Farydak: Package Insert / Prescribing Info
Package insert / product label
Generic name: panobinostat
Dosage form: capsule
Drug class: Histone deacetylase inhibitors
Medically reviewed by Drugs.com. Last updated on Feb 26, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
FARYDAK® (panobinostat) capsules, for oral use
Initial U.S. Approval: 2015
WARNING: FATAL AND SERIOUS TOXICITIES: SEVERE DIARRHEA AND CARDIAC TOXICITIES
See full prescribing information for complete boxed warning.
- Severe diarrhea occurred in 25% of FARYDAK treated patients. Monitor for symptoms, institute anti-diarrheal treatment, interrupt FARYDAK and then reduce dose or discontinue FARYDAK. (5.1)
- Severe and fatal cardiac ischemic events, severe arrhythmias, and ECG changes have occurred in patients receiving FARYDAK. Arrhythmias may be exacerbated by electrolyte abnormalities. Obtain ECG and electrolytes at baseline and periodically during treatment as clinically indicated. (5.2)
Recent Major Changes
|
Warnings and Precautions, Embryo-Fetal Toxicity (5.7) |
6/2016 |
Indications and Usage for Farydak
FARYDAK, a histone deacetylase inhibitor, in combination with bortezomib and dexamethasone, is indicated for the treatment of patients with multiple myeloma who have received at least 2 prior regimens, including bortezomib and an immunomodulatory agent. This indication is approved under accelerated approval based on progression free survival. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials. (1)
Farydak Dosage and Administration
- 20 mg, taken orally once every other day for 3 doses per week (on Days 1, 3, 5, 8, 10, and 12) of Weeks 1 and 2 of each 21-day cycle for 8 cycles (2.1)
- Consider continuing treatment for an additional 8 cycles for patients with clinical benefit, unless they have unresolved severe or medically significant toxicity (2.1)
Dosage Forms and Strengths
Capsules: 10 mg, 15 mg, and 20 mg panobinostat (equivalent to 12.58 mg, 18.86 mg, and 25.15 mg respectively of panobinostat lactate) (3)
Contraindications
None (4)
Warnings and Precautions
- Hemorrhage: Fatal and serious cases of gastrointestinal and pulmonary hemorrhage. Monitor platelet counts and transfuse as needed. (5.3)
- Hepatotoxicity: Monitor hepatic enzymes and adjust dosage if abnormal liver function tests are observed during FARYDAK therapy. (5.6)
- Embryo-Fetal Toxicity: can cause fetal harm. Advise women of the potential hazard to the fetus and to avoid pregnancy while taking FARYDAK. (5.7)
Adverse Reactions/Side Effects
The most common adverse reactions (incidence of at least 20%) in clinical studies are diarrhea, fatigue, nausea, peripheral edema, decreased appetite, pyrexia, and vomiting. (6.1)
The most common non-hematologic laboratory abnormalities (incidence ≥40%) are hypophosphatemia, hypokalemia, hyponatremia, and increased creatinine. The most common hematologic laboratory abnormalities (incidence ≥60%) are thrombocytopenia, lymphopenia, leukopenia, neutropenia, and anemia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Secura Bio, Inc. at 1-844-973-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
Use In Specific Populations
Hepatic Impairment: Hepatic impairment can increase panobinostat exposure. Reduce FARYDAK dose in patients with mild or moderate hepatic impairment. Avoid use in patients with severe hepatic impairment. (8.6)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2021
Full Prescribing Information
WARNING: FATAL AND SERIOUS TOXICITIES: SEVERE DIARRHEA AND CARDIAC TOXICITIES
Severe diarrhea occurred in 25% of FARYDAK treated patients. Monitor for symptoms, institute anti-diarrheal treatment, interrupt FARYDAK and then reduce dose or discontinue FARYDAK. (5.1)
Severe and fatal cardiac ischemic events, severe arrhythmias, and ECG changes have occurred in patients receiving FARYDAK. Arrhythmias may be exacerbated by electrolyte abnormalities. Obtain ECG and electrolytes at baseline and periodically during treatment as clinically indicated. (5.2)
1. Indications and Usage for Farydak
FARYDAK, a histone deacetylase inhibitor, in combination with bortezomib and dexamethasone, is indicated for the treatment of patients with multiple myeloma who have received at least 2 prior regimens, including bortezomib and an immunomodulatory agent. This indication is approved under accelerated approval based on progression free survival [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trials.
2. Farydak Dosage and Administration
2.1 Recommended Dosing
The recommended starting dose of FARYDAK is 20 mg, taken orally once every other day for 3 doses per week in Weeks 1 and 2 of each 21-day cycle for up to 8 cycles. Consider continuing treatment for an additional 8 cycles for patients with clinical benefit who do not experience unresolved severe or medically significant toxicity. The total duration of treatment may be up to 16 cycles (48 weeks). FARYDAK is administered in combination with bortezomib and dexamethasone as shown in Table 1 and Table 2.
The recommended dose of bortezomib is 1.3 mg/m2 given as an injection. The recommended dose of dexamethasone is 20 mg taken orally per scheduled day, on a full stomach.
| 21-Day Cycle | |||||||||||||||
| Cycles 1 to 8 (3-Week cycles) | Week 1 Days | Week 2 Days | Week 3 | ||||||||||||
| FARYDAK | 1 | 3 | 5 | 8 | 10 | 12 | Rest period | ||||||||
| Bortezomib | 1 | 4 | 8 | 11 | Rest period | ||||||||||
| Dexamethasone | 1 | 2 | 4 | 5 | 8 | 9 | 11 | 12 | Rest period | ||||||
| 21-Day Cycle | |||||||||||||||
| Cycles 9 to 16 (3-Week cycles) | Week 1 Days | Week 2 Days | Week 3 | ||||||||||||
| FARYDAK | 1 | 3 | 5 | 8 | 10 | 12 | Rest period | ||||||||
| Bortezomib | 1 | 8 | Rest period | ||||||||||||
| Dexamethasone | 1 | 2 | 8 | 9 | Rest period | ||||||||||
2.2 Administration and Monitoring Instructions
FARYDAK should be taken orally once on each scheduled day at about the same time, either with or without food [see Clinical Pharmacology (12.3)].
FARYDAK capsules should be swallowed whole with a cup of water. Do not open, crush, or chew the capsules [see How Supplied/Storage and Handling (16)].
If a dose is missed it can be taken up to 12 hours after the specified dose time. If vomiting occurs the patient should not repeat the dose, but should take the next usual scheduled dose.
Counsel patients on the correct dosing schedule, technique of administration of FARYDAK, and when to take FARYDAK if dosing adjustments are made.
Prior to the start of FARYDAK treatment and during treatment, monitoring should include:
- Complete Blood Count (CBC): Obtain a CBC before initiating treatment. Verify that the baseline platelet count is at least 100 x 109/L and the baseline absolute neutrophil count (ANC) is at least 1.5 x 109/L. Monitor the CBC weekly (or more often as clinically indicated) during treatment. [see Warnings and Precautions (5.4) Adverse Reactions (6.1)].
- ECG: Perform an ECG prior to the start of therapy and repeat periodically during treatment as clinically indicated. Verify that the QTcF is less than 450 msec prior to initiation of treatment with FARYDAK. If during treatment with FARYDAK, the QTcF increases to ≥480 msec, interrupt treatment. Correct any electrolyte abnormalities. If QT prolongation does not resolve, permanently discontinue treatment with FARYDAK [see Warnings and Precautions (5.2), Adverse Reactions (6.1)]. During the clinical trial, ECGs were performed at baseline and prior to initiation of each cycle for the first 8 cycles.
- Serum Electrolytes: Obtain electrolytes, including potassium and magnesium, at baseline and monitor during therapy. Correct abnormal electrolyte values before treatment [see Warnings and Precautions (5.2), Adverse Reactions (6.1)]. During the trial, monitoring was conducted prior to the start of each cycle, at Day 11 of cycles 1 to 8, and at the start of each cycle for cycles 9 to 16.
For additional information please refer to the bortezomib and dexamethasone prescribing information.
2.3 Dose Adjustments and Modifications for Toxicity
Dose and/or schedule modification of FARYDAK may be required based on toxicity. Management of adverse drug reactions may require treatment interruption and/or dose reductions. If dose reduction is required, the dose of FARYDAK should be reduced in increments of 5 mg (i.e., from 20 mg to 15 mg, or from 15 mg to 10 mg). If the dosing of FARYDAK is reduced below 10 mg given 3 times per week, discontinue FARYDAK. Keep the same treatment schedule (3-week treatment cycle) when reducing dose. The table also lists Bortezomib (BTZ) dose modification procedures from the clinical trials.
| BTZ = bortezomib ANC = absolute neutrophil count Hb = hemoglobin IV = intravenous |
||||
| Thrombocytopenia | Platelets <50 x 109/L CTCAE Grade 3 | Platelets <50 x 109/L with bleeding CTCAE Grade 3 | Platelets <25 x 109/L CTCAE Grade 4 |
|
| Maintain FARYDAK dose. Monitor platelet counts at least weekly. | Interrupt FARYDAK. Monitor platelet counts at least weekly until ≥50 x 109/L, then restart at reduced dose | Interrupt FARYDAK. Monitor platelet counts at least weekly until ≥50 x 109/L, then restart at reduced dose |
||
| Maintain BTZ dose | - Interrupt BTZ until thrombocytopenia resolves to ≥ 50 x 109/L - If only 1 dose was omitted prior to correction to these levels, restart BTZ at same dose - If 2 or more doses were omitted consecutively, or within the same cycle, BTZ should be restarted at a reduced dose |
|||
| Neutropenia | ANC 0.75 to 1.0 x 109/L CTCAE Grade 3 | ANC 0.5 to 0.75 x 109/L CTCAE Grade 3 (2 or more occurrences) | ANC <1.0 x 109/L (CTCAE Grade 3) with febrile Neutropenia (any grade) | ANC <0.5 x 109/L CTCAE Grade 4 |
| Maintain FARYDAK dose. | Interrupt FARYDAK until ANC ≥1.0 x 109/L, then restart at same dose | Interrupt FARYDAK until febrile neutropenia resolves and ANC ≥1.0 x 109/L, then restart at reduced dose | Interrupt FARYDAK until ANC ≥1.0 x 109/L, then restart at reduced dose |
|
| Maintain BTZ dose | - Interrupt BTZ until febrile neutropenia resolves and ANC ≥1.0 x 109/L - If only 1 dose was omitted prior to correction to these levels, restart BTZ at same dose - If 2 or more doses were omitted consecutively, or within the same cycle, BTZ should be restarted at a reduced dose |
|||
| Anemia | Hb <8 g/dL CTCAE Grade 3 |
|||
| Interrupt FARYDAK until Hb ≥10 g/dL Restart at reduced dose. |
||||
| Diarrhea | Moderate Diarrhea 4 to 6 stools/day CTCAE Grade 2 | Severe Diarrhea (≥7 stools/day) IV fluids or hospitalization required CTCAE Grade 3 | Life-threatening Diarrhea CTCAE Grade 4 |
|
| Interrupt FARYDAK until resolved. Restart at same dose. | Interrupt FARYDAK until resolved. Restart at reduced dose. | Permanently discontinue FARYDAK | ||
| Consider Interruption of BTZ until resolved. Restart at same dose. | Interrupt BTZ until resolved. Restart at reduced dose. | Permanently discontinue BTZ | ||
| Nausea or Vomiting | Severe Nausea CTCAE Grade 3/4 | Severe / Life-threatening Vomiting CTCAE Grade 3/4 |
||
| Interrupt FARYDAK until resolved, then restart at reduced dose. | Interrupt FARYDAK until resolved, then restart at reduced dose. |
|||
Myelosuppression
Interrupt or reduce the dose of FARYDAK in patients who have thrombocytopenia, neutropenia or anemia according to instructions in Table 3. For patients with severe thrombocytopenia, consider platelet transfusions [see Warnings and Precautions (5.4), Adverse Reactions (6.1)]. Discontinue FARYDAK treatment if thrombocytopenia does not improve despite the recommended treatment modifications or if repeated platelet transfusions are required.
In the event of Grade 3 or 4 neutropenia, consider dose reduction and/or the use of growth factors (e.g., G-CSF). Discontinue FARYDAK if neutropenia does not improve despite dose modifications, colony-stimulating factors, or in case of severe infection.
Gastrointestinal Toxicity
Gastrointestinal toxicity is common in patients treated with FARYDAK. Patients who experience diarrhea, nausea, or vomiting may require treatment interruption or dose reduction (Table 3). At the first sign of abdominal cramping, loose stools, or onset of diarrhea, patients should be treated with anti-diarrheal medication (e.g., loperamide). Consider and administer prophylactic anti-emetics as clinically indicated.
Other Adverse Drug Reactions
For patients experiencing Grade 3/4 adverse drug reactions other than thrombocytopenia, neutropenia, or gastrointestinal toxicity, the recommendation is the following:
- CTC Grade 2 toxicity recurrence and CTC Grade 3 and 4 - omit the dose until recovery to CTC Grade 1 or less and restart treatment at a reduced dose
- CTC Grade 3 or 4 toxicity recurrence, a further dose reduction may be considered once the adverse events have resolved to CTC Grade 1 or less.
2.4 Dose Modifications for Use in Hepatic Impairment
Reduce the starting dose of FARYDAK to 15 mg in patients with mild hepatic impairment and 10 mg in patients with moderate hepatic impairment. Avoid use in patients with severe hepatic impairment. Monitor patients frequently for adverse events and adjust dose as needed for toxicity [see Dosing and Administration (2.2), Warnings and Precautions (5.6), Hepatic Impairment (8.6), Clinical Pharmacology (12.3)].
2.5 Dose Modifications for Use with Strong CYP3A Inhibitors
Reduce the starting dose of FARYDAK to 10 mg when coadministered with strong CYP3A inhibitors (e.g., boceprevir, clarithromycin, conivaptan, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir) [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
Capsules: 10 mg, 15 mg, and 20 mg panobinostat (equivalent to 12.58 mg, 18.86 mg, and 25.15 mg respectively of panobinostat lactate)
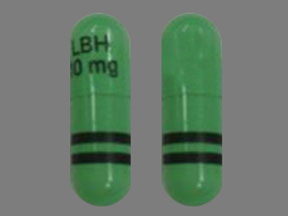
10 mg: Size #3 light green opaque capsule, radial markings on cap with black ink “LBH 10 mg” and two radial bands with black ink on body, containing white to almost white powder.
15 mg: Size #1 orange opaque capsule, radial markings on cap with black ink “LBH 15 mg” and two radial bands with black ink on body, containing white to almost white powder.
20 mg: Size #1 red opaque capsule, radial markings on cap with black ink “LBH 20 mg” and two radial bands with black ink on body, containing white to almost white powder.
5. Warnings and Precautions
5.1 Diarrhea
Severe diarrhea occurred in 25% of patients treated with FARYDAK [see Adverse Reactions (6.1)]. Diarrhea of any grade occurred in 68% of patients treated with FARYDAK compared to 42% of patients in the control arm. Diarrhea can occur at any time. Monitor patient hydration status and electrolyte blood levels, including potassium, magnesium and phosphate, at baseline and weekly (or more frequently as clinically indicated) during therapy and correct to prevent dehydration and electrolyte disturbances. Initiate anti-diarrheal medication at the onset of diarrhea. Interrupt FARYDAK at the onset of moderate diarrhea (4 to 6 stools per day) [see Dosage and Administration (2.3)]. Ensure that patients initiating therapy with FARYDAK have anti-diarrheal medications on hand.
5.2 Cardiac Toxicities
Severe and fatal cardiac ischemic events, as well as severe arrhythmias, and electrocardiogram (ECG) changes occurred in patients receiving FARYDAK. Arrhythmias occurred in 12% of patients receiving FARYDAK, compared to 5% of patients in the control arm. Cardiac ischemic events occurred in 4% of patients treated with FARYDAK compared with 1% of patients in the control arm. Do not initiate FARYDAK treatment in patients with history of recent myocardial infarction or unstable angina.
Electrocardiographic abnormalities such as ST-segment depression and T-wave abnormalities also occurred more frequently in patients receiving FARYDAK compared to the control arm: 22% versus 4% and 40% versus 18%, respectively. FARYDAK may prolong cardiac ventricular repolarization (QT interval). Do not initiate treatment with FARYDAK in patients with a QTcF >450 msec or clinically significant baseline ST-segment or T-wave abnormalities. Arrhythmias may be exacerbated by electrolyte abnormalities. If during treatment with FARYDAK, the QTcF increases to ≥480 msec, interrupt treatment. Correct any electrolyte abnormalities. If QT prolongation does not resolve, permanently discontinue treatment with FARYDAK.
Obtain ECG at baseline and periodically during treatment as clinically indicated. Monitor electrolytes during treatment with FARYDAK and correct abnormalities as clinically indicated.
5.3 Hemorrhage
Fatal and serious hemorrhage occurred during treatment with FARYDAK. In the clinical trial in patients with relapsed multiple myeloma, 5 patients receiving FARYDAK compared to 1 patient in the control arm died due to a hemorrhagic event. All 5 patients had grade ≥3 thrombocytopenia at the time of the event. Grade 3/4 hemorrhage was reported in 4% of patients treated with the FARYDAK arm and 2% of patients in the control arm.
5.4 Myelosuppression
FARYDAK causes myelosuppression, including severe thrombocytopenia, neutropenia and anemia. In the clinical trial in patients with relapsed multiple myeloma, 67% of patients treated with FARYDAK developed Grade 3 to 4 thrombocytopenia compared with 31% in the control arm. Thrombocytopenia led to treatment interruption and or dose modification in 31% of patients receiving FARYDAK compared to 11% of patients in the control arm. For patients receiving FARYDAK, 33% required platelet transfusion compared to 10% of patients in the control arm [see Dosage and Administration (2.2)].
Severe neutropenia occurred in 34% of patients treated with FARYDAK, compared to 11% of patients in the control arm. Neutropenia led to treatment interruption and or dose modification in 10% of patients receiving FARYDAK. The use of granulocyte-colony stimulating factor (G-CSF) was higher in patients treated with FARYDAK compared to the control arm, 13% compared to 4%, respectively.
Obtain a baseline CBC and monitor the CBC weekly during treatment (or more frequently if clinically indicated). Dose modifications are recommended for Myelosuppression [see Dosage and Administration (2.2)]. Monitor CBCs more frequently in patients over 65 years of age due to the increased frequency of myelosuppression in these patients [see Use in Specific Populations (8.5)].
5.5 Infections
Localized and systemic infections, including pneumonia, bacterial infections, invasive fungal infections, and viral infections have been reported in patients taking FARYDAK. Severe infections occurred in 31% of patients (including 10 deaths) treated with FARYDAK compared with 24% of patients (including 6 deaths) in the control arm. Infections of all grades occurred at a similar rate between arms. FARYDAK treatment should not be initiated in patients with active infections. Monitor patients for signs and symptoms of infections during treatment; if a diagnosis of infection is made, institute appropriate anti-infective treatment promptly and consider interruption or discontinuation of FARYDAK.
5.6 Hepatotoxicity
Hepatic dysfunction, primarily elevations in aminotransferases and total bilirubin, occurred in patients treated with FARYDAK. Liver function should be monitored prior to treatment and regularly during treatment. If abnormal liver function tests are observed dose adjustments may be considered and the patient should be followed until values return to normal or pretreatment levels [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
5.7 Embryo-Fetal Toxicity
FARYDAK can cause fetal harm when administered to a pregnant woman. Panobinostat was teratogenic in rats and rabbits. If FARYDAK is used during pregnancy, or if the patient becomes pregnant while taking FARYDAK, the patient should be apprised of the potential hazard to the fetus [see Use in Specific Populations (8.1, 8.3)].
Advise females of reproductive potential to avoid becoming pregnant while taking FARYDAK. Advise sexually-active females of reproductive potential to use effective contraception while taking FARYDAK and for at least 3 months after the last dose of FARYDAK.
Advise sexually active men to use condoms while on treatment and for 6 months after their last dose of FARYDAK [see Use in Specific Populations (8.3)].
6. Adverse Reactions/Side Effects
The following adverse reactions are described in detail in other sections of the label:
- Diarrhea [see Warnings and Precautions (5.1)]
- Cardiac Toxicities [see Warnings and Precautions (5.2)]
- Hemorrhage [see Warnings and Precautions (5.3)]
- Myelosuppression [see Warnings and Precautions (5.4)]
- Infections [see Warnings and Precautions (5.5)]
- Hepatotoxicity [see Warnings and Precautions (5.6)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1 Clinical Trials Experience
The safety data reflect subject exposure to FARYDAK from a clinical trial, in which 758 subjects with relapsed multiple myeloma received FARYDAK in combination with bortezomib and dexamethasone or placebo in combination with bortezomib and dexamethasone (referred to as the control arm). The median duration of exposure to FARYDAK was 5 months with 16% of patients exposed to study treatment for ≥48 weeks.
Serious adverse events (SAEs) occurred in 60% of patients in the FARYDAK, bortezomib, and dexamethasone compared to 42% of patients in the control arm. The most frequent (≥5%) treatment-emergent SAEs reported for patients treated with FARYDAK were pneumonia (18%), diarrhea, (11%), thrombocytopenia (7%), fatigue (6%), and sepsis (6%).
Adverse reactions that led to discontinuation of FARYDAK occurred in 36% of patients. The most common adverse reactions leading to treatment discontinuations were diarrhea, fatigue, and pneumonia.
Deaths occurred in 8% of patients in the FARYDAK arm versus 5% on the control arm. The most frequent causes of death were infection and hemorrhage.
Table 4 summarizes the adverse reactions occurring in at least 10% of patients with ≥ 5% greater incidence in the FARYDAK arm, and Table 5 summarizes the treatment-emergent laboratory abnormalities.
| [1] BTZ = bortezomib | ||||
| [2] Dex = dexamethasone | ||||
| [3] Arrhythmia includes the terms: arrhythmia, arrhythmia supraventricular, atrial fibrillation, atrial flutter, atrial tachycardia, bradycardia, cardiac arrest, cardio-respiratory arrest, sinus bradycardia, sinus tachycardia, supraventricular extra-systoles, tachycardia, ventricular arrhythmia, and ventricular tachycardia | ||||
| [4] Fatigue includes the terms: fatigue, malaise, asthenia, and lethargy | ||||
| Primary System Organ Class Preferred term | FARYDAK, BTZ [1], Dex [2] N=381 All grades % | FARYDAK, BTZ [1], Dex [2] N=381 Grade 3/4 % | Placebo, BTZ [1], Dex [2] N=377 All grades % | Placebo, BTZ [1], Dex [2] N=377 Grade 3/4 % |
| Cardiac disorders | ||||
| Arrhythmia[3] | 12 | 3 | 5 | 2 |
| Gastrointestinal disorders | ||||
| Diarrhea | 68 | 25 | 42 | 8 |
| Nausea | 36 | 6 | 21 | 1 |
| Vomiting | 26 | 7 | 13 | 1 |
| General disorders and administration site conditions | ||||
| Fatigue[4] | 60 | 25 | 42 | 12 |
| Peripheral edema | 29 | 2 | 19 | <1 |
| Pyrexia | 26 | 1 | 15 | 2 |
| Investigations | ||||
| Weight decreased | 12 | 2 | 5 | 1 |
| Metabolism and nutrition disorders | ||||
| Decreased appetite | 28 | 3 | 12 | 1 |
Other Adverse Reactions
Other notable adverse drug reactions of FARYDAK not described above, which were either clinically significant, or occurred with a frequency less than 10% but had a frequency in the FARYDAK arm greater than 2% over the control arm in the multiple myeloma clinical trial are listed below:
Infections and infestations: hepatitis B.
Endocrine disorders: hypothyroidism.
Metabolism and nutrition disorders: hyperglycemia, dehydration, fluid retention, hyperuricemia, hypomagnesemia.
Nervous system disorders: dizziness, headache, syncope, tremor, dysgeusia.
Cardiac disorders: palpitations.
Vascular disorders: hypotension, hypertension, orthostatic hypotension.
Respiratory, thoracic and mediastinal disorders: cough, dyspnea, respiratory failure, rales, wheezing.
Gastrointestinal disorders: abdominal pain, dyspepsia, gastritis, cheilitis, abdominal distension, dry mouth, flatulence, colitis, gastrointestinal pain.
Skin and subcutaneous disorders: skin lesions, rash, erythema.
Musculoskeletal and connective tissue disorders: joint swelling.
Renal and urinary disorders: renal failure, urinary incontinence.
General disorders and administration site conditions: chills.
Investigations: blood urea increased, glomerular filtration rate decreased, blood alkaline phosphatase increased.
Psychiatric disorders: insomnia.
| [1] BTZ = bortezomib | ||||
| [2] Dex = dexamethasone | ||||
| Investigations | FARYDAK, BTZ[1], Dex[2] N=381 Any grade % | FARYDAK, BTZ[1], Dex[2] N=381 Grade 3/4 % | Placebo, BTZ[1], Dex[2] N=377 Any grade % | Placebo, BTZ[1], Dex[2] N=377 Grade 3/4 % |
| Hematology | ||||
| Thrombocytopenia | 97 | 67 | 83 | 31 |
| Anemia | 62 | 18 | 52 | 19 |
| Neutropenia | 75 | 34 | 36 | 11 |
| Leukopenia | 81 | 23 | 48 | 8 |
| Lymphopenia | 82 | 53 | 74 | 40 |
| Chemistry | ||||
| Blood creatinine increased | 41 | 1 | 23 | 2 |
| Hypokalemia | 52 | 18 | 36 | 7 |
| Hypophosphatemia | 63 | 20 | 45 | 12 |
| Hyponatremia | 49 | 13 | 36 | 7 |
| Hyperbilirubinemia | 21 | 1 | 13 | <1 |
| Hypocalcemia | 67 | 5 | 55 | 2 |
| Hypoalbuminemia | 63 | 2 | 38 | 2 |
| Hyperphosphatemia | 29 | 2 | 20 | <1 |
| Hypermagnesemia | 27 | 5 | 14 | 1 |
Fatigue and Asthenia
Grade 1 to Grade 4 asthenic conditions (fatigue, malaise, asthenia, and lethargy) were reported in 60% of the patients in the FARYDAK arm compared to 42% of patients in the control arm. Grade ≥3 asthenic conditions were reported in 25% of the patients in the FARYDAK arm compared to 12% of patients in the control arm. Asthenic conditions led to treatment discontinuation in 6% of patients in the FARYDAK arm versus 3% of patients in the control arm.
The prespecified sub-group upon which the efficacy and safety of FARYDAK was based had a similar adverse reaction profile to the entire safety population of patients treated with FARYDAK, bortezomib, and dexamethasone.
Related/similar drugs
7. Drug Interactions
Panobinostat is a CYP3A substrate and inhibits CYP2D6. Panobinostat is a P-glycoprotein (P-gp) transporter system substrate.
7.1 Agents that May Increase FARYDAK Blood Concentrations
CYP3A Inhibitors: Coadministration of FARYDAK with a strong CYP3A inhibitor increased the Cmax and AUC of panobinostat by 62% and 73% respectively, compared to when FARYDAK was given alone [see Clinical Pharmacology (12.3)].
Reduce dose to 10 mg when coadministered with strong CYP3A inhibitors (e.g., boceprevir, clarithromycin, conivaptan, indinavir, itraconazole, ketoconazole, lopinavir/ritonavir, nefazodone, nelfinavir, posaconazole, ritonavir, saquinavir, telaprevir, telithromycin, voriconazole) [see Dosage and Administration (2.5)]. Instruct patients to avoid star fruit, pomegranate or pomegranate juice, and grapefruit or grapefruit juice because these foods are known to inhibit CYP3A enzymes.
7.2 Agents that May Decrease FARYDAK Plasma Concentrations
CYP3A Inducers: Coadministration of FARYDAK with strong CYP3A inducers was not evaluated in vitro or in a clinical trial however, a reduction in panobinostat exposure is likely. An approximately 70% decrease in the systemic exposure of panobinostat in the presence of strong inducers of CYP3A was observed in simulations using mechanistic models. Therefore, the concomitant use of strong CYP3A inducers should be avoided [see Clinical Pharmacology (12.3)].
7.3 Agents whose Plasma Concentrations May be Increased by FARYDAK
CYP2D6 Substrates: FARYDAK increased the median Cmax and AUC of a sensitive substrate of CYP2D6 by approximately 80% and 60%, respectively; however this was highly variable [see Clinical Pharmacology (12.3)]. Avoid coadministrating FARYDAK with sensitive CYP2D6 substrates (i.e., atomoxetine, desipramine, dextromethorphan, metoprolol, nebivolol, perphenazine, tolterodine, and venlafaxine) or CYP2D6 substrates that have a narrow therapeutic index (i.e., thioridazine, pimozide). If concomitant use of CYP2D6 substrates is unavoidable, monitor patients frequently for adverse reactions.
7.4 Drugs that Prolong QT interval
Concomitant use of anti-arrhythmic medicines (including, but not limited to amiodarone, disopyramide, procainamide, quinidine and sotalol) and other drugs that are known to prolong the QT interval (including, but not limited to chloroquine, halofantrine, clarithromycin, methadone, moxifloxacin, bepridil and pimozide) is not recommended. Anti-emetic drugs with known QT prolonging risk, such as dolasetron, ondansetron, and tropisetron can be used with frequent ECG monitoring [see Warnings and Precautions (5.2)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
FARYDAK can cause fetal harm when administered to a pregnant woman. Panobinostat was teratogenic in rats and rabbits. If FARYDAK is used during pregnancy or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the fetus.
Data
Animal Data
In embryofetal development studies, panobinostat was administered orally 3 times per week during the period of organogenesis to pregnant rats (30, 100, and 300 mg/kg) and rabbits (10, 40, and 80 mg/kg). In rats, maternal toxicity including death was observed at doses greater than or equal to 100 mg/kg/day. Embryofetal toxicities occurred at 30 mg/kg (the only dose with live fetuses) and consisted of fetal malformations and anomalies, such as cleft palate, short tail, extra presacral vertebrae, and extra ribs. The dose of 30 mg/kg resulted in exposures (AUCs) approximately 3-fold the human exposure at the human dose of 20 mg. In rabbits, maternal toxicity including death was observed at doses greater than or equal to 80 mg/kg. Increased pre- and/or post-implantation loss occurred at all doses tested. Embryofetal toxicities included decreased fetal weights at doses greater than or equal to 40 mg/kg and malformations (absent digits, cardiac interventricular septal defects, aortic arch interruption, missing gallbladder, and irregular ossification of skull) at 80 mg/kg. The dose of 40 mg/kg in rabbits results in systemic exposure approximately 4-fold the human exposure and the dose of 80 mg/kg results in exposure 7-fold the human exposure, at the human dose of 20 mg.
8.2 Lactation
Risk Summary
It is not known whether FARYDAK is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse drug reactions in nursing infants, decide whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.3 Females and Males of Reproductive Potential
Embryofetal toxicity including malformations occurred in embryofetal development studies in rats [see Pregnancy (8.1)].
Pregnancy Testing
Perform pregnancy testing in women of childbearing potential prior to starting treatment with FARYDAK and intermittently during treatment with FARYDAK.
Contraception
Females
FARYDAK can cause fetal harm. Advise females of reproductive potential to avoid becoming pregnant while taking FARYDAK. Advise sexually-active females of reproductive potential to use effective contraception while taking FARYDAK and for at least 3 months after the last dose of FARYDAK. Advise patients to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking FARYDAK [see Use in Specific Populations (8.1)].
Males
Advise sexually active men to use condoms while on treatment and for at least 6 months after their last dose of FARYDAK.
8.5 Geriatric Use
In clinical trials of FARYDAK in patients with multiple myeloma, 42% of patients were 65 years of age or older.
Patients over 65 years of age had a higher frequency of selected adverse events and of discontinuation of treatment due to adverse events. In patients over 65 years of age, the incidence of deaths not related to disease progression was 9% in patients ≥65 years of age compared to 5 % in patients <65.
In the randomized clinical trial in patients with relapsed multiple myeloma, no major differences in effectiveness were observed in older patients compared to younger patients. Adverse reactions leading to permanent discontinuation occurred in 45% of patients ≥65 years of age in the FARYDAK treatment arm compared to 30% of patients <65 years age in the FARYDAK treatment arm. Monitor for toxicity more frequently in patients over 65 years of age, especially for gastrointestinal toxicity, myelosuppression, and cardiac toxicity [see Warnings and Precautions (5.1, 5.4)].
8.6 Hepatic Impairment
The safety and efficacy of FARYDAK in patients with hepatic impairment has not been evaluated.
In a pharmacokinetic trial, patients with mild (bilirubin ≤1xULN and AST >1xULN, or bilirubin >1.0 to 1.5x ULN and any AST) or moderate (bilirubin >1.5x to 3.0x ULN, any AST) hepatic impairment (NCI-ODWG criteria) had increased AUC of panobinostat by 43% and 105%, respectively. Reduce the starting dose of FARYDAK in patients with mild or moderate hepatic impairment. Avoid use in patients with severe hepatic impairment. Monitor patients with hepatic impairment frequently for adverse events [see Dosage and Administration (2.4), Warnings and Precautions (5.6), Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Mild [creatinine clearance (CrCl) ≥50 to <80 mL/min] to severe renal impairment (CrCl <30 mL/min) did not impact the plasma exposure of panobinostat. FARYDAK has not been studied in patients with end stage renal disease (ESRD) or patients on dialysis. The dialyzability of panobinostat is unknown [see Clinical Pharmacology (12.3)].
10. Overdosage
There is limited experience with overdosage. Expect exaggeration of adverse reactions observed during the clinical trial, including hematologic and gastrointestinal reactions such as thrombocytopenia, pancytopenia, diarrhea, nausea, vomiting and anorexia. Monitor cardiac status including ECGs, and assess and correct electrolytes. Consider platelet transfusions for thrombocytopenic bleeding. It is not known if FARYDAK is dialyzable.
11. Farydak Description
FARYDAK (panobinostat lactate) is a histone deacetylase inhibitor.
The chemical name of panobinostat lactate is 2-Hydroxypropanoic acid, compd. with 2-(E)-N-hydroxy-3-[4-[[[2-(2-methyl-1H-indol-3-yl)ethyl]amino]methyl]phenyl]-2-propenamide (1:1).
The structural formula is:
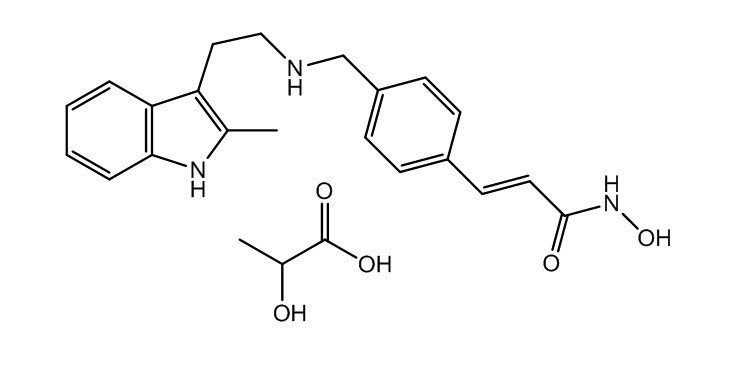
Panobinostat lactate anhydrous is a white to slightly yellowish or brownish powder. The molecular formula is C21H23N3O2•C3H6O3 (lactate); its molecular weight is 439.51 (as a lactate), equivalent to 349.43 (free base). Panobinostat lactate anhydrous is light sensitive. Panobinostat lactate anhydrous is both chemically and thermodynamically a stable crystalline form with no polymorphic behavior. Panobinostat free base is not chiral and shows no specific optical rotation. Panobinostat lactate anhydrous is slightly soluble in water. Solubility of panobinostat lactate anhydrous is pH-dependent, with the highest solubility in buffer pH 3.0 (citrate).
FARYDAK capsules contain 10 mg, 15 mg, or 20 mg panobinostat free base (equivalent to 12.58 mg, 18.86 mg, and 25.15 mg respectively of panobinostat lactate). The inactive ingredients are magnesium stearate, mannitol, microcrystalline cellulose and pregelatinized starch. The capsules contain gelatin, FD&C Blue 1 (10 mg capsules), yellow iron oxide (10 mg and 15 mg capsules), red iron oxide (15 mg and 20 mg capsules) and titanium dioxide.
12. Farydak - Clinical Pharmacology
12.1 Mechanism of Action
FARYDAK is a histone deacetylase (HDAC) inhibitor that inhibits the enzymatic activity of HDACs at nanomolar concentrations. HDACs catalyze the removal of acetyl groups from the lysine residues of histones and some non-histone proteins. Inhibition of HDAC activity results in increased acetylation of histone proteins, an epigenetic alteration that results in a relaxing of chromatin, leading to transcriptional activation. In vitro, panobinostat caused the accumulation of acetylated histones and other proteins, inducing cell cycle arrest and/or apoptosis of some transformed cells. Increased levels of acetylated histones were observed in xenografts from mice that were treated with panobinostat. Panobinostat shows more cytotoxicity towards tumor cells compared to normal cells.
12.2 Pharmacodynamics
Cardiac Electrophysiology
FARYDAK may prolong cardiac ventricular repolarization (QT interval) [see Warnings and Precautions (5.2)]. In the randomized multiple myeloma trial, QTc prolongation with values between 451 msec to 480 msec occurred in 10.8% of FARYDAK treated patients. Events with values of 481 msec to 500 msec occurred in 1.3% of FARYDAK treated patients. A maximum QTcF increase from baseline of between 31 msec and 60 msec was reported in 14.5% of FARYDAK treated patients. A maximum QTcF increase from baseline of >60 msec was reported in 0.8% of FARYDAK treated patients. No episodes of QTcF prolongation >500 msec have been reported with the dose of 20 mg FARYDAK in the randomized multiple myeloma trial conducted in combination with bortezomib and dexamethasone. Pooled clinical data from over 500 patients treated with single agent FARYDAK in multiple indications and at different dose levels has shown that the incidence of CTC Grade 3 QTc prolongation (QTcF >500 msec) was approximately 1% overall and 5% or more at a dose of 60 mg or higher.
12.3 Pharmacokinetics
Absorption
The absolute oral bioavailability of FARYDAK is approximately 21%. Peak concentrations of panobinostat are observed within 2 hours (Tmax) of oral administration in patients with advanced cancer. FARYDAK exhibits an approximate dose proportional increase in both Cmax and AUC over the dosing range.
Plasma panobinostat Cmax and AUC0–48 were approximately 44% and 16% lower compared to fasting conditions, respectively, following ingestion of an oral FARYDAK dose 30 minutes after a high-fat meal by 36 patients with advanced cancer. The median Tmax was also delayed by 2.5 hours in these patients.
The aqueous solubility of panobinostat is pH dependent, with higher pH resulting in lower solubility [see Description (11)]. Coadministration of FARYDAK with drugs that elevate the gastric pH was not evaluated in vitro or in a clinical trial; however, altered panobinostat absorption was not observed in simulations using physiologically-based pharmacokinetic (PBPK) models.
Distribution
Panobinostat is approximately 90% bound to human plasma proteins in vitro and is independent of concentration. Panobinostat is a P-gp substrate.
Metabolism
Panobinostat is extensively metabolized. Pertinent metabolic pathways involved in the biotransformation of panobinostat are reduction, hydrolysis, oxidation, and glucuronidation processes. The fraction metabolized through CYP3A accounts for approximately 40% of the total hepatic panobinostat elimination. In vitro, additional contributions from the CYP2D6 and CYP2C19 pathways are minor. In vitro, UGT1A1, UGT1A3, UGT1A7, UGT1A8, UGT1A9, and UGT2B4 contribute to the glucuronidation of panobinostat.
Elimination
Twenty-nine percent to 51% of administered radioactivity is excreted in urine and 44% to 77% in the feces after a single oral dose of [14C] panobinostat in 4 patients with advanced cancer. Unchanged panobinostat accounted for <2.5% of the dose in urine and <3.5% of the dose in feces with the remainder consisting of metabolites.
An oral clearance (CL/F) and terminal elimination half-life (t1/2) of approximately 160 L/hr and 37 hours, respectively, was estimated using a population based pharmacokinetic (pop-PK) model in patients with advanced cancer. An inter-subject variability 65% on the clearance estimate was also reported. Up to 2-fold accumulation was observed with chronic oral dosing in patients with advanced cancer.
Specific Populations
Population pharmacokinetic (PK) analyses of FARYDAK indicated that body surface area, gender, age, and race do not have a clinically meaningful influence on clearance.
Hepatic Impairment: The effect of hepatic impairment on the pharmacokinetics of panobinostat was evaluated in a phase 1 study in 24 patients with advanced cancer with varying degrees of hepatic impairment. In patients with NCI-CTEP class mild (i.e., Group B) and moderate (i.e., Group C) hepatic impairment, AUC0-inf increased 43% and 105% compared to the group with normal hepatic function, respectively. The relative change in Cmax followed a similar pattern. The effect of severe hepatic impairment was indeterminate in this study due to the small sample size (n=1). A dose modification is recommended for patients with mild and moderate hepatic impairment [see Use in Specific Populations (8.6)].
Renal Impairment: The effect of renal impairment on the pharmacokinetics of panobinostat was assessed in a phase 1 trial of 37 patients with advanced cancer and varying degrees of renal impairment. Panobinostat AUC0–inf in the mild, moderate and severe renal impairment groups were 64%, 99% and 59%, of the normal group, respectively. The relative change in Cmax followed a similar pattern [see Use in Specific Populations (8.7)].
Drug Interactions:
Strong CYP3A Inhibitors: Coadministration of a single 20 mg FARYDAK dose with ketoconazole (200 mg twice daily for 14 days) increased the Cmax and AUC0–48 of panobinostat by 62% and 73% respectively, compared to when FARYDAK was given alone in 14 patients with advanced cancer. Tmax was unchanged. A modified starting dose is recommended [see Dose and Administration (2.4), Drug Interactions (7.1)].
Strong CYP3A Inducers: The human oxidative metabolism of panobinostat via the cytochrome P450 system primarily involves CYP3A isozymes. Simulations using PBPK models, predicted an approximately 70% decrease in the systemic exposure of panobinostat in the presence of strong inducers of CYP3A. Avoid coadministration of FARYDAK with strong CYP3A inducers [see Drug Interactions (7.2)].
CYP2D6 Substrates: Coadministration of a single 60 mg dextromethorphan (DM) dose with FARYDAK (20 mg once per day, on Days 3, 5, and 8) increased the Cmax and AUC0–∞ of DM by 20% to 200% and 20% to 130% (interquartile ranges), respectively, compared to when DM was given alone in 14 patients with advanced cancer. These DM exposures were extremely variable (CV% >150%). Avoid coadministration of FARYDAK with sensitive CYP2D6 substrates or CYP2D6 substrates that have a narrow therapeutic index [see Drug Interactions (7.3)].
CYP3A Substrates: Simulations using PBPK models predict that an exposure increase of less than 10% for the sensitive CYP3A substrate midazolam is likely following coadministration with panobinostat. The clinical implications of this finding are not known.
In vitro studies with CYP or UDPglucuronosyltransferase (UGT) substrates:
Panobinostat inhibits CYP2D6, CYP2C19 and CYP3A4 (time-dependent), but does not inhibit CYP1A2, CYP2C8, CYP2C9, and CYP2E. Panobinostat does not induce CYP1A1/2, CYP2B6, CYP2C8/9/19, CYP3A and UGT1A1.
In vitro studies with drug transporter system substrates:
Panobinostat inhibits OAT3, OCT1, OCT2, OATP1B1 and OATP1B3, but does not inhibit P-gp and breast cancer resistant protein (BCRP), or OAT1.
Panobinostat does not induce P-gp and multidrug resistance protein 2 (MRP2) transporters.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with panobinostat.
Panobinostat was mutagenic in the Ames assay, and caused endo-reduplication (increased number of chromosomes) in human peripheral blood lymphocytes in vitro and DNA damage in an in vitro COMET assay in mouse lymphoma L5178Y cells.
FARYDAK may impair male and female fertility. In an oral fertility study conducted in rats, 10, 30, or 100 mg/kg doses of panobinostat were administered to females 3 times weekly (Days 1, 3, and 5) for 2 weeks prior to mating, then during the mating period, and on gestation Days 0, 3, and 6. An increase in early resorption and/or post-implantation loss in female rats were observed at doses ≥10 mg/kg. Number of pregnancies was reduced at doses ≥30 mg/kg. Prostate atrophy accompanied by reduced secretory granules, and testicular degeneration, oligospermia and increased epididymal debris were observed in repeated dose oral toxicity studies in dogs, e.g., in the 4-week study at the dose of 1.5 mg/kg. These effects were not completely reversed following a 4-week nondosing period.
13.2 Animal Toxicology and/or Pharmacology
Adverse findings observed in animals and not reported (or reported with low incidence) in patients treated with panobinostat include thyroid, bone marrow, and skin findings. Thyroid hormone changes in oral studies in rats and dogs included decreases in triodothyronine (T3), tetraiodothyronine (T4) and thyroid stimulating hormone (TSH). Histopathology changes of the thyroid included decreases in follicular colloid and epithelial vacuolation, and increases in thyroid follicular hypertrophy. A benign thyroid follicular cell adenoma was also seen in 1 rat in the 26-week study. Bone marrow findings in one or both species included hyperostosis, plasmacytosis, increased number of granulocytic cells, and presence of abnormal cytoplasmic granulation. Osseous metaplasia of the lung and skin hyperplasia and papilloma were observed in dogs in the 39-week study.
14. Clinical Studies
14.1 Relapsed Multiple Myeloma
The efficacy and safety of FARYDAK in combination with bortezomib and dexamethasone was evaluated in a randomized, double-blind, placebo-controlled, multicenter study in patients with relapsed multiple myeloma who had received 1 to 3 prior lines of therapy.
Patients received bortezomib (1.3 mg/m2 injected intravenously) with dexamethasone (20 mg) in addition to FARYDAK 20 mg (or placebo), taken orally every other day, for 3 doses per week in Weeks 1 and 2 of each 21-day cycle. Treatment was administered for a maximum of 16 cycles (48 weeks).
A total of 768 patients were randomized in a 1:1 ratio to receive either the combination of FARYDAK, bortezomib, dexamethasone (n=387) or placebo, bortezomib, dexamethasone (n=381), stratified by prior use of bortezomib and the number of prior lines of anti-myeloma therapy. Demographics and baseline disease characteristics were balanced between arms. The median age was 63 years (range 28 to 84); 42% of patients were older than 65 years; 53% of patients were male; Caucasians comprised 65% of the study population, Asians 30%, and blacks 3%. The ECOG performance status was 0 to 1 in 93% of patients. The median number of prior therapies was 1; 48% of patients received 2 or 3 prior lines of therapy. More than half (57%) of the patients had prior stem cell transplantation. The most common prior antineoplastic therapies were corticosteroids (90%), melphalan (80%), thalidomide (53%), cyclophosphamide (47%), bortezomib (44%), and lenalidomide (19%). The median duration of follow-up was 29 months in both arms.
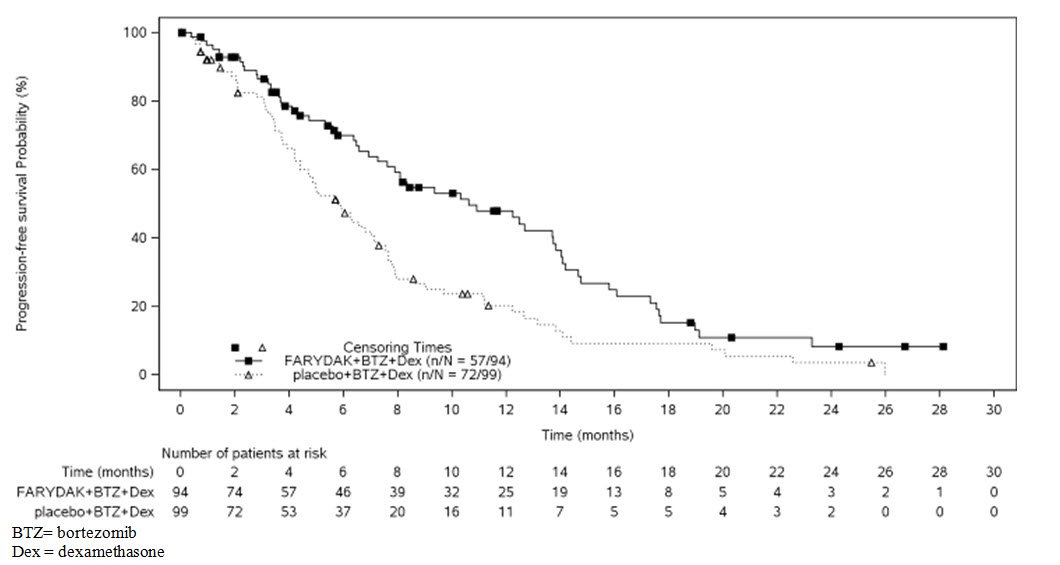
The primary endpoint was progression-free survival (PFS), using modified European Bone Marrow Transplant Group (EBMT) criteria, as assessed by the investigators. In the overall trial population, the median PFS (95% CI) was 12 months (10.3, 12.9) in the FARYDAK, bortezomib, dexamethasone arm and 8.1 months (7.6, 9.2) in the placebo, bortezomib, dexamethasone arm, [HR: 0.63 (95% CI: 0.52, 0.76)]. At the time of interim analysis, overall survival was not statistically different between arms. The approval of FARYDAK was based upon the efficacy and safety in a prespecified subgroup analysis of 193 patients who had received prior treatment with both bortezomib and an immunomodulatory agent and a median of 2 prior therapies as the benefit:risk appeared to be greater in this more heavily pretreated population than in the overall trial population. Of these 193 patients, 76% of them had received ≥2 prior lines of therapy. The median PFS (95% CI) was 10.6 months (7.6, 13.8) in the FARYDAK, bortezomib, and dexamethasone arm and 5.8 months (4.4, 7.1) in the placebo, bortezomib, and dexamethasone arm [HR: 0.52 (0.36, 0.76]. Efficacy results are summarized in Table 6 and the Kaplan- Meier curves for PFS are provided in Figure 1.
| 1 Hazard ratio obtained from stratified Cox model | ||
| FARYDAK bortezomib and dexamethasone N=94 | Placebo bortezomib and dexamethasone N=99 |
|
| Progression-free Survival | ||
| Median, months [95% CI] | 10.6 [7.6, 13.8] | 5.8 [4.4, 7.1] |
| Hazard ratio [95% CI]1 | 0.52 (0.36, 0.76) | |
Figure 1: Kaplan-Meier Plot of Progression-Free Survival in Patients with Multiple Myeloma who Received Prior Treatment with Both Bortezomib and an Immunomodulatory Agent
In the subgroup of patients who had received prior treatment with both bortezomib and an immunomodulatory agent (n=193), the overall response rate using modified EBMT criteria was 59% in the FARYDAK, bortezomib, and dexamethasone arm and 41% in the placebo, bortezomib, and dexamethasone arm. Response rates are summarized in Table 7.
| FARYDAK bortezomib and dexamethasone N=94 | Placebo bortezomib and dexamethasone N=99 |
|
| Overall response | 55 (58.5%) | 41 (41.4%) |
| [95% CI] | (47.9, 68.6) | (31.6, 51.8) |
| Complete response | 8 (8.5%) | 2 (2.0%) |
| Near complete response | 13 (13.8%) | 7 (7.1%) |
| Partial response | 34 (36.2%) | 32 (32.3%) |
16. How is Farydak supplied
How Supplied
FARYDAK 10 mg panobinostat (equivalent to 12.58 mg of panobinostat lactate): Size # 3 light green opaque capsule, radial markings on cap with black ink “LBH 10 mg” and two radial bands with black ink on body, containing white to almost white powder.
FARYDAK 15 mg panobinostat (equivalent to 18.86 mg of panobinostat lactate): Size #1 orange opaque capsule, radial markings on cap with black ink “LBH 15 mg” and two radial bands with black ink on body, containing white to off-white powder.
FARYDAK 20 mg panobinostat (equivalent to 25.15 mg of panobinostat lactate): Size #1 red opaque capsule, radial markings on cap with black ink “LBH 20 mg” and two radial bands with black ink on body, containing white to off-white powder.
FARYDAK capsules are packaged in PVC/PCTFE blister packs.
10 mg
Blister packs containing 6 capsules ………………………….…….NDC 73116-100-06
15 mg
Blister packs containing 6 capsules ………………………….…….NDC 73116-101-06
20 mg
Blister packs containing 6 capsules ………………………….…….NDC 73116-102-06
Storage and Handling
Store at 20°C to 25°C (68°F to 77°F), excursions permitted between 15°C and 30°C (59°F and 86°F). Store blister pack in original carton to protect from light. FARYDAK capsules should not be opened, crushed, or chewed. Direct contact of the powder in FARYDAK capsules with the skin or mucous membranes should be avoided. If such contact occurs wash thoroughly. Personnel should avoid exposure to crushed and/or broken capsules.
FARYDAK is a cytotoxic drug. Follow special handling and disposal procedures [see References (15)1].
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Dosing and Administration
Instruct patients to take FARYDAK exactly as prescribed and not to change their dose or to stop taking FARYDAK unless they are told to do so by their healthcare provider. If a patient misses a dose, advise them to take their dose as soon possible and up to 12 hours after the specified dose time. If vomiting occurs advise the patient not to repeat the dose, but to take the next usual prescribed dose on schedule.
Cardiac Toxicity/Electrocardiographic Changes
Inform patients to report chest pain or discomfort, changes in heart beat (fast or slow), palpitations, lightheadedness, fainting, dizziness, blue discoloration of lips, shortness of breath, and swelling of lower limbs or skin as these may be warning signs of a heart problem.
Bleeding Risk
Inform patients that FARYDAK is associated with thrombocytopenia. Advise patients to contact their healthcare provider right away if they experience any signs of bleeding and inform patients that it might take longer than usual for them to stop bleeding. Advise patients of the need to monitor blood chemistry and hematology prior to the start of FARYDAK therapy and periodically thereafter.
Infections
Inform patients of the risk of neutropenia and severe and life-threatening infections. Instruct patients to contact their physician immediately if they develop a fever and/or any exhibit any signs of infection.
Gastrointestinal Toxicities
Inform patients that FARYDAK can cause severe nausea, vomiting and diarrhea which may require medication for treatment. Advise patients to contact their physician at the start of diarrhea, for persistent vomiting, or signs of dehydration. Inform patients to consult with their physicians prior to using medications with laxative properties.
Pregnancy
Inform patients that FARYDAK can cause fetal harm. Advise women of reproductive potential to avoid pregnancy while taking FARYDAK. Advise women of reproductive potential to use effective contraception while taking FARYDAK and for at least 3 months after the last dose of the drug.
Advise sexually active men to use condoms while receiving FARYDAK and for at least 6 months following the last dose of the drug.
Lactation
Advise women not to breastfeed while taking FARYDAK.
Distributed by:
Secura Bio, Inc.
Las Vegas, NV 89134
©Secura Bio
USFPR1900101
September 2019
Medication Guide
| This Medication Guide has been approved by the U.S. Food and Drug Administration. | Revised: September 2019 | ||
| MEDICATION GUIDE
FARYDAK® (FAYR-ah-dak) (panobinostat) capsules |
|||
| What is the most important information I should know about FARYDAK?
FARYDAK can cause serious side effects, including: • Diarrhea is common with FARYDAK and can be severe. Tell your healthcare provider right away if you have abdominal (stomach) cramps, loose stool, diarrhea, or if you feel like you are becoming dehydrated. Your healthcare provider may prescribe medicines to help prevent or treat these side effects. Taking or using stool softeners or laxative medicines may worsen diarrhea, talk to your healthcare provider before taking or using these medicines. Your healthcare provider will do regular tests to check the levels of fluid and electrolytes in your blood during treatment with FARYDAK. • Heart problems. FARYDAK can cause severe heart problems which can lead to death. Your risk of heart problems may be increased if you have a condition called “long QT syndrome” or other heart problems. Your healthcare provider will do blood tests to check your electrolytes and do an electrocardiogram (ECG) tests before and during treatment with FARYDAK. Call your healthcare provider and get emergency medical help right away if you have any of the following symptoms of heart problems: |
|||
| o chest pain | o dizziness | ||
| o faster or slower heart beat | o blue colored lips | ||
| o palpitations (feel like your heart is racing) | o shortness of breath | ||
| o feel lightheaded or faint | o swelling in your legs | ||
| • Bleeding. FARYDAK can cause severe bleeding which can lead to death. It may take you longer than usual to stop bleeding while you are taking FARYDAK. Your healthcare provider will check your platelet counts before you start FARYDAK and during your treatment with FARYDAK. Tell your healthcare provider right away if you get any of the following signs of bleeding: | |||
| o blood in your stools or black stools (look like tar) | o increased bruising | ||
| o pink or brown urine | o feeling dizzy or weak | ||
| o unexpected bleeding or bleeding that is severe or that you cannot control | o confusion | ||
| o vomit blood or vomit looks like coffee grounds | o change in your speech | ||
| o cough up blood or blood clots | o headache that lasts a long time | ||
| What is FARYDAK?
FARYDAK is a prescription medicine used, in combination with bortezomib and dexamethasone, to treat people with a type of cancer called multiple myeloma after at least 2 other types of treatment have been tried. It is not known if FARYDAK is safe and effective in children. |
|||
| What should I tell my healthcare provider before taking FARYDAK?
Before you take FARYDAK, tell your healthcare provider about all of your medical conditions, including if you: • have diarrhea • have heart problems • have a history of bleeding problems • have an infection. You should not take FARYDAK if you have an infection. • have liver problems • are pregnant or plan to become pregnant. FARYDAK can harm your unborn baby. You should not become pregnant during treatment with FARYDAK. • If you become pregnant while taking FARYDAK, or think you may be pregnant, tell your healthcare provider right away. o Females who are able to become pregnant should use effective birth control during treatment with FARYDAK and for at least 3 months after the last dose of FARYDAK. o Males who are sexually active should use a condom during treatment with FARYDAK and for at least 6 months after the last dose of FARYDAK. • are breastfeeding or plan to breastfeed. It is not known if FARYDAK will pass into your breast milk. You and your healthcare provider should decide if you will take FARYDAK or breastfeed. You should not do both. Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine. |
|||
| How should I take FARYDAK?
• Take FARYDAK exactly as your healthcare provider tells you to take it. • Your healthcare provider will tell you how much FARYDAK to take and when to take it. • Your healthcare provider may change your dose or stop treatment temporarily if you have side effects. Do not change your dose or stop taking FARYDAK without first talking with your healthcare provider. • Take FARYDAK 1 time on each scheduled day at about the same time. • FARYDAK can be taken with or without food. • FARYDAK capsule should be swallowed whole with a cup of water. Do not open, crush, or chew FARYDAK. • Avoid contact of the powder in the FARYDAK capsules. If you accidentally get powder from the FARYDAK capsule on your skin, wash the area with soap and water. If you accidentally get powder from the FARYDAK capsule in your eyes, flush your eyes with water. • If you miss a dose of FARYDAK, take it as soon possible, up to 12 hours after the time the dose should have been taken. • If you vomit after taking FARYDAK, do not take another capsule. Stay on your dosing schedule and take your next dose as usual. • If you take too much FARYDAK, call your healthcare provider. |
|||
| What should I avoid while taking FARYDAK?
• Avoid eating star fruit, pomegranate or pomegranate juice, and grapefruit or grapefruit juice while taking FARYDAK. These foods may affect the amount of FARYDAK in your blood. |
|||
| What are the possible side effects of FARYDAK?
FARYDAK may cause serious side effects, including: • See “What is the most important information I should know about FARYDAK?” • Low blood cell counts are common with FARYDAK and can be severe. Your healthcare provider will check your blood counts before you start FARYDAK and during your treatment with FARYDAK. o Low platelet count (thrombocytopenia) can cause unusual bleeding or bruising under your skin. o Low white cell count (neutropenia) can cause you to get infections. o Low red blood cell count (anemia) may make you feel weak, tired, or you may get tired easily, you look pale, or you feel short of breath. • Infections. There is an increased risk of infection while taking FARYDAK. Contact your healthcare provider right away if you have a fever or have any signs of an infection: |
|||
| o sweats or chills | o blood in your phlegm | ||
| o cough | o sores on your body | ||
| o flu-like symptoms | o warm or painful areas on your body | ||
| o shortness of breath | o feeling very tired | ||
| • Liver problems (hepatotoxicity). Your healthcare provider should do blood tests to check your liver before you start taking FARYDAK and if you have symptoms of liver problems while you take FARYDAK. Call your healthcare provider right away if you have the following symptoms of liver problems: | |||
| o feel tired or weak | o upper abdominal (stomach) pain | ||
| o loss of appetite | o yellowing of your skin or the white of your eyes | ||
| o Dark amber colored urine | |||
| The most common side effects of FARYDAK include tiredness, nausea, swelling in your arms or legs, decreased appetite, fever, and vomiting. Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of FARYDAK. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|||
| How should I store FARYDAK?
• Store FARYDAK between 68°F to 77°F (20°C to 25°C). Store blister pack in original carton to protect from light. Keep FARYDAK and all medicines out of the reach of children. |
|||
| General information about the safe and effective use of FARYDAK
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FARYDAK for a condition for which it was not prescribed. Do not give FARYDAK to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about FARYDAK that is written for health professionals. |
|||
| What are the ingredients in FARYDAK?
Active ingredient: panobinostat Inactive ingredients: magnesium stearate, mannitol, microcrystalline cellulose, and pre-gelatinized starch Capsule shell contains: gelatin, FD&C Blue 1 (10 mg capsules), yellow iron oxide (10 mg and 15 mg capsules), red iron oxide (15 mg and 20 mg capsules), and titanium dioxide Distributed by: Secura Bio, Inc., Las Vegas, NV 89134 For more information, go to www.FARYDAK.com or call 1-844-9-SECURA (1-844-973-2872). © Secura BioUSFPR1900101 |
|||
PRINCIPAL DISPLAY PANEL
NDC 73116-100-06
6 capsules
FARYDAK®
(panobinostat)
capsules
10 mg
Equivalent to 12.58 mg of panobinostat lactate
Swallow Whole with Water and Do Not Open, Crush or Chew.
Store blister pack in original carton to protect from light.
ATTENTION: Dispense with enclosed Medication Guide
Rx only
PRINCIPAL DISPLAY PANEL
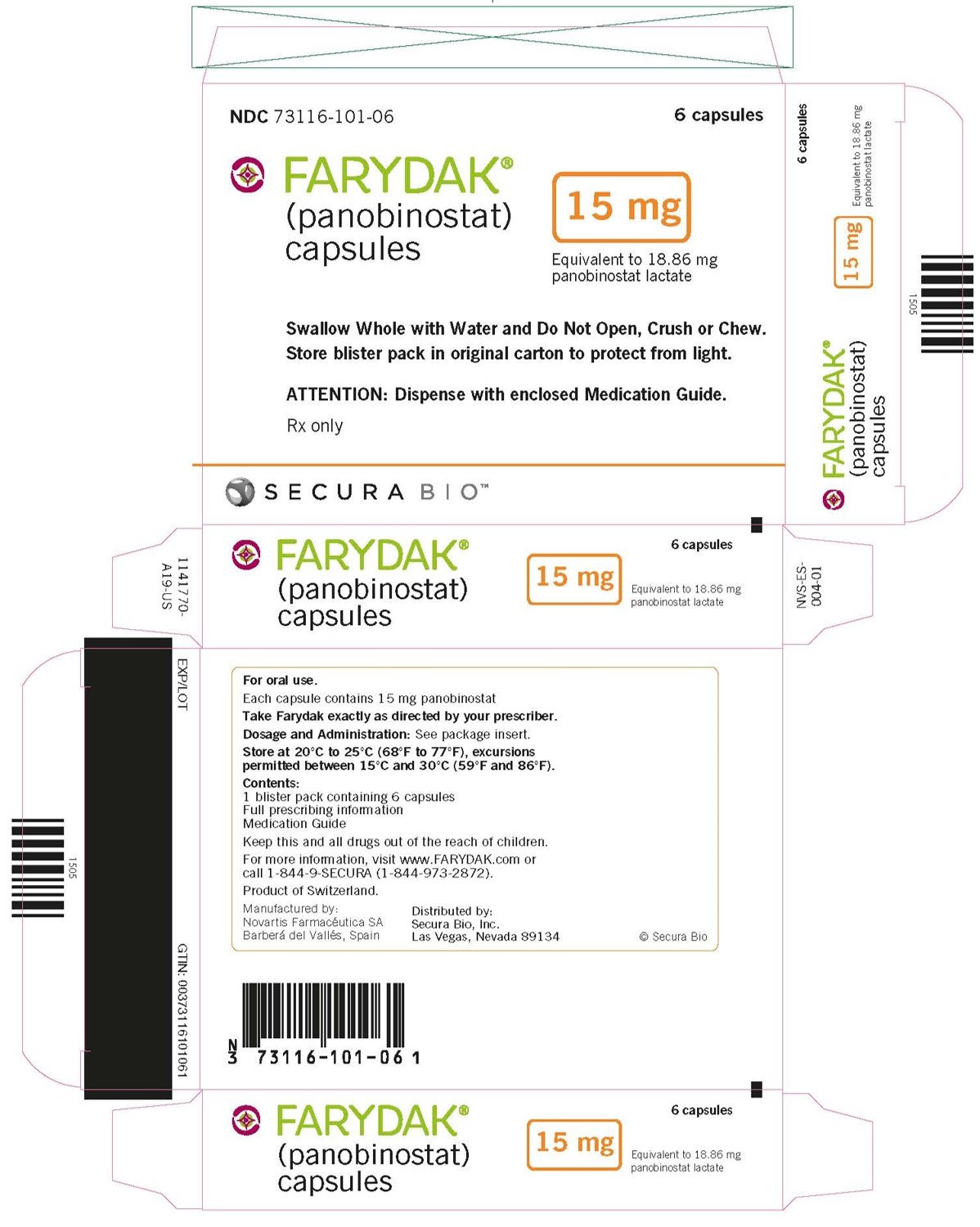
NDC 73116-101-06
6 capsules
FARYDAK®
(panobinostat)
capsules
15 mg
Equivalent to 18.86 mg of panobinostat lactate
Swallow Whole with Water and Do Not Open, Crush or Chew.
Store blister pack in original carton to protect from light.
ATTENTION: Dispense with enclosed Medication Guide
Rx only
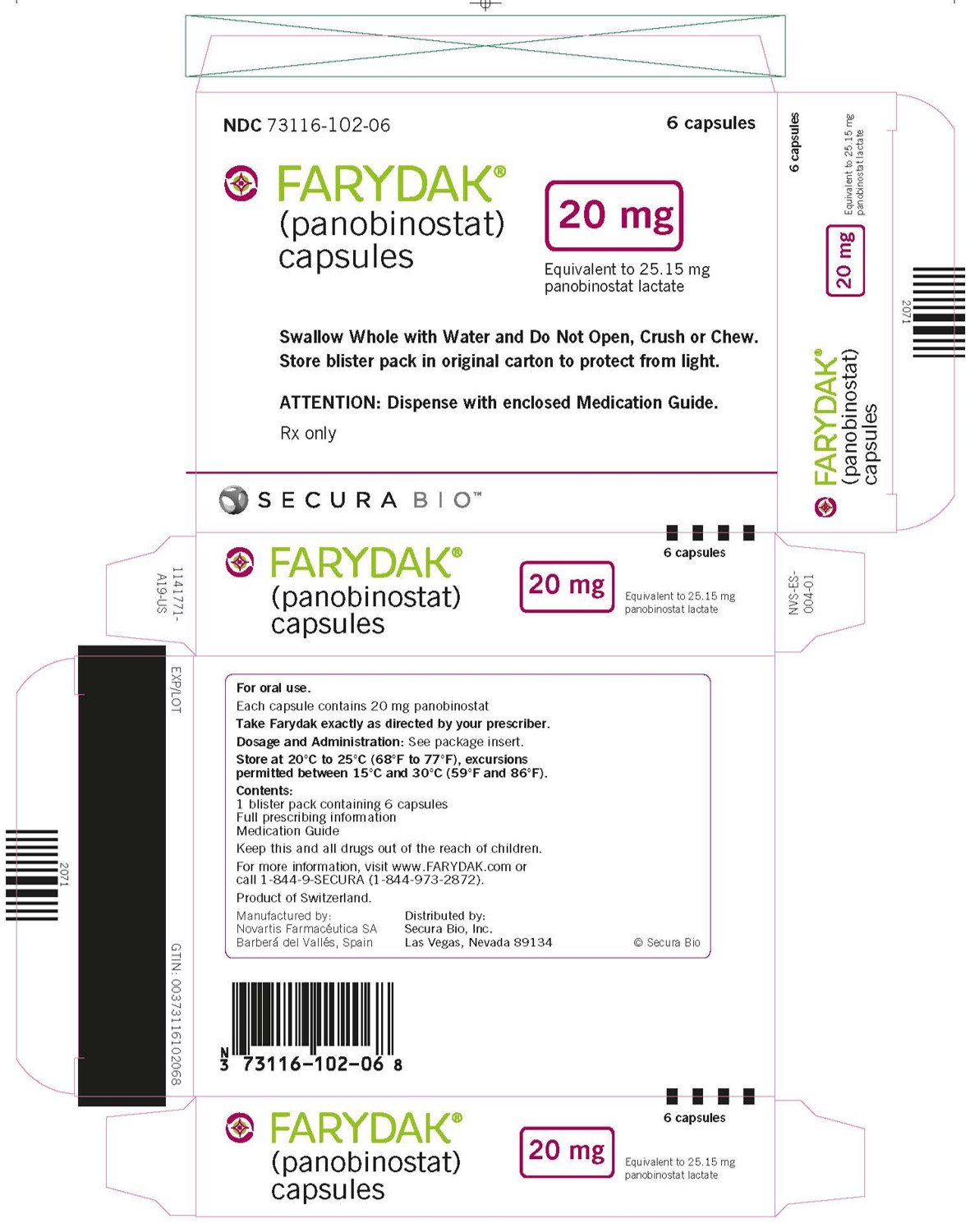
PRINCIPAL DISPLAY PANEL
NDC 73116-102-06
6 capsules
FARYDAK®
(panobinostat)
capsules
20 mg
Equivalent to 25.15 mg of panobinostat lactate
Swallow Whole with Water and Do Not Open, Crush or Chew.
Store blister pack in original carton to protect from light.
ATTENTION: Dispense with enclosed Medication Guide.
Rx only
| FARYDAK
panobinostat capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FARYDAK
panobinostat capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| FARYDAK
panobinostat capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Secura Bio, Inc. (117026236) |
| Registrant - Secura Bio, Inc. (117026236) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Siegfried Barbera, S.L. | 469834249 | ANALYSIS(73116-100, 73116-101, 73116-102) , LABEL(73116-100, 73116-101, 73116-102) , MANUFACTURE(73116-100, 73116-101, 73116-102) , PACK(73116-100, 73116-101, 73116-102) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Novartis Pharma Schweizerhalle AG | 481963890 | API MANUFACTURE(73116-100, 73116-101, 73116-102) , MANUFACTURE(73116-100, 73116-101, 73116-102) | |
More about Farydak (panobinostat)
- Check interactions
- Compare alternatives
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: histone deacetylase inhibitors
- En español