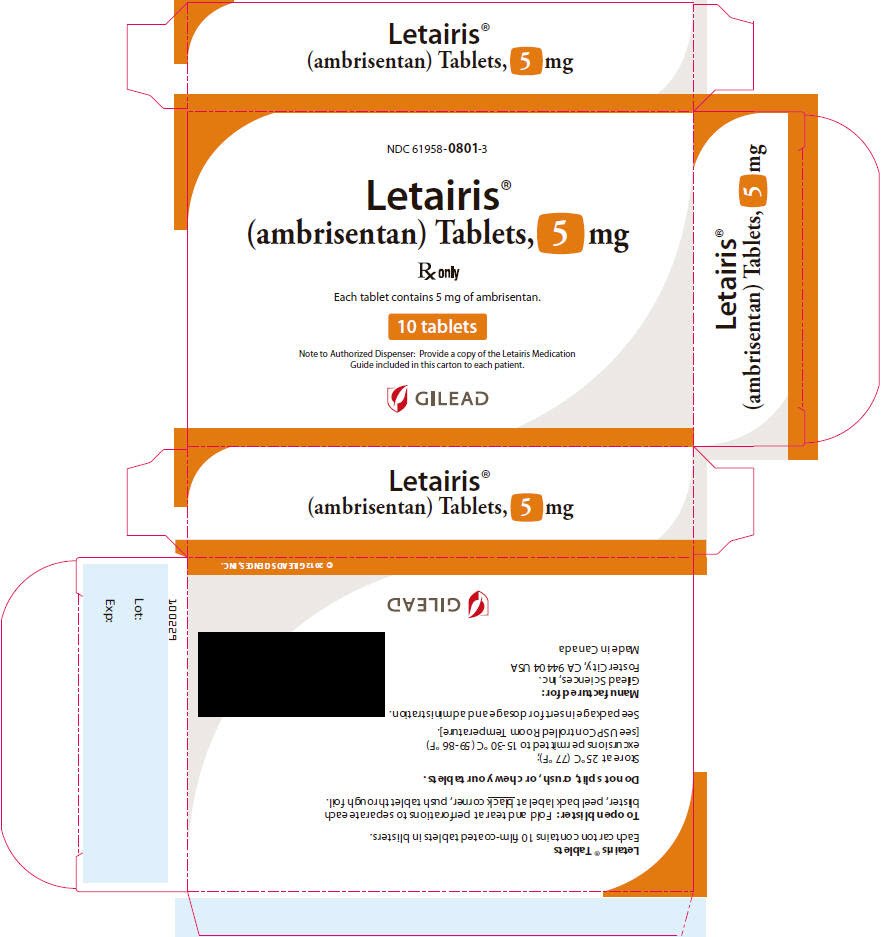
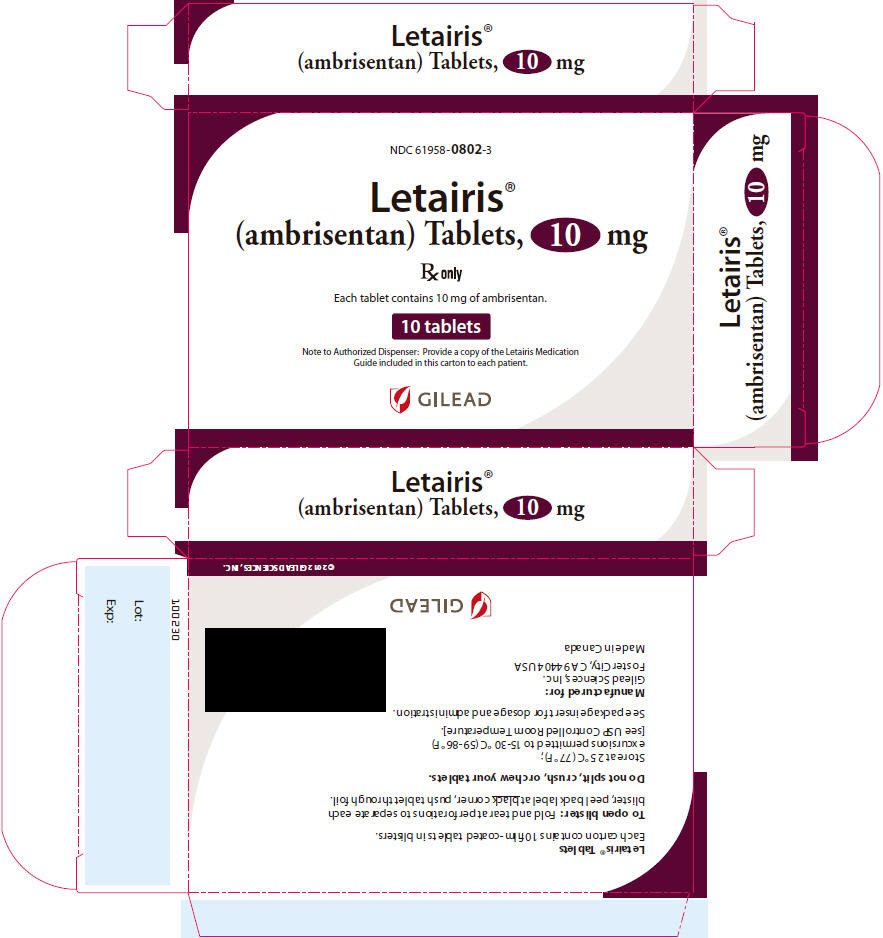
16. How is Letairis supplied
Letairis film-coated tablets are supplied as follows:
| Tablet Strength | Package Configuration | NDC No. | Description of Tablet;
Debossed on Tablet; Size |
|---|---|---|---|
| 5 mg | 30 count blister | 61958-0801-2 | Square convex; pale pink; "5" on side 1 and "GSI" on side 2; 6.6 mm Square |
| 30 count bottle | 61958-0801-1 | ||
| 10 count blister | 61958-0801-3 | ||
| 10 count bottle | 61958-0801-5 | ||
| 10 mg | 30 count blister | 61958-0802-2 | Oval convex; deep pink; "10" on side 1 and "GSI" on side 2; 9.8 mm × 4.9 mm Oval |
| 30 count bottle | 61958-0802-1 | ||
| 10 count blister | 61958-0802-3 | ||
| 10 count bottle | 61958-0802-5 |
17. Patient Counseling Information
Advise patients to read the FDA-approved patient labeling (Medication Guide).
Embryo-fetal Toxicity
Instruct patients on the risk of fetal harm when Letairis is used in pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)]. Instruct females of reproductive potential to immediately contact their physician if they suspect they may be pregnant.
Educate and counsel patients who can become pregnant about the need to use effective contraception prior to treatment with Letairis, during treatment, and for one month after treatment discontinuation [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1, 8.3)]. Patients who can become pregnant should have a negative pregnancy test prior to treatment with Letairis [see Dosage and Administration (2.2), Contraindications (4.1), Warnings and Precautions (5.1), Use in Specific Populations (8.1, 8.3)].
Counsel patients who can become pregnant about pregnancy planning and prevention, including emergency contraception, or designate counseling by another healthcare provider trained in contraceptive counseling [see Boxed Warning].
Hepatic Effects
Advise patients of the symptoms of potential liver injury and instruct them to report any of these symptoms to their physician.
Gilead Sciences, Inc., Foster City, CA 94404
Letairis is a registered trademark of Gilead Sciences, Inc. Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc. Other brands noted herein are the property of their respective owners.
© 2025 Gilead Sciences, Inc.
GS22-081-018
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised April 2025 | |
| Medication Guide Letairis® (le-TAIR-is) (ambrisentan) tablets |
||
| Read this Medication Guide before you start taking Letairis and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking with your doctor about your medical condition or your treatment. | ||
|
What is the most important information I should know about Letairis?
|
||
What is Letairis?
|
||
| Who should not take Letairis?
Do not take Letairis if:
|
||
| What should I tell my doctor before taking Letairis?
Before you take Letairis, tell your doctor if you:
Especially tell your doctor if you take the medicine cyclosporine (Gengraf, Neoral, Sandimmune). Your doctor may need to change your dose of Letairis. |
||
How should I take Letairis?
|
||
What should I avoid while taking Letairis?
|
||
| What are the possible side effects of Letairis?
Letairis can cause serious side effects including:
|
||
|
|
|
| Some medicines that are like Letairis can cause liver problems. Tell your doctor if you get any of these symptoms of a liver problem while taking Letairis: | ||
|
|
|
| Tell your doctor if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of Letairis. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
| How should I store Letairis?
Store Letairis at room temperature between 68°F to 77°F (20°C to 25°C), in the package it comes in. Keep Letairis and all medicines out of the reach of children. |
||
| General information about the safe and effective use of Letairis
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use Letairis for a condition for which it was not prescribed. Do not give Letairis to other people, even if they have the same symptoms that you have. It may harm them. This Medication Guide summarizes the most important information about Letairis. If you would like more information, ask your doctor. You can ask your doctor or pharmacist for information about Letairis that is written for health professionals. |
||
| What are the ingredients in Letairis?
Active ingredient: ambrisentan Inactive Ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate and microcrystalline cellulose. The tablets are film-coated with a coating material containing FD&C Red #40 aluminum lake, lecithin, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. Gilead Sciences, Inc., Foster City, CA 94404 Letairis is a registered trademark of Gilead Sciences, Inc. Gilead and the Gilead logo are trademarks of Gilead Sciences, Inc. Other brands noted herein are the property of their respective owners. © 2025 Gilead Sciences, Inc. GS22-081-018 For more information call 1-866-664-5327 or go to www.letairis.com or www.gilead.com. |
||
| LETAIRIS
ambrisentan tablet, film coated |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LETAIRIS
ambrisentan tablet, film coated |
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||
| Labeler - Gilead Sciences, Inc. (185049848) |
More about Letairis (ambrisentan)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (12)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- FDA approval history
- Drug class: agents for pulmonary hypertension
- Breastfeeding
- En español
