Zaltrap: Package Insert / Prescribing Info
Package insert / product label
Generic name: ziv-aflibercept
Dosage form: injection solution, concentrate
Drug class: VEGF/VEGFR inhibitors
J Code (medical billing code): J9400 (1 mg, injection)
Medically reviewed by Drugs.com. Last updated on May 26, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
ZALTRAP® (ziv-aflibercept) injection, for intravenous use
Initial U.S. Approval: 2012
Indications and Usage for Zaltrap
ZALTRAP, a vascular endothelial growth factor inhibitor, in combination with fluorouracil, leucovorin, irinotecan (FOLFIRI), is indicated for the treatment of patients with metastatic colorectal cancer that is resistant to or has progressed following an oxaliplatin-containing regimen. (1)
Zaltrap Dosage and Administration
Dosage Forms and Strengths
Injection: 100 mg/4 mL (25 mg/mL) and 200 mg/8 mL (25 mg/mL) solution in a single-dose vial (3)
Contraindications
None (4)
Warnings and Precautions
- Hemorrhage: Severe and sometimes fatal hemorrhage, including gastrointestinal (GI) hemorrhage, has been reported in patients who have received ZALTRAP. Do not administer ZALTRAP to patients with severe hemorrhage. (5.1)
- Gastrointestinal Perforation: Discontinue ZALTRAP therapy in patients who experience GI perforation. (5.2)
- Impaired Wound Healing: Withhold ZALTRAP for at least 4 weeks prior to elective surgery. Do not administer for at least 4 weeks following major surgery and until wounds have adequately healed. Discontinue ZALTRAP in patients with impaired wound healing. The safety of resumption of ZALTRAP after resolution of wound healing complications has not been established. (5.3)
- Fistula Formation: Discontinue ZALTRAP if fistula occurs. (2.2, 5.4)
- Hypertension: Monitor blood pressure and treat hypertension. Temporarily suspend ZALTRAP if hypertension is not controlled. Discontinue ZALTRAP if hypertensive crisis develops. (2.2, 5.5)
- Arterial Thromboembolic Events (ATE): Discontinue ZALTRAP if ATE develops. (5.6)
- Proteinuria: Monitor urine protein. Suspend ZALTRAP for proteinuria of 2 grams per 24 hours or more. Discontinue ZALTRAP if nephrotic syndrome or thrombotic microangiopathy (TMA) develops. (2.2, 5.7)
- Neutropenia and Neutropenic Complications: Delay administration of ZALTRAP/FOLFIRI until neutrophil count is 1.5 × 109/L or higher. (5.8)
- Diarrhea and Dehydration: Incidence of severe diarrhea and dehydration is increased. Monitor elderly patients more closely. (5.9, 8.5)
- Reversible Posterior Leukoencephalopathy Syndrome: Discontinue ZALTRAP. (5.10)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of potential risk to fetus and need for use of effective contraception. (5.11, 8.1, 8.3)
Adverse Reactions/Side Effects
Most common adverse reactions (≥20% incidence) were leukopenia, diarrhea, neutropenia, proteinuria, AST increased, stomatitis, fatigue, thrombocytopenia, ALT increased, hypertension, weight decreased, decreased appetite, epistaxis, abdominal pain, dysphonia, serum creatinine increased, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 5/2025
Full Prescribing Information
1. Indications and Usage for Zaltrap
ZALTRAP, in combination with fluorouracil, leucovorin, irinotecan-(FOLFIRI), is indicated for the treatment of patients with metastatic colorectal cancer (mCRC) that is resistant to or has progressed following an oxaliplatin-containing regimen.
2. Zaltrap Dosage and Administration
2.1 Recommended Dose and Schedule
The recommended dosage of ZALTRAP is 4 mg per kg of actual body weight as an intravenous infusion over 1 hour every two weeks in combination with FOLFIRI until disease progression or unacceptable toxicity. Administer ZALTRAP prior to any component of the FOLFIRI regimen on the day of treatment.
Refer to prescribing information for irinotecan, fluorouracil, and leucovorin for the recommended dosage and dosage modifications for these drugs.
2.2 Dosage Modifications for Adverse Reactions
Discontinue ZALTRAP for:
- Severe hemorrhage [see Warnings and Precautions (5.1)]
- Gastrointestinal perforation [see Warnings and Precautions (5.2)]
- Impaired wound healing [see Warnings and Precautions (5.3)]
- Fistula formation [see Warnings and Precautions (5.4)]
- Hypertensive crisis or hypertensive encephalopathy [see Warnings and Precautions (5.5)]
- Arterial thromboembolic events (ATE) [see Warnings and Precautions (5.6)]
- Nephrotic syndrome or thrombotic microangiopathy (TMA) [see Warnings and Precautions (5.7)]
- Reversible posterior leukoencephalopathy syndrome (RPLS) [see Warnings and Precautions (5.10)]
Temporarily suspend ZALTRAP:
- At least 4 weeks prior to elective surgery [see Warnings and Precautions (5.3)].
- For uncontrolled hypertension until controlled. Upon resumption, permanently reduce the ZALTRAP dose to 2 mg per kg [see Warnings and Precautions (5.5)].
- For proteinuria of 2 grams per 24 hours or more. Resume when proteinuria is less than 2 grams per 24 hours. For recurrent proteinuria, suspend ZALTRAP until proteinuria is less than 2 grams per 24 hours and then permanently reduce the ZALTRAP dose to 2 mg per kg [see Warnings and Precautions (5.7)].
2.3 Preparation and Administration
Preparation
Inspect vials visually prior to use. ZALTRAP is a clear, colorless to pale yellow solution. Do not use vial if the solution is discolored or cloudy or if the solution contains particles.
Withdraw the prescribed dose of ZALTRAP and dilute in 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP to achieve a final concentration of 0.6 mg/mL to 8 mg/mL.
Do not re-enter the vial after the initial puncture. Discard any unused portion left in the vial.
Use polyvinyl chloride (PVC) infusion bags containing bis (2-ethylhexyl) phthalate (DEHP) or polyolefin infusion bags.
Store diluted ZALTRAP refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours or at controlled room temperature of 20°C to 25°C (68°F to 77°F) for up to 8 hours. Discard any unused portion left in the infusion bag.
Administration
Administer the diluted ZALTRAP solution as an intravenous infusion over 1 hour through a 0.2-micron polyethersulfone filter. Do not use filters made of polyvinylidene fluoride (PVDF) or nylon.
Do not administer as an intravenous push or bolus.
Do not combine ZALTRAP with other drugs in the same infusion bag or intravenous line.
Administer ZALTRAP using an infusion set made of one of the following materials:
- PVC containing DEHP
- DEHP free PVC containing trioctyl-trimellitate (TOTM)
- polypropylene
- polyethylene lined PVC
- polyurethane
3. Dosage Forms and Strengths
ZALTRAP is a clear, colorless to pale-yellow solution available as:
- Injection: 100 mg/4 mL (25 mg/mL) solution in a single-dose vial
- Injection: 200 mg/8 mL (25 mg/mL) solution in a single-dose vial
5. Warnings and Precautions
5.1 Hemorrhage
Patients treated with ZALTRAP have an increased risk of hemorrhage, including severe and sometimes fatal hemorrhagic events. In patients with mCRC, bleeding/hemorrhage (all grades) was reported in 38% of patients treated with ZALTRAP/FOLFIRI compared to 19% of patients treated with placebo/FOLFIRI. Grade 3–4 hemorrhagic events, including gastrointestinal hemorrhage, hematuria, and post-procedural hemorrhage, were reported in 3% of patients receiving ZALTRAP/FOLFIRI compared with 1% of patients receiving placebo/FOLFIRI. Severe intracranial hemorrhage and pulmonary hemorrhage/hemoptysis including fatal events have also occurred in patients receiving ZALTRAP.
Monitor patients for signs and symptoms of bleeding. Do not initiate ZALTRAP in patients with severe hemorrhage. Discontinue ZALTRAP in patients who develop severe hemorrhage [see Dosage and Administration (2.2)].
5.2 Gastrointestinal Perforation
Gastrointestinal (GI) perforation including fatal GI perforation can occur in patients receiving ZALTRAP. Across three placebo-controlled clinical studies (colorectal, pancreatic, and lung cancer populations), the incidence of GI perforation (all grades) was 0.8% for patients treated with ZALTRAP and 0.3% for patients treated with placebo. Grade 3–4 GI perforation occurred in 0.8% of patients treated with ZALTRAP and 0.2% of patients treated with placebo.
Monitor patients for signs and symptoms of GI perforation. Discontinue ZALTRAP therapy in patients who experience GI perforation [see Dosage and Administration (2.2)].
5.3 Impaired Wound Healing
Grade 3 impaired wound healing was reported in 2 patients (0.3%) treated with ZALTRAP/FOLFIRI regimen.
Withhold ZALTRAP for at least 4 weeks prior to elective surgery. Do not administer ZALTRAP for at least 4 weeks after major surgery and until wounds have adequately healed. For minor surgery such as central venous access port placement, biopsy, and tooth extraction, ZALTRAP may be initiated/resumed once the surgical wound is fully healed. Discontinue ZALTRAP in patients with impaired wound healing. The safety of resumption of ZALTRAP after resolution of wound healing complications has not been established [see Dosage and Administration (2.2)].
5.4 Fistula Formation
Fistula formation involving gastrointestinal and non-gastrointestinal sites occurs at a higher incidence in patients treated with ZALTRAP. In patients with mCRC, fistulas (anal, enterovesical, enterocutaneous, colovaginal, intestinal sites) were reported in 9 of 611 patients (1.5%) treated with ZALTRAP/FOLFIRI regimen and 3 of 605 patients (0.5%) treated with placebo/FOLFIRI regimen. Grade 3 GI fistula formation occurred in 2 patients treated with ZALTRAP (0.3%) and in 1 patient treated with placebo (0.2%).
Discontinue ZALTRAP therapy in patients who develop fistula [see Dosage and Administration (2.2)].
5.5 Hypertension
ZALTRAP increases the risk of Grade 3–4 hypertension. There is no clinical trial experience administering ZALTRAP to patients with NYHA class III or IV heart failure. In patients with mCRC, Grade 3 hypertension (defined as requiring adjustment in existing antihypertensive therapy or treatment with more than one drug) was reported in 1.5% of patients treated with placebo/FOLFIRI and 19% of patients treated with ZALTRAP/FOLFIRI. Grade 4 hypertension (hypertensive crisis) was reported in 1 patient (0.2%) treated with ZALTRAP/FOLFIRI. Among those patients treated with ZALTRAP/FOLFIRI developing Grade 3–4 hypertension, 54% had onset during the first two cycles of treatment.
Monitor blood pressure every two weeks or more frequently as clinically indicated during treatment with ZALTRAP. Treat with appropriate antihypertensive therapy and continue monitoring blood pressure regularly. Temporarily suspend ZALTRAP in patients with uncontrolled hypertension until controlled and permanently reduce the ZALTRAP dose to 2 mg per kg for subsequent cycles. Discontinue ZALTRAP in patients with hypertensive crisis or hypertensive encephalopathy [see Dosage and Administration (2.2)].
5.6 Arterial Thromboembolic Events
ATE, including transient ischemic attack, cerebrovascular accident, and angina pectoris, occurred more frequently in patients who have received ZALTRAP. In patients with mCRC, ATE was reported in 2.6% of patients treated with ZALTRAP/FOLFIRI and 1.7% of patients treated with placebo/FOLFIRI. Grade 3–4 events occurred in 11 patients (1.8%) treated with ZALTRAP/FOLFIRI and 4 patients (0.7%) treated with placebo/FOLFIRI.
Discontinue ZALTRAP in patients who experience an ATE [see Dosage and Administration (2.2)].
5.7 Proteinuria
Severe proteinuria, nephrotic syndrome, and thrombotic microangiopathy (TMA) occurred more frequently in patients treated with ZALTRAP. In patients with mCRC, proteinuria was reported in 62% patients treated with ZALTRAP/FOLFIRI compared to 41% patients treated with placebo/FOLFIRI. Grade 3–4 proteinuria occurred in 8% of patients treated with ZALTRAP/FOLFIRI compared to 1% of patients treated with placebo/FOLFIRI [see Adverse Reactions (6.1)]. Nephrotic syndrome occurred in 2 patients (0.5%) treated with ZALTRAP/FOLFIRI compared to none of the patients treated with placebo/FOLFIRI. TMA was reported in 3 of 2258 patients with cancer enrolled across completed studies.
Monitor proteinuria by urine dipstick analysis and/or urinary protein creatinine ratio (UPCR) for the development or worsening of proteinuria during ZALTRAP therapy. Patients with a dipstick of ≥2+ for protein or a UPCR greater than 1 should undergo a 24-hour urine collection.
Suspend ZALTRAP administration for proteinuria 2 grams per 24 hours or more, and resume when proteinuria is less than 2 grams per 24 hours. If recurrent, suspend until proteinuria is less than 2 grams per 24 hours and then permanently reduce the ZALTRAP dose to 2 mg per kg. Discontinue ZALTRAP in patients who develop nephrotic syndrome or TMA [see Dosage and Administration (2.2)].
5.8 Neutropenia and Neutropenic Complications
A higher incidence of neutropenic complications (febrile neutropenia and neutropenic infection) occurred in patients receiving ZALTRAP. In patients with mCRC, Grade 3–4 neutropenia occurred in 37% of patients treated with ZALTRAP/FOLFIRI compared to 30% patients treated with placebo/FOLFIRI [see Adverse Reactions (6.1)]. Grade 3–4 febrile neutropenia occurred in 4% of patients treated with ZALTRAP/FOLFIRI compared to 2% of patients treated with placebo/FOLFIRI. Grade 3–4 neutropenic infection/sepsis occurred in 1.5% of patients treated with ZALTRAP/FOLFIRI and 1.2% of patients treated with placebo/FOLFIRI.
Monitor CBC with differential count at baseline and prior to initiation of each cycle of ZALTRAP. Delay ZALTRAP/FOLFIRI until neutrophil count is at or above 1.5 × 109/L.
5.9 Diarrhea and Dehydration
The incidence of severe diarrhea is increased in patients treated with ZALTRAP/FOLFIRI. In patients with mCRC, Grade 3–4 diarrhea was reported in 19% of patients treated with ZALTRAP/FOLFIRI compared to 8% of patients treated with placebo/FOLFIRI. Grade 3–4 dehydration was reported in 4% of patients treated with ZALTRAP/FOLFIRI compared to 1% of patients treated with placebo/FOLFIRI [see Adverse Reactions (6.1)]. The incidence of diarrhea is increased in patients who are age 65 years or older as compared to patients younger than 65 years of age [see Use in Specific Populations (8.5)]. Monitor elderly patients closely for diarrhea.
5.10 Reversible Posterior Leukoencephalopathy Syndrome
RPLS (also known as posterior reversible encephalopathy syndrome) was reported in 0.5% of 3795 patients treated with ZALTRAP monotherapy or in combination with chemotherapy.
Confirm the diagnosis of RPLS with magnetic resonance imaging (MRI) and discontinue ZALTRAP in patients who develop RPLS. Symptoms usually resolve or improve within days, although some patients have experienced ongoing neurologic sequelae or death [see Dosage and Administration (2.2)].
5.11 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, ZALTRAP can cause fetal harm when administered to pregnant women. Administration of ziv-aflibercept during the period of organogenesis was embryotoxic and teratogenic in rabbits at exposure levels approximately 0.3 times the human exposure at the 4 mg per kg dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with ZALTRAP and for 3 months following the last dose [see Use in Specific Populations (8.1, 8.3)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hemorrhage [see Warnings and Precautions (5.1)]
- Gastrointestinal Perforation [see Warnings and Precautions (5.2)]
- Impaired Wound Healing [see Warnings and Precautions (5.3)]
- Fistula Formation [see Warnings and Precautions (5.4)]
- Hypertension [see Warnings and Precautions (5.5)]
- Arterial Thromboembolic Events [see Warnings and Precautions (5.6)]
- Proteinuria [see Warnings and Precautions (5.7)]
- Neutropenia and Neutropenic Complications [see Warnings and Precautions (5.8)]
- Diarrhea and Dehydration [see Warnings and Precautions (5.9)]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of ZALTRAP in combination with FOLFIRI was evaluated in VELOUR (EFC102621) [see Clinical Studies (14)]. Patients received ZALTRAP 4 mg per kg (N=611) or placebo (N=605) intravenously every two weeks (one cycle) in combination with FOLFIRI. Patients received a median of 9 cycles of ZALTRAP/FOLFIRI.
The most common Grade 3–4 adverse reactions (≥5%) in the ZALTRAP/FOLFIRI arm were neutropenia, diarrhea, hypertension, leukopenia, stomatitis, fatigue, proteinuria, and asthenia.
The most frequent adverse reactions leading to permanent discontinuation in ≥1% of patients treated with ZALTRAP/FOLFIRI regimen were asthenia/fatigue, infections, diarrhea, dehydration, hypertension, stomatitis, venous thromboembolic events, neutropenia, and proteinuria.
The ZALTRAP dose was reduced and/or omitted in 17% of patients. Cycle delays >7 days occurred in 60% of patients treated with ZALTRAP/FOLFIRI.
The most common adverse reactions (≥20%) in the ZALTRAP/FOLFIRI arm were leukopenia, diarrhea, neutropenia, proteinuria, AST increased, stomatitis, fatigue, thrombocytopenia, ALT increased, hypertension, weight decreased, decreased appetite, epistaxis, abdominal pain, dysphonia, serum creatinine increased, and headache.
Adverse reactions and laboratory abnormalities that occurred in ≥5% (all grades) of patients receiving ZALTRAP in combination with FOLFIRI and which occurred at ≥2% higher frequency in patients who received ZALTRAP/FOLFIRI compared to those who received placebo/FOLFIRI in VELOUR are shown in Table 1. VELOUR was not designed to demonstrate a statistically significant difference in adverse reaction rates for ZALTRAP/FOLFIRI as compared to placebo/FOLFIRI for any adverse reactions listed below.
| Primary System Organ Class Preferred Term | ZALTRAP/ FOLFIRI (N=611) | Placebo/ FOLFIRI (N=605) |
||
|---|---|---|---|---|
| All grades (%) | Grades 3–4 (%) | All grades (%) | Grades 3–4 (%) | |
| Note: Adverse Reactions are reported using MedDRA version 13.1 and graded using NCI CTC version 3.0 | ||||
|
||||
| Blood and lymphatic system disorders | ||||
| Leukopenia | 78 | 16 | 72 | 12 |
| Neutropenia | 67 | 37 | 57 | 30 |
| Thrombocytopenia | 48 | 3 | 35 | 2 |
| Gastrointestinal disorders | ||||
| Diarrhea | 69 | 19 | 57 | 8 |
| Stomatitis | 50 | 13 | 33 | 5 |
| Abdominal Pain | 27 | 4 | 24 | 2 |
| Abdominal Pain Upper | 11 | 1 | 8 | 1 |
| Hemorrhoids | 6 | 0 | 2 | 0 |
| Rectal Hemorrhage | 5 | 0.7 | 2 | 0.5 |
| Proctalgia | 5 | 0.3 | 2 | 0.3 |
| Investigations | ||||
| AST increased | 62 | 3 | 54 | 2 |
| ALT increased | 50 | 3 | 39 | 2 |
| Weight decreased | 32 | 3 | 14 | 0.8 |
| Renal and urinary disorders | ||||
| Proteinuria* | 62 | 8 | 41 | 1 |
| Serum creatinine increased | 23 | 0 | 19 | 0.5 |
| General disorders and administration site conditions | ||||
| Fatigue | 48 | 13 | 39 | 8 |
| Asthenia | 18 | 5 | 13 | 3 |
| Vascular disorders | ||||
| Hypertension | 41 | 19 | 11 | 1.5 |
| Metabolism and nutrition disorders | ||||
| Decreased Appetite | 32 | 3 | 24 | 2 |
| Dehydration | 9 | 4 | 3 | 1 |
| Respiratory, thoracic and mediastinal disorders | ||||
| Epistaxis | 28 | 0.2 | 7 | 0 |
| Dysphonia | 25 | 0.5 | 3 | 0 |
| Dyspnea | 12 | 0.8 | 9 | 0.8 |
| Oropharyngeal Pain | 8 | 0.2 | 3 | 0 |
| Rhinorrhea | 6 | 0 | 2 | 0 |
| Nervous system disorders | ||||
| Headache | 22 | 2 | 9 | 0.3 |
| Skin and subcutaneous tissue disorders | ||||
| Palmar-Plantar Erythrodysesthesia Syndrome | 11 | 3 | 4 | 0.5 |
| Skin Hyperpigmentation | 8 | 0 | 3 | 0 |
| Infections | ||||
| Urinary Tract Infection | 9 | 0.8 | 6 | 0.8 |
Infections occurred at a higher frequency in patients receiving ZALTRAP/FOLFIRI (46%, all grades; 12%, Grade 3–4) than in patients receiving placebo/FOLFIRI (33%, all grades; 7%, Grade 3–4), including urinary tract infection, nasopharyngitis, upper respiratory tract infection, pneumonia, catheter site infection, and tooth infection.
In patients with mCRC, severe hypersensitivity reactions have been reported with ZALTRAP/FOLFIRI (0.3%) and placebo/FOLFIRI (0.5%).
In patients with mCRC, venous thromboembolic events (VTE), consisting primarily of deep venous thrombosis and pulmonary embolism, occurred in 9% of patients treated with ZALTRAP/FOLFIRI and 7% of patients treated with placebo/FOLFIRI. Grade 3–4 VTE occurred in 8% of patients treated with ZALTRAP/FOLFIRI and in 6% of patients treated with placebo/FOLFIRI. Pulmonary embolism occurred in 5% of patients treated with ZALTRAP/FOLFIRI and 3.4% of patients treated with placebo/FOLFIRI.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In patients with various cancers across 15 studies, 1.4% (41/2862) of patients tested positive for antiproduct antibody (APA) at baseline. The incidence of APA development was 3.1% (53/1687) in patients receiving intravenous ziv-aflibercept and 1.7% (19/1134) in patients receiving placebo. Among patients who tested positive for APA and had sufficient samples for further testing, neutralizing antibodies were detected in 17 of 48 ziv-aflibercept-treated patients and in 2 of 40 patients receiving placebo.
The mean free ziv-aflibercept trough concentrations were lower in patients with positive neutralizing antibodies than in the overall population. The impact of neutralizing antibodies on efficacy and safety could not be assessed based on limited available data.
6.3 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ZALTRAP. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Musculoskeletal and connective tissue disorders: Osteonecrosis of the jaw
Cardiac disorders: Cardiac failure, ejection fraction decreased
Vascular disorders: Arterial (including aortic) aneurysms, dissections, and rupture
Related/similar drugs
7. Drug Interactions
No dedicated drug-drug interaction studies have been conducted for ZALTRAP. No clinically important pharmacokinetic interactions were found between ziv-aflibercept and irinotecan/SN-38 or fluorouracil [see Clinical Pharmacology (12.3)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies and its mechanism of action [see Clinical Pharmacology (12.1)], ZALTRAP can cause fetal harm when administered to pregnant women. There is insufficient data in pregnant women exposed to ZALTRAP to assess the risks. Administration of ziv-aflibercept during the period of organogenesis was embryotoxic and teratogenic in rabbits at exposure levels approximately 0.3 times the human exposure at the 4 mg per kg dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal data
In pregnant rabbits, administration of ziv-aflibercept during the period of organogenesis resulted in an increase in postimplantation loss and external (including anasarca, umbilical hernia, diaphragmatic hernia and gastroschisis, cleft palate, ectrodactyly, and anal atresia), visceral (heart, great vessels, and arteries), and skeletal fetal malformations (including fused vertebrae, sternebrae, and ribs, supernumerary arches and ribs, and incomplete ossification) at doses greater than or equal to 3 mg per kg, administered once every 3 days (approximately 0.3 times the human exposure at the 4 mg per kg dose based on AUC).
8.2 Lactation
Risk Summary
There are no data on the presence of ziv-aflibercept in human milk, or the effects of ziv-aflibercept on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants, advise women not to breastfeed during treatment with ZALTRAP and for 1 month following the last dose.
8.3 Females and Males of Reproductive Potential
ZALTRAP can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status in females of reproductive potential prior to initiating ZALTRAP [see Use in Specific Populations (8.1)].
Contraception
Females
Based on data from animal studies and its mechanism of action, ZALTRAP can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Advise female patients of reproductive potential to use effective contraception during treatment with ZALTRAP and for 3 months following the last dose.
Infertility
Advise female and male patients of reproductive potential that ZALTRAP may impair reproductive function and fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness in pediatric patients have not been established. Safety and efficacy were assessed, but not established in a dose-escalation, safety, and tolerability study (NCT00622414) in 21 patients with solid tumors 2 to 21 years of age (median age 12.9). The mean elimination half-life of free ziv-aflibercept determined after the first dose in 8 pediatric patients aged 5 to 17 years was within the range of values previously observed in adults. The maximum tolerated dose based on body weight in these pediatric patients was lower than the dose known to be safe and effective in adults with mCRC.
Juvenile Animal Toxicity Data
Weekly/every-two-weeks intravenous administration of ziv-aflibercept at dose of 3 mg per kg (approximately 0.6 times the human exposure at the 4 mg per kg dose based on AUC) to growing young adult (sexually mature) cynomolgus monkeys for up to 6 months resulted in changes in the bone (effects on growth plate and the axial and appendicular skeleton), nasal cavity (atrophy/loss of the septum and/or turbinates), kidney (glomerulopathy with inflammation), ovary (decreased number of maturing follicles, granulosa cells, and/or theca cells), and adrenal gland (decreased vacuolation with inflammation). In another study in sexually immature cynomolgus monkeys (treated intravenously for 3 months), there were similar effects. The skeletal and nasal cavity effects were not reversible after a post-dosing recovery period.
8.5 Geriatric Use
Of the 611 patients with mCRC, patients treated with ZALTRAP/FOLFIRI, 205 (34%) were 65 years or older, and 33 (5%) were 75 years or older. Elderly patients (≥65 years of age) experienced higher incidences (≥5%) of diarrhea, dizziness, asthenia, weight decrease, and dehydration when compared to younger patients. Monitor elderly patients more closely for diarrhea and dehydration [see Warnings and Precautions (5.9)].
The effect of ZALTRAP on overall survival was similar in patients <65 years old and ≥65 years old who received ZALTRAP/FOLFIRI.
8.6 Renal Impairment
No dosage modification is recommended for patients with renal impairment [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage modification is recommended for patients with mild (total bilirubin >1 to 1.5 times upper limit normal [ULN] and any aspartate transaminase [AST]) and moderate (total bilirubin >1.5 to 3 times ULN and any AST) hepatic impairment [see Clinical Pharmacology (12.3)]. ZALTRAP has not been studied in patients with severe hepatic impairment (total bilirubin >3 times ULN and any AST).
11. Zaltrap Description
Ziv-aflibercept is a vascular endothelial growth factor inhibitor. It is a recombinant fusion protein consisting of Vascular Endothelial Growth Factor (VEGF)-binding portions from the extracellular domains of human VEGF Receptors 1 and 2 fused to the Fc portion of the human IgG1. Ziv-aflibercept is produced by recombinant DNA technology in a Chinese hamster ovary (CHO) K-1 mammalian expression system. Ziv-aflibercept is a dimeric glycoprotein with a protein molecular weight of 97 kilodaltons (kDa) and contains glycosylation, constituting an additional 15% of the total molecular mass, resulting in a total molecular weight of 115 kDa.
ZALTRAP (ziv-aflibercept) injection is a sterile, clear, colorless to pale-yellow, non-pyrogenic, preservative-free, solution for intravenous use. ZALTRAP is supplied in single-dose vials of 100 mg/4 mL and 200 mg/8 mL formulated as 25 mg/mL ziv-aflibercept in polysorbate 20 (1 mg/mL), sodium chloride (5.84 mg/mL), sodium citrate (1.45 mg/mL), sodium phosphate (0.8 mg/mL), and sucrose (200 mg/mL), in Water for Injection, USP, at a pH of 6.2.
12. Zaltrap - Clinical Pharmacology
12.1 Mechanism of Action
Ziv-aflibercept acts as a soluble receptor that binds to human VEGF-A (equilibrium dissociation constant KD of 0.5 pM for VEGF-A165 and 0.36 pM for VEGF-A121), to human VEGF-B (KD of 1.92 pM), and to human PlGF (KD of 39 pM for PlGF-2). By binding to these endogenous ligands, ziv-aflibercept can inhibit the binding and activation of their cognate receptors. This inhibition can result in decreased neovascularization and decreased vascular permeability.
In animals, ziv-aflibercept was shown to inhibit the proliferation of endothelial cells, thereby inhibiting the growth of new blood vessels. Ziv-aflibercept inhibited the growth of xenotransplanted colon tumors in mice.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of 6 mg per kg intravenous ZALTRAP every three weeks on QTc interval was evaluated in 87 patients with solid tumors in a randomized, placebo-controlled study. No large changes in the mean QT interval from baseline (i.e., greater than 20 ms as corrected for placebo) based on Fridericia correction method were detected in the study. However, a small increase in the mean QTc interval (i.e., less than 10 ms) cannot be excluded due to limitations of the study design.
12.3 Pharmacokinetics
Plasma concentrations of free and VEGF-bound ziv-aflibercept were measured using specific enzyme-linked immunosorbent assays (ELISA). Free ziv-aflibercept concentrations appear to exhibit linear pharmacokinetics in the dose range of 2 mg per kg to 9 mg per kg. Steady state concentrations of free ziv-aflibercept were reached by the second dose. The accumulation ratio for free ziv-aflibercept was approximately 1.2 after administration of 4 mg per kg every two weeks.
Elimination
Following a dose of 4 mg per kg every two weeks administered intravenously, the elimination half-life of free ziv-aflibercept was approximately 6 days (range 4–7 days).
Specific Populations
Based on a population pharmacokinetic analysis, age, race, and sex did not have a clinically important effect on the exposure of free ziv-aflibercept. Patients weighing ≥100 kg had a 29% increase in systemic exposure compared to patients weighing 50 to 100 kg.
Patients with hepatic impairment
Based on a population pharmacokinetic analysis which included patients with mild (total bilirubin >1 to 1.5 times ULN and any AST, n=63) and moderate (total bilirubin >1.5 to 3 times ULN and any AST, n=5) hepatic impairment, there was no effect of total bilirubin, AST, and alanine aminotransferase on the clearance of free ziv-aflibercept. There are no data available for patients with severe hepatic impairment (total bilirubin >3 times ULN and any AST).
Patients with renal impairment
Based on a population pharmacokinetic analysis which included patients with mild (CLCR 50–80 mL/min, n=549), moderate (CLCR 30–50 mL/min, n=96), and severe renal impairment (CLCR <30 mL/min, n=5), there was no clinically important effect of creatinine clearance on the clearance of free ziv-aflibercept.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No studies have been conducted to evaluate carcinogenicity or mutagenicity of ziv-aflibercept.
Ziv-aflibercept impaired reproductive function and fertility in monkeys. In a 6-month repeat-dose toxicology study in sexually mature monkeys, ziv-aflibercept inhibited ovarian function and follicular development, as evidenced by: decreased ovary weight, decreased amount of luteal tissue, decreased number of maturing follicles, atrophy of uterine endometrium and myometrium, vaginal atrophy, abrogation of progesterone peaks and menstrual bleeding. Alterations in sperm morphology and decreased sperm motility were present in male monkeys. These effects were observed at all doses tested including the lowest dose tested, 3 mg per kg. Reversibility was observed within 18 weeks after cessation of treatment. Systemic exposure (AUC) with a 3 mg per kg per dose in monkeys was approximately 0.6 times the AUC in patients at the 4 mg per kg dose.
13.2 Animal Toxicology and/or Pharmacology
Repeated administration of ziv-aflibercept resulted in a delay in wound healing in rabbits. In full-thickness excisional and incisional skin wound models, ziv-aflibercept administration reduced fibrous response, neovascularization, epidermal hyperplasia/re-epithelialization, and tensile strength.
14. Clinical Studies
The efficacy of ZALTRAP was evaluated in VELOUR (NCT00561470), a randomized (1:1), double-blind, placebo-controlled study in patients with mCRC who are resistant to or have progressed during or within 6 months of receiving oxaliplatin-based combination chemotherapy, with or without prior bevacizumab. Patients were randomized to receive either ZALTRAP 4 mg per kg intravenously over 1 hour on day 1 or placebo in combination with FOLFIRI (irinotecan 180 mg/m2 intravenously over 90 minutes and leucovorin [dl racemic] 400 mg/m2 intravenously over 2 hours at the same time on day 1 using a Y-line, followed by fluorouracil 400 mg/m2 as an intravenous bolus and then by fluorouracil 2400 mg/m2 as a continuous intravenous infusion over 46 hours). The treatment cycles on both arms were repeated every 2 weeks. Patients were treated until disease progression or unacceptable toxicity. Randomization was stratified by the Eastern Cooperative Oncology Group performance status (PS) (0 versus 1 versus 2) and according to prior therapy with bevacizumab (yes or no). The major efficacy outcome measure was overall survival (OS). Additional efficacy outcome measures were progression-free survival (PFS) and overall response rate (ORR).
Demographics characteristics were similar between treatment arms. A total of 1226 patients were randomized, 612 to the ZALTRAP arm and 614 to the placebo arm. The median age was 61 years, 59% were men, 87% were White, 7% were Asian, 3.5% were Black, and 98% had a baseline ECOG PS of 0 or 1. Among the 1226 randomized patients, 89% and 90% of patients treated with placebo/FOLFIRI and ZALTRAP/FOLFIRI, respectively, received prior oxaliplatin-based combination chemotherapy in the metastatic/advanced setting. A total of 346 patients (28%) received bevacizumab in combination with the prior oxaliplatin-based treatment.
Efficacy results are summarized in Figure 1 and Table 2.
Figure 1: Kaplan-Meier Curves of Overall Survival for VELOUR
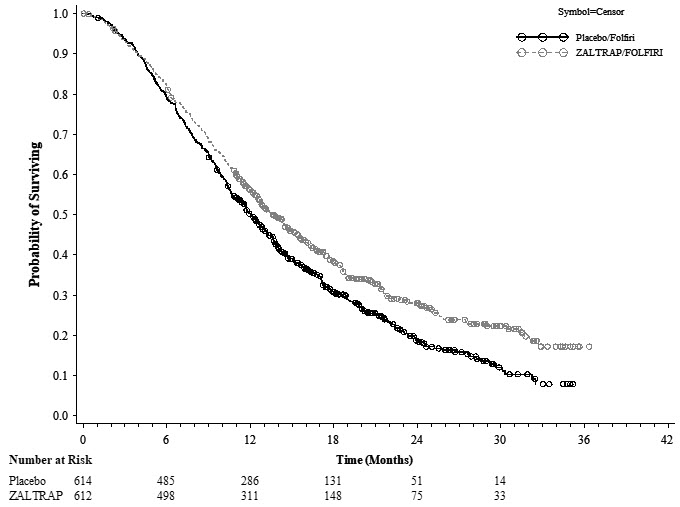
| ZALTRAP/FOLFIRI N=612 | Placebo/FOLFIRI N=614 |
|
|---|---|---|
| Overall Survival | ||
| Number of deaths, n (%) | 403 (65.8%) | 460 (74.9%) |
| Median overall survival (95% CI) (months) | 13.50 (12.52, 14.95) | 12.06 (11.07, 13.08) |
| Stratified Hazard ratio (95% CI) | 0.817 (0.714, 0.935) | |
| Stratified Log-Rank test p-value | 0.0032 | |
| Progression Free Survival (PFS)* | ||
| Number of events, n (%) | 393 (64.2%) | 454 (73.9%) |
| Median PFS (95% CI) (months) | 6.90 (6.51, 7.20) | 4.67 (4.21, 5.36) |
| Stratified Hazard ratio (95% CI) | 0.758 (0.661, 0.869) | |
| Stratified Log-Rank test p-value† | 0.00007 | |
| Overall Response Rate (ORR) | ||
| ORR (CR+PR) (95% CI)‡ | 19.8% (16.4%, 23.2%) | 11.1% (8.5%, 13.8%) |
| Stratified Cochran-Mantel-Haenszel test p-value | 0.0001 | |
Planned subgroup analyses for overall survival based on stratification factors at randomization yielded an HR of 0.86 (95% CI: 0.68, 1.1) in patients who received prior bevacizumab and an HR of 0.79 (95% CI: 0.67, 0.93) in patients without prior bevacizumab exposure.
16. How is Zaltrap supplied
ZALTRAP (ziv-aflibercept) injection is a clear, colorless to pale-yellow solution supplied in single-dose vials with a concentration of 25 mg/mL.
NDC 0024-5840-01: carton containing one single-dose vial of 100 mg/4 mL (25 mg/mL)
NDC 0024-5841-01: carton containing one single-dose vial of 200 mg/8 mL (25 mg/mL)
17. Patient Counseling Information
Hemorrhage
- Inform patients that ZALTRAP can cause severe bleeding and advise patients to contact their healthcare provider for bleeding or symptoms of bleeding, including lightheadedness [see Warnings and Precautions (5.1)].
Gastrointestinal Perforation and Fistula Formation
- Advise patients to immediately contact their healthcare provider for signs and symptoms of gastrointestinal perforation or fistula [see Warnings and Precautions (5.2, 5.4)].
Impaired Wound Healing
- Advise patients that ZALTRAP may impair wound healing. Instruct patients to discuss any planned surgical procedure (including tooth extractions) with all their healthcare providers [see Warnings and Precautions (5.3)].
Hypertension
- Inform patients that ZALTRAP can cause or exacerbate existing hypertension. Advise patients to undergo routine blood pressure monitoring and to contact their healthcare provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms [see Warnings and Precautions (5.5)].
Arterial Thromboembolic Events
- Inform patients of an increased risk of ATE [see Warnings and Precautions (5.6)].
Proteinuria
- Advise patients that they will need to undergo regular laboratory tests to monitor protein in their urine [see Warnings and Precautions (5.7)].
Neutropenia and Neutropenic Complications
- Advise patients to notify their healthcare provider of fever or other signs of infection [see Warnings and Precautions (5.8)].
Diarrhea and Dehydration
- Advise patients to notify their healthcare provider of severe diarrhea, vomiting, or severe abdominal pain [see Warnings and Precautions (5.9)].
Posterior Reversible Leukoencephalopathy Syndrome
- Advise patients to immediately contact their healthcare provider for new onset or worsening neurological function [see Warnings and Precautions (5.10)].
Embryo-Fetal Toxicity
Advise females of reproductive potential:
- of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.11), Use in Specific Populations (8.1)].
- to use effective contraception during treatment with ZALTRAP and for 3 months following the last dose [see Use in Specific Populations (8.3)].
Lactation
- Advise women not to breastfeed during treatment with ZALTRAP and for 1 month following the last dose [see Use in Specific Populations (8.2)].
Aneurysms and Artery Dissections
- Advise patients to notify their healthcare provider if they have or have had an aneurysm (swelling/enlargement and weakening of part of a blood vessel) or artery dissection (tear in a blood vessel wall) [see Adverse Reactions (6.3)].
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License No. 1752
ZALTRAP is a registered trademark of Regeneron Pharmaceuticals, Inc.
©2025 sanofi-aventis U.S. LLC
PRINCIPAL DISPLAY PANEL - 100 mg/4 mL Vial Carton
NDC 0024-5840-01
ZALTRAP®
(ziv-aflibercept)
Injection for
Intravenous Infusion
100 mg/4 mL
(25 mg/mL)
For intravenous infusion only.
Not to be administered by
other routes.
Hyperosmotic, must be diluted.
Single-dose vial.
Discard unused portion
Rx ONLY
SANOFI

PRINCIPAL DISPLAY PANEL - 200 mg/8 mL Vial Carton
NDC 0024-5841-01
ZALTRAP®
(ziv-aflibercept)
Injection for
Intravenous Infusion
200 mg/8 mL
(25 mg/mL)
For intravenous infusion only.
Not to be administered by
other routes.
Hyperosmotic, must be diluted.
Single-dose vial.
Discard unused portion
Rx ONLY
SANOFI

| ZALTRAP
ziv-aflibercept solution, concentrate |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| ZALTRAP
ziv-aflibercept solution, concentrate |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
||||||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Regeneron Pharmaceuticals, Inc. | 945589711 | ANALYSIS(0024-5840, 0024-5841) , API MANUFACTURE(0024-5840, 0024-5841) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi-Aventis Deutschland GmbH | 313218430 | ANALYSIS(0024-5840, 0024-5841) , LABEL(0024-5840, 0024-5841) , MANUFACTURE(0024-5840, 0024-5841) , PACK(0024-5840, 0024-5841) | |
Biological Products Related to Zaltrap
Find detailed information on biosimilars for this medication.
More about Zaltrap (ziv-aflibercept)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: VEGF/VEGFR inhibitors
- Breastfeeding
- En español