Stadol: Package Insert / Prescribing Info
Package insert / product label
Generic name: butorphanol tartrate
Dosage form: Injection, USP and Nasal Spray
Drug class: Opioids (narcotic analgesics)
Medically reviewed by Drugs.com. Last updated on Mar 25, 2025.
The Stadol brand name has been discontinued in the U.S. If generic versions of this product have been approved by the FDA, there may be generic equivalents available.
On This Page
Stadol Description
Butorphanol tartrate is a synthetically derived opioid agonist-antagonist analgesic of the phenanthrene series. The chemical name is (-)-17-(cyclobutylmethyl) morphinan-3, 14-diol [S-(R*,R*)] - 2,3 - dihydroxybutanedioate (1:1) (salt). The molecular formula is C21H29NO2,C4H6O6, which corresponds to a molecular weight of 477.55 and the following structural formula:
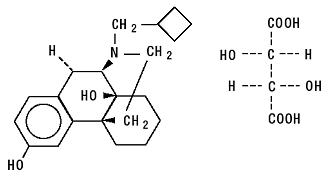
Butorphanol tartrate is a white crystalline substance. The dose is expressed as the tartrate salt. One milligram of the salt is equivalent to 0.68 mg of the free base. The n-octanol/aqueous buffer partition coefficient of butorphanol is 180:1 at pH 7.5.
STADOL (butorphanol tartrate) Injection, USP, is a sterile, parenteral, aqueous solution of butorphanol tartrate for intravenous or intramuscular administration. In addition to 1 or 2 mg of butorphanol tartrate, each mL of solution contains 3.3 mg of citric acid, 6.4 mg sodium citrate, and 6.4 mg sodium chloride, and 0.1 mg benzethonium chloride (in multiple dose vial only) as a preservative.
STADOL NS (butorphanol tartrate) Nasal Spray is an aqueous solution of butorphanol tartrate for administration as a metered spray to the nasal mucosa. Each bottle of STADOL NS contains 2.5 mL of a 10 mg/mL solution of butorphanol tartrate with sodium chloride, citric acid, and benzethonium chloride in purified water with sodium hydroxide and/or hydrochloric acid added to adjust the pH to 5.0. The pump reservoir must be fully primed (see PATIENT INSTRUCTIONS) prior to initial use. After initial priming each metered spray delivers an average of 1.0 mg of butorphanol tartrate and the 2.5-mL bottle will deliver an average of 14–15 doses of STADOL NS. If not used for 48 hours or longer, the unit must be reprimed (see PATIENT INSTRUCTIONS). With intermittent use requiring repriming before each dose, the 2.5-mL bottle will deliver an average of 8–10 doses of STADOL NS depending on how much repriming is necessary.
Stadol - Clinical Pharmacology
General Pharmacology and Mechanism of Action
Butorphanol is a mixed agonist-antagonist with low intrinsic activity at receptors of the µ-opioid type (morphine-like). It is also an agonist at κ-opioid receptors.
Its interactions with these receptors in the central nervous system apparently mediate most of its pharmacologic effects, including analgesia.
In addition to analgesia, CNS effects include depression of spontaneous respiratory activity and cough, stimulation of the emetic center, miosis, and sedation. Effects possibly mediated by non-CNS mechanisms include alteration in cardiovascular resistance and capacitance, bronchomotor tone, gastrointestinal secretory and motor activity, and bladder sphincter activity.
In an animal model, the dose of butorphanol tartrate required to antagonize morphine analgesia by 50% was similar to that for nalorphine, less than that for pentazocine and more than that for naloxone.
The pharmacological activity of butorphanol metabolites has not been studied in humans; in animal studies, butorphanol metabolites have demonstrated some analgesic activity.
In human studies of butorphanol (see Clinical Trials), sedation is commonly noted at doses of 0.5 mg or more. Narcosis is produced by 10–12 mg doses of butorphanol administered over 10–15 minutes intravenously.
Butorphanol, like other mixed agonist-antagonists with a high affinity for the κ-receptor, may produce unpleasant psychotomimetic effects in some individuals.
Nausea and/or vomiting may be produced by doses of 1 mg or more administered by any route.
In human studies involving individuals without significant respiratory dysfunction, 2 mg of butorphanol IV and 10 mg of morphine sulfate IV depressed respiration to a comparable degree. At higher doses, the magnitude of respiratory depression with butorphanol is not appreciably increased; however, the duration of respiratory depression is longer. Respiratory depression noted after administration of butorphanol to humans by any route is reversed by treatment with naloxone, a specific opioid antagonist (see OVERDOSAGE: Treatment).
Butorphanol tartrate demonstrates antitussive effects in animals at doses less than those required for analgesia.
Hemodynamic changes noted during cardiac catheterization in patients receiving single 0.025 mg/kg intravenous doses of butorphanol have included increases in pulmonary artery pressure, wedge pressure and vascular resistance, increases in left ventricular end diastolic pressure, and in systemic arterial pressure.
Pharmacodynamics
The analgesic effect of butorphanol is influenced by the route of administration. Onset of analgesia is within a few minutes for intravenous administration, within 15 minutes for intramuscular injection, and within 15 minutes for the nasal spray doses.
Peak analgesic activity occurs within 30–60 minutes following intravenous and intramuscular administration and within 1–2 hours following the nasal spray administration.
The duration of analgesia varies depending on the pain model as well as the route of administration, but is generally 3–4 hours with IM and IV doses as defined by the time 50% of patients required remedication. In postoperative studies, the duration of analgesia with IV or IM butorphanol was similar to morphine, meperidine, and pentazocine when administered in the same fashion at equipotent doses (see Clinical Trials). Compared to the injectable form and other drugs in this class, STADOL NS has a longer duration of action (4–5 hours) (see Clinical Trials).
Pharmacokinetics
STADOL Injection is rapidly absorbed after IM injection and peak plasma levels are reached in 20–40 minutes.
After nasal administration, mean peak blood levels of 0.9–1.04 ng/mL occur at 30–60 minutes after a 1 mg dose (see Table 1). The absolute bioavailability of STADOL NS is 60–70% and is unchanged in patients with allergic rhinitis. In patients using a nasal vasoconstrictor (oxymetazoline) the fraction of the dose absorbed was unchanged, but the rate of absorption was slowed. The peak plasma concentrations were approximately half those achieved in the absence of the vasoconstrictor.
Following its initial absorption/distribution phase, the single dose pharmacokinetics of butorphanol by the intravenous, intramuscular, and nasal routes of administration are similar (see Figure 1).
Figure 1—Butorphanol Plasma Levels After IV, IM, and Nasal Spray Administration of 2-mg Dose
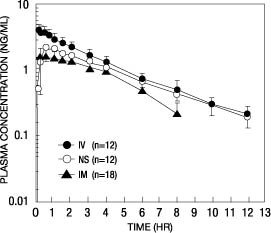
Serum protein binding is independent of concentration over the range achieved in clinical practice (up to 7 ng/mL) with a bound fraction of approximately 80%.
The volume of distribution of butorphanol varies from 305–901 liters and total body clearance from 52–154 liters/hour (see Table 1).
| (a) Young subjects (n=24) are from 20 to 40 years old and elderly (n=24) are greater than 65 years of age. | ||||
| (b) Time to peak plasma concentration. | ||||
| (c) Peak plasma concentration normalized to 1-mg dose. | ||||
| (d) Area under the plasma concentration-time curve after a 1-mg dose. | ||||
| (e) Mean (1 S.D.) | ||||
| (f) Derived from IV data. | ||||
| (g) (range of observed values) | ||||
| Table 1: | Mean Pharmacokinetic Parameters of Butorphanol in Young and Elderly Subjectsa | |||
|---|---|---|---|---|
| Intravenous | Nasal | |||
| Parameters | Young | Elderly | Young | Elderly |
| Tmaxb (h) | 0.62 (0.32)e
(0.15-1.50)g | 1.03 (0.74) (0.25-3.00) |
||
| Cmaxc (ng/mL) | 1.04 (0.40) (0.35-1.97) | 0.90 (0.57) (0.10-2.68) |
||
| AUC (inf)d
(h•ng/mL) | 7.24 (1.57) (4.40-9.77) | 8.71 (2.02) (4.76-13.03) | 4.93 (1.24) (2.16-7.27) | 5.24 (2.27) (0.30-10.34) |
| Half-life (h) | 4.56 (1.67) (2.06-8.70) | 5.61 (1.36) (3.25-8.79) | 4.74 (1.57) (2.89-8.79) | 6.56 (1.51) (3.75-9.17) |
| Absolute Bioavailability (%) | 69 (16) (44-13) | 61 (25) (3-121) |
||
| Volume of Distributionf (L) | 487 (155) (305-901) | 552 (124) (305-737) | ||
| Total Body Clearance (L/h) | 99 (23) (70-154) | 82 (21) (52-143) | ||
Dose proportionality for STADOL NS has been determined at steady state in doses up to 4 mg at 6 hour intervals. Steady state is achieved within 2 days. The mean peak plasma concentration at steady state was 1.8-fold (maximal 3-fold) following a single dose.
The drug is transported across the blood brain and placental barriers and into human milk (see PRECAUTIONS: Labor and Delivery and Nursing Mothers).
Butorphanol is extensively metabolized in the liver. Metabolism is qualitatively and quantitatively similar following intravenous, intramuscular, or nasal administration. Oral bioavailability is only 5–17% because of extensive first pass metabolism of butorphanol.
The major metabolite of butorphanol is hydroxybutorphanol, while norbutorphanol is produced in small amounts. Both have been detected in plasma following administration of butorphanol, with norbutorphanol present at trace levels at most time points. The elimination half-life of hydroxybutorphanol is about 18 hours and, as a consequence, considerable accumulation (~5-fold) occurs when butorphanol is dosed to steady state (1 mg transnasally q6h for 5 days).
Elimination occurs by urine and fecal excretion. When 3H labelled butorphanol is administered to normal subjects, most (70–80%) of the dose is recovered in the urine, while approximately 15% is recovered in the feces.
About 5% of the dose is recovered in the urine as butorphanol. Forty-nine percent is eliminated in the urine as hydroxybutorphanol. Less than 5% is excreted in the urine as norbutorphanol.
Butorphanol pharmacokinetics in the elderly differ from younger patients (see Table 1). The mean absolute bioavailability of STADOL NS in elderly women (48%) was less than that in elderly men (75%), young men (68%), or young women (70%). Elimination half-life is increased in the elderly (6.6 hours as opposed to 4.7 hours in younger subjects).
In renally impaired patients with creatinine clearances <30 mL/min, the elimination half-life was approximately doubled and the total body clearance was approximately one half (10.5 hours [clearance 150 L/h] compared to 5.8 hours [clearance 260 L/h] in healthy subjects). No effect on Cmax or Tmax was observed after a single dose.
After intravenous administration to patients with hepatic impairment, the elimination half-life of butorphanol was approximately tripled and total body clearance was approximately one half (half-life 16.8 hours, clearance 92 L/h) compared to healthy subjects (half-life 4.8 hours, clearance 175 L/h). The exposure of hepatically impaired patients to butorphanol was significantly greater (about 2-fold) than that in healthy subjects. Similar results were seen after nasal administration. No effect on Cmax or Tmax was observed after a single intranasal dose.
For further recommendations refer to PRECAUTIONS: Hepatic and Renal Disease, Drug Interactions, and Geriatric Use and CLINICAL PHARMACOLOGY: Individualization of Dosage.
Clinical Trials
The effectiveness of opioid analgesics varies in different pain syndromes. Studies with STADOL Injection have been performed in postoperative (primarily abdominal and orthopedic) pain and pain during labor and delivery, as preoperative and preanesthetic medication, and as a supplement to balanced anesthesia (see below).
Studies with STADOL NS have been performed in postoperative (general, orthopedic, oral, cesarean section) pain, in postepisiotomy pain, in pain of musculoskeletal origin, and in migraine headache pain (see below).
Use in the Management of Pain
Postoperative pain: The analgesic efficacy of STADOL Injection in postoperative pain was investigated in several double-blind active-controlled studies involving 958 butorphanol-treated patients. The following doses were found to have approximately equivalent analgesic effect: 2 mg butorphanol, 10 mg morphine, 40 mg pentazocine and 80 mg meperidine.
After intravenous administration of STADOL Injection, onset and peak analgesic effect occurred by the time of first observation (30 minutes). After intramuscular administration, pain relief onset occurred at 30 minutes or less, and peak effect occurred between 30 minutes and 1 hour. The duration of action of STADOL Injection was 3–4 hours when defined as the time necessary for pain intensity to return to pretreatment level or the time to retreatment.
The analgesic efficacy of STADOL NS was evaluated (approximately 35 patients per treatment group) in a general and orthopedic surgery trial. Single doses of STADOL NS (1 or 2 mg) and IM meperidine (37.5 or 75 mg) were compared. Analgesia provided by 1 and 2 mg doses of STADOL NS was similar to 37.5 and 75 mg meperidine, respectively, with onset of analgesia within 15 minutes and peak analgesic effect within 1 hour. The median duration of pain relief was 2.5 hours with 1 mg STADOL NS, 3.5 hours with 2 mg STADOL NS and 3.3 hours with either dose of meperidine.
In a postcesarean section trial, STADOL NS administered to 35 patients as two 1-mg doses 60 minutes apart was compared with a single 2-mg dose of STADOL NS or a single 2-mg IV dose of STADOL Injection (37 patients each). Onset of analgesia was within 15 minutes for all STADOL regimens. Peak analgesic effects of 2 mg intravenous STADOL Injection and STADOL NS were similar in magnitude. The duration of pain relief provided by both 2-mg STADOL NS regimens was approximately 4.5 hours and was greater than intravenous STADOL Injection (2.6 hours).
Migraine headache pain: The analgesic efficacy of two 1-mg doses 1 hour apart of STADOL NS in migraine headache pain was compared with a single dose of 10 mg IM methadone (31 and 32 patients, respectively). Significant onset of analgesia occurred within 15 minutes for both STADOL NS and IM methadone. Peak analgesic effect occurred at 2 hours for STADOL NS and 1.5 hours for methadone. The median duration of pain relief was 6 hours with STADOL NS and 4 hours with methadone as judged by the time when approximately half of the patients remedicated.
In two other trials in patients with migraine headache pain, a 2-mg initial dose of STADOL NS followed by an additional 1-mg dose 1 hour later (76 patients) was compared with either 75 mg IM meperidine (24 patients) or placebo (72 patients). Onset, peak activity, and duration were similar with both active treatments; however, the incidence of adverse experiences (nausea, vomiting, dizziness) was higher in these two trials with the 2-mg initial dose of STADOL NS than in the trial with the 1-mg initial dose.
Preanesthetic Medication
STADOL Injection (2 mg and 4 mg) and meperidine (80 mg) were studied for use as preanesthetic medication in hospitalized surgical patients. Patients received a single intramuscular dose of either STADOL Injection or meperidine approximately 90 minutes prior to anesthesia. The anesthesia regimen included barbiturate induction, followed by nitrous oxide and oxygen with halothane or enflurane, with or without a muscle relaxant.
Anesthetic preparation was rated as satisfactory in all 42 STADOL Injection patients regardless of the type of surgery.
Balanced Anesthesia
STADOL Injection administered intravenously (mean dose 2 mg) was compared to intravenous morphine sulfate (mean dose 10 mg) as premedication shortly before thiopental induction, followed by balanced anesthesia in 50 ASA Class 1 and 2 patients. Anesthesia was then maintained by repeated intravenous doses, averaging 4.6 mg STADOL Injection and 22.8 mg morphine per patient.
Anesthetic induction and maintenance were generally rated as satisfactory with both STADOL Injection (25 patients) and morphine (25 patients) regardless of the type of surgery performed. Emergence from anesthesia was comparable with both agents.
Labor
(see PRECAUTIONS)
The analgesic efficacy of intravenous STADOL Injection was studied in pain during labor. In a total of 145 patients, STADOL Injection (1 mg and 2 mg) was as effective as 40 mg and 80 mg of meperidine (144 patients) in the relief of pain in labor with no effect on the duration or progress of labor. Both drugs readily crossed the placenta and entered fetal circulation. The condition of the infants in these studies, determined by Apgar scores at 1 and 5 minutes (8 or above) and time to sustained respiration, showed that STADOL Injection had the same effects on the infants as meperidine.
In these studies, neurobehavioral testing in infants exposed to STADOL Injection at a mean of 18.6 hours after delivery showed no significant differences between treatment groups.
Individualization of Dosage
Use of butorphanol in geriatric patients, patients with renal impairment, patients with hepatic impairment, and during labor requires extra caution (see below and the appropriate sections in PRECAUTIONS).
STADOL Injection
For pain relief the recommended initial dosage regimen of STADOL Injection is 1 mg IV or 2 mg IM with repeated doses every 3 to 4 hours, as necessary. This dosage regimen is likely to be effective for the majority of patients. Dosage adjustments of STADOL Injection should be based on observations of its beneficial and adverse effects. The initial dose in the elderly and in patients with renal or hepatic impairment should generally be half the recommended adult dose (0.5 mg IV and 1.0 mg IM). Repeat doses in these patients should be determined by the patient’s response rather than at fixed intervals but will generally be no less than 6 hours (see PRECAUTIONS).
The usual preoperative dose is 2 mg IM given 60–90 minutes before surgery or 2 mg IV shortly before induction. This is approximately equivalent in sedative effect to 10 mg morphine or 80 mg of meperidine. This single preoperative dose should be individualized based on age, body weight, physical status, underlying pathological condition, use of other drugs, type of anesthesia to be used, and the surgical procedure involved.
During maintenance in balanced anesthesia the usual incremental dose of STADOL Injection is 0.5 to 1.0 mg IV. The incremental dose may be higher, up to 0.06 mg/kg (4 mg/70 kg), depending on previous sedative, analgesic, and hypnotic drugs administered. The total dose of STADOL Injection will vary; however, patients seldom require less than 4 mg or more than 12.5 mg (approximately 0.06 to 0.18 mg/kg).
As with other opioids of this class, STADOL Injection may not provide adequate intraoperative analgesia in every patient or under all conditions. A failure to achieve successful analgesia during balanced anesthesia is commonly reflected by increases in general sympathetic tone. Consequently, if blood pressure or heart rate continues to rise, consideration should be given to adding a potent volatile liquid inhalation anesthetic or another intravenous medication.
In labor, the recommended initial dose of STADOL Injection is 1 or 2 mg IM or IV in mothers with fetuses of 37 weeks gestation or beyond and without signs of fetal distress. Dosage adjustments of STADOL Injection in labor should be based on initial response with consideration given to concomitant analgesic or sedative drugs and the expected time of delivery. A dose should not be repeated in less than 4 hours nor administered less than 4 hours prior to the anticipated delivery (see PRECAUTIONS).
STADOL NS
The usual recommended dose for initial nasal administration is 1 mg (1 spray in one nostril). If adequate pain relief is not achieved within 60–90 minutes, an additional 1-mg dose may be given.
The initial dose sequence outlined above may be repeated in 3–4 hours as required after the second dose of the sequence.
For the management of severe pain, an initial dose of 2 mg (1 spray in each nostril) may be used in patients who will be able to remain recumbent in the event drowsiness or dizziness occurs. In such patients additional doses should not be given for 3-4 hours. The incidence of adverse events is higher with an initial 2-mg dose (see Clinical Trials).
The initial dose sequence in elderly patients and patients with renal or hepatic impairment should be limited to 1 mg followed, if needed, by 1 mg in 90-120 minutes. The repeat dose sequence in these patients should be determined by the patient’s response rather than at fixed times but will generally be no less than at 6-hour intervals (see PRECAUTIONS).
Indications and Usage for Stadol
STADOL (butorphanol tartrate) Injection and STADOL NS (butorphanol tartrate) Nasal Spray are indicated for the management of pain when the use of an opioid analgesic is appropriate.
STADOL Injection is also indicated as a preoperative or preanesthetic medication, as a supplement to balanced anesthesia, and for the relief of pain during labor.
Contraindications
STADOL Injection and STADOL NS are contraindicated in patients hypersensitive to butorphanol tartrate or the preservative benzethonium chloride in STADOL NS or STADOL Injection in the multi-dose vial.
Warnings
Patients Dependent on Narcotics
Because of its opioid antagonist properties, butorphanol is not recommended for use in patients dependent on narcotics. Such patients should have an adequate period of withdrawal from opioid drugs prior to beginning butorphanol therapy. In patients taking opioid analgesics chronically, butorphanol has precipitated withdrawal symptoms such as anxiety, agitation, mood changes, hallucinations, dysphoria, weakness, and diarrhea.
Because of the difficulty in assessing opioid tolerance in patients who have recently received repeated doses of narcotic analgesic medication, caution should be used in the administration of butorphanol to such patients.
Drug Abuse and Dependence
Drug Abuse—Butorphanol tartrate, by all routes of administration, has been associated with episodes of abuse. Of the cases received, there were more reports of abuse with the nasal spray formulation than with the injectable formulation.
Physical Dependence, Tolerance, and Withdrawal—Prolonged, continuous use of butorphanol tartrate may result in physical dependence or tolerance (a decrease in response to a given dose). Abrupt cessation of use by patients with physical dependence may result in symptoms of withdrawal.
Note—Proper patient selection, dose and prescribing limitations, appropriate directions for use, and frequent monitoring are important to minimize the risk of abuse and physical dependence. (See DRUG ABUSE AND DEPENDENCE.)
Precautions
General
Hypotension associated with syncope during the first hour of dosing with STADOL NS has been reported rarely, particularly in patients with past history of similar reactions to opioid analgesics. Therefore, patients should be advised to avoid activities with potential risks.
Head Injury and Increased Intracranial Pressure
As with other opioids, the use of butorphanol in patients with head injury may be associated with carbon dioxide retention and secondary elevation of cerebrospinal fluid pressure, drug-induced miosis, and alterations in mental state that would obscure the interpretation of the clinical course of patients with head injuries. In such patients, butorphanol should be used only if the benefits of use outweigh the potential risks.
Disorders of Respiratory Function or Control
Butorphanol may produce respiratory depression, especially in patients receiving other CNS active agents, or patients suffering from CNS diseases or respiratory impairment.
Hepatic and Renal Disease
In patients with hepatic or renal impairment, the initial dose of STADOL Injection should generally be half the recommended adult dose (0.5 mg IV and 1.0 mg IM). Repeat doses in these patients should be determined by the patient’s response rather than at fixed intervals but will generally be no less than 6 hours apart. The initial dose sequence of STADOL NS should be limited to 1 mg followed, if needed, by 1 mg in 90–120 minutes. The repeat dose sequence in these patients should be determined by the patient’s response rather than at fixed times but will generally be at intervals of no less than 6 hours (see CLINICAL PHARMACOLOGY: Pharmacokinetics and Individualization of Dosage).
Cardiovascular Effects
Because butorphanol may increase the work of the heart, especially the pulmonary circuit, the use of butorphanol in patients with acute myocardial infarction, ventricular dysfunction, or coronary insufficiency should be limited to those situations where the benefits clearly outweigh the risk (see CLINICAL PHARMACOLOGY).
Severe hypertension has been reported rarely during butorphanol therapy. In such cases, butorphanol should be discontinued and the hypertension treated with antihypertensive drugs. In patients who are not opioid dependent, naloxone has also been reported to be effective.
Use in Ambulatory Patients
- Opioid analgesics, including butorphanol, impair the mental and physical abilities required for the performance of potentially dangerous tasks such as driving a car or operating machinery. Effects such as drowsiness or dizziness can appear, usually within the first hour after dosing. These effects may persist for varying periods of time after dosing. Patients who have taken butorphanol should not drive or operate dangerous machinery for at least 1 hour and until the effects of the drug are no longer present.
- Alcohol should not be consumed while using butorphanol. Concurrent use of butorphanol with drugs that affect the central nervous system (eg, alcohol, barbiturates, tranquilizers, and antihistamines) may result in increased central nervous system depressant effects such as drowsiness, dizziness, and impaired mental function.
- Butorphanol is one of a class of drugs known to be abused and thus should be handled accordingly (see DRUG ABUSE AND DEPENDENCE).
- Patients should be instructed on the proper use of STADOL NS (see PATIENT INSTRUCTIONS).
Drug Interactions
Concurrent use of butorphanol with central nervous system depressants (eg, alcohol, barbiturates, tranquilizers, and antihistamines) may result in increased central nervous system depressant effects. When used concurrently with such drugs, the dose of butorphanol should be the smallest effective dose and the frequency of dosing reduced as much as possible when administered concomitantly with drugs that potentiate the action of opioids.
In healthy volunteers, the pharmacokinetics of a 1-mg dose of butorphanol administered as STADOL NS were not affected by the coadministration of a single 6-mg subcutaneous dose of sumatriptan. However, in another study in healthy volunteers, the pharmacokinetics of butorphanol were significantly altered (29% decrease in AUC and 38% decrease in Cmax) when a 1-mg dose of STADOL NS was administered 1 minute after a 20-mg dose of sumatriptan nasal spray. (The two drugs were administered in opposite nostrils.) When the STADOL NS was administered 30 minutes after the sumatriptan nasal spray, the AUC of butorphanol increased 11% and Cmax decreased 18%. In neither case were the pharmacokinetics of sumatriptan affected by coadministration with STADOL NS. These results suggest that the analgesic effect of STADOL NS may be diminished when it is administered shortly after sumatriptan nasal spray, but by 30 minutes any such reduction in effect should be minimal.
The safety of using STADOL NS and IMITREX® (sumatriptan) Nasal Spray during the same episode of migraine has not been established. However, it should be noted that both products are capable of producing transient increases in blood pressure.
The pharmacokinetics of a 1-mg dose of butorphanol administered as STADOL NS were not affected by the coadministration of cimetidine (300 mg QID). Conversely, the administration of STADOL NS (1 mg butorphanol QID) did not alter the pharmacokinetics of a 300-mg dose of cimetidine.
It is not known if the effects of butorphanol are altered by other concomitant medications that affect hepatic metabolism of drugs (erythromycin, theophylline, etc.), but physicians should be alert to the possibility that a smaller initial dose and longer intervals between doses may be needed.
The fraction of STADOL NS absorbed is unaffected by the concomitant administration of a nasal vasoconstrictor (oxymetazoline), but the rate of absorption is decreased. Therefore, a slower onset can be anticipated if STADOL NS is administered concomitantly with, or immediately following, a nasal vasoconstrictor.
No information is available about the use of butorphanol concurrently with MAO inhibitors.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted in mice and rats given butorphanol tartrate in the diet up to 60 mg/kg/day (180 mg/m2 for mice and 354 mg/m2 for rats). There was no evidence of carcinogenicity in either species in these studies.
Butorphanol was not genotoxic in S. typhimurium or E. coli assays or in unscheduled DNA synthesis and repair assays conducted in cultured human fibroblast cells.
Rats treated orally with 160 mg/kg/day (944 mg/m2) had a reduced pregnancy rate. However, a similar effect was not observed with a 2.5 mg/kg/day (14.75 mg/m2) subcutaneous dose.
Pregnancy
Pregnancy Category C: Reproduction studies in mice, rats, and rabbits during organogenesis did not reveal any teratogenic potential to butorphanol. However, pregnant rats treated subcutaneously with butorphanol at 1 mg/kg (5.9 mg/m2) had a higher frequency of stillbirths than controls. Butorphanol at 30 mg/kg/oral (360 mg/m2) and 60 mg/kg/oral (720 mg/m2) also showed higher incidences of post-implantation loss in rabbits.
There are no adequate and well-controlled studies of STADOL in pregnant women before 37 weeks of gestation. STADOL should be used during pregnancy only if the potential benefit justifies the potential risk to the infant.
Labor and Delivery
There have been rare reports of infant respiratory distress/apnea following the administration of STADOL Injection during labor. The reports of respiratory distress/apnea have been associated with administration of a dose within 2 hours of delivery, use of multiple doses, use with additional analgesic or sedative drugs, or use in preterm pregnancies (see OVERDOSAGE: Treatment).
In a study of 119 patients, the administration of 1 mg of IV STADOL Injection during labor was associated with transient (10–90 minutes) sinusoidal fetal heart rate patterns, but was not associated with adverse neonatal outcomes. In the presence of an abnormal fetal heart rate pattern, STADOL Injection should be used with caution.
STADOL NS is not recommended during labor or delivery because there is no clinical experience with its use in this setting.
Nursing Mothers
Butorphanol has been detected in milk following administration of STADOL Injection to nursing mothers. The amount an infant would receive is probably clinically insignificant (estimated 4 µg/L of milk in a mother receiving 2 mg IM four times a day).
Although there is no clinical experience with the use of STADOL NS in nursing mothers, it should be assumed that butorphanol will appear in the milk in similar amounts following the nasal route of administration.
Pediatric Use
Butorphanol is not recommended for use in patients below 18 years of age because safety and efficacy have not been established in this population.
Geriatric Use
Of the approximately 1500 patients treated with STADOL Injection in clinical studies, 15% were 61 years of age or older and 1% were 76 years or older. Of the approximately 1700 patients treated with STADOL NS in clinical studies, 8% were 65 years of age or older and 2% were 75 years or older.
Due to changes in clearance, the mean half-life of butorphanol is increased by 25% (to over 6 hours) in patients over the age of 65 years (see CLINICAL PHARMACOLOGY: Pharmacokinetics). Elderly patients may be more sensitive to the side effects of butorphanol. In clinical studies of STADOL NS, elderly patients had an increased frequency of headache, dizziness, drowsiness, vertigo, constipation, nausea and/or vomiting, and nasal congestion compared with younger patients. There are insufficient efficacy data for patients ≥65 years to determine whether they respond differently from younger patients.
The initial dose of STADOL Injection recommended for elderly patients should generally be half the recommended adult dose (0.5 mg IV and 1.0 mg IM). Repeat doses should be determined by the patient’s response rather than at fixed intervals, but will generally be no less than 6 hours apart (see CLINICAL PHARMACOLOGY: Individualization of Dosage).
Initially a 1-mg dose of STADOL NS should generally be used in geriatric patients and 90–120 minutes should elapse before administering a second 1-mg dose, if needed (see CLINICAL PHARMACOLOGY: Individualization of Dosage).
Butorphanol and its metabolites are known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection.
Adverse Reactions/Side Effects
Clinical Trial Experience
A total of 2446 patients were studied in premarketing clinical trials of butorphanol. Approximately half received STADOL Injection with the remainder receiving STADOL NS. In nearly all cases the type and incidence of side effects with butorphanol by any route were those commonly observed with opioid analgesics.
The adverse experiences described below are based on data from short-term and long-term clinical trials in patients receiving butorphanol by any route. There has been no attempt to correct for placebo effect or to subtract the frequencies reported by placebo-treated patients in controlled trials.
The most frequently reported adverse experiences across all clinical trials with STADOL Injection and STADOL NS were somnolence (43%), dizziness (19%), nausea and/or vomiting (13%). In long-term trials with STADOL NS only, nasal congestion (13%) and insomnia (11%) were frequently reported.
The following adverse experiences were reported at a frequency of 1% or greater in clinical trials and were considered to be probably related to the use of butorphanol.
Body as a Whole: asthenia/lethargy, headache, sensation of heat.
Cardiovascular: vasodilation, palpitations.
Digestive: anorexia, constipation, dry mouth, nausea and/or vomiting, stomach pain.
Nervous: anxiety, confusion, dizziness, euphoria, floating feeling, insomnia, nervousness, paresthesia, somnolence, tremor.
Respiratory: bronchitis, cough, dyspnea, epistaxis, nasal congestion, nasal irritation, pharyngitis, rhinitis, sinus congestion, sinusitis, upper respiratory infection.
Skin and Appendages: sweating/clammy, pruritus.
Special Senses: blurred vision, ear pain, tinnitus, unpleasant taste.
The following adverse experiences were reported with a frequency of less than 1% in clinical trials and were considered to be probably related to the use of butorphanol.
Cardiovascular: hypotension, syncope.
Nervous: abnormal dreams, agitation, dysphoria, hallucinations, hostility, withdrawal symptoms.
Skin and Appendages: rash/hives.
Urogenital: impaired urination.
The following infrequent additional adverse experiences were reported in a frequency of less than 1% of the patients studied in short-term STADOL NS trials and under circumstances where the association between these events and butorphanol administration is unknown. They are being listed as alerting information for the physician.
Body as a Whole: edema.
Cardiovascular: chest pain, hypertension, tachycardia.
Nervous: depression.
Respiratory: shallow breathing.
Postmarketing Experience
Postmarketing experience with STADOL NS and STADOL Injection has shown an adverse event profile similar to that seen during the premarketing evaluation of butorphanol by all routes of administration. Adverse experiences that were associated with the use of STADOL NS or STADOL Injection and that are not listed above have been chosen for inclusion below because of their seriousness, frequency of reporting, or probable relationship to butorphanol. Because they are reported voluntarily from a population of unknown size, estimates of frequency cannot be made. These adverse experiences include apnea, convulsion, delusion, drug dependence, excessive drug effect associated with transient difficulty speaking and/or executing purposeful movements, overdose, and vertigo. Reports of butorphanol overdose with a fatal outcome have usually but not always been associated with ingestion of multiple drugs.
Related/similar drugs
Drug Abuse and Dependence
STADOL (butorphanol tartrate) Injection and STADOL NS (butorphanol tartrate) Nasal Spray are listed in Schedule IV of the Controlled Substances Act (CSA).
Proper patient selection, dose and prescribing limitations, appropriate directions for use, and frequent monitoring are important to minimize the risk of abuse and physical dependence with butorphanol tartrate. Special care should be exercised in administering butorphanol to patients with a history of drug abuse or to patients receiving the drug on a continuous basis for an extended period.
Clinical Trial Experience
In all clinical trials, less than 1% of patients using STADOL NS had experiences that suggested the development of physical dependence or tolerance. Much of this information is based on experience with patients who did not have prolonged continuous exposure to STADOL NS. However, in one controlled clinical trial where patients with chronic pain from nonmalignant disease were treated with STADOL NS (n=303) or placebo (n=99) for up to 6 months, overuse (which may suggest the development of tolerance) was reported in nine (2.9%) patients receiving STADOL NS and no patients receiving placebo. Probable withdrawal symptoms were reported in eight (2.6%) patients using STADOL NS and no patients receiving placebo in the chronic nonmalignant pain study. Most of these patients abruptly discontinued STADOL NS after extended use or high doses. Symptoms suggestive of withdrawal included anxiety, agitation, tremulousness, diarrhea, chills, sweats, insomnia, confusion, incoordination, and hallucinations.
Overdosage
Clinical Manifestations
The clinical manifestations of butorphanol overdose are those of opioid drugs in general. Consequences of overdose vary with the amount of butorphanol ingested and individual response to the effects of opiates. The most serious symptoms are hypoventilation, cardiovascular insufficiency, coma, and death. Butorphanol overdose may be associated with ingestion of multiple drugs (see ADVERSE REACTIONS: Postmarketing Experience).
Overdose can occur due to accidental or intentional misuse of butorphanol, especially in young children who may gain access to the drug in the home.
Treatment
The management of suspected butorphanol overdosage includes maintenance of adequate ventilation, peripheral perfusion, normal body temperature, and protection of the airway. Patients should be under continuous observation with adequate serial measures of mental state, responsiveness, and vital signs. Oxygen and ventilatory assistance should be available with continual monitoring by pulse oximetry if indicated. In the presence of coma, placement of an artificial airway may be required. An adequate intravenous portal should be maintained to facilitate treatment of hypotension associated with vasodilation.
The use of a specific opioid antagonist such as naloxone should be considered. As the duration of butorphanol action usually exceeds the duration of action of naloxone, repeated dosing with naloxone may be required.
In managing cases of suspected butorphanol overdosage, the possibility of multiple drug ingestion should always be considered.
Stadol Dosage and Administration
Factors to be considered in determining the dose are age, body weight, physical status, underlying pathological condition, use of other drugs, type of anesthesia to be used, and surgical procedure involved. Use in the elderly, in patients with hepatic or renal disease, or in labor requires extra caution (see PRECAUTIONS and CLINICAL PHARMACOLOGY: Individualization of Dosage). The following doses are for patients who do not have impaired hepatic or renal function and who are not on CNS active agents.
Use for Pain
STADOL Injection
Intravenous: The usual recommended single dose for IV administration is 1 mg repeated every 3 to 4 hours as necessary. The effective dosage range, depending on the severity of pain, is 0.5 to 2 mg repeated every 3 to 4 hours.
Intramuscular: The usual recommended single dose for IM administration is 2 mg in patients who will be able to remain recumbent, in the event drowsiness or dizziness occurs. This may be repeated every 3 to 4 hours, as necessary. The effective dosage range depending on the severity of pain is 1 to 4 mg repeated every 3 to 4 hours. There are insufficient clinical data to recommend single doses above 4 mg.
STADOL NS
The usual recommended dose for initial nasal administration is 1 mg (1 spray in one nostril). Adherence to this dose reduces the incidence of drowsiness and dizziness. If adequate pain relief is not achieved within 60–90 minutes, an additional 1-mg dose may be given.
The initial dose sequence outlined above may be repeated in 3–4 hours as required after the second dose of the sequence.
Depending on the severity of the pain, an initial dose of 2 mg (1 spray in each nostril) may be used in patients who will be able to remain recumbent in the event drowsiness or dizziness occurs. In such patients single additional 2-mg doses should not be given for 3–4 hours.
Use as Preoperative/Preanesthetic Medication
The preoperative medication dosage of STADOL Injection should be individualized (see CLINICAL PHARMACOLOGY: Individualization of Dosage). The usual adult dose is 2 mg IM, administered 60–90 minutes before surgery. This is approximately equivalent in sedative effect to 10 mg morphine or 80 mg meperidine.
Use in Balanced Anesthesia
The usual dose of STADOL Injection is 2 mg IV shortly before induction and/or 0.5 to 1.0 mg IV in increments during anesthesia. The increment may be higher, up to 0.06 mg/kg (4 mg/70 kg), depending on previous sedative, analgesic, and hypnotic drugs administered. The total dose of STADOL Injection will vary; however, patients seldom require less than 4 mg or more than 12.5 mg (approximately 0.06 to 0.18 mg/kg).
The use of STADOL NS is not recommended because it has not been studied in induction or maintenance of anesthesia.
Labor
In patients at full term in early labor a 1–2 mg dose of STADOL Injection IV or IM may be administered and repeated after 4 hours. Alternative analgesia should be used for pain associated with delivery or if delivery is expected to occur within 4 hours.
If concomitant use of STADOL with drugs that may potentiate its effects is deemed necessary (see PRECAUTIONS: Drug Interactions), the lowest effective dose should be employed.
The use of STADOL NS is not recommended as it has not been studied in labor.
Safety and Handling
STADOL Injection is supplied in sealed delivery systems that have a low risk of accidental exposure to health care workers. Ordinary care should be taken to avoid aerosol generation while preparing a syringe for use. Following skin contact, rinsing with cool water is recommended.
STADOL NS is an open delivery system with increased risk of exposure to health care workers.
In the priming process, a certain amount of butorphanol may be aerosolized; therefore, the pump sprayer should be aimed away from the patient or other people or animals.
The disposal of Schedule IV controlled substances must be consistent with State and Federal Regulations. The unit should be disposed of by unscrewing the cap, rinsing the bottle, and placing the parts in a waste container.
How is Stadol supplied
STADOL® (butorphanol tartrate) Injection, USP, for IM or IV use is available as follows:
- NDC 0015-5644-20—2 mg per mL, 2–mL vial
- NDC 0015-5645-20—1 mg per mL, 1–mL vial
- NDC 0015-5646-20—2 mg per mL, 1–mL vial
- NDC 0015-5648-20—2 mg per mL, 10–mL multi-dose vial
STADOL NS® (butorphanol tartrate) Nasal Spray
STADOL NS is supplied in a child-resistant prescription vial containing a metered-dose spray pump with protective clip and dust cover, a bottle of nasal spray solution, and a patient instruction leaflet. On average, one bottle will deliver 14–15 doses if no repriming is necessary.
- NDC 0087-5650-41—10 mg per mL, 2.5–mL bottle.
-
U.S. Patent No. 4,464,378
PHARMACIST ASSEMBLY INSTRUCTIONS FOR STADOL NS NASAL SPRAY
The pharmacist will assemble STADOL NS prior to dispensing to the patient, according to the following instructions:
- Open the child-resistant prescription vial and remove the spray pump and solution bottle.
- Assemble STADOL NS by first unscrewing the white cap from the solution bottle and screwing the pump unit tightly onto the bottle. Make sure the clear cover is on the pump unit.
- Return the STADOL NS bottle to the child-resistant prescription vial for dispensing to the patient.
Storage Conditions
Store at 25° C (77° F) controlled room temperature. See USP. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
IMITREX® is the registered trademark of the GlaxoSmithKline Group of Companies.
| STADOL
butorphanol tartrate injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| STADOL
butorphanol tartrate injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| STADOL
butorphanol tartrate injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| STADOL
butorphanol tartrate injection, solution |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| STADOL NS
butorphanol tartrate spray |
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
|
|||||||||||||||
| Labeler - Bristol-Myers Squibb |
More about Stadol (butorphanol)
- Check interactions
- Compare alternatives
- Reviews (79)
- Side effects
- Dosage information
- During pregnancy
- Drug class: Opioids (narcotic analgesics)
- Breastfeeding