Monjuvi: Package Insert / Prescribing Info
Package insert / product label
Generic name: tafasitamab-cxix injection
Dosage form: injection, powder, lyophilized, for solution
Drug class: CD19 monoclonal antibodies
J Code (medical billing code): J9349 (2 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jul 13, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
MONJUVI® (tafasitamab-cxix) for injection, for intravenous use
Initial U.S. Approval: 2020
Indications and Usage for Monjuvi
MONJUVI is a CD19-directed cytolytic antibody indicated:
Diffuse Large B-cell Lymphoma
- in combination with lenalidomide for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, including DLBCL arising from low grade lymphoma, and who are not eligible for autologous stem cell transplant (ASCT).
This indication is approved under accelerated approval based on overall response rate. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s). (1.1)
Follicular Lymphoma
- in combination with a rituximab product and lenalidomide for the treatment of adult patients with previously treated follicular lymphoma (FL). (1.2)
Monjuvi Dosage and Administration
- Administer premedications prior to starting MONJUVI. (2.4)
- See Full Prescribing Information for instructions on preparation and administration. (2.6)
Diffuse Large B-cell Lymphoma
- The recommended dosage of MONJUVI is 12 mg/kg as an intravenous infusion according to the following dosing schedule: (2.2)
- Cycle 1: Days 1, 4, 8, 15, and 22 of each 28-day cycle.
- Cycles 2 and 3: Days 1, 8, 15, and 22 of each 28-day cycle.
- Cycle 4 and beyond: Days 1 and 15 of each 28-day cycle.
- Administer MONJUVI in combination with lenalidomide for a maximum of 12 cycles and then continue MONJUVI as monotherapy until disease progression or unacceptable toxicity. (2.2)
- See Full Prescribing Information for instructions on preparation and administration. (2.3, 2.4)
Follicular Lymphoma
- The recommended dosage of MONJUVI is 12 mg/kg as an intravenous infusion according to the following dosing schedule: (2.3)
- Cycles 1 to 3: Days 1, 8, 15 and 22 of each 28-day cycle.
- Cycles 4 to 12: Days 1 and 15 of each 28-day cycle.
- Administer MONJUVI in combination with rituximab (Cycles 1 to 5) and lenalidomide (Cycles 1 to 12). (2.3)
Dosage Forms and Strengths
For injection: 200 mg of tafasitamab-cxix as lyophilized powder in single-dose vial for reconstitution.(3)
Contraindications
None. (4)
Warnings and Precautions
- Infusion-Related Reactions: Monitor patients frequently during infusion. Interrupt or discontinue infusion based on severity. (2.5, 5.1)
- Myelosuppression: Monitor complete blood counts. Manage using dose modifications and growth factor support. Interrupt or discontinue MONJUVI based on severity. (2.5, 5.2)
- Infections: Bacterial, fungal and viral infections can occur during and following MONJUVI. Monitor patients for infections. (2.5, 5.3)
- Embryo-Fetal Toxicity: May cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and use of effective contraception. (5.4)
Adverse Reactions/Side Effects
The most common adverse reactions (≥ 20%) in patients with relapsed or refractory DLBCL are neutropenia, fatigue, anemia, diarrhea, thrombocytopenia, cough, pyrexia, peripheral edema, respiratory tract infection, and decreased appetite. (6.1)
The most common adverse reactions (≥ 20%) in patients with previously treated FL are viral infections, diarrhea, rash, fatigue, constipation, and bacterial infections.
To report SUSPECTED ADVERSE REACTIONS, contact MORPHOSYS US INC. at 1-844-667-1992 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
Lactation: Advise not to breastfeed. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 7/2025
Full Prescribing Information
1. Indications and Usage for Monjuvi
1.1 Diffuse Large B-cell Lymphoma
MONJUVI, in combination with lenalidomide, is indicated for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not otherwise specified, including DLBCL arising from low grade lymphoma, and who are not eligible for autologous stem cell transplant (ASCT).
This indication is approved under accelerated approval based on overall response rate [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
2. Monjuvi Dosage and Administration
2.1 Important Dosing Information
MONJUVI should be administered by a healthcare professional with immediate access to emergency equipment and appropriate medical support to manage infusion-related reactions [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage for Diffuse Large B-cell Lymphoma
The recommended dose of MONJUVI is 12 mg/kg based on actual body weight administered as an intravenous infusion in combination with lenalidomide, according to the dosing schedule in Table 1.
| Cycle* | Dosing Schedule |
|---|---|
|
|
| Cycle 1 | Days 1, 4, 8, 15, and 22 |
| Cycles 2 and 3 | Days 1, 8, 15, and 22 |
| Cycle 4 and beyond | Days 1 and 15 |
Administer MONJUVI in combination with lenalidomide 25 mg for a maximum of 12 cycles, then continue MONJUVI as monotherapy until disease progression or unacceptable toxicity [see Clinical Studies (14.1)]. Refer to the lenalidomide prescribing information for lenalidomide dosage recommendations.
2.3 Recommended Dosage for Follicular Lymphoma
The recommended dose of MONJUVI is 12 mg/kg based on actual body weight administered as an intravenous infusion in combination with rituximab and lenalidomide.
MONJUVI dosing schdedules are provided in Table 2.
| Cycle* | Dosing Schedule |
| Cycles 1 to 3 | Days 1, 8, 15, and 22 |
| Cycles 4 to 12 | Days 1 and 15 |
Administer MONJUVI in combination with rituximab 375 mg/m2 (Cycles 1 to 5) and lenalidomide 20 mg (Cycles 1 to 12) [see Clinical Studies (14.2)]. Refer to the lenalidomide prescribing information and the rituximab prescribing information for the respective dosage recommendations.
2.4 Recommended Premedications
Administer premedications 30 minutes to 2 hours prior to starting MONJUVI infusion to minimize infusion-related reactions [see Warnings and Precautions (5.1)]. Premedications may include acetaminophen, histamine H1 receptor antagonists, histamine H2 receptor antagonists, and/or glucocorticosteroids.
For patients not experiencing infusion-related reactions during the first 3 infusions, premedication is optional for subsequent infusions.
If a patient experiences an infusion-related reaction, administer premedications before each subsequent infusion.
2.5 Dosage Modifications for Adverse Reactions
The recommended dosage modifications for adverse reactions are summarized in Table 3.
| Adverse Reaction | Severity | Dosage Modification |
|---|---|---|
| Infusion-related reactions[seeWarnings and Precautions (5.1)] | Grade 2 (moderate) |
|
| Grade 3 (severe) |
|
|
| Grade 4 (life-threatening) |
|
|
| Myelosuppression[seeWarnings and Precautions (5.2)] | Platelet count of 50,000/ mcL or less |
|
| Neutrophil count of 1,000/ mcL or less for at least 7 days OR Neutrophil count of 1,000/ mcL or less with an increase of body temperature to 100.4°F (38°C) or higher OR Neutrophil count less than 500/mcL |
|
2.6 Preparation and Administration
Reconstitute and dilute MONJUVI prior to infusion.
Reconstitution
- Calculate the dose (mg) and determine the number of vials needed.
- Reconstitute each 200 mg MONJUVI vial with 5 mL Sterile Water for Injection, USP with the stream directed toward the wall of each vial to obtain a final concentration of 40 mg/mL tafasitamab-cxix.
- Gently swirl the vial(s) until completely dissolved. Do not shake or swirl vigorously. Complete dissolution may take up to 5 minutes.
- Visually inspect the reconstituted solution for particulate matter or discoloration. The reconstituted solution should appear as a colorless to slightly yellow solution. Discard the vial(s) if the solution is cloudy, discolored, or contains visible particles.
- Use the reconstituted MONJUVI solution immediately. If needed, store the reconstituted solution in the vial for a maximum of 12 hours either refrigerated at 36°F to 46°F (2°C to 8°C) or room temperature at 68°F to 77°F (20°C to 25°C) before dilution. Protect from light during storage.
Dilution
- Determine the volume (mL) of the 40 mg/mL reconstituted MONJUVI solution needed based on the required dose.
- Remove a volume equal to the required MONJUVI solution from a 250 mL 0.9% Sodium Chloride Injection, USP infusion bag and discard it.
- Withdraw the necessary amount of MONJUVI and slowly dilute in the infusion bag that contains the 0.9% Sodium Chloride Injection, USP to a final concentration of 2 mg/mL to 8 mg/mL. Discard any unused portion of MONJUVI remaining in the vial.
- Gently mix the intravenous bag by slowly inverting the bag. Do not shake. Visually inspect the infusion bag with the diluted MONJUVI infusion solution for particulate matter and discoloration prior to administration.
- If not used immediately, store the diluted MONJUVI infusion solution refrigerated for up to 18 hours at 36°F to 46°F (2°C to 8°C) and/or at room temperature for up to 12 hours at 68°F to 77°F (20°C to 25°C). The room temperature storage includes time for infusion. Protect from light during storage.
Do not shake or freeze the reconstituted or diluted infusion solutions.
Administration
- Administer MONJUVI as an intravenous infusion.
- For the first infusion, use an infusion rate of 70 mL/h for the first 30 minutes, then, increase the rate so that the infusion is administered within 1.5 to 2.5 hours.
- Administer all subsequent infusions within 1.5 to 2 hours.
- Infuse the entire contents of the bag containing MONJUVI.
- Do not co-administer other drugs through the same infusion line.
- No incompatibilities have been observed between MONJUVI with infusion containers made of polypropylene (PP), polyvinylchloride (PVC), polyethylene (PE), polyethylenterephthalate (PET), or glass and infusion sets made of polyurethane (PUR) or PVC.
3. Dosage Forms and Strengths
For injection: 200 mg of tafasitamab-cxix as white to slightly yellowish lyophilized powder in single-dose vial for reconstitution and further dilution.
5. Warnings and Precautions
5.1 Infusion-Related Reactions
MONJUVI can cause infusion-related reactions [see Adverse Reactions (6.1)]. In L-MIND, infusion-related reactions occurred in 6% of the 81 patients with DLBCL who received MONJUVI in combination with lenalidomide. Eighty percent of infusion-related reactions occurred during cycle 1 or 2. In inMIND, infusion-related reactions occurred in 16% of the 274 patients with FL who received MONJUVI in combination with rituximab and lenalidomide. Signs and symptoms included fever, chills, rash, flushing, dyspnea, and hypertension. These reactions were managed with temporary interruption of the infusion and/or with supportive medication.
Premedicate patients prior to starting MONJUVI infusion [see Dosage and Administration (2.4)]. Monitor patients frequently during infusion. Based on the severity of the infusion-related reaction, interrupt or discontinue MONJUVI [see Dosage and Administration (2.5)]. Institute appropriate medical management.
5.2 Myelosuppression
MONJUVI can cause serious or severe myelosuppression, including neutropenia, thrombocytopenia, and anemia [see Adverse Reactions (6.1)]. In L-MIND, Grade 3 neutropenia occurred in 25% of the 81 patients with DLBCL who received MONJUVI in combination with lenalidomide, Grade 3 thrombocytopenia in 12%, and Grade 3 anemia in 7%. Grade 4 neutropenia occurred in 25% and Grade 4 thrombocytopenia in 6%. Neutropenia led to treatment discontinuation in 3.7% of the 81 patients with DLBCL.
In inMIND, Grade 3 neutropenia occurred in 28% of the 274 patients with FL who received MONJUVI in combination with rituximab and lenalidomide, Grade 3 thrombocytopenia in 4%, and Grade 3 anemia in 4%. Grade 4 neutropenia occurred in 18% and Grade 4 thrombocytopenia in 2%. Neutropenia and thrombocytopenia led to treatment discontinuation in 2 patients each.
Monitor CBC prior to administration of each treatment cycle and throughout treatment. Monitor patients with neutropenia for signs of infection. Consider granulocyte colony-stimulating factor administration. Withhold MONJUVI based on the severity of the adverse reaction [see Dosage and Administration (2.3)].Refer to the lenalidomide prescribing information and the rituximab prescribing information for the respective dosage modifications.
5.3 Infections
Fatal and serious infections, including opportunistic infections, occurred in patients during treatment with MONJUVI and following the last dose [see Adverse Reactions (6.1)].
In L-MIND, 73% of the 81 patients with DLBCL who received MONJUVI in combination with lenalidomide, developed an infection. The most frequent infections were respiratory tract infection (24%), urinary tract infection (17%), bronchitis (16%), nasopharyngitis (10%) and pneumonia (10%). Grade 3 or higher infection occurred in 30% of the 81 patients. The most frequent Grade 3 or higher infection was pneumonia (7%). Infection-related deaths were reported in 2.5% of the 81 patients.
In inMIND, 68% of the 274 patients with FL who received MONJUVI in combination with rituximab and lenalidomide, developed an infection. The most frequent infections were COVID-19 (31%), pneumonia (12%), upper respiratory tract infection (9%), urinary tract infection (7%), respiratory tract infection (7%), and nasopharyngitis (6%). Grade 3 or 4 infection occurred in 24% of the 274 patients. The most frequent Grade 3 or 4 infection was pneumonia (8%) and COVID-19 (6%). Infection-related deaths were reported in 3 of the 274 patients.
Monitor patients for signs and symptoms of infection and manage infections as appropriate.
5.4 Embryo-Fetal Toxicity
Based on its mechanism of action, MONJUVI may cause fetal B-cell depletion when administered to a pregnant woman. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with MONJUVI and for at least 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)].
The combination of MONJUVI with lenalidomide and of MONJUVI with rituximab and lenalidomide is contraindicated in pregnant women because lenalidomide can cause birth defects and death of the unborn child. Refer to the lenalidomide prescribing information on use during pregnancy.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infusion-related reactions [see Warnings and Precautions (5.1)]
- Myelosuppression [see Warnings and Precautions (5.2)]
- Infections [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in other clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed or Refractory Diffuse Large B-Cell Lymphoma
The safety of MONJUVI in patients with DLBCL was evaluated in L-MIND [see Clinical Studies (14.1)]. Patients (N = 81) received MONJUVI 12 mg/kg intravenously in combination with lenalidomide for a maximum of 12 cycles, followed by MONJUVI as monotherapy until disease progression or unacceptable toxicity as follows:
- Cycle 1: Days 1, 4, 8, 15, and 22 of the 28-day cycle;
- Cycles 2 and 3: Days 1, 8, 15, and 22 of each 28-day cycle;
- Cycles 4 and beyond: Days 1 and 15 of each 28-day cycle.
Among patients who received MONJUVI, 57% were exposed for 6 months or longer, 42% were exposed for greater than one year, and 24% were exposed for greater than two years.
Serious adverse reactions occurred in 52% of patients who received MONJUVI. Serious adverse reactions in ≥ 6% of patients included infections (26%), including pneumonia (7%), and febrile neutropenia (6%). Fatal adverse reactions occurred in 5% of patients who received MONJUVI, including cerebrovascular accident (1.2%), respiratory failure (1.2%), progressive multifocal leukoencephalopathy (1.2%), and sudden death (1.2%).
Permanent discontinuation of MONJUVI or lenalidomide due to an adverse reaction occurred in 25% of patients and permanent discontinuation of MONJUVI due to an adverse reaction occurred in 15%. The most frequent adverse reactions which resulted in permanent discontinuation of MONJUVI were infections (5%), nervous system disorders (2.5%), respiratory, thoracic and mediastinal disorders (2.5%).
Dosage interruptions of MONJUVI or lenalidomide due to an adverse reaction occurred in 69% of patients and dosage interruption of MONJUVI due to an adverse reaction occurred in 65%. The most frequent adverse reactions which required a dosage interruption of MONJUVI were blood and lymphatic system disorders (41%), and infections (27%).
The most common adverse reactions (≥ 20%) were neutropenia, fatigue, anemia, diarrhea, thrombocytopenia, cough, pyrexia, peripheral edema, respiratory tract infection, and decreased appetite.
Table 4 summarizes the adverse reactions in L-MIND.
| Adverse Reaction | MONJUVI (N=81) |
|
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) |
|
|
||
| Blood and lymphatic system disorders
|
||
| Neutropenia | 51 | 49 |
| Anemia | 36 | 7 |
| Thrombocytopenia | 31 | 17 |
| Febrile neutropenia | 12 | 12 |
| General disorders and administration site conditions
|
||
| Fatigue* | 38 | 3.7 |
| Pyrexia | 24 | 1.2 |
| Peripheral edema | 24 | 0 |
| Gastrointestinal disorders
|
||
| Diarrhea | 36 | 1.2 |
| Constipation | 17 | 0 |
| Abdominal pain† | 15 | 1.2 |
| Nausea | 15 | 0 |
| Vomiting | 15 | 0 |
| Respiratory, thoracic and mediastinal disorders
|
||
| Cough | 26 | 1.2 |
| Dyspnea | 12 | 1.2 |
| Infections
|
||
| Respiratory tract infection‡ | 24 | 4.9 |
| Urinary tract infection§ | 17 | 4.9 |
| Bronchitis | 16 | 1.2 |
| Metabolism and nutrition disorders
|
||
| Decreased appetite | 22 | 0 |
| Hypokalemia | 19 | 6 |
| Musculoskeletal and connective tissue disorders
|
||
| Back pain | 19 | 2.5 |
| Muscle spasms | 15 | 0 |
| Skin and subcutaneous tissue disorders
|
||
| Rash¶ | 15 | 2.5 |
| Pruritus | 10 | 1.2 |
Clinically relevant adverse reactions in < 10% of patients with relapsed or refractory DLBCL who received MONJUVI in L-MIND were:
- Blood and lymphatic system disorders: lymphopenia (6%)
- General disorders and administration site conditions: infusion-related reaction (6%)
- Infections: sepsis (4.9%)
- Investigations: weight decreased (4.9%)
- Musculoskeletal and connective tissue disorders: arthralgia (9%), pain in extremity (9%), musculoskeletal pain (2.5%)
- Neoplasms benign, malignant and unspecified: basal cell carcinoma (1.2%)
- Nervous system disorders: headache (9%), paresthesia (7%), dysgeusia (6%)
- Respiratory, thoracic and mediastinal disorders: nasal congestion (4.9%), exacerbation of chronic obstructive pulmonary disease (1.2%)
- Skin and subcutaneous tissue disorders: erythema (4.9%), alopecia (2.5%), hyperhidrosis (2.5%)
Table 5 summarizes the laboratory abnormalities in L-MIND.
| Laboratory Abnormality | MONJUVI* | |
|---|---|---|
| All Grades (%) | Grade 3 or 4 (%) |
|
|
||
| Chemistry
|
||
| Glucose increased | 49 | 5 |
| Calcium decreased | 47 | 1.4 |
| Gamma glutamyl transferase increased | 34 | 5 |
| Albumin decreased | 26 | 0 |
| Magnesium decreased | 22 | 0 |
| Urate increased | 20 | 7 |
| Phosphate decreased | 20 | 5 |
| Creatinine increased | 20 | 1.4 |
| Aspartate aminotransferase increased | 20 | 0 |
| Coagulation
|
||
| Activated partial thromboplastin time increased | 46 | 4.1 |
Previously Treated Follicular Lymphoma
The safety of MONJUVI in patients with FL was evaluated in 546 patients in the inMIND trial [see Clinical Studies (14.2)]. Patients received MONJUVI 12 mg/kg (N = 274) or placebo (N = 272) intravenously for a maximum of 12 cycles in combination with rituximab 375 mg/m2 (Cycles 1 to 5) and lenalidomide 20 mg (Cycles 1 to 12), as follows:
- Cycle 1 to 3: Days 1, 8, 15, and 22 of each 28-day cycle;
- Cycles 4 to 12: Days 1 and 15 of each 28-day cycle.
Among patients who received MONJUVI in combination with rituximab and lenalidomide, the median number of treatment cycles was 12 (range, 1 - 12).
Serious adverse reactions occurred in 19% of patients who received MONJUVI in combination with rituximab and lenalidomide. Serious adverse reactions in ≥ 2% of patients included viral infections (12%), bacterial infections (3%), and febrile neutropenia (3%). Fatal adverse reactions occurred in 3 patients who received MONJUVI in combination with rituximab and lenalidomide, including COVID-19 (2 patients) and sepsis (1 patient).
Permanent discontinuation of MONJUVI due to an adverse reaction occurred in 8% of patients who received MONJUVI in combination with rituximab and lenalidomide. The most frequent adverse reactions which resulted in permanent discontinuation of MONJUVI were viral infections (3%) and pyrexia (1%).
Dosage interruptions of MONJUVI due to an adverse reaction occurred in 67% of patients who received MONJUVI in combination with rituximab and lenalidomide. The most frequent adverse reactions which required a dosage interruption were neutropenia (37%), viral infections (24%), and bacterial infections (5%).
The most common adverse reactions (≥ 20%) were viral infections, diarrhea, rash, fatigue, constipation, and bacterial infections.
Table 6 summarizes the adverse reactions in inMIND.
|
||||||
| Adverse Reaction | All Grades (%) | Grade 3 or 4 (%) | ||||
|
MONJUVI in Combination with Rituximab and Lenalidomide (N = 274) |
Placebo in Combination with Rituximab and Lenalidomide (N = 272) |
MONJUVI in Combination with Rituximab and Lenalidomide (N = 274) |
Placebo in Combination with Rituximab and Lenalidomide (N = 272) |
|||
| Infections | ||||||
| Viral infections* | 41 | 32 | 11 | 4.4 | ||
| Bacterial infections* | 25 | 24 | 6 | 7 | ||
| Gastrointestinal disorders
|
||||||
| Diarrhea | 38 | 28 | 0.7 | 1.8 | ||
| Constipation | 29 | 25 | 0.7 | 0 | ||
| Abdominal pain*
| 12 | 17 | 0 | 2.2 | ||
| Skin and subcutaneous tissue disorders
|
||||||
| Rash*
| 37 | 32 | 2.9 | 1.5 | ||
| Pruritus | 16 | 10 | 0.4 | 0 | ||
| General disorders and administration site conditions | ||||||
| Fatigue*
| 35 | 26 | 2.9 | 0.7 | ||
| Pyrexia | 19 | 16 | 1.5 | 2.2 | ||
| Injury, poisoning and procedural complications | ||||||
| Infusion-related reaction | 16 | 15 | 0.7 | 0.4 | ||
| Nervous system disorders
|
||||||
| Headache | 10 | 7 | 0.4 | 0 | ||
Clinically relevant adverse reactions in < 10% of patients with previously treated FL who received MONJUVI in inMIND were:
- Blood and lymphatic system disorders: febrile neutropenia (4.4%)
- General disorders and administration site conditions: chills (4.7%)
- Metabolism and nutrition disorders: tumor lysis syndrome (0.7%)
Table 7 summarizes the laboratory abnormalities in inMIND.
|
||||
| Laboratory Abnormality | All Grades (%)* | Grade 3 or 4 (%)* | ||
| MONJUVI in Combination with Rituximab and Lenalidomide | Placebo in Combination with Rituximab and Lenalidomide | MONJUVI in Combination with Rituximab and Lenalidomide
| Placebo in Combination with Rituximab and Lenalidomide | |
| Hematology | ||||
| Decreased hemoglobin | 60 | 54 | 9 | 7 |
| Decreased leukocytes | 71 | 68 | 21 | 15 |
| Increased leukocytes | 22 | 21 | 0 | 0 |
| Decreased lymphocytes | 57 | 50 | 22 | 19 |
| Decreased neutrophils | 75 | 71 | 48 | 44 |
| Decreased platelets | 40 | 43 | 8 | 9 |
| Chemistry | ||||
| Glucose increased | 27 | 28 | 0.4 | 1.1 |
| Creatinine increased | 29 | 30 | 1.5 | 0.7 |
| Aspartate aminotransferase increased | 29 | 32 | 0 | 0.4 |
| Alanine aminotransferase increased | 47 | 42 | 1.5 | 1.5 |
| Alkaline phosphatase increased | 33 | 27 | 0 | 0 |
| Lactate dehydrogenase increased | 26 | 29 | 0 | 0 |
| Potassium decreased | 24 | 24 | 3.3 | 2.9 |
| Sodium decreased | 24 | 22 | 1.5 | 0.7 |
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, MONJUVI may cause fetal B-cell depletion when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. There are no available data on MONJUVI use in pregnant women to evaluate for a drug-associated risk. Animal reproductive toxicity studies have not been conducted with tafasitamab-cxix.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
MONJUVI is administered in combination with lenalidomide for up to 12 cycles. Lenalidomide can cause embryo-fetal harm and is contraindicated for use in pregnancy. Refer to the lenalidomide prescribing information for additional information. Lenalidomide is only available through a REMS program.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Immunoglobulin G (IgG) monoclonal antibodies are transferred across the placenta. Based on its mechanism of action, MONJUVI may cause depletion of fetal CD19 positive immune cells. Defer administering live vaccines to neonates and infants exposed to tafasitamab-cxix in utero until a hematology evaluation is completed.
Data
Animal Data
Animal reproductive studies have not been conducted with tafasitamab-cxix. Tafasitamab-cxix is an IgG antibody and thus has the potential to cross the placental barrier permitting direct fetal exposure and depleting fetal B lymphocytes.
8.2 Lactation
Risk Summary
There are no data on the presence of tafasitamab-cxix in human milk or the effects on the breastfed child or milk production. Maternal immunoglobulin G is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to MONJUVI are unknown. Because of the potential for serious adverse reactions in the breastfed child, advise women not to breastfeed during treatment with MONJUVI and for at least 3 months after the last dose. Refer to lenalidomide prescribing information for additional information.
8.3 Females and Males of Reproductive Potential
MONJUVI can cause fetal B-cell depletion when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Refer to the prescribing information for lenalidomide for pregnancy testing requirements prior to initiating the combination of MONJUVI with lenalidomide or the combination of MONJUVI with rituximab and lenalidomide.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with MONJUVI and for at least 3 months after the last dose. Additionally, refer to the lenalidomide prescribing information for additional recommendations for contraception.
Males
Refer to the lenalidomide prescribing information for recommendations.
8.4 Pediatric Use
The safety and effectiveness of MONJUVI in pediatric patients have not been established.
8.5 Geriatric Use
Relapsed or Refractory Diffuse Large B-Cell Lymphoma
Among 81 patients who received MONJUVI and lenalidomide in L-MIND, 72% were 65 years and older, while 38% were 75 years and older. Clinical studies of MONJUVI did not include sufficient numbers of patients aged 65 and older to determine whether effectiveness differs compared to that of younger subjects. Patients 65 years and older had more serious adverse reactions (57%) than younger patients (39%).
Previously Treated Follicular Lymphoma
Among the 274 patients with FL who received MONJUVI in combination with rituximab and lenalidomide in inMIND, 50% were 65 years and older and 20% were 75 years and older. No clinically meaningful differences in safety or effectiveness were observed between these patients and younger patients but greater sensitivity of some older individuals cannot be ruled out.
11. Monjuvi Description
Tafasitamab-cxix is a humanized CD19-directed cytolytic monoclonal antibody that contains an IgG1/2 hybrid Fc-domain with 2 amino acid substitutions to modify the Fc-mediated functions of the antibody. It is produced by recombinant DNA technology in mammalian cells (Chinese hamster ovary). Tafasitamab-cxix has a molecular weight of approximately 150 kDa.
MONJUVI (tafasitamab-cxix) for injection is supplied as a sterile, preservative-free, white to slightly yellowish lyophilized powder in a single-dose vial for intravenous use after reconstitution and further dilution. After reconstitution with 5 mL of Sterile Water for Injection, USP, the resulting concentration is 40 mg/mL with a pH of 6.0. Each single-dose vial contains 200 mg tafasitamab-cxix, citric acid monohydrate (3.7 mg), polysorbate 20 (1 mg), sodium citrate dihydrate (31.6 mg) and trehalose dihydrate (378.3 mg).
12. Monjuvi - Clinical Pharmacology
12.1 Mechanism of Action
Tafasitamab-cxix is an Fc-modified monoclonal antibody that binds to CD19 antigen expressed on the surface of pre-B and mature B lymphocytes and on several B-cell malignancies, including diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL).
Upon binding to CD19, tafasitamab-cxix mediates B-cell lysis through apoptosis and immune effector mechanisms, including antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP).
In studies conducted in vitro in DLBCL tumor cells, tafasitamab-cxix in combination with lenalidomide resulted in increased ADCC activity compared to tafasitamab-cxix or lenalidomide alone. In a preclinical study, the combination of tafasitamab and rituximab improved efficacy compared to single-agent treatments in models of aggressive B-cell lymphoma in vitro and in vivo.
12.2 Pharmacodynamics
Tafasitamab-cxix reduced peripheral blood B cell counts by 97% after eight days of treatment in patients with relapsed or refractory DLBCL. Nadir, with a reduction of 100%, was reached within 16 weeks of treatment.
Circulating B-cells decreased to undetectable levels (nearly 0 cells/microliter) after administration of the recommended dosage of MONJUVI in patients with FL who had detectable B-cells at treatment initiation by Cycle 1 Day 8 and the depletion was sustained while patients remained on treatment.
12.3 Pharmacokinetics
Mean trough concentrations (± standard deviation) were 177 (± 66) μg/mL following administration of MONJUVI at 12 mg/kg on Days 1, 8, 15, and 22 in Cycles 1-3 , and 166 (± 75) μg/mL following administration of MONJUVI at 12 mg/kg on Days 1 and 15 in Cycles 4 - 6. Mean maximum tafasitamab-cxix serum concentrations were 491 (±128) μg/mL.
Distribution
The total volume of distribution at steady state for tafasitamab-cxix was 6.7 L (95% CI: 2.9, 10.5 L).
Elimination
The clearance of tafasitamab-cxix was 0.48 L/day (CV: 28.2%) and terminal elimination half-life was 11.3 days (95% CI: 4.9, 17.8 days).
Specific Populations
Bodyweight (37.6 to 163 kg) has a significant effect on the pharmacokinetics of tafasitamab-cxix, with higher clearance and volume of distribution expected with higher body weight. No clinically meaningful differences in the pharmacokinetics of tafasitamab-cxix were observed based on age (16 to 90 years), sex, race (Asian versus non-Asian), renal impairment (CLcr 15 to < 90 mL/min estimated by the Cockcroft-Gault equation), or hepatic impairment (total bilirubin 1 to > 3 times ULN and any AST).
Drug Interaction Studies
No clinically meaningful differences in tafasitamab-cxix pharmacokinetics were observed when used concomitantly with lenalidomide and with the combination of rituximab and lenalidomide.
12.6 Immunogenicity
As with all therapeutic proteins, there is the potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assays. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other tafasitamab products may be misleading.
Overall, no treatment-emergent or treatment-boosted anti-tafasitamab antibodies were observed. No clinically meaningful differences in the pharmacokinetics, efficacy, or safety profile of tafasitamab-cxix were observed in 2.5% of 81 patients with relapsed or refractory DLBCL with pre-existing anti-tafasitamab antibodies in L-MIND.
Anti-drug antibodies (ADAs) were evaluated in 327 patients who received MONJUVI in inMIND. The incidence of tafasitamab-cxix treatment-emergent ADAs was 0.9% (3/327) using a bridging enzyme-linked immunosorbent assay. No neutralizing antibodies were detected. There was no apparent clinically meaningful effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, or effectiveness of MONJUVI over the treatment duration of 322.5 days.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity and genotoxicity studies have not been conducted with tafasitamab-cxix.
Fertility studies have not been conducted with tafasitamab-cxix.
In the 13-week repeat-dose general toxicity study in cynomolgus monkeys, no adverse effects on male and female reproductive organs were observed up to the highest dose tested, 100 mg/kg/week (approximately 9 times the human exposure based on AUC at the clinical dose of 12 mg/kg/week).
14. Clinical Studies
14.1 Diffuse Large B-cell Lymphoma
The efficacy of MONJUVI in combination with lenalidomide followed by MONJUVI as monotherapy was evaluated in L-MIND,an open-label, multicenter, single arm trial (NCT02399085). Eligible patients had relapsed or refractory DLBCL after 1 to 3 prior systemic therapies, including a CD20-directed cytolytic antibody, and were not candidates for high dose chemotherapy (HDC) followed by autologous stem cell transplantation (ASCT). Patients received MONJUVI 12 mg/kg intravenously in combination with lenalidomide (25 mg orally on Days 1 to 21 of each 28-day cycle) for a maximum of 12 cycles, followed by MONJUVI as monotherapy until disease progression or unacceptable toxicity as follows:
- Cycle 1: Days 1, 4, 8, 15 and 22 of the 28-day cycle;
- Cycles 2 and 3: Days 1, 8, 15 and 22 of each 28-day cycle;
- Cycles 4 and beyond: Days 1 and 15 of each 28-day cycle.
Of the 71 patients with DLBCL confirmed by central laboratory who received the combination therapy, the median age was 71 years (range: 41 to 86 years); 55% were males, and 100% had received a prior CD20-containing therapy. Race was collected in 92% of patients; of these, 95% were White, and 3% were Asian. The median number of prior therapies was two; 49% had one prior line of treatment, and 51% had 2 to 4 prior lines. Thirty-two patients (45%) were refractory to their last prior therapy and 30 (42%) were refractory to rituximab. Nine patients (13%) had received prior ASCT. The primary reasons patients were not candidates for ASCT included age (47%), refractoriness to salvage chemotherapy (27%), comorbidities (13%) and refusal of high dose chemotherapy/ASCT (13%).
Efficacy was established based on best overall response rate, defined as the proportion of complete and partial responders, and duration of response, as assessed by an Independent Review Committee using the International Working Group Response Criteria (Cheson, 2007). Results are summarized in Table 8.
|
|
| N = 71 | |
|
Best overall response rate, n (%) (95% CI) Complete response rate Partial response rate |
39 (55%) (43%, 67%) 37% 18% |
|
Duration of Response Median (range) in months* |
21.7 (0, 24) |
14.2 Follicular Lymphoma
The efficacy of MONJUVI in combination with rituximab and lenalidomide in patients with previously treated FL was evaluated in inMIND, a randomized, double-blind, placebo-controlled trial (NCT04680052). A total of 548 patients at least 18 years of age with histologically confirmed Grade 1, 2, or 3a relapsed or refractory FL were enrolled in the study. Eligible patients had received at least 1 prior systemic therapy, including an anti-CD20 antibody, had at least one measurable nodal or extranodal lesion by CT or MRI scan, had adequate bone marrow, liver, and renal function, and had Eastern Cooperative Oncology Group (ECOG) performance status of 0 – 2.
Patients were randomized in a 1:1 ratio to receive MONJUVI or placebo in combination with rituximab and lenalidomide. Randomization was stratified by progression of disease within 24 months after initial diagnosis (POD24) (yes versus no), refractoriness to prior CD20-directed antibody therapy (yes versus no), and the number of prior lines of therapy (< 2 versus ≥ 2). Dosing in each treatment arm was as follows:
- MONJUVI 12 mg/kg intravenously (Days 1, 8, 15, and 22 of Cycles 1 to 3 and on Days 1 and 15 of Cycles 4 to 12) in combination with rituximab 375 mg/m⊃; intravenously (Days 1, 8, 15, and 22 of Cycle 1 and on Day 1 of Cycles 2 to 5) and lenalidomide 20 mg orally once daily (Days 1 to 21 of Cycles 1 to 12).
- Placebo intravenously (Days 1, 8, 15, and 22 of Cycles 1 to 3 and on Days 1 and 15 of Cycles 4 to 12) in combination with rituximab 375 mg/m⊃; intravenously (Days 1, 8, 15, and 22 of Cycle 1 and on Day 1 of Cycles 2 to 5) and lenalidomide 20 mg orally once daily (Days 1 to 21 of Cycles 1 to 12).
The baseline demographic and disease characteristics in the inMIND trial are summarized in Table 9.
|
||
| Parameter |
MONJUVI in Combination with Rituximab and Lenalidomide (N = 273) |
Placebo in Combination with Rituximab and Lenalidomide (N = 275) |
| Age (years) | ||
|
Median (minimum, maximum) | 64 (36, 88) | 64 (31, 85) |
| Age group, n (%) | ||
|
< 65 years | 137 (50) | 139 (51) |
|
≥ 65 years | 136 (50) | 136 (50) |
| Sex, n (%) | ||
|
Male | 150 (55) | 149 (54) |
|
Female | 123 (45) | 126 (46) |
| Race, n (%) | ||
|
White | 219 (80) | 219 (80) |
|
Asian | 40 (15) | 42 (15) |
|
Other races | 3 (1) | 4 (2) |
|
Not reported | 11 (4) | 10 (4) |
| FL grade at trial entry, n (%) | ||
|
Grade 1 | 61 (22) | 51 (19) |
|
Grade 2 | 142 (52) | 152 ( 55) |
|
Grade 3a | 67 (25) | 71 (26) |
| Relapsed or refractory status to the most recent prior therapy* , n (%) | ||
|
Relapsed | 148 (54) | 164 (60) |
|
Refractory | 112 (41) | 97 (35) |
|
Indeterminate† | 13 (5) | 14 (5) |
| FLIPI score at baseline, n (%) | ||
|
Low (0 – 1) | 57 (21) | 57 (21) |
|
Intermediate (2) | 79 (29) | 67 (25) |
|
High (≥ 3) | 137 (50) | 150 (55) |
|
Missing | 0 (0.0) | 1 (0.4) |
| ECOG score at baseline, n (%) | ||
|
0 | 181 (66) | 192 (70) |
|
1 | 85 (31) | 75 (27) |
|
2 | 7 (3) | 8 (3) |
| POD24 status at trial entry, n (%) | ||
|
Yes | 109 (40) | 110 (40) |
|
No | 164 (60) | 165 (60) |
| Refractoriness to prior anti-CD20 therapy, n (%) | ||
|
Yes | 94 (34) | 94 (34) |
|
No | 179 (66) | 181 (66) |
| Number of prior lines of therapy, n (%) | ||
|
< 2 | 141 (52) | 143 (52) |
|
≥ 2 | 132 (48) | 132 (48) |
|
FLIPI = follicular lymphoma international prognostic index; ECOG = Eastern Cooperative Oncology Group; POD24 = progression of disease within 24 months after initial diagnosis |
||
The major efficacy outcome measure of the inMIND trial was investigator-assessed progression-free survival (PFS), defined as the time from randomization to first documented disease progression, or death from any cause, whichever occurs first. Additional efficacy outcome measures included investigator-assessed positron emission tomography (PET)–complete response (CR) rate, defined as a complete metabolic response at any time after start of treatment, overall survival (OS), overall response rate (ORR), and duration of response (DOR).The median duration of PFS follow-up was 14.3 months (95% CI: 11.8, 15) in the MONJUVI group and 14.1 months (95% CI: 11.5, 15) in the placebo group. Efficacy results are summarized in Table 9 and Figure 1.
| Endpoint |
MONJUVI in Combination with Rituximab and Lenalidomide (N = 273) |
Placebo in Combination with Rituximab and Lenalidomide (N = 275) |
| Progression-free survival* † | ||
| Patients with event, n (%) | 75 (27.5) | 131 (47.6) |
|
Disease progression | 67 (24.5) | 124 (45.1) |
|
Death | 8 (2.9) | 7 (2.5) |
| Median PFS (months) (95% CI)‡ | 22.4 (19.2, NE) | 13.9 (11.5, 16.4) |
| Hazard ratio§(95% CI) | 0.43 (0.32, 0.58) | |
| p-value | < 0.0001 | |
| Positron Emission Tomography–Complete Response Rate* | (N = 251) | (N = 254) |
| PET-CR rate (95% CI)¶ | 49.4 (43.1, 55.8) | 39.8 (33.7, 46.1) |
| Odds ratio (95% CI)# | 1.5 (1, 2.1) | |
| p-value | 0.0286 | |
|
PFS = progression-free survival; CI = confidence interval; NE = not evaluable; CMR = complete metabolic response. |
||
Figure 1: Kaplan-Meier Curve for Progression-Free Survival by Investigator Assessment in inMIND
Figure 1: Kaplan-Meier Curve for Progression-Free Survival by Investigator Assessment in inMIND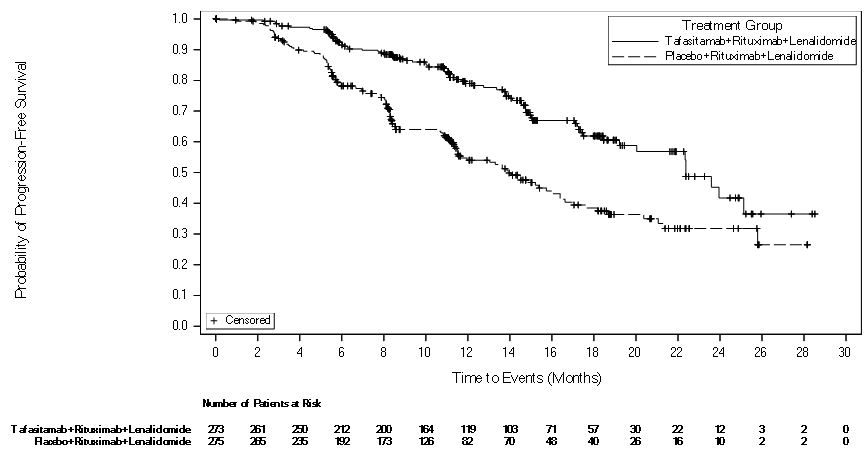
At the interim analysis, the key secondary endpoint of OS was immature and median OS was not reached in either treatment group [hazard ratio (95% CI) = 0.59 (0.31, 1.13); p-value = 0.1061). ORR was 83.5% (95% CI: 78.6%, 87.7%) in the MONJUVI group and 72.4% (95% CI: 66.7%, 77.6%) in the placebo group. Median DOR was 21.2 months (95% CI: 19.5, NE) in the MONJUVI group and 13.6 months (95% CI: 12.4, 18.6) in the placebo group [hazard ratio (95% CI) = 0.47 (0.33, 0.68).
PFS by Independent Review Committee (IRC) assessment was consistent with PFS by investigator assessment, with an estimated hazard ratio of 0.41 (95% CI: 0.29, 0.56). The median PFS by IRC assessment was not reached (95% CI: 19.3, not evaluable) in the MONJUVI group and 16 months (95% CI: 13.9, 21.1) in the placebo group.
16. How is Monjuvi supplied
MONJUVI (tafasitamab-cxix) for injection is a sterile, preservative-free, white to slightly yellowish lyophilized powder for reconstitution supplied as a 200 mg single-dose vial.
Each 200 mg vial is individually packaged in a carton (NDC 73535–208–01).
Store refrigerated at 36°F to 46°F (2°C to 8°C) in the original carton to protect from light. Do not shake. Do not freeze.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Infusion-Related Reactions
Advise patients to contact their healthcare provider if they experience signs and symptoms of infusion-related reactions [see Warnings and Precautions (5.1)].
Myelosuppression
Inform patients about the risk of myelosuppression. Advise patients to immediately contact their healthcare provider for a fever of 100.4°F (38°C) or greater or signs or symptoms of bruising or bleeding. Advise patients of the need for periodic monitoring of blood counts [see Warnings and Precautions (5.2)].
Infections
Inform patients about the risk of infections. Advise patients to immediately contact their healthcare provider for a fever of 100.4°F (38°C) or greater or signs or symptoms of infection [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
- Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.4), Use in Specific Population (8.1)].
- Advise females of reproductive potential to use effective contraception during treatment with MONJUVI and for at least 3 months after the last dose [see Use in Specific Populations (8.3)].
- Advise patients that lenalidomide has the potential to cause fetal harm and has specific requirements regarding contraception, pregnancy testing, blood and sperm donation, and transmission in sperm. Lenalidomide is only available through a REMS program [see Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment with MONJUVI and for at least 3 months after the last dose [see Use in Specific Populations (8.2)].
Manufactured by:
Incyte Corporation
Wilmington, DE 19803
U.S. License No. 228
MONJUVI and the MONJUVI logo are registered trademarks of Incyte.
Patent Information: www.incyte.com/patents
© 20YY Incyte Corporation. All rights reserved.
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Issued: M/20YY | |
| PATIENT INFORMATION
MONJUVI® (mon-JOO-vee) (tafasitamab-cxix) for injection |
||
|
What is MONJUVI?
It is not known if MONJUVI is safe and effective in children. |
||
Before you receive MONJUVI, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. |
||
How will I receive MONJUVI?
|
||
| What are the possible side effects of MONJUVI? MONJUVI may cause serious side effects, including:
|
||
|
|
|
| The most common side effects of MONJUVI when given with rituximab and lenalidomide in people with FL include: | ||
|
|
|
| These are not all the possible side effects of MONJUVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
||
| General information about the safe and effective use of MONJUVI.
Medicines are sometimes prescribed for purposes other than those listed in this Patient Information. If you would like more information about MONJUVI, talk with your healthcare provider. You can ask your healthcare provider for information about MONJUVI that is written for health professionals. |
||
|
What are the ingredients in MONJUVI?
U.S. License No. 2228 MONJUVI and the MONJUVI logo are registered trademarks of Incyte © 20YY Incyte Corporation. All rights reserved. |
||

| MONJUVI
tafasitamab-cxix injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Incyte Corporation (556967347) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| MILLMOUNT HEALTHCARE LIMITED | 986018132 | label(73535-208) , pack(73535-208) | |
Biological Products Related to Monjuvi
Find detailed information on biosimilars for this medication.
Frequently asked questions
More about Monjuvi (tafasitamab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (1)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: CD19 monoclonal antibodies
- Breastfeeding
- En español
