Thalomid: Package Insert / Prescribing Info
Package insert / product label
Generic name: thalidomide
Dosage form: capsule
Drug classes: Leprostatics, Miscellaneous antineoplastics, Other immunosuppressants
Medically reviewed by Drugs.com. Last updated on Mar 31, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Drug Abuse and Dependence
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
- Medication Guide
Highlights of Prescribing Information
THALOMID (thalidomide) capsules, for oral use
Initial U.S. Approval: 1998
WARNING: EMBRYO-FETAL TOXICITY AND VENOUS THROMBOEMBOLISM
See full prescribing information for complete boxed warning.
EMBRYO -FETAL TOXICITY
- •
- If THALOMID is taken during pregnancy, it can cause severe birth defects or embryo-fetal death. THALOMID should never be used by females who are pregnant or who could be pregnant while taking the drug. Even a single dose [1 capsule (regardless of strength)] taken by a pregnant woman during her pregnancy can cause severe birth defects.
- •
- Pregnancy must be excluded before start of treatment. Prevent pregnancy thereafter by the use of two reliable methods of contraception. (5.1, 8.3)
THALOMID is only available through a restricted distribution program, the THALOMID REMS® program (5.2).
VENOUS THROMBOEMBOLISM
- •
- Significant increased risk of deep vein thrombosis (DVT) and pulmonary embolism (PE) in patients with multiple myeloma receiving THALOMID with dexamethasone (5.3).
Indications and Usage for Thalomid
- •
- THALOMID in combination with dexamethasone is indicated for the treatment of patients with newly diagnosed multiple myeloma (MM). (1.1)
- •
- THALOMID is indicated for the acute treatment of the cutaneous manifestations of moderate to severe erythema nodosum leprosum (ENL).
THALOMID is not indicated as monotherapy for such ENL treatment in the presence of moderate to severe neuritis.
THALOMID is also indicated as maintenance therapy for prevention and suppression of the cutaneous manifestations of ENL recurrence. (1.2)
Thalomid Dosage and Administration
Dosage Forms and Strengths
Capsules: 50 mg, 100 mg, 150 mg and 200 mg. (3)
Contraindications
Warnings and Precautions
- •
- Ischemic heart disease (including myocardial infarction) and stroke have been observed in patients treated with THALOMID in combination with dexamethasone. (5.3)
- •
- Increased Mortality: Observed in patients with MM when pembrolizumab was added to dexamethasone and a thalidomide analogue. (5.4)
- •
- Drowsiness and Somnolence: Instruct patients to avoid situations where drowsiness may be a problem and not to take other medications that may cause drowsiness. (5.5)
- •
- Peripheral Neuropathy: Monitor patients for signs or symptoms of peripheral neuropathy during treatment. Discontinue THALOMID if symptoms of drug-induced peripheral neuropathy occur, if clinically appropriate. (5.6)
- •
- Dizziness and Orthostatic Hypotension: Advise patients to sit upright for a few minutes prior to standing up from a recumbent position. (5.7)
- •
- Neutropenia and Thrombocytopenia: Patients may require dose interruption and/or dose reduction. (5.8, 5.9)
- •
- Increased HIV Viral Load: Measure viral load during treatment. (5.10)
- •
- Bradycardia: Monitor patients for bradycardia and possible syncope. Dose reduction or discontinuation may be required. (5.11)
- •
- Severe Cutaneous Reactions: Discontinue THALOMID for severe reactions. (5.12)
- •
- Seizures: Monitor patients with a history of seizures or other risk factors for acute seizure activity. (5.13)
- •
- Tumor Lysis Syndrome: Monitor patients at risk (e.g., those with high tumor burden prior to treatment) and take appropriate precautions. (5.14)
- •
- Hypersensitivity: Monitor patients for potential hypersensitivity. Discontinue THALOMID for angioedema and anaphylaxis. (5.16)
Adverse Reactions/Side Effects
- •
- MM: The most common adverse reactions (≥ 20%) are fatigue, hypocalcemia, edema, constipation, neuropathy-sensory, dyspnea, muscle weakness, leukopenia, neutropenia, rash/desquamation, confusion, anorexia, nausea, anxiety/agitation, asthenia, tremor, fever, weight loss, thrombosis/embolism, neuropathy-motor, weight gain, dizziness, and dry skin. (6.1)
- •
- ENL: The most common adverse reactions (≥ 10%) are somnolence, rash, and headache. (6.1)
To report SUSPECTED ADVERSE REACTIONS or embryo-fetal exposure: contact Bristol Myers Squibb at 1-800-721-5072 or 1-888-423-5436, respectively, or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- •
- Use caution if other drugs which have sedative and hypnotic properties, slow cardiac conduction and/or cause peripheral neuropathy must be used. (7.1, 7.2, 7.3)
- •
- It is not known whether concomitant use of hormonal contraceptives further increases the risk of thromboembolism with THALOMID. (5.15, 7.4)
- •
- Patients taking concomitant therapies such as erythropoietin stimulating agents or estrogen containing therapies may have an increased risk of thromboembolism. (7.7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2023
Full Prescribing Information
WARNING: EMBRYO-FETAL TOXICITY AND VENOUS THROMBOEMBOLISM
EMBRYO-FETAL TOXICITY
If THALOMID is taken during pregnancy, it can cause severe birth defects or embryo-fetal death. THALOMID should never be used by females who are pregnant or who could become pregnant while taking the drug. Even a single dose [1 capsule (regardless of strength)] taken by a pregnant woman during her pregnancy can cause severe birth defects.
Because of this toxicity and in an effort to make the chance of embryo-fetal exposure to THALOMID as negligible as possible, THALOMID is approved for marketing only through a special restricted distribution program: THALOMID REMS program, approved by the Food and Drug Administration.
Information about THALOMID and the THALOMID REMS program is available at www.thalomidrems.com or by calling the REMS Call Center at 1-888-423-5436.
VENOUS THROMBOEMBOLISM
The use of THALOMID in multiple myeloma results in an increased risk of venous thromboembolism, such as deep venous thrombosis and pulmonary embolism. This risk increases significantly when THALOMID is used in combination with standard chemotherapeutic agents including dexamethasone. In one controlled trial, the rate of venous thromboembolism was 22.5% in patients receiving THALOMID in combination with dexamethasone compared to 4.9% in patients receiving dexamethasone alone (p = 0.002). Patients and physicians are advised to be observant for the signs and symptoms of thromboembolism. Instruct patients to seek medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling. Consider thromboprophylaxis based on an assessment of individual patients' underlying risk factors.
1. Indications and Usage for Thalomid
1.1 Multiple Myeloma
THALOMID in combination with dexamethasone is indicated for the treatment of patients with newly diagnosed multiple myeloma (MM) [see Clinical Studies (14.1)].
1.2 Erythema Nodosum Leprosum
THALOMID is indicated for the acute treatment of the cutaneous manifestations of moderate to severe erythema nodosum leprosum (ENL).
THALOMID is not indicated as monotherapy for such ENL treatment in the presence of moderate to severe neuritis.
THALOMID is also indicated as maintenance therapy for prevention and suppression of the cutaneous manifestations of ENL recurrence [see Clinical Studies (14.2)].
2. Thalomid Dosage and Administration
2.1 Required Baseline Testing
Drug prescribing to females of reproductive potential is contingent upon initial and continued negative results of pregnancy testing [see Warnings and Precautions (5.1 and 5.2)].
THALOMID must only be administered in compliance with all of the terms outlined in the THALOMID REMS program. THALOMID may only be prescribed by prescribers certified with the THALOMID REMS program and may only be dispensed by pharmacists certified with the THALOMID REMS program.
2.2 Recommended Dosage for Multiple Myeloma
The recommended dose of THALOMID in combination with dexamethasone is 200 mg once daily (in 28-day treatment cycles) orally with water, preferably at bedtime and at least 1 hour after the evening meal. The dose of dexamethasone is 40 mg daily administered orally on days 1-4, 9-12, and 17-20 every 28 days.
2.3 Recommended Dosage for Erythema Nodosum Leprosum
The recommended dose of THALOMID for an episode of cutaneous ENL is 100 to 300 mg/day once daily orally with water, preferably at bedtime and at least 1 hour after the evening meal. Initiate dosing for patients weighing less than 50 kilograms at the low end of the dose range.
Consider dosing in the higher dosage range for patients with a severe cutaneous ENL reaction, or in those who have previously required higher doses to control the reaction (possibly up to 400 mg/day) once daily at bedtime or in divided doses with water, at least 1 hour after meals.
Consider concomitant use of corticosteroids in patients with moderate to severe neuritis associated with a severe ENL reaction. Steroid usage can be tapered and discontinued when the neuritis has ameliorated.
Continue dosing with THALOMID until signs and symptoms of active reaction have subsided, usually a period of at least 2 weeks. Patients may then be tapered off medication in 50 mg decrements every 2 to 4 weeks.
Patients who have a documented history of requiring prolonged maintenance treatment to prevent the recurrence of cutaneous ENL or who flare during tapering should be maintained on the minimum dose necessary to control the reaction. Tapering off medication should be attempted every 3 to 6 months, in decrements of 50 mg every 2 to 4 weeks.
2.4 Dosage Modifications for Adverse Reactions
Interrupt THALOMID for constipation, somnolence, or peripheral neuropathy. Consider a reduced dose upon resumption of treatment.
Consider dose reduction, delay, or discontinuation in patients who develop National Cancer Institute Common Toxicity Criteria (NCI CTC) Grade 3 or 4 adverse reactions and/or based on clinical judgment.
Permanently discontinue THALOMID for angioedema, anaphylaxis, Grade 4 rash, skin exfoliation, bullae, or any other severe dermatologic reactions [see Warnings and Precautions (5.12 and 5.16)].
3. Dosage Forms and Strengths
Capsules:
- •
- 50 mg white, printed with "BMS/50 mg" on the body and a "Do Not Get Pregnant" logo on the cap.
- •
- 100 mg tan, printed with "BMS/100 mg" on the body and a "Do Not Get Pregnant" logo on the cap.
- •
- 150 mg tan body printed with "BMS/150 mg" and blue cap printed with a "Do Not Get Pregnant" logo.
- •
- 200 mg blue, printed with "BMS/200 mg" on the body and a "Do not Get Pregnant" logo on the cap.
4. Contraindications
4.1 Pregnancy
THALOMID is contraindicated in females who are pregnant. THALOMID can cause fetal harm when administered to a pregnant female [see Boxed Warning, Warnings and Precautions (5.1) and Use in Specific Populations (8.1)]. THALOMID is a powerful human teratogen, inducing a high frequency of severe and life-threatening birth defects, even after a single dose [see Boxed Warning]. Mortality at or shortly after birth has been reported in about 40% of infants. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential risk to a fetus. If pregnancy occurs during THALOMID treatment, the drug should be discontinued immediately.
4.2 Hypersensitivity
THALOMID is contraindicated in patients who have demonstrated hypersensitivity to the drug or its components [see Warnings and Precautions (5.16)].
5. Warnings and Precautions
5.1 Embryo-Fetal Toxicity
THALOMID is a powerful human teratogen that induces a high frequency of severe and life-threatening birth defects, even after a single dose. Mortality at or shortly after birth has been reported in about 40% of infants. When there is no satisfactory alternative treatment, females of reproductive potential may be treated with THALOMID provided adequate precautions are taken to avoid pregnancy. THALOMID is only available through the THALOMID REMS program [see Warnings and Precautions (5.2)].
Oral ingestion is the only type of maternal THALOMID exposure known to result in drug-associated birth defects. There are no specific data available regarding the reproductive risks of cutaneous absorption or inhalation of THALOMID; however, females of reproductive potential should avoid contact with THALOMID Capsules. THALOMID Capsules should be stored in blister packs until ingestion. If there is contact with non-intact THALOMID capsules or the powder contents, the exposed area should be washed with soap and water.
If healthcare providers or other care givers are exposed to body fluids from patients receiving THALOMID, the exposed area should be washed with soap and water. Appropriate precautions should be utilized, such as wearing gloves to prevent the potential cutaneous exposure to THALOMID.
Females of Reproductive Potential
Females of reproductive potential must avoid pregnancy for at least 4 weeks before beginning THALOMID therapy, during therapy, during dose interruptions and for at least 4 weeks after completing therapy.
Females must commit either to abstain continuously from heterosexual sexual intercourse or to use two methods of reliable birth control, beginning 4 weeks prior to initiating treatment with THALOMID, during therapy, during dose interruptions and continuing for 4 weeks following discontinuation of THALOMID therapy.
Two negative pregnancy tests must be obtained prior to initiating therapy. The first test should be performed within 10-14 days and the second test within 24 hours prior to prescribing THALOMID therapy and then weekly during the first month, then monthly thereafter in females with regular menstrual cycles or every 2 weeks in females with irregular menstrual cycles [see Use in Specific Populations (8.3)].
Males
Thalidomide is present in the semen of patients receiving THALOMID. Therefore, males must always use a latex or synthetic condom during any sexual contact with females of reproductive potential while taking THALOMID and for up to 4 weeks after discontinuing THALOMID, even if they have undergone a successful vasectomy. Male patients taking THALOMID must not donate sperm [see Use in Specific Populations (8.3)].
5.2 THALOMID REMS Program
Because of the embryo-fetal risk [see Warnings and Precautions (5.1)], THALOMID is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS), the THALOMID REMS program.
Required components of the THALOMID REMS program include the following:
- •
- Prescribers must be certified with the THALOMID REMS program by enrolling and complying with the REMS requirements.
- •
- Patients must sign a Patient-Physician Agreement Form and comply with the REMS requirements. In particular, female patients of reproductive potential who are not pregnant must comply with the pregnancy testing and contraception requirements [see Use in Specific Populations (8.3)] and males must comply with contraception requirements [see Use in Specific Populations (8.3)].
- •
- Pharmacies must be certified with the THALOMID REMS program, must only dispense to patients who are authorized to receive THALOMID and comply with REMS requirements.
Further information about the THALOMID REMS program is available at www.thalomidrems.com or by telephone at 1-888-423-5436.
5.3 Venous and Arterial Thromboembolism
The use of THALOMID in patients with MM results in an increased risk of venous thromboembolism, such as deep venous thrombosis and pulmonary embolism. This risk increases significantly when THALOMID is used in combination with standard chemotherapeutic agents including dexamethasone. In one controlled trial, the rate of venous thromboembolism was 22.5% in patients receiving THALOMID in combination with dexamethasone compared to 4.9% in patients receiving dexamethasone alone (p = 0.002).
Ischemic heart disease (11.1%), including myocardial infarction (1.3%), and stroke (cerebrovascular accident, 2.6%) have also occurred in patients with previously untreated MM treated with THALOMID and dexamethasone compared to placebo and dexamethasone (4.7%, 1.7%, and 0.9%, respectively) in one clinical trial [see Adverse Reactions (6.1)].
Consider thromboprophylaxis based on an assessment of individual patients' underlying risk factors. Patients and physicians should be observant for the signs and symptoms of thromboembolism. Advise patients to seek immediate medical care if they develop symptoms such as shortness of breath, chest pain, or arm or leg swelling [see Boxed Warning]. Agents that also may increase the risk of thromboembolism should be used with caution in patients receiving THALOMID [see Drug Interactions (7.7)].
5.4 Increased Mortality in Patients with MM When Pembrolizumab Is Added to a Thalidomide Analogue and Dexamethasone
In two randomized clinical trials in patients with MM, the addition of pembrolizumab to a thalidomide analogue plus dexamethasone, a use for which no PD-1 or PD-L1 blocking antibody is indicated, resulted in increased mortality. Treatment of patients with MM with a PD-1 or PD-L1 blocking antibody in combination with a thalidomide analogue plus dexamethasone is not recommended outside of controlled clinical trials.
5.5 Drowsiness and Somnolence
THALOMID frequently causes drowsiness and somnolence. Patients should be instructed to avoid situations where drowsiness may be a problem and not to take other medications that may cause drowsiness without adequate medical advice [see Drug Interactions (7.1)]. Advise patients as to the possible impairment of mental and/or physical abilities required for the performance of hazardous tasks, such as driving a car or operating other complex or dangerous machinery. Dose reductions may be required.
5.6 Peripheral Neuropathy
THALOMID is known to cause nerve damage that may be permanent. Peripheral neuropathy is a common (≥10%) and potentially severe adverse reaction of treatment with THALOMID that may be irreversible. Peripheral neuropathy generally occurs following chronic use over a period of months; however, peripheral neuropathy following relatively short-term use has been reported. The correlation with cumulative dose is unclear. Symptoms may occur some time after THALOMID treatment has been stopped and may resolve slowly or not at all.
Few reports of neuropathy have arisen in the treatment of ENL despite long-term THALOMID treatment. However, the inability clinically to differentiate THALOMID neuropathy from the neuropathy often seen in ENL makes it difficult to determine accurately the incidence of THALOMID-related neuropathy in patients with ENL treated with THALOMID.
Patients should be examined at monthly intervals for the first 3 months of THALOMID therapy to enable the clinician to detect early signs of neuropathy, which include numbness, tingling or pain in the hands and feet. Patients should be evaluated periodically thereafter during treatment. Patients should be regularly counseled, questioned, and evaluated for signs or symptoms of peripheral neuropathy. Consideration should be given to electrophysiological testing, consisting of measurement of sensory nerve action potential (SNAP) amplitudes at baseline and thereafter every 6 months in an effort to detect asymptomatic neuropathy. If symptoms of drug-induced neuropathy develop, THALOMID should be discontinued immediately to limit further damage, if clinically appropriate. Usually, treatment with THALOMID should only be reinitiated if the neuropathy returns to baseline status.
Medications known to be associated with neuropathy should be used with caution in patients receiving THALOMID [see Drug Interactions (7.3)].
5.7 Dizziness and Orthostatic Hypotension
Patients should also be advised that THALOMID may cause dizziness and orthostatic hypotension and that, therefore, they should sit upright for a few minutes prior to standing up from a recumbent position.
5.8 Neutropenia
Decreased white blood cell counts, including neutropenia, have been reported in association with the clinical use of THALOMID. Treatment should not be initiated with an absolute neutrophil count (ANC) of <750/mm3. White blood cell count and differential should be monitored on an ongoing basis, especially in patients who may be more prone to neutropenia, such as patients who are HIV-seropositive. If ANC decreases to below 750/mm3 while on treatment, the patient's medication regimen should be re-evaluated and, if the neutropenia persists, consideration should be given to withholding THALOMID if clinically appropriate.
5.9 Thrombocytopenia
Thrombocytopenia, including Grade 3 or 4 occurrences, has been reported in association with the clinical use of THALOMID. Monitor blood counts, including platelet counts. Dose reduction, delay, or discontinuation may be required. Monitor for signs and symptoms of bleeding including petechiae, epistaxis, and gastrointestinal bleeding, especially if concomitant medication may increase the risk of bleeding.
5.10 Increased HIV Viral Load
In a randomized, placebo-controlled trial of thalidomide in an HIV-seropositive patient population, plasma HIV RNA levels were found to increase (median change = 0.42 log10 copies HIV RNA/mL, p = 0.04 compared to placebo). A similar trend was observed in a second, unpublished study conducted in patients who were HIV-seropositive. The clinical significance of this increase is unknown. Both studies were conducted prior to availability of highly active antiretroviral therapy. Until the clinical significance of this finding is further understood, in HIV-seropositive patients, viral load should be measured after the first and third months of treatment and every 3 months thereafter.
5.11 Bradycardia
Bradycardia in association with THALOMID use has been reported. Cases of bradycardia have been reported, some required medical interventions. The clinical significance and underlying etiology of the bradycardia noted in some THALOMID-treated patients are presently unknown. Monitor patients for bradycardia and syncope. Dose reduction or discontinuation may be required.
Medications known to decrease heart rate should be used with caution in patients receiving THALOMID [see Drug Interactions (7.2)].
5.12 Severe Cutaneous Reactions
Severe cutaneous reactions including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported with THALOMID use. DRESS may present with a cutaneous reaction (such as rash or exfoliative dermatitis), eosinophilia, fever, and/or lymphadenopathy with systemic complications such as hepatitis, nephritis, pneumonitis, myocarditis, and/or pericarditis. These events can be fatal. THALOMID interruption or discontinuation should be considered for Grade 2-3 skin rash. Discontinue THALOMID for Grade 4 rash, exfoliative or bullous rash, or for other severe cutaneous reactions such as SJS, TEN, or DRESS, and do not resume therapy [see Dosage and Administration (2.4)].
5.13 Seizures
Although not reported from pre-marketing controlled clinical trials, seizures, including grand mal convulsions, have been reported during post-approval use of THALOMID in clinical practice. Because these events are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. Most patients had disorders that may have predisposed them to seizure activity, and it is not currently known whether THALOMID has any epileptogenic influence. During therapy with THALOMID, patients with a history of seizures or with other risk factors for the development of seizures should be monitored closely for clinical changes that could precipitate acute seizure activity.
5.14 Tumor Lysis Syndrome
Monitor patients at risk of tumor lysis syndrome (e.g., patients with high tumor burden prior to treatment) and take appropriate precautions.
5.15 Contraceptive Risks
Some contraceptive methods may pose a higher risk of adverse effects or may be medically contraindicated in some patients treated with THALOMID. Because some patients may develop sudden, severe neutropenia and/or thrombocytopenia, use of an intrauterine device (IUD) or implantable contraception in these patients may carry an increased risk for infection or bleeding either at insertion, removal or during use. Treatment with THALOMID, the presence of an underlying malignancy, and/or use of an estrogen-containing contraceptive can each increase the risk of thromboembolism. It is not known if these risks of thromboembolism are additive. However, they should be taken into consideration when choosing contraceptive methods.
5.16 Hypersensitivity
Hypersensitivity, including angioedema and anaphylactic reactions to THALOMID has been reported. Signs and symptoms have included the occurrence of erythematous macular rash, possibly associated with fever, tachycardia, and hypotension, and if severe, may necessitate interruption of therapy. If the reaction recurs when dosing is resumed, THALOMID should be discontinued. Do not resume THALOMID treatment after angioedema and anaphylaxis [see Dosage and Administration (2.4)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described in detail in other labeling sections:
- •
- Teratogenicity [see Boxed Warning, Warnings and Precautions (5.1, 5.2), and Patient Counseling Information (17)]
- •
- Venous and Arterial Thromboembolism [see Boxed Warning, Warnings and Precautions (5.3), and Patient Counseling Information (17)]
- •
- Increased Mortality in Patients with MM When Pembrolizumab Is Added to a Thalidomide Analogue and Dexamethasone [see Warnings and Precautions (5.4)]
- •
- Drowsiness and Somnolence [see Warnings and Precautions (5.5)]
- •
- Peripheral Neuropathy [see Warnings and Precautions (5.6)]
- •
- Dizziness and Orthostatic Hypotension [see Warnings and Precautions (5.7)]
- •
- Neutropenia [see Warnings and Precautions (5.8)]
- •
- Thrombocytopenia [see Warnings and Precautions (5.9)]
- •
- Increased HIV Viral Load [see Warnings and Precautions (5.10)]
- •
- Bradycardia [see Warnings and Precautions (5.11)]
- •
- Severe Cutaneous Reactions [see Warnings and Precautions (5.12)]
- •
- Seizures [see Warnings and Precautions (5.13)]
- •
- Tumor Lysis Syndrome [see Warnings and Precautions (5.14)]
- •
- Hypersensitivity [see Warnings and Precautions (5.16)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Most patients taking THALOMID can be expected to experience adverse reactions.
Adverse Reactions in Multiple Myeloma Controlled Clinical Trials
The safety analyses were conducted in two controlled clinical studies (Study 1 and Study 2). The safety analysis in Study 1 was conducted on 204 patients who received treatment. Table 1 lists the most common adverse reactions (≥ 10%). The most frequently reported adverse reactions were fatigue, hypocalcemia, edema, constipation, sensory neuropathy, dyspnea, muscle weakness, leukopenia, neutropenia, rash/desquamation, confusion, anorexia, nausea, anxiety/agitation, tremor, fever, weight loss, thrombosis/embolism, neuropathy-motor, weight gain, dizziness, and dry skin.
Twenty-three percent of patients (47/204) discontinued due to adverse reactions; 30% (31/102) from the THALOMID/dexamethasone arm and 16% (16/102) from the dexamethasone alone arm.
| Body System
Adverse Reaction | Thal + Dex*
(N=102) | Dex Alone*
(N=102) |
||
|---|---|---|---|---|
| All Grades
n (%) | Grade 3/4
n (%) | All Grades
n (%) | Grade 3/4
n (%) |
|
| * Treatment-emergent adverse reactions reported in ≥10% of patients in THALOMID/dexamethasone arm and with a ≥1% difference in the THALOMID/dexamethasone arm compared to the dexamethasone alone arm. | ||||
|
Metabolic/Laboratory |
97 (95) |
33 (32) |
96 (94) |
30 (29) |
|
Hypocalcemia |
73 (72) |
11 (11) |
60 (59) |
5 (5) |
|
Neurology |
92 (90) |
30 (29) |
76 (74) |
18 (18) |
|
Neuropathy-sensory |
55 (54) |
4 (4) |
28 (28) |
1 (1) |
|
Confusion |
29 (28) |
9 (9) |
12 (12) |
3 (3) |
|
Anxiety/agitation |
26 (26) |
1 (1) |
14 (14) |
3 (3) |
|
Tremor |
26 (26) |
1 (1) |
6 (6) |
0 (0) |
|
Neuropathy-motor |
22 (22) |
8 (8) |
16 (16) |
5 (5) |
|
Dizziness/ lightheadedness |
20 (20) |
1 (1) |
14 (14) |
0 (0) |
|
Depressed level of consciousness |
16 (16) |
3 (3) |
3 (3) |
3 (3) |
|
Constitutional Symptoms |
91 (89) |
19 (19) |
84 (82) |
16 (16) |
|
Fatigue |
81 (79) |
17 (17) |
72 (71) |
13 (13) |
|
Fever |
24 (24) |
1 (1) |
20 (20) |
3 (3) |
|
Weight loss |
23 (23) |
1 (1) |
21 (21) |
2 (2) |
|
Weight gain |
22 (22) |
1 (1) |
13 (13) |
0 (0) |
|
Blood/Bone Marrow |
88 (86) |
29 (29) |
96 (94) |
19 (19) |
|
Leukocytes (decreased) |
36 (35) |
6 (6) |
30 (29) |
3 (3) |
|
Neutrophils (decreased) |
32 (31) |
10 (10) |
24 (24) |
10 (10) |
|
Gastrointestinal |
83 (81) |
22 (22) |
70 (69) |
8 (8) |
|
Constipation |
56 (55) |
8 (8) |
29 (28) |
1 (1) |
|
Anorexia |
29 (28) |
4 (4) |
25 (24) |
2 (2) |
|
Nausea |
29 (28) |
5 (5) |
23 (22) |
1 (1) |
|
Mouth dryness |
12 (12) |
1 (1) |
6 (6) |
0 (0) |
|
Cardiovascular |
70 (69) |
37 (36) |
60 (59) |
21 (21) |
|
Edema |
58 (56) |
6 (6) |
47 (46) |
4 (4) |
|
Thrombosis/embolism |
23 (22) |
21 (21) |
5 (5) |
5 (5) |
|
Pain |
64 (63) |
10 (10) |
66 (65) |
15 (15) |
|
Myalgia |
17 (17) |
0 (0) |
14 (14) |
1 (1) |
|
Arthralgia |
13 (13) |
0 (0) |
10 (10) |
2 (2) |
|
Pulmonary |
52 (51) |
19 (19) |
51 (50) |
20 (20) |
|
Dyspnea |
43 (42) |
13 (13) |
32 (31) |
15 (15) |
|
Dermatology/Skin |
48 (47) |
5 (5) |
35 (34) |
2 (2) |
|
Rash/desquamation |
31 (30) |
4 (4) |
18 (18) |
2 (2) |
|
Dry skin |
21 (21) |
0 (0) |
11 (11) |
0 (0) |
|
Hepatic |
47 (46) |
7 (7) |
45 (44) |
4 (4) |
|
Bilirubin |
14 (14) |
2 (2) |
10 (10) |
2 (2) |
|
Musculoskeletal |
42 (41) |
9 (9) |
41 (40) |
14 (14) |
|
Muscle weakness |
41 (40) |
6 (6) |
38 (37) |
13 (13) |
The safety analysis in Study 2 was conducted on 466 patients who received treatment. Table 2 lists the most common adverse reactions (≥10%) that were observed. Table 3 lists the most common Grade 3/4 adverse reactions (occurring at >2%) that were observed. The adverse reactions most often reported by patients treated with THALOMID/dexamethasone were constipation, peripheral edema, tremor, asthenia, dizziness and fatigue. Adverse reactions with a frequency at least 2-fold higher in the THALOMID/dexamethasone group than in the placebo/dexamethasone group include constipation, tremor, deep vein thrombosis and peripheral sensory neuropathy.
Twenty-six percent of patients (121/466) discontinued due to adverse reactions; 37% (86/234) from the THALOMID/dexamethasone arm and 15% (35/232) from the placebo/dexamethasone arm.
| Body System
Adverse Reaction | Thal/Dex (N=234)*
n (%) | Placebo/Dex (N=232)*
n (%) |
|---|---|---|
| *All adverse reactions reported in ≥10% of patients in THALOMID/dexamethasone arm and with a ≥1% difference in proportion of patients between the THALOMID/dexamethasone arm compared to the placebo/dexamethasone arm. NOS = not otherwise specified. |
||
|
Patients with at least 1 Adverse Reaction |
233 (99) |
230 (99) |
|
General Disorders and Administration Site Conditions |
176 (75) |
149 (64) |
|
Edema peripheral |
80 (34) |
57 (25) |
|
Asthenia |
56 (24) |
47 (20) |
|
Fatigue |
50 (21) |
36 (16) |
|
Edema NOS |
31 (13) |
19 (8) |
|
Gastrointestinal Disorders |
162 (69) |
149 (64) |
|
Constipation |
116 (50) |
49 (21) |
|
Nausea |
30 (13) |
27 (12) |
|
Dyspepsia |
27 (11) |
21 (9) |
|
Nervous System Disorders |
161 (69) |
138 (60) |
|
Tremor |
62 (26) |
29 (12) |
|
Dizziness |
51 (23) |
32 (14) |
|
Paresthesia |
27 (12) |
15 (6) |
|
Peripheral sensory neuropathy |
24 (10) |
12 (5) |
|
Infections and Infestations |
139 (59) |
138 (60) |
|
Pneumonia NOS |
35 (15) |
28 (12) |
|
Psychiatric Disorders |
90 (38) |
97 (42) |
|
Anxiety |
27 (12) |
22 (10) |
|
Depression |
24 (10) |
19 (8) |
|
Metabolism and Nutrition Disorders |
96 (41) |
89 (38) |
|
Hyperglycemia NOS |
36 (15) |
32 (14) |
|
Vascular Disorders |
92 (39) |
53 (23) |
|
Deep vein thrombosis |
30 (13) |
4 (2) |
| Body System
Adverse Reaction | THALOMID/Dex (N=234)*
n (%) | Placebo/Dex (N=232)*
n (%) |
|---|---|---|
| * All Grade 3/4 adverse reactions with >2% of patients in THALOMID/dexamethasone arm and with a higher frequency in the THALOMID/dexamethasone arm compared to the placebo/dexamethasone arm. NOS = not otherwise specified. |
||
|
Infections and Infestations |
50 (21) |
36 (16) |
|
Pneumonia NOS |
17 (7) |
14 (6) |
|
Bronchopneumonia NOS |
7 (3) |
3 (1) |
|
General Disorders and
|
44 (19) |
26 (11) |
|
Asthenia |
11 (5) |
4 (2) |
|
Metabolism and Nutrition
|
33 (14) |
34 (15) |
|
Hypokalemia |
7 (3) |
3 (1) |
|
Nervous System Disorders |
47 (20) |
20 (9) |
|
Syncope |
8 (3) |
1 (<1) |
|
Peripheral neuropathy NOS |
8 (3) |
0 (0) |
|
Cerebrovascular accident |
6 (3) |
1 (<1) |
|
Cardiac Disorders |
35 (15) |
27 (11) |
|
Atrial fibrillation |
11 (5) |
8 (3) |
|
Myocardial ischemia |
6 (3) |
2 (1) |
|
Vascular Disorders |
42 (18) |
14 (6) |
|
Deep vein thrombosis |
27 (12) |
4 (2) |
|
Gastrointestinal Disorders |
26 (11) |
22 (10) |
|
Constipation |
7 (3) |
2 (1) |
|
Investigations |
21 (9) |
21 (9) |
|
Weight increased |
8 (3) |
4 (2) |
|
Blood and Lymphatic System
|
24 (10) |
17 (7) |
|
Neutropenia |
8 (3) |
6 (3) |
|
Respiratory, Thoracic, and
|
27 (12) |
13 (6) |
|
Pulmonary embolism |
16 (7) |
4 (2) |
|
Psychiatric Disorders |
19 (8) |
8 (3) |
|
Anxiety |
5 (2) |
3 (1) |
|
Confusional state |
5 (2) |
2 (1) |
|
Ear and Labyrinth Disorders |
6 (3) |
0 (0) |
|
Vertigo |
5 (2) |
0 (0) |
Less Common Adverse Reactions in Multiple Myeloma Controlled Clinical Trials
In Study 2, THALOMID in combination with dexamethasone in patients with multiple myeloma, the following adverse reactions not described above were reported*:
Gastrointestinal disorders: Vomiting NOS, dry mouth, peritonitis, diverticular perforation
Nervous system disorders: Somnolence, hypoesthesia, polyneuropathy NOS, transient ischemic attack
Respiratory, thoracic, and mediastinal disorders: Bronchitis NOS
Psychiatric disorders: Mood alteration NOS
Vascular disorders: Hypotension NOS, orthostatic hypotension
Cardiac disorders: Bradycardia NOS
Eye disorders: Blurred vision
* All adverse reactions with ≥3% of patients in THALOMID/dexamethasone arm and with a ≥1% difference in proportion of patients between the THALOMID/dexamethasone arm compared to the placebo/dexamethasone arm. All grade 3/4 and serious adverse reactions reported >2 patients in THALOMID/dexamethasone arm and with a percentage higher in the THALOMID/dexamethasone arm compared to the placebo/dexamethasone arm have been considered for possible inclusion. In any cases medical judgment has been applied for consideration of causality assessment.
Adverse Reactions in Erythema Nodosum Leprosum (ENL) Clinical Trials
Table 4 lists treatment-emergent signs and symptoms that occurred in THALOMID-treated patients in clinical trials in ENL. The most common adverse reactions (≥10%) reported in patients with ENL were somnolence, rash, headache. Doses ranged from 50 to 300 mg/day. All adverse reactions were mild to moderate in severity, and none resulted in discontinuation.
| All ARs Reported in Patients with ENL | ARs Reported in ≥3 HIV-seropositive Patients | |||
|---|---|---|---|---|
| Body System/ Adverse Reaction | THALOMID
50 to 300 mg/day (N=24) n (%) | THALOMID
100 mg/day (N=36) n (%) | THALOMID
200 mg/day (N=32) n (%) | Placebo
(N=35) n (%) |
|
Blood and Lymphatic |
0 |
8 (22) |
13 (41) |
10 (29) |
|
Anemia |
0 |
2 (6) |
4 (13) |
3 (9) |
|
Leukopenia |
0 |
6 (17) |
8 (25) |
3 (9) |
|
Lymphadenopathy |
0 |
2 (6) |
4 (13) |
3 (9) |
|
Body as a Whole |
16 (67) |
18 (50) |
19 (59) |
13 (37) |
|
Abdominal pain |
1 (4) |
1 (3) |
1 (3) |
4 (11) |
|
Accidental injury |
1 (4) |
2 (6) |
0 |
1 (3) |
|
Asthenia |
2 (8) |
2 (6) |
7 (22) |
1 (3) |
|
Back pain |
1 (4) |
2 (6) |
0 |
0 |
|
Chills |
1 (4) |
0 |
3 (9) |
4 (11) |
|
Facial edema |
1 (4) |
0 |
0 |
0 |
|
Fever |
0 |
7 (19) |
7 (22) |
6 (17) |
|
Headache |
3 (13) |
6 (17) |
6 (19) |
4 (11) |
|
Infection |
0 |
3 (8) |
2 (6) |
1 (3) |
|
Malaise |
2 (8) |
0 |
0 |
0 |
|
Neck pain |
1 (4) |
0 |
0 |
0 |
|
Neck rigidity |
1 (4) |
0 |
0 |
0 |
|
Pain |
2 (8) |
0 |
1 (3) |
2 (6) |
|
Digestive System |
5 (21) |
16 (44) |
16 (50) |
15 (43) |
|
Anorexia |
0 |
1 (3) |
3 (9) |
2 (6) |
|
Constipation |
1 (4) |
1 (3) |
3 (9) |
0 |
|
Diarrhea |
1 (4) |
4 (11) |
6 (19) |
6 (17) |
|
Dry mouth |
0 |
3 (8) |
3 (9) |
2 (6) |
|
Flatulence |
0 |
3 (8) |
0 |
2 (6) |
|
Liver function tests multiple abnormalities |
0 |
0 |
3 (9) |
0 |
|
Nausea |
1 (4) |
0 |
4 (13) |
1 (3) |
|
Oral moniliasis |
1 (4) |
4 (11) |
2 (6) |
0 |
|
Tooth pain |
1 (4) |
0 |
0 |
0 |
|
Metabolic and Endocrine Disorders |
1 (4) |
8 (22) |
12 (38) |
8 (23) |
|
Edema peripheral |
1 (4) |
3 (8) |
1 (3) |
0 |
|
Hyperlipidemia |
0 |
2 (6) |
3 (9) |
1 (3) |
|
SGOT increased |
0 |
1 (3) |
4 (13) |
2 (6) |
|
Nervous System |
13 (54) |
19 (53) |
18 (56) |
12 (34) |
|
Agitation |
0 |
0 |
3 (9) |
0 |
|
Dizziness |
1 (4) |
7 (19) |
6 (19) |
0 |
|
Insomnia |
0 |
0 |
3 (9) |
2 (6) |
|
Nervousness |
0 |
1 (3) |
3 (9) |
0 |
|
Neuropathy |
0 |
3 (8) |
0 |
0 |
|
Paresthesia |
0 |
2 (6) |
5 (16) |
4 (11) |
|
Somnolence |
9 (38) |
13 (36) |
12 (38) |
4 (11) |
|
Tremor |
1 (4) |
0 |
0 |
0 |
|
Vertigo |
2 (8) |
0 |
0 |
0 |
|
Respiratory System |
3 (13) |
9 (25) |
6 (19) |
9 (26) |
|
Pharyngitis |
1 (4) |
3 (8) |
2 (6) |
2 (6) |
|
Rhinitis |
1 (4) |
0 |
0 |
4 (11) |
|
Sinusitis |
1 (4) |
3 (8) |
1 (3) |
2 (6) |
|
Skin and Appendages |
10 (42) |
17 (47) |
18 (56) |
19 (54) |
|
Acne |
0 |
4 (11) |
1 (3) |
0 |
|
Dermatitis fungal |
1 (4) |
2 (6) |
3 (9) |
0 |
|
Nail disorder |
1 (4) |
0 |
1 (3) |
0 |
|
Pruritus |
2 (8) |
1 (3) |
2 (6) |
2 (6) |
|
Rash |
5 (21) |
9 (25) |
8 (25) |
11 (31) |
|
Rash maculopapular |
1 (4) |
6 (17) |
6 (19) |
2 (6) |
|
Sweating |
0 |
0 |
4 (13) |
4 (11) |
|
Urogenital System |
2 (8) |
6 (17) |
2 (6) |
4 (11) |
|
Albuminuria |
0 |
3 (8) |
1 (3) |
2 (6) |
|
Hematuria |
0 |
4 (11) |
0 |
1 (3) |
|
Impotence |
2 (8) |
1 (3) |
0 |
0 |
Other Adverse Reactions Observed in ENL Patients
THALOMID in doses up to 400 mg/day has been administered investigationally in the United States over a 19-year period in 1465 patients with ENL. The published literature describes the treatment of an additional 1678 patients. To provide a meaningful estimate of the proportion of the individuals having adverse reactions, similar types of events were grouped into a smaller number of standardized categories using a modified COSTART dictionary/terminology. These categories are used in the listing below. All reported events are included except those already listed in the previous table. Due to the fact that these data were collected from uncontrolled studies, the incidence rate cannot be determined. No causal relationship between THALOMID and these events can be conclusively determined at this time. These are reports of all adverse events noted by investigators in patients to whom they had administered THALOMID.
Blood and Lymphatic: ESR decrease, eosinophilia, granulocytopenia, hypochromic anemia, leukemia, leukocytosis, leukopenia, MCV elevated, RBC abnormal, spleen palpable, thrombocytopenia.
Body as a Whole: Abdomen enlarged, fever, photosensitivity, upper extremity pain.
Cardiovascular System: Bradycardia, hypertension, hypotension, peripheral vascular disorder, tachycardia, vasodilation.
Digestive System: Anorexia, appetite increase/weight gain, dry mouth, dyspepsia, enlarged liver, eructation, flatulence, increased liver function tests, intestinal obstruction, vomiting.
Metabolic and Endocrine: ADH inappropriate, amyloidosis, bilirubinemia, BUN increased, creatinine increased, cyanosis, diabetes, edema, electrolyte abnormalities, hyperglycemia, hyperkalemia, hyperuricemia, hypocalcemia, hypoproteinemia, LDH increased, phosphorus decreased, SGPT increased.
Muscular Skeletal: Arthritis, bone tenderness, hypertonia, joint disorder, leg cramps, myalgia, myasthenia, periosteal disorder.
Nervous System: Abnormal thinking, agitation, amnesia, anxiety, causalgia, circumoral paresthesia, confusion, depression, euphoria, hyperesthesia, insomnia, nervousness, neuralgia, neuritis, neuropathy, paresthesia, peripheral neuritis, psychosis.
Respiratory System: Cough, emphysema, epistaxis, pulmonary embolus, rales, upper respiratory infection, voice alteration.
Skin and Appendages: Acne, alopecia, dry skin, eczematous rash, exfoliative dermatitis, ichthyosis, perifollicular thickening, skin necrosis, seborrhea, sweating, urticaria, vesiculobullous rash.
Special Senses: Amblyopia, deafness, dry eye, eye pain, tinnitus.
Urogenital: Decreased creatinine clearance, hematuria, orchitis, proteinuria, pyuria, urinary frequency.
Other Adverse Reactions Observed in HIV-seropositive Patients
In addition to controlled clinical trials, THALOMID has been used in uncontrolled studies in 145 patients. Less frequent adverse reactions that have been reported in these HIV-seropositive patients treated with THALOMID were grouped into a smaller number of standardized categories using modified COSTART dictionary/terminology and these categories are used in the listing below. Adverse reactions that have already been included in the tables and narrative above, or that are too general to be informative are not listed.
Blood and Lymphatic: Aplastic anemia, macrocytic anemia, megaloblastic anemia, microcytic anemia.
Body as a Whole: Ascites, AIDS, allergic reaction, cellulitis, chest pain, chills and fever, cyst, decreased CD4 count, facial edema, flu syndrome, hernia, thyroid hormone level altered, moniliasis, photosensitivity reaction, sarcoma, sepsis, viral infection.
Cardiovascular System: Angina pectoris, arrhythmia, atrial fibrillation, bradycardia, cerebral ischemia, cerebrovascular accident, congestive heart failure, deep thrombophlebitis, heart arrest, heart failure, hypertension, hypotension, murmur, myocardial infarct, palpitation, pericarditis, peripheral vascular disorder, postural hypotension, syncope, tachycardia, thrombophlebitis, thrombosis.
Digestive System: Cholangitis, cholestatic jaundice, colitis, dyspepsia, dysphagia, esophagitis, gastroenteritis, gastrointestinal disorder, gastrointestinal hemorrhage, gum disorder, hepatitis, pancreatitis, parotid gland enlargement, periodontitis, stomatitis, tongue discoloration, tooth disorder.
Metabolic and Endocrine: Avitaminosis, bilirubinemia, dehydration, hypercholesterolemia, hypoglycemia, increased alkaline phosphatase, increased lipase, increased serum creatinine, peripheral edema.
Muscular Skeletal: Myalgia, myasthenia.
Nervous System: Abnormal gait, ataxia, decreased libido, decreased reflexes, dementia, dysesthesia, dyskinesia, emotional lability, hostility, hypalgesia, hyperkinesia, incoordination, meningitis, neurologic disorder, tremor, vertigo.
Respiratory System: Apnea, bronchitis, lung disorder, lung edema, pneumonia (including Pneumocystis carinii pneumonia), rhinitis.
Skin and Appendages: Angioedema, benign skin neoplasm, eczema, herpes simplex, incomplete Stevens-Johnson syndrome, nail disorder, pruritus, psoriasis, skin discoloration, skin disorder.
Special Senses: Conjunctivitis, eye disorder, lacrimation disorder, retinitis, taste perversion.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post approval use of THALOMID and are not already included in Clinical Trials Experience [see Adverse Reactions (6.1)]. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic: Decreased white blood cell counts including febrile neutropenia, changes in prothrombin time, pancytopenia, chronic myelogenous leukemia, nodular sclerosing Hodgkin's disease, erythroleukemia, lymphedema, lymphopenia.
Body as a Whole: Hangover effect
Cardiovascular System: Sick sinus syndrome, EKG abnormalities, pulmonary hypertension.
Digestive System: Intestinal perforation, gastrointestinal perforations, bile duct obstruction, stomach ulcer, aphthous, stomatitis.
Ear and Labyrinthine Disorders: Hearing impairment.
Immune System Disorders: Hypersensitivity including anaphylaxis, solid organ transplant rejection.
Infections and infestations: Severe infections (e.g., fatal sepsis including septic shock), viral infections (including varicella zoster virus, cytomegalovirus, and hepatitis B virus reactivation) and progressive multifocal leukoencephalopathy (PML).
Metabolic and Endocrine: Electrolyte imbalance including hypercalcemia, hyponatremia and hypomagnesemia, hypothyroidism, increased alkaline phosphatase, tumor lysis syndrome, myxedema.
Nervous System: Changes in mental status or mood including suicide attempts, disturbances in consciousness including lethargy, loss of consciousness or stupor, seizures including grand mal convulsions and status epilepticus, Parkinson's disease, stroke, carpal tunnel, Raynaud's syndrome, migraine, foot drop.
Renal and Urinary Disorders: Renal failure, acute renal failure, oliguria, enuresis.
Reproductive System and Breast Disorders: amenorrhea, sexual dysfunction, galactorrhea, gynecomastia, metrorrhagia.
Respiratory System: Pleural effusion, interstitial lung disease.
Skin and Appendages: Erythema multiforme, erythema nodosum, toxic epidermal necrolysis (TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), purpura, petechiae.
Special Senses: Diplopia, nystagmus
Related/similar drugs
7. Drug Interactions
7.1 Opioids, Antihistamines, Antipsychotics, Anti-anxiety Agents, or Other CNS Depressants (Including Alcohol)
The use of opioids, antihistamines, antipsychotics, anti-anxiety agents, or other CNS depressants concomitantly with THALOMID may cause an additive sedative effect and should be avoided.
7.2 Drugs which Cause Bradycardia
The use of drugs which slow cardiac conduction concomitantly with THALOMID may cause an additive bradycardic effect and should be used with caution. Cardiovascular medications which may cause bradycardia include calcium channel blockers, beta blockers, alpha/beta-adrenergic blockers, and digoxin. Non-cardiac drugs that may cause bradycardia include H2 blockers (e.g., famotidine, cimetidine), lithium, tricyclic antidepressants and neuromuscular blockers (succinylcholine).
In 16 healthy men, the pharmacokinetic profile of a single 0.5 mg digoxin dose was similar with and without the coadministration of THALOMID 200 mg/day at steady state levels. The single dose of digoxin had no effect on the pharmacokinetic profile of THALOMID. The safety of long-term concomitant use of THALOMID and digoxin has not been evaluated.
7.3 Drugs which Cause Peripheral Neuropathy
The use of drugs which cause peripheral neuropathy (e.g., bortezomib, amiodarone, cisplatin, docetaxel, paclitaxel, vincristine, disulfiram, phenytoin, metronidazole, alcohol) can cause an additive effect and should be used with caution.
7.4 Hormonal Contraceptives
Hormonal contraceptives increase the risk of thromboembolism. It is not known whether concomitant use of hormonal contraceptives further increases the risk of thromboembolism with THALOMID.
In 10 healthy women, the pharmacokinetic profiles of norethindrone and ethinyl estradiol following administration of a single dose containing 1.0 mg of norethindrone acetate and 75 mcg of ethinyl estradiol were studied. The results were similar with and without coadministration of THALOMID 200 mg/day to steady-state levels.
7.5 Warfarin
In 13 healthy men, the pharmacokinetic profile and international normalized ratio (INR) of prothrombin time for warfarin, following a single oral dose of 25 mg, were similar with and without the coadministration of THALOMID 200 mg/day at steady-state levels. The single dose of warfarin had no effect on the pharmacokinetic profile of thalidomide.
7.6 Drugs that Interfere with Hormonal Contraceptives
Concomitant use of HIV-protease inhibitors, griseofulvin, modafinil, penicillins, rifampin, rifabutin, phenytoin, carbamazepine, or certain herbal supplements such as St. John's Wort with hormonal contraceptive agents may reduce the effectiveness of the contraception up to one month after discontinuation of these concomitant therapies. Therefore, females requiring treatment with one or more of these drugs must use two OTHER effective or highly effective methods of contraception while taking THALOMID.
7.7 Concomitant Therapies that may Increase the Risk of Thromboembolism
Erythropoietic agents, or other agents that may increase the risk of thromboembolism, such as estrogen containing therapies, should be used with caution in multiple myeloma patients receiving THALOMID with dexamethasone [see Warnings and Precautions (5.3)].
8. Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in females exposed to THALOMID during pregnancy as well as female partners of male patients who are exposed to THALOMID. This registry is also used to understand the root cause for the pregnancy. Report any suspected fetal exposure to THALOMID to the FDA via the MedWatch program at 1-800-FDA-1088 and to the REMS Call Center at 1-888-423-5436.
Risk Summary
Based on the mechanism of action [see Clinical Pharmacology (12.1)], human and animal data (see Data), THALOMID can cause embryo-fetal harm when administered to a pregnant female and is contraindicated during pregnancy [see Boxed Warning, Contraindications (4.1), and Warnings and Precautions (5.1)].
THALOMID is a human teratogen, inducing a high frequency of severe and life-threatening birth defects such as amelia (absence of limbs), phocomelia (short limbs), hypoplasticity of the bones, absence of bones, external ear abnormalities (including anotia, micropinna, small or absent external auditory canals), facial palsy, eye abnormalities (anophthalmos, microphthalmos), and congenital heart defects. Alimentary tract, urinary tract, and genital malformations have also been documented and mortality at or shortly after birth has been reported in about 40% of infants. Even a single dose taken by a pregnant woman can cause birth defects. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential risk to a fetus.
If pregnancy does occur during treatment, immediately discontinue the drug. Under these conditions, refer the patient to an obstetrician/gynecologist experienced in reproductive toxicity for further evaluation and counseling. Report any suspected fetal exposure to THALOMID to the FDA via the MedWatch program at 1-800-FDA-1088 and also the REMS Call Center at 1-888-423-5436.
Thalidomide crossed the placenta after administration to pregnant hamsters (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk in the U.S. general population of major birth defects is 2%-4% and of miscarriage is 15%-20% of clinically recognized pregnancies.
Data
Animal Data
A pre- and postnatal reproductive toxicity study was conducted in pregnant female rabbits. Compound-related increased abortion incidences and elevated fetotoxicity were observed at the lowest oral dose level of 30 mg/kg/day (approximately 1.5-fold the maximum human dose based upon BSA) and all higher dose levels. Neonatal mortality was elevated at oral dose levels to the lactating female rabbits ≥150 mg/kg/day (approximately 7.5-fold the maximum human dose based upon BSA). No delay in postnatal development, including learning and memory functions, were noted at the oral dose level to the lactating female rabbits of 150 mg/kg/day (average thalidomide concentrations in milk ranged from 22 to 36 mcg per mL).
In a study conducted in pregnant rabbits, thalidomide levels in fetal plasma were approximately 11% to 73% of the maternal Cmax. In a study conducted with 14C-thalidomide (150 mg/kg orally) in pregnant hamsters, radioactivity was detected in the embryo, and the relative concentrations of radioactivity in the embryo and maternal plasma were about the same at 4, 12 and 24 hours after dosing. Based on the radioactivity data, thalidomide crossed the placental barrier, and the fetal levels of drug-related material were approximately similar to those of maternal levels.
8.2 Lactation
Risk Summary
There is no information regarding the presence of thalidomide in human milk, the effects of THALOMID on the breastfed child, or the effects of THALOMID on milk production. Thalidomide is excreted in the milk of lactating rabbits (see Data). Because many drugs are excreted in human milk and because of the potential for adverse reactions in a breastfed child from THALOMID, advise women not to breastfeed during treatment with THALOMID.
Data
Animal Data
In lactating female rabbits at an oral dose of 150 mg/kg/day, the average thalidomide concentrations in milk ranged from 22 to 36 mcg per mL. In the study of lactating female rabbits, high concentrations of thalidomide (7741 – 71425 ng per mL) were noted in milk during four weeks of pre-weaning period. Milk concentrations were 1.16 - 2.11, 1.05 – 2.43, and 0.64 – 3.63 times that of plasma at 30, 150 and 500 mg/kg thalidomide doses, respectively; thalidomide, as a lipophilic compound, distributed into milk, with concentrations attained similar to or slightly higher than those of systemic concentrations.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
THALOMID can cause fetal harm when administered during pregnancy [see Use in Specific Populations (8.1)]. Verify the pregnancy status of females of reproductive potential prior to initiating THALOMID therapy and during therapy. Advise females of reproductive potential that they must avoid pregnancy 4 weeks before therapy, while taking THALOMID, during dose interruptions and for at least 4 weeks after completing therapy.
Females of reproductive potential must have 2 negative pregnancy tests before initiating THALOMID. The first test should be performed within 10-14 days, and the second test within 24 hours prior to prescribing THALOMID. Once treatment has started and during dose interruptions, pregnancy testing for females of reproductive potential should occur weekly during the first 4 weeks of use, then pregnancy testing should be repeated every 4 weeks in females with regular menstrual cycles. If menstrual cycles are irregular, the pregnancy testing should occur every 2 weeks. Pregnancy testing and counseling should be performed if a patient misses her period or if there is any abnormality in her menstrual bleeding. THALOMID treatment must be discontinued during this evaluation.
Contraception
Females
Females of reproductive potential must commit either to abstain continuously from heterosexual sexual intercourse or to use 2 methods of reliable birth control simultaneously: one highly effective form of contraception – tubal ligation, IUD, hormonal (birth control pills, injections, hormonal patches, vaginal rings, or implants), or partner's vasectomy, and 1 additional effective contraceptive method – male latex or synthetic condom, diaphragm, or cervical cap. Contraception must begin 4 weeks prior to initiating treatment with THALOMID, during therapy, during dose interruptions, and continuing for 4 weeks following discontinuation of THALOMID therapy. Reliable contraception is indicated even where there has been a history of infertility, unless due to hysterectomy. Females of reproductive potential should be referred to a qualified provider of contraceptive methods, if needed.
Males
Thalidomide is present in the semen of males who take THALOMID. Therefore, males must always use a latex or synthetic condom during any sexual contact with females of reproductive potential while taking THALOMID, during dose interruptions and for up to 28 days after discontinuing THALOMID, even if they have undergone a successful vasectomy. Male patients taking THALOMID must not donate sperm.
Infertility
Based on findings in animals, male fertility may be compromised by treatment with THALOMID [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients below the age of 12 years have not been established.
8.5 Geriatric Use
One hundred and seventy-six (52%) of 336 patients treated with THALOMID in combination with dexamethasone were ≥65 of age while 50 (15%) were ≥75. Patients ≥65 years of age on Study 2 had higher incidences of atrial fibrillation, constipation, fatigue, nausea, hypokalemia, deep venous thrombosis, hyperglycemia, pulmonary embolism, and asthenia compared to patients <65.
9. Drug Abuse and Dependence
Physical and psychological dependence has not been reported in patients taking THALOMID; however, as with other tranquilizers/hypnotics, thalidomide has been reported to result in habituation to its soporific effects.
10. Overdosage
There is no specific antidote for a THALOMID overdose. In the event of an overdose, the patient's vital signs should be monitored and appropriate supportive care given to maintain blood pressure and respiratory status.
11. Thalomid Description
THALOMID, α-(N-phthalimido) glutarimide, is an immunomodulatory agent. The empirical formula for thalidomide is C13H10N2O4 and the gram molecular weight is 258.2. The CAS number of thalidomide is 50-35-1.
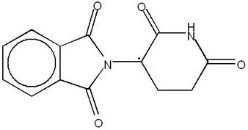
Thalidomide is an off-white to white, odorless, crystalline powder that is soluble at 25°C in dimethyl sulfoxide and sparingly soluble in water and ethanol. The glutarimide moiety contains a single asymmetric center and, therefore, may exist in either of two optically active forms designated S-(-) or R-(+). THALOMID is an equal mixture of the S-(-) and R-(+) forms and, therefore, has a net optical rotation of zero.
THALOMID is available in 50 mg, 100 mg, 150 mg and 200 mg capsules for oral administration. Active ingredient: thalidomide. Inactive ingredients: pregelatinized starch and magnesium stearate. The 50 mg capsule shell contains gelatin, titanium dioxide, and black ink. The 100 mg capsule shell contains black iron oxide, yellow iron oxide, titanium dioxide, gelatin, and black ink. The 150 mg capsule shell contains FD&C blue #2, black iron oxide, yellow iron oxide, titanium dioxide, gelatin, and black and white ink. The 200 mg capsule shell contains FD&C blue #2, titanium dioxide, gelatin, and white ink.
12. Thalomid - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism of action of THALOMID is not fully understood. Cellular activities of thalidomide are mediated through its target cereblon, a component of a cullin ring E3 ubiquitin ligase enzyme complex. THALOMID possesses immunomodulatory, anti-inflammatory and antiangiogenic properties. Available data from in vitro studies and clinical trials suggest that the immunologic effects of this compound can vary substantially under different conditions, but may be related to suppression of excessive tumor necrosis factor-alpha (TNF-α) production and down-modulation of selected cell surface adhesion molecules involved in leukocyte migration. For example, administration of thalidomide has been reported to decrease circulating levels of TNF-α in patients with erythema nodosum leprosum (ENL); however, it has also been shown to increase plasma TNF-α levels in HIV-seropositive patients. Other anti-inflammatory and immunomodulatory properties of thalidomide may include suppression of macrophage involvement in prostaglandin synthesis, and modulation of interleukin-10 and interleukin-12 production by peripheral blood mononuclear cells. Thalidomide treatment of multiple myeloma patients is accompanied by an increase in the number of circulating natural killer cells, and an increase in plasma levels of interleukin-2 and interferon-gamma (T cell-derived cytokines associated with cytotoxic activity). Thalidomide was found to inhibit angiogenesis in a human umbilical artery explant model in vitro. The cellular processes of angiogenesis inhibited by thalidomide may include the proliferation of endothelial cells.
12.3 Pharmacokinetics
Absorption
Absorption of THALOMID is slow after oral administration. The maximum plasma concentrations are reached approximately 2-5 hours after administration. The absolute bioavailability of thalidomide from thalidomide capsules has not yet been characterized in human subjects due to its poor aqueous solubility. Based on the 14C-radiolabel thalidomide study in human, greater than 90% of the total radioactivity is recovered in urine suggesting good oral absorption. While the extent of absorption (as measured by area under the curve [AUC]) is proportional to dose in healthy subjects, the observed peak concentration (Cmax) increased in a less than proportional manner (see Table 5 below). This lack of Cmax dose proportionality, coupled with the observed increase in Tmax values, suggests that the poor solubility of thalidomide in aqueous media may be hindering the rate of absorption.
| Population/ Single Dose | AUC0-∞
mcg∙hr/mL | Cmax
mcg/mL | Tmax
(hrs) | Half-life
(hrs) |
|---|---|---|---|---|
|
Healthy Subjects (n=14) |
||||
|
50 mg |
4.9 (16%) |
0.62 (52%) |
2.9 (66%) |
5.52 (37%) |
|
200 mg |
18.9 (17%) |
1.76 (30%) |
3.5 (57%) |
5.53 (25%) |
|
400 mg |
36.4 (26%) |
2.82 (28%) |
4.3 (37%) |
7.29 (36%) |
|
Patients with Hansen's Disease (n=6) |
||||
|
400 mg |
46.4 (44.1%) |
3.44 (52.6%) |
5.7 (27%) |
6.86 (17%) |
Coadministration of THALOMID (thalidomide) with a high-fat meal causes minor (<10%) changes in the observed AUC and Cmax values; however, it causes an increase in Tmax to approximately 6 hours.
Distribution
In human plasma, the geometric mean plasma protein binding was 55% and 66%, respectively, for (+)-(R)- and (-)-(S)-thalidomide. In a pharmacokinetic study of thalidomide in HIV-seropositive adult male subjects receiving thalidomide 100 mg/day, thalidomide was detectable in the semen.
Metabolism
In a 14C-radiolabel ADME study in humans, unchanged drug is the predominant circulating component. Thalidomide is not a substrate of the cytochrome P450 system. At therapeutic concentrations, thalidomide is not an inhibitor or inducer of human cytochrome P450 enzymes in vitro. Pharmacokinetic drug-drug interactions with substrates, inhibitors or inducers of CYP450 are not anticipated.
Elimination
The mean elimination half-life of thalidomide in plasma following single oral doses between 50 mg and 400 mg was 5.5 to 7.3 hours. Following a single 400 mg oral dose of radiolabeled thalidomide, the total mean recovery was 93.6% of the administered dose by Day 8. The majority of the radioactive dose was excreted within 48 hours following dose administration. In humans, 14C-thalidomide is primarily excreted in urine (91.9% of the radioactive dose) mainly as hydrolytic metabolites while fecal excretion is minor (<2% of the dose). Unchanged thalidomide is not eliminated by the kidney to a notable degree (<3.5% of the dose).
Effects of Weight
There is a linear relationship between body weight and estimated thalidomide clearance. In MM patients with body weight from 47-133 kg, thalidomide clearance ranged from approximately 6-12 L/h, representing an increase in thalidomide clearance of 0.605 L/h per 10 kg body weight increase.
Effects of Age, Gender and Race
Analysis of the data from pharmacokinetic studies in healthy volunteers and patients with Hansen's disease ranging in age from 20 to 69 years does not reveal any age-related changes.
While a comparative trial of the effects of gender on thalidomide pharmacokinetics has not been conducted, examination of the data for thalidomide does not reveal any significant gender differences in pharmacokinetic parameter values.
Pharmacokinetic differences due to race have not been studied.
Pharmacokinetic Data in Special Populations
HIV-seropositive Subjects: There is no apparent significant difference in measured pharmacokinetic parameter values between healthy human subjects and HIV-seropositive subjects following single-dose administration of THALOMID Capsules.
Patients with Hansen's Disease: Analysis of data from a small study in Hansen's patients suggests that these patients, relative to healthy subjects, may have an increased bioavailability of THALOMID. The increase is reflected both in an increased area under the curve and in increased peak plasma levels. The clinical significance of this increase is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted in male and female rats and mice. No compound-related tumorigenic effects were observed at the highest dose levels of 3,000 mg/kg/day to male and female mice (38-fold greater than the highest recommended daily human dose of 400 mg based upon body surface area [BSA]), 3,000 mg/kg/day to female rats (75-fold the maximum human dose based upon BSA), and 300 mg/kg/day to male rats (7.5-fold the maximum human dose based upon BSA).
Thalidomide was neither mutagenic nor genotoxic in the following assays: the Ames bacterial (S. typhimurium and E. coli) reverse mutation assay, a Chinese hamster ovary cell (AS52/XPRT) forward mutation assay, and an in vivo mouse micronucleus test.
Fertility studies were conducted in male and female rabbits; no compound-related effects in mating and fertility indices were observed at any oral thalidomide dose level including the highest of 100 mg/kg/day to female rabbits and 500 mg/kg/day to male rabbits (approximately 5- and 25- fold the maximum human dose, respectively, based upon BSA). Testicular pathological and histopathological effects (classified as slight) were seen in male rabbits at dose levels ≥30 mg/kg/day (approximately 1.5-fold the maximum human dose based upon BSA).
14. Clinical Studies
14.1 Multiple Myeloma (MM)
The efficacy and safety of THALOMID in patients with multiple myeloma were evaluated in two randomized, multi-center studies (Study 1 and Study 2). Study 1 was an open-label study which randomized 207 symptomatic patients with newly diagnosed MM to THALOMID plus dexamethasone (N = 103) versus dexamethasone alone (N=104). The THALOMID dose was 200 mg daily and the dexamethasone dose was 40 mg orally once daily on days 1-4, 9-12, and 17-20 every 28-days. Each group was treated for four 28-day cycles.
Study 2 randomized 470 newly diagnosed patients with MM to THALOMID plus dexamethasone (N=235) versus placebo plus dexamethasone (N=235). In the THALOMID/dexamethasone arm, a starting dose of thalidomide 50 mg was escalated to 200 mg/day (cycle 2) once daily for 28 days. Patients in both treatment groups took 40 mg of dexamethasone once daily given on days 1-4, 9-12, and 17-20 (every 28 days). Beginning with Cycle 5, the dose of dexamethasone was reduced to 40 mg once daily on Days 1 to 4 of each cycle. Treatment continued as tolerated until disease progression.
Baseline demographics for both studies are presented in Table 6 and disease characteristics for the study population are summarized in Tables 7 (Study 1) and 8 (Study 2).
| Characteristic | Study 1 | Study 2 | ||
|---|---|---|---|---|
| THALOMID/ Dexamethasone (N=103) | Dexamethasone
(N=104) | THALOMID/ Dexamethasone (N=235) | Placebo/ Dexamethasone (N=235) |
|
| 1 Missing information in Study 1 for 1 patient in the Dex alone group 2 Missing information in Study 1 for 1 patient per arm 3 Black/Hispanic [1 (0.4%)], Hispanic [2 (0.9%)], Hispanic/White [1 (0.4%)], Other [0 (0.0%)] 4 Hispanic [1 (0.4%)], Asian/Pacific Islander [2 (0.9%)], Other [1 (0.4%)] |
||||
|
Age (years) |
||||
|
Median |
65 |
68 |
65 |
66 |
|
Range |
37 - 83 |
38 - 83 |
39 - 86 |
31 - 84 |
|
Gender1, N (%) |
||||
|
Male |
53 (51) |
61 (59) |
118 (50) |
120 (51) |
|
Female |
50 (49) |
42 (40) |
117 (50) |
115 (49) |
|
Race2, N (%) |
||||
|
Caucasian |
90 (87) |
90 (87) |
224 (95) |
221 (94) |
|
Black |
11 (11) |
11 (11) |
7 (3) |
10 (4) |
|
Other |
1 (1) |
2 (2) |
4 (2)3 |
4 (2)4 |
| Disease Characteristic | THALOMID/Dexamethasone
(N=103) | Dexamethasone alone
(N=104) |
|---|---|---|
| 1 Missing information for 1 patient in Thal + Dex arm 2 Missing information for 19 patients in Thal + Dex arm and 20 patients in Dex alone arm 3 Missing information for 17 patients in Thal + Dex arm and 30 patients in Dex alone arm 4 Missing information for 16 patients in Thal + Dex arm and 11 patients in Dex alone arm |
||
|
Stage (Durie-Salmon), N (%)1 | ||
|
I |
14 (13.6%) |
17 (16.3%) |
|
II |
47 (45.6%) |
44 (42.3%) |
|
III |
41 (39.8%) |
43 (41.3%) |
|
Immunoglobulin Type, N (%)2 | ||
|
IgA |
21 (20.4%) |
22 (21.2%) |
|
IgG |
63 (61.2%) |
60 (57.7%) |
|
IgM |
0 (0.0%) |
1 (1.0%) |
|
Biclonal |
0 (0.0%) |
1 (1.0%) |
|
Lytic Lesions3 | ||
|
None |
28 (27.1%) |
14 (13.5%) |
|
1-3 lesions |
24 (23.3%) |
19 (18.3%) |
|
>3 lesions |
34 (33.0%) |
41 (39.4%) |
|
Serum Light Chain4 | ||
|
Kappa |
59 (57.3%) |
53 (51.0%) |
|
Lambda |
28 (27.2%) |
40 (38.5%) |
| Disease Characteristic | THALOMID/Dexamethasone
(N=235) | Placebo/Dexamethasone
(N=235) |
|---|---|---|
| KEY: ECOG=Eastern Cooperative Oncology Group | ||
|
Baseline MM Stage (Durie-Salmon), n (%) | ||
|
I |
2 (1) |
2 (1) |
|
II |
76 (32) |
88 (37) |
|
III |
157 (69) |
145 (62) |
|
ECOG Performance Status, n (%) | ||
|
0 |
40 (17) |
54 (23) |
|
1 |
124 (53) |
112 (48) |
|
2 |
70 (30) |
68 (29) |
|
3 |
0 (0) |
1 (<1) |
|
Missing |
1 (<1) |
0 (0) |
|
Lytic Bone Lesions, n (%) | ||
|
Present |
185 (79) |
188 (80) |
|
Absent |
49 (21) |
46 (20) |
|
Missing |
1 (<1) |
1 (<1) |
|
Bone Marrow Aspirate/Biopsy Cellularity, n (%) | ||
|
Normal |
102 (43) |
108 (46) |
|
Hyperplasia |
77 (33) |
76 (32) |
|
Hypoplasia |
53 (23) |
50 (21) |
|
Missing |
3 (1) |
1 (<1) |
|
Baseline β-2 Microglobulin, n (%) | ||
|
≤2.5 mg/L |
33 (14) |
35 (15) |
|
>2.5 mg/L |
200 (85) |
199 (85) |
|
Missing |
2 (1) |
1 (<1) |
In Study 1, response rate was the primary endpoint. Response rates based on serum or urine paraprotein measurements were significantly higher in the combination arm (52% vs. 36%). The primary efficacy endpoint in Study 2 was time to progression (TTP), defined as the time from randomization to the first documentation of disease progression, based on the myeloma response criteria. A preplanned interim analysis for Study 2 demonstrated that the combination of THALOMID plus dexamethasone was superior to placebo plus dexamethasone with respect to TTP (Table 9).
| Thalidomide/Dexamethasone
(N=235) | Placebo/Dexamethasone
(N=235) |
|
|---|---|---|
| a The 95% confidence intervals about the median overall TTP, or median overall survival. CI: confidence interval; NR: not reached. b Based on a proportional hazards model comparing the hazard functions associated with treatment groups (thalidomide/dexamethasone:placebo/dexamethasone). c P-value based on the interim analysis was compared with the nominal significance level of 0.0027. Based on a one-sided unstratified log rank test of survival curve differences between treatment groups. d Disease response assessments were determined according to the Bladé criteria. Response is the highest assessment of response during the treatment phase of the study. |
||
|
Time to Progression | ||
|
Progressed – n (%) |
72 (31) |
126 (54) |
|
Median (Weeks) (95% CIa) |
97.7 (61.86, NR) |
28.3 (27.71, 36.43) |
|
Hazard Ratio (95% CI)b |
0.43 (0.32, 0.58) |
|
|
P-valuec |
< 0.0001 |
|
|
Overall Survival | ||
|
Death – n (%) |
57 (24) |
68 (29) |
|
Median (Weeks) (95% CIa) |
NR (112.14, NR) |
128.6 (113.43, NR) |
|
Hazard Ratio (95% CI)b |
0.82 (0.57, 1.16) |
|
|
Myeloma Response Rated – n (%) | ||
|
Complete Response (CR) |
18 (8) |
6 (3) |
|
Partial Response (PR) |
130 (55) |
102 (43) |
|
Overall Response (CR + PR) |
148 (63) |
108 (46) |
|
95% CI (%) |
(56, 69) |
(39, 53) |
The Kaplan-Meier plot of the time to progression by treatment group is presented in Figure 1.
Figure 1: Kaplan-Meier Plot of Time to Disease Progression
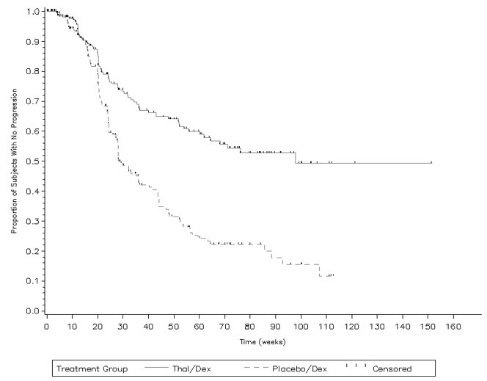
KEY: Placebo/Dex=placebo/dexamethasone; Thal/Dex=THALOMID/dexamethasone
14.2 Erythema Nodosum Leprosum (ENL)
The primary data demonstrating the efficacy of thalidomide in the treatment of the cutaneous manifestations of moderate to severe ENL are derived from the published medical literature and from a retrospective study of 102 patients treated by the U.S. Public Health Service.
Two double-blind, randomized, controlled trials reported the dermatologic response to a 7-day course of 100 mg thalidomide (four times daily) or control. Dosage was lower for patients under 50 kg in weight.
| Reference | No. of Patients | No. Treatment Courses* | Percent Responding** | |
|---|---|---|---|---|
| * In patients with cutaneous lesions **Iyer: Complete response or lesions absent **Sheskin: Complete improvement + "striking" improvement (i.e., >50% improvement) |
||||
|
Iyer et al.
|
92 |
204 |
Thalidomide |
Aspirin |
|
Sheskin et al.
|
52 |
173 |
Thalidomide |
Placebo |
Waters reported the results of two studies, both double-blind, randomized, placebo-controlled, crossover trials in a total of 10 hospitalized, steroid-dependent patients with chronic ENL treated with 100 mg thalidomide or placebo (three times daily). All patients also received dapsone. The primary endpoint was reduction in weekly steroid dosage.
| Reference | Duration of Treatment | No. of Patients | Number Responding | |
|---|---|---|---|---|
| Thalidomide | Placebo | |||
|
Waters |
4 weeks |
9 |
4/5 |
0/4 |
|
Lep Rev 1971;42:26 |
6 weeks |
8 |
8/8 |
1/8 |
Data on the efficacy of thalidomide in prevention of ENL relapse were derived from a retrospective evaluation of 102 patients treated under the auspices of the U.S. Public Health Service. A subset of patients with ENL controlled on thalidomide demonstrated repeated relapse upon drug withdrawal and remission with reinstitution of therapy.
Twenty U.S. patients between the ages of 11 and 17 years were treated with thalidomide, generally at 100 mg daily. Response rates and safety profiles were similar to that observed in the adult population.
Thirty-two other published studies containing over 1600 patients consistently report generally successful treatment of the cutaneous manifestations of moderate to severe ENL with thalidomide.
15. References
- 1.
- OSHA Hazardous Drugs. OSHA [Accessed on 02 July 2014, from http://www.osha.gov/SLTC/hazardousdrugs/index.html].
16. How is Thalomid supplied
(THIS PRODUCT IS ONLY SUPPLIED TO PHARMACIES CERTIFIED IN THE THALOMID REMS PROGRAM - See BOXED WARNING)
16.1 How Supplied
50 mg capsules [white opaque], imprinted "BMS/50 mg" with a "Do Not Get Pregnant" logo.
Individual blister packs of 1 capsule (NDC 59572-205-17).
Individual blister packs of 28 capsules (NDC 59572-205-14).
100 mg capsules [tan], imprinted "BMS/100 mg" with a "Do Not Get Pregnant" logo.
Individual blister packs of 28 capsules (NDC 59572-210-15).
150 mg capsules [tan and blue], imprinted "BMS/150 mg" with a "Do Not Get Pregnant" logo.
Individual blister packs of 28 capsules (NDC 59572-215-13).
200 mg capsules [blue], imprinted "BMS/200 mg" with a "Do Not Get Pregnant" logo.
Individual blister packs of 28 capsules (NDC 59572-220-16).
16.2 Storage
This drug must not be repackaged.
Store at 20°C- 25°C (68°F -77°F); excursions permitted to 15-30º C (59-86º F). [See USP Controlled Room Temperature]. Protect from light.
16.3 Handling and Disposal
Care should be exercised in handling of THALOMID. THALOMID capsules should not be opened or crushed. If powder from THALOMID contacts the skin, wash the skin immediately and thoroughly with soap and water. If THALOMID contacts the mucous membranes, flush thoroughly with water.
Procedures for proper handling and disposal of anticancer drugs should be considered. Several guidelines on the subject have been published1.
Rx only and only able to be prescribed and dispensed under the terms of the THALOMID REMS Restricted Distribution Program.
17. Patient Counseling Information
Advise the patient to read the FDA-approved Patient Labeling (Medication Guide)
Embryo-Fetal Toxicity
Advise patients that THALOMID is contraindicated in pregnancy and can cause serious birth defects or death to a developing baby [see Boxed Warning, Contraindications (4.1), Warnings and Precautions (5.1), and Use in Specific Populations (8.1)].
- •
- Advise females of reproductive potential that they must avoid pregnancy while taking THALOMID and for at least 4 weeks after completing therapy.
- •
- Initiate THALOMID treatment in females of reproductive potential only following a negative pregnancy test.
- •
- Advise females of reproductive potential of the importance of monthly pregnancy tests and the need to use 2 different forms of contraception, including at least 1 highly effective form, simultaneously during THALOMID therapy, during therapy interruption and for 4 weeks after she has completely finished taking THALOMID. Highly effective forms of contraception other than tubal ligation include IUD and hormonal (birth control pills, injections, patch or implants) and a partner's vasectomy. Additional effective contraceptive methods include latex or synthetic condom, diaphragm and cervical cap.
- •
- Instruct patient to immediately stop taking THALOMID and contact her healthcare provider if she becomes pregnant while taking this drug, if she misses her menstrual period, or experiences unusual menstrual bleeding, if she stops taking birth control, or if she thinks FOR ANY REASON that she may be pregnant.
- •
- Advise patient that if her healthcare provider is not available, she should call the REMS Call Center at 1-888-423-5436 [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
- •
- Advise males to always use a latex or synthetic condom during any sexual contact with females of reproductive potential while taking THALOMID and for up to 4 weeks after discontinuing THALOMID, even if they have undergone a successful vasectomy.
- •
- Advise male patients taking THALOMID that they must not donate sperm [see Warnings and Precautions (5.1) and Use in Specific Populations (8.3)].
- •
- All patients must be instructed to not donate blood while taking THALOMID and for 4 weeks following discontinuation of THALOMID [see Warnings and Precautions (5.1)].
THALOMID REMS Program
Because of the risk of embryo-fetal toxicity, THALOMID is only available through a restricted program called the THALOMID REMS program [see Warnings and Precautions (5.2)].
- •
- Patients must sign a Patient-Physician Agreement Form and comply with the requirements to receive THALOMID. In particular, females of reproductive potential must comply with the pregnancy testing, contraception requirements and participate in monthly telephone surveys. Males must comply with the contraception requirements [see Use in Specific Populations (8.3)].
- •
- THALOMID is available only from pharmacies that are certified in THALOMID REMS program. Provide patients with the telephone number and website for information on how to obtain the product.
Pregnancy Exposure Registry
Inform females there is a Pregnancy Exposure Registry that monitors pregnancy outcomes in females exposed to THALOMID during pregnancy and that they can contact the Pregnancy Exposure Registry by calling 1-888-423-5436 [see Use in Specific Populations (8.1)].
Venous and Arterial Thromboembolism
Inform patients of the potential risk of developing venous thromboembolism (such as DVT and PE), ischemic heart disease (including myocardial infarction), and stroke, and discuss the need for appropriate prophylactic treatment [see Boxed Warning and Warnings and Precautions (5.3)].
Drowsiness and Somnolence
Inform patients of the risk of drowsiness and somnolence with the drug and to avoid situations where drowsiness or somnolence may be a problem and not to take other medications that may cause drowsiness or somnolence without adequate medical advice [see Warnings and Precautions (5.5)].
Peripheral Neuropathy
Inform patients of the risk of peripheral neuropathy and report the signs and symptoms associated with this event to their health care provider for further evaluation [see Warnings and Precautions (5.6)].
Dizziness and Orthostatic Hypotension
Inform patients of the risk of dizziness and orthostatic hypotension with the drug. Inform patients to sit upright for a few minutes prior to standing [see Warnings and Precautions (5.7)].
Neutropenia
Inform patients on the risk of developing neutropenia and the need to monitor their white blood cell count [see Warnings and Precautions (5.8)].
Thrombocytopenia
Inform patients of the risk of developing thrombocytopenia and the need to monitor their platelet count [see Warnings and Precautions (5.9)].
Increased HIV Viral Load
Inform HIV seropositive patients of the risk of increased viral load and the need to monitor viral load [see Warnings and Precautions (5.10)].
Bradycardia
Inform patients of the risk of bradycardia and report signs and symptoms associated with this event to their healthcare provider for evaluation [see Warnings and Precautions (5.11)].
Severe Cutaneous Reactions
Inform patients of the potential risk for severe skin reactions such as SJS, TEN and DRESS and report any signs and symptoms associated with these events to their healthcare provider for evaluation [see Warnings and Precautions (5.12)].
Seizures
Inform patients of the risk of seizures and report any seizure while taking THALOMID [see Warnings and Precautions (5.13)].
Tumor Lysis Syndrome
Inform patients of the potential risk of tumor lysis syndrome and report any signs and symptoms associated with this event to their healthcare provider for evaluation [see Warnings and Precautions (5.14)].
Contraceptive Risks
Inform patients that some contraceptive methods may pose a higher risk of adverse effects or may be medically contraindicated in some patients treated with THALOMID [see Warnings and Precautions (5.15), Drug Interactions (7.4, 7.7)].
Hypersensitivity
Inform patients of the potential for severe hypersensitivity reactions such as angioedema and anaphylaxis to THALOMID. Instruct patients to contact their healthcare provider right away for signs of a skin reaction. Advise patients to seek emergency medical attention for signs or symptoms of severe hypersensitivity reactions [see Warnings and Precautions (5.16)].
Marketed by:
Bristol-Myers Squibb Company
Princeton, NJ 08543 USA
THALOMID® and THALOMID REMS® are trademarks of Celgene Corporation, a Bristol-Myers Squibb company.
THALPLYPI.029/MG.027
| This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: March 2023 |
|||||
|
MEDICATION GUIDE
|
|||||
|
What is the most important information I should know about THALOMID? Before you begin taking THALOMID, you must read and agree to all of the instructions in the THALOMID REMS® program. For more information, call 1-888-423-5436 or go to www.thalomidrems.com. Before prescribing THALOMID, your healthcare provider will explain the THALOMID REMS program to you and have you sign the Patient-Physician Agreement Form. THALOMID can cause serious side effects including:
There is a pregnancy exposure registry that monitors the outcomes of females who take THALOMID during pregnancy, or if their male partner takes THALOMID and they are exposed during pregnancy. You can enroll in this registry by calling the REMS Call Center at the phone number listed above.
|
|||||
|
What is THALOMID?
It is not known if THALOMID is safe and effective in children under 12 years of age. |
|||||
|
Who should not take THALOMID?
|
|||||
|
What should I tell my healthcare provider before taking THALOMID?
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. THALOMID and other medicines may affect each other, causing serious side effects. Talk with your healthcare provider before taking any new medicines. Certain medicines can affect the way that birth control pills, injections, transdermal systems (patches), or implants work. You could become pregnant. See "What is the most important information I should know about THALOMID?"
|
|||||
|
How should I take THALOMID?
|
|||||
|
What should I avoid while taking THALOMID?
|
|||||
|
What are the possible side effects of THALOMID?
|
|||||
|
|
|
|
||
|
|||||
|
|
||||
|
|||||
|
|
||||
|
Get emergency medical help right away if you develop any of the following signs or symptoms during treatment with THALOMID: |
|||||
|
|
||||
Your healthcare provider may tell you to decrease your dose, temporarily stop or permanently stop taking THALOMID if you develop certain serious side effects during treatment with THALOMID. |
|||||
|
The most common side effects of THALOMID for treatment of multiple myeloma include: |
|||||
|
|
||||
|
The most common side effects of THALOMID for treatment of leprosy include: |
|||||
|
|
|
|||
|
These are not all the possible side effects of THALOMID. |
|||||
|
How should I store THALOMID?
Keep THALOMID and all medicines out of the reach of children. |
|||||
|
General information about the safe and effective use of THALOMID.
|
|||||
|
What are the ingredients in THALOMID?
Marketed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USA |
|||||




| THALOMID
thalidomide capsule |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| THALOMID
thalidomide capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| THALOMID
thalidomide capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| THALOMID
thalidomide capsule |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Celgene Corporation (174201137) |
Frequently asked questions
More about Thalomid (thalidomide)
- Check interactions
- Compare alternatives
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Drug class: leprostatics
- Breastfeeding
- En español