Frova: Package Insert / Prescribing Info
Package insert / product label
Generic name: frovatriptan succinate
Dosage form: tablet, film coated
Drug class: Antimigraine agents
Medically reviewed by Drugs.com. Last updated on Sep 15, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
FROVA® (frovatriptan succinate) tablets, for oral use
Initial U.S. Approval: 2001
Indications and Usage for Frova
FROVA is a serotonin (5-HT1B/1D) receptor agonist (triptan) indicated for the acute treatment of migraine with or without aura in adults (1)
Limitations of Use
Frova Dosage and Administration
- 1 tablet taken with fluids. Second tablet may be taken 2 hours after initial dose if headache recurs following initial relief. Total dose not to exceed 3 tablets in any 24-hour period (2)
Dosage Forms and Strengths
Tablets: 2.5 mg (3)
Contraindications
- History of coronary artery disease or coronary artery vasospasm (4)
- Wolff-Parkinson-White syndrome or other cardiac accessory conduction pathway disorders (4)
- History of stroke, transient ischemic attack, or hemiplegic or basilar migraine (4)
- Peripheral vascular disease (4)
- Ischemic bowel disease (4)
- Uncontrolled hypertension (4)
- Recent (within 24 hours) use of treatment with another 5-HT1 agonist, or an ergotamine-containing medication (4)
- Hypersensitivity to FROVA (angioedema and anaphylaxis seen) (4)
Warnings and Precautions
-
Myocardial ischemia/infarction or Prinzmetal’s angina: Perform cardiac evaluation in patients with multiple cardiovascular risk factors (5.1)
-
Arrhythmias:Discontinue FROVA if occurs (5.2)
-
Chest/throat/neck/jaw pain, tightness, pressure, or heaviness:Generally not associated with myocardial ischemia; evaluate high risk patients for coronary artery disease (5.3)
-
Cerebral hemorrhage, subarachnoid hemorrhage, and stroke: Discontinue FROVA if occurs (5.4)
-
Gastrointestinal ischemic reactions and peripheral vasospastic reactions: Discontinue FROVA if occurs (5.5)
-
Medication overuse headache:Detoxification may be necessary (5.6)
Adverse Reactions/Side Effects
-
Most common adverse reactions (≥2% and >placebo) were dizziness, headache, paresthesia, dry mouth, dyspepsia, fatigue, hot or cold sensation, chest pain, skeletal pain, and flushing (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Endo Pharmaceuticals Inc. at 1-800-462-3636 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm (8.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 8/2018
Full Prescribing Information
1. Indications and Usage for Frova
FROVA is indicated for the acute treatment of migraine with or without aura in adults.
Limitations of Use
-
Use only if a clear diagnosis of migraine has been established. If a patient has no response for the first migraine attack treated with FROVA, reconsider the diagnosis of migraine before FROVA is administered to treat any subsequent attacks.
-
FROVA is not indicated for the prevention of migraine attacks.
-
Safety and effectiveness of FROVA have not been established for cluster headache.
2. Frova Dosage and Administration
Dosing Information
The recommended dose is a single tablet of FROVA (frovatriptan 2.5 mg) taken orally with fluids.
If the migraine recurs after initial relief, a second tablet may be taken, providing there is an interval of at least 2 hours between doses. The total daily dose of FROVA should not exceed 3 tablets (3 x 2.5 mg per 24 hour period).
There is no evidence that a second dose of FROVA is effective in patients who do not respond to a first dose of the drug for the same headache.
The safety of treating an average of more than 4 migraine attacks in a 30-day period has not been established.
3. Dosage Forms and Strengths
2.5 mg Tablets: Round, white, film-coated tablets debossed with 2.5 on one side and "E" on the other side.
4. Contraindications
FROVA is contraindicated in patients with:
-
Ischemic coronary artery disease (CAD) (e.g., angina pectoris, history of myocardial infarction, or documented silent ischemia), or coronary artery vasospasm, including Prinzmetal’s angina [see Warnings and Precautions (5.1)].
-
Wolff-Parkinson-White Syndrome or arrhythmias associated with other cardiac accessory conduction pathway disorders [see Warnings and Precautions (5.2)].
-
History of stroke, transient ischemic attack (TIA), or history of hemiplegic or basilar migraine because these patients are at a higher risk of stroke [see Warnings and Precautions (5.4)].
-
Peripheral vascular disease [see Warnings and Precautions (5.5)].
-
Ischemic bowel disease [see Warnings and Precautions (5.5)].
-
Uncontrolled hypertension [see Warnings and Precautions (5.8)].
-
Recent use (i.e., within 24 hours) of another 5-HT1 agonist, an ergotamine containing or ergot-type medication such as dihydroergotamine (DHE) or methysergide [see Drug Interactions (7.1, 7.2)].
-
Hypersensitivity to frovatriptan succinate (angioedema and anaphylaxis seen) [see Warnings and Precautions (5.9)].
5. Warnings and Precautions
5.1 Myocardial Ischemia, Myocardial Infarction, and Prinzmetal’s Angina
FROVA is contraindicated in patients with ischemic or vasospastic CAD. There have been rare reports of serious cardiac adverse reactions, including acute myocardial infarction, occurring within a few hours following administration of FROVA. Some of these reactions occurred in patients without known CAD. FROVA may cause coronary artery vasospasm (Prinzmetal’s angina), even in patients without a history of CAD.
Perform a cardiovascular evaluation in triptan-naïve patients who have multiple cardiovascular risk factors (e.g., increased age, diabetes, hypertension, smoking, obesity, strong family history of CAD) prior to receiving FROVA. Do not administer FROVA if there is evidence of CAD or coronary artery vasospasm [see Contraindications (4)]. For patients with multiple cardiovascular risk factors who have a negative cardiovascular evaluation, consider administrating the first FROVA dose in a medically-supervised setting and performing an electrocardiogram (ECG) immediately following FROVA administration. For such patients, consider periodic cardiovascular evaluation in intermittent long-term users of FROVA.
5.2 Arrhythmias
Life-threatening disturbances of cardiac rhythm including ventricular tachycardia and ventricular fibrillation leading to death have been reported within a few hours following the administration of 5-HT1 agonists. Discontinue FROVA if these disturbances occur. FROVA is contraindicated in patients with Wolff-Parkinson-White syndrome or arrhythmias associated with other cardiac accessory conduction pathway disorders [see Contraindications (4)].
5.3 Chest, Throat, Neck, and Jaw Pain/Tightness/Pressure
Sensations of pain, tightness, pressure, and heaviness have been reported in the chest, throat, neck, and jaw after treatment with FROVA and are usually non-cardiac in origin. However, perform a cardiac evaluation if these patients are at high cardiac risk. The use of FROVA is contraindicated in patients with CAD and those with Prinzmetal’s angina [see Contraindications (4)].
5.4 Cerebrovascular Events
Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with 5-HT1 agonists, and some have resulted in fatalities. In a number of cases, it appears possible that the cerebrovascular events were primary, the agonist having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine, when they were not.
Before treating headaches in patients not previously diagnosed as migraineurs, and in migraineurs who present with symptoms atypical of migraine, other potentially serious neurological conditions need to be excluded. FROVA is contraindicated in patients with a history of stroke or TIA [see Contraindications (4)].
5.5 Other Vasospasm Reactions
FROVA may cause non-coronary vasospastic reactions, such as peripheral vascular ischemia, gastrointestinal vascular ischemia and infarction (presenting with abdominal pain and bloody diarrhea), splenic infarction, and Raynaud’s syndrome. In patients who experience symptoms or signs suggestive of a vasospastic reaction following the use of any 5-HT1 agonist, rule out a vasospastic reaction before using FROVA [see Contraindications (4)].
Reports of transient and permanent blindness and significant partial vision loss have been reported with the use of 5-HT1 agonists. Since visual disorders may be part of a migraine attack, a causal relationship between these events and the use of 5-HT1 agonists have not been clearly established.
5.6 Medication Overuse Headache
Overuse of acute migraine drugs (e.g., ergotamine, triptans, opioids, or combination of these drugs for 10 or more days per month) may lead to exacerbation of headache (medication overuse headache). Medication overuse headache may present as migraine-like daily headaches or as a marked increase in frequency of migraine attacks. Detoxification of patients, including withdrawal of the overused drugs, and treatment of withdrawal symptoms (which often includes a transient worsening of headache) may be necessary.
5.7 Serotonin Syndrome
Serotonin syndrome may occur with FROVA, particularly during co-administration with selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs), and monoamine oxidase (MAO) inhibitors [see Drug Interactions (7.3)]. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination), and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). The onset of symptoms usually occurs within minutes to hours of receiving a new or a greater dose of a serotonergic medication. Discontinue FROVA if serotonin syndrome is suspected.
5.8 Increase in Blood Pressure
Significant elevation in blood pressure, including hypertensive crisis with acute impairment of organ systems, has been reported on rare occasions in patients treated with 5-HT1 agonists, including patients without a history of hypertension. Monitor blood pressure in patients treated with FROVA. FROVA is contraindicated in patients with uncontrolled hypertension [see Contraindications (4)].
5.9 Hypersensitivity Reactions
There have been reports of reactions, including anaphylaxis and angioedema, in patients receiving FROVA. Such reactions can be life threatening or fatal. In general, anaphylactic reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens. FROVA is contraindicated in patients with a history of hypersensitivity reaction to FROVA [see Contraindications (4)].
6. Adverse Reactions/Side Effects
The following serious adverse reactions are described elsewhere in other sections of the labeling:
- Myocardial Ischemia, Myocardial Infarction, and Prinzmetal’s Angina [see Warnings and Precautions (5.1)]
- Arrhythmias [see Warnings and Precautions (5.2)]
- Chest, Throat, Neck, and/or Jaw Pain/Tightness/Pressure [see Warnings and Precautions (5.3)]
- Cerebrovascular Events [see Warnings and Precautions (5.4)]
- Other Vasospasm Reactions [see Warnings and Precautions (5.5)]
- Medication Overuse Headache [see Warnings and Precautions (5.6)]
- Serotonin Syndrome [see Warnings and Precautions (5.7)]
- Increases in Blood Pressure [see Warnings and Precautions (5.8)]
- Hypersensitivity Reactions [see Contraindications (4) and Warnings and Precautions (5.9)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
FROVA was evaluated in four randomized, double-blind, placebo-controlled, short-term trials. These trials involved 2392 patients (1554 on FROVA 2.5 mg and 838 on placebo). In these short-term trials, patients were predominately female (88%) and Caucasian (94%) with a mean age of 42 years (range 18 to 69).The treatment-emergent adverse events that occurred most frequently following administration of FROVA 2.5 mg (i.e., in at least 2% of patients), and at an incidence ≥1% greater than with placebo, were dizziness, paresthesia, headache, dry mouth, fatigue, flushing, hot or cold sensation, dyspepsia, skeletal pain, and chest pain. In a long-term, open-label study where 496 patients were allowed to treat multiple migraine attacks with FROVA 2.5 mg for up to 1 year, 5% of patients (n=26) discontinued due to treatment-emergent adverse events.
Table 1 lists treatment-emergent adverse events reported within 48 hours of drug administration that occurred with FROVA 2.5 mg at an incidence of ≥2% and more often than on placebo, in the four placebo-controlled trials. The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, these incidence estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Table 1
Patients in Four Pooled Placebo-Controlled Migraine Trials
|
Adverse Reactions |
FROVA 2.5 mg (n=1554) |
Placebo (n=838) |
|
Central & peripheral nervous system Dizziness Headache Paresthesia |
4% 4% |
3% 2% |
|
Gastrointestinal system disorders Dry mouth Dyspepsia |
2% |
1% |
|
Body as a whole – general disorders Fatigue Hot or cold sensation Chest pain |
3% 2% |
2% 1% |
|
Musculo-skeletal Skeletal pain |
|
|
|
Vascular Flushing |
|
|
The incidence of adverse events in clinical trials did not increase when up to 3 doses were used within 24 hours. The incidence of adverse events in placebo-controlled clinical trials was not affected by gender, age, or concomitant medications commonly used by migraine patients. There were insufficient data to assess the impact of race on the incidence of adverse events.
Other Events Observed in Association with the Administration of FROVA
The incidence of frequently reported adverse events in four placebo-controlled trials is presented below. Events are further classified within body system categories. Frequent adverse events are those occurring in at least 1/100 patients.
Central and peripheral nervous system: dysesthesia and hypoesthesia.
Gastrointestinal: vomiting, abdominal pain and diarrhea.
Body as a whole: pain.
Psychiatric: insomnia and anxiety.
Respiratory: sinusitis and rhinitis.
Vision disorders: vision abnormal.
Skin and appendages: sweating increased.
Hearing and vestibular disorders: tinnitus.
Heart rate and rhythm: palpitation.
6.2 Postmarketing Experience
The following adverse reactions were identified during post approval use of FROVA. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Central and peripheral nervous system: Seizure.
Related/similar drugs
7. Drug Interactions
7.1 Ergot-containing Drugs
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because these effects may be additive, use of ergotamine-containing or ergot-type medications (like dihydroergotamine or methysergide) and FROVA within 24 hours of each other is contraindicated [see Contraindications (4)].
7.2 5-HT1B/1D Agonists
Because their vasospastic effects may be additive, co-administration of FROVA and other 5-HT1 agonists (e.g., triptans) within 24 hours of each other is contraindicated [see Contraindications (4)].
7.3 Selective Serotonin Reuptake Inhibitors / Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome
Cases of serotonin syndrome have been reported during combined use of triptans and SSRIs, SNRIs, TCAs, and MAO inhibitors [see Warnings and Precautions (5.7)].
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risk associated with the use of FROVA in pregnant women. In animal studies, frovatriptan produced developmental toxicity (embryofetal lethality, fetal abnormalities, and decreased embryofetal growth) when administered to pregnant rats and rabbits at doses greater than those used clinically [see Animal Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The reported rate of major birth defects among deliveries to women with migraine ranged from 2.2% to 2.9% and the reported rate of miscarriage was 17%, which were similar to rates reported in women without migraine.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Published data have suggested that women with migraines may be at increased risk of preeclampsia during pregnancy.
Data
Animal Data
When pregnant rats were administered frovatriptan (oral doses of 0, 100, 500, or 1000 mg/kg/day) throughout organogenesis, increased embryofetal mortality and an increased incidence of fetal malformations were observed at the high dose; decreased fetal body weights and increased incidences of fetal structural variations associated with growth impairment were observed at all doses. The lowest effect dose for embryofetal developmental toxicity in rats (100 mg/kg/day) is approximately 130 times the maximum recommended human dose (MRHD) of 7.5 mg/day on a body surface area (mg/m2) basis.
When pregnant rabbits were administered frovatriptan (oral doses of 0, 5, 20, or 80 mg/kg/day) throughout organogenesis, embryofetal mortality was increased at the mid and high doses. The no-effect dose for embryofetal developmental toxicity in rabbits (5 mg/kg/day) is approximately 13 times the MRHD on a mg/m2 basis.
Oral administration of frovatriptan (0, 100, 500, or 1000 mg/kg/day) to female rats throughout pregnancy and lactation resulted in increased embryofetal mortality at the high dose. The no effect dose for pre- and postnatal developmental toxicity in rats (500 mg/kg/day) is approximately 650 times the MRHD on a mg/m2 basis.
8.2 Lactation
Risk Summary
There are no data on the presence of frovatriptan in human milk, the effects of frovatriptan on the breastfed infant, or the effects of frovatriptan on milk production. In rats, oral dosing with frovatriptan resulted in levels of frovatriptan and/or its metabolites in milk up to four times higher than in plasma.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for FROVA and any potential adverse effects on the breastfed infant from frovatriptan or from the underlying maternal condition.
8.4 Pediatric use
The safety and effectiveness in pediatric patients have not been established. Therefore, FROVA is not recommended for use in patients under 18 years of age. There are no additional adverse reactions identified in pediatric patients based on postmarketing experience that were not previously identified in adults.
8.5 Geriatric use
Mean blood concentrations of frovatriptan in elderly patients were 1.5 to 2 times higher than those seen in younger adults [see Clinical Pharmacology (12.3)]. No dosage adjustment is necessary.
8.6 Patients with Hepatic Impairment
No dosage adjustment is necessary when FROVA is given to patients with mild to moderate hepatic impairment.
There is no clinical or pharmacokinetic experience with FROVA in patients with severe hepatic impairment. Because a greater than two-fold increase in AUC is predicted in patients with severe hepatic impairment, there is a greater potential for adverse events in these patients, and FROVA should therefore be used with caution in that population.
10. Overdosage
The elimination half-life of frovatriptan is 26 hours [see Clinical Pharmacology (12.3)]. Therefore, monitoring of patients after overdose with frovatriptan should continue for at least 48 hours or while symptoms or signs persist. There is no specific antidote to frovatriptan. It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentrations of frovatriptan.
11. Frova Description
FROVA (frovatriptan succinate) tablets contain frovatriptan succinate, a selective 5-hydroxy-tryptamine1 (5-HT1B/1D) receptor subtype agonist (triptan), as the active ingredient. Frovatriptan succinate is chemically designated as R-(+) 3-methylamino-6-carboxamido-1,2,3,4-tetrahydrocarbazole monosuccinate monohydrate and it has the following structure:
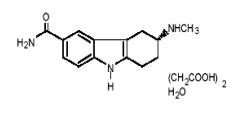
The empirical formula is C14H17N3O.C4H6O4.H2O, representing a molecular weight of 379.4. Frovatriptan succinate is a white to off-white powder that is soluble in water.
Each FROVA tablet for oral administration contains 3.91 mg frovatriptan succinate, equivalent to 2.5 mg of frovatriptan base. Each tablet also contains the inactive ingredients lactose NF, microcrystalline cellulose NF, colloidal silicon dioxide NF, sodium starch glycolate NF, magnesium stearate NF, hypromellose USP, polyethylene glycol 3000 USP, triacetin USP, and titanium dioxide USP.
12. Frova - Clinical Pharmacology
12.1 Frova Mechanism of Action
Frovatriptan binds with high affinity to 5-HT1B/1D receptors. The therapeutic activity of FROVA is thought to be due to the agonist effects at the 5-HT1B/1D receptors on intracranial blood vessels (including the arterio-venous anastomoses) and sensory nerves of the trigeminal system which result in cranial vessel constriction and inhibition of pro-inflammatory neuropeptide release.
12.3 Pharmacokinetics
The pharmacokinetics of frovatriptan are similar in migraine patients and healthy subjects.
Absorption
Mean maximum blood concentrations (Cmax) in patients are achieved approximately 2 to 4 hours after administration of a single oral dose of frovatriptan 2.5 mg. The absolute bioavailability of an oral dose of frovatriptan 2.5 mg in healthy subjects is about 20% in males and 30% in females. Food has no significant effect on the bioavailability of frovatriptan, but delays tmax by one hour.
Distribution
Binding of frovatriptan to serum proteins is low (approximately 15%). Reversible binding to blood cells at equilibrium is approximately 60%, resulting in a blood: plasma ratio of about 2:1 in both males and females. The mean steady state volume of distribution of frovatriptan following intravenous administration of 0.8 mg is 4.2 L/kg in males and 3.0 L/kg in females.
Metabolism
In vitro, cytochrome P450 1A2 appears to be the principal enzyme involved in the metabolism of frovatriptan. Following administration of a single oral dose of radiolabeled frovatriptan 2.5 mg to healthy male and female subjects, 32% of the dose was recovered in urine and 62% in feces. Radiolabeled compounds excreted in urine were unchanged frovatriptan, hydroxylated frovatriptan, N-acetyl desmethyl frovatriptan, hydroxylated N-acetyl desmethyl frovatriptan and desmethyl frovatriptan, together with several other minor metabolites. Desmethyl frovatriptan has lower affinity for 5-HT1B/1D receptors compared to the parent compound. The N-acetyl desmethyl metabolite has no significant affinity for 5-HT receptors. The activity of the other metabolites is unknown.
Elimination
After an intravenous dose, mean clearance of frovatriptan was 220 and 130 mL/min in males and females, respectively. Renal clearance accounted for about 40% (82 mL/min) and 45% (60 mL/min) of total clearance in males and females, respectively. The mean terminal elimination half-life of frovatriptan in both males and females is approximately 26 hours.
Special Populations
Hepatic Impairment
The AUC of frovatriptan in patients with mild (Child-Pugh 5-6) to moderate (Child-Pugh 7-9) hepatic impairment was about twice that of young, healthy subjects, but within the range observed in healthy elderly subjects and was considerably lower than the values attained with higher doses of frovatriptan (up to 40 mg), which were not associated with any serious adverse effects. There is no clinical or pharmacokinetic experience with FROVA in patients with severe hepatic impairment.
Renal Impairment
The pharmacokinetics of frovatriptan following a single oral dose of 2.5 mg was not different in patients with renal impairment (5 males and 6 females, creatinine clearance 16 - 73 mL/min) compared to subjects with normal renal function.
Age
Mean AUC of frovatriptan was 1.5- to 2-fold higher in healthy elderly subjects (age 65 to 77 years) compared to those in healthy younger subjects (age 21 to 37 years). There was no difference in tmax or t1/2 between the two populations.
Sex
There was no difference in the mean terminal elimination half-life of frovatriptan in males and females. Bioavailability was higher, and systemic exposure to frovatriptan was approximately 2-fold greater, in females than males, irrespective of age.
Race
The effect of race on the pharmacokinetics of frovatriptan has not been examined.
Drug Interaction Studies
Frovatriptan is not an inhibitor of human monoamine oxidase (MAO) enzymes or cytochrome P450 (isozymes 1A2, 2C9, 2C19, 2D6, 2E1, 3A4) in vitro at concentrations up to 250- to 500- fold higher than the highest blood concentrations observed in man at a dose of 2.5 mg. No induction of drug metabolizing enzymes was observed following multiple dosing of frovatriptan to rats or on addition to human hepatocytes in vitro. Although no clinical trials have been performed, it is unlikely that frovatriptan will affect the metabolism of co-administered drugs metabolized by these mechanisms.
Oral Contraceptives
Retrospective analysis of pharmacokinetic data from females across trials indicated that the mean Cmax and AUC of frovatriptan are 30% higher in those subjects taking oral contraceptives compared to those not taking oral contraceptives.
Ergotamine
The AUC and Cmax of frovatriptan (2 x 2.5 mg dose) were reduced by approximately 25% when co-administered with ergotamine tartrate [see Contraindications (4), Drug Interactions (7.1)].
Propranolol
Propranolol increased the AUC of frovatriptan 2.5 mg in males by 60% and in females by 29%. The Cmax of frovatriptan was increased 23% in males and 16% in females in the presence of propranolol. The tmax as well as half-life of frovatriptan, though slightly longer in the females, were not affected by concomitant administration of propranolol.
Moclobemide
The pharmacokinetic profile of frovatriptan was unaffected when a single oral dose of frovatriptan 2.5 mg was administered to healthy female subjects receiving the MAO-A inhibitor, moclobemide, at an oral dose of 150 mg twice a day for 8 days.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
The carcinogenic potential of orally administered frovatriptan was evaluated in an 84-week study in mice (4, 13, and 40 mg/kg/day), a 104-week study in rats (8.5, 27, and 85 mg/kg/day), and a 26-week study in p53(+/-) transgenic mice (20, 62.5, 200, and 400 mg/kg/day). Although a maximum tolerated dose was not achieved in the 84-week mouse study and in female rats, plasma exposures at the highest doses studied were higher than that achieved in humans at the MRHD of 7.5 mg/day. There were no increases in tumor incidence in the 84-week mouse study at doses producing plasma exposures (AUC) 140 times that in humans at the MRHD. In the rat study, there was a statistically significant increase in the incidence of pituitary adenomas in males only at 85 mg/kg/day, a dose associated with a plasma AUC 250 times that in humans at the MRHD. In the 26-week p53(+/-) transgenic mouse study, the incidence of subcutaneous sarcomas was increased in females at doses of 200 and 400 mg/kg/day.
These sarcomas were associated with subcutaneously implanted animal identification transponders, and are not considered to be relevant to humans. There were no other increases in tumor incidence of any type in any dose group.
Mutagenesis
Frovatriptan was clastogenic in human lymphocyte cultures, in the absence of metabolic activation. In the bacterial reverse mutation assay (Ames test), frovatriptan produced an equivocal response in the absence of metabolic activation. Frovatriptan was negative in an in vitro mouse lymphoma tk assay and an in vivo mouse bone marrow micronucleus test.
Impairment of Fertility
Male and female rats were dosed orally with frovatriptan prior to and during mating and in females up to implantation, at doses of 100, 500, and 1000 mg/kg/day (equivalent to approximately 130, 650, and 1300 times the MRHD on a mg/m2 basis). At all dose levels, there was an increase in the number of females that mated on the first day of pairing compared to control animals. This occurred in conjunction with a prolongation of the estrous cycle. In addition, females had a decreased mean number of corpora lutea, and consequently a lower number of live fetuses per litter, which suggested a partial impairment of ovulation. There were no other fertility-related effects.
14. Clinical Studies
The efficacy of FROVA in the acute treatment of migraine headaches was demonstrated in four randomized, double-blind, placebo-controlled, short-term outpatient trials. In these trials, patients received doses of frovatriptan from 0.5 mg to 40 mg. In these controlled short-term trials, patients were predominately female (88%) and Caucasian (94%) with a mean age of 42 years (range 18 to 69). Patients were instructed to treat a moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed for up to 24 hours after dosing. The associated symptoms nausea, vomiting, photophobia, and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours post dose. In two of the trials a second dose of FROVA was provided after the initial treatment, to treat recurrence of the headache within 24 hours. Other medication, excluding other 5-HT1 agonists and ergotamine containing compounds, was permitted from 2 hours after the first dose of FROVA. The frequency and time to use of additional medications were also recorded.
In all four placebo-controlled trials, the percentage of patients achieving a headache response 2 hours after treatment was significantly greater for those taking FROVA 2.5 mg compared to those taking placebo (Table 2).
Lower doses of frovatriptan (1 mg or 0.5 mg) were not effective at 2 hours. Higher doses (5 mg to 40 mg) of frovatriptan showed no added benefit over 2.5 mg but did cause a greater incidence of adverse events.
Table 2
Percentage of Patients with Headache Response (Mild or No Headache) 2 Hours Following Treatmenta
|
Trial |
FROVA 2.5 mg |
Placebo |
|
1 |
42%* (n=90) |
22% (n=91) |
|
2 |
39%* (n=187) |
21% (n=99) |
|
3 |
46%** (n=672) |
27% (n=347) |
|
4 |
37%** (n=438) |
23% (n=225) |
aITT observed data, excludes patients who had missing data or were asleep.
*p<0.05.
**p<0.001 in comparison with placebo.
The estimated probability of achieving an initial headache response by 2 hours following treatment is depicted in Figure 1.
Figure 1
Estimated Probability of Achieving Initial Headache Response Within 2 Hours
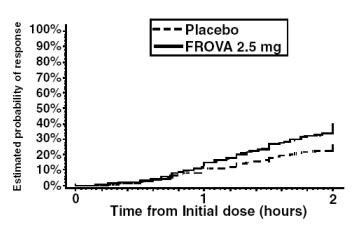
Figure 1 shows a Kaplan-Meier plot of the probability over time of obtaining headache response (no or mild pain) following treatment with FROVA 2.5 mg or placebo. The probabilities displayed are based on pooled data from the four placebo-controlled trials described in Table 2. Patients who did not achieve a response were censored at 24 hours.
In patients with migraine-associated nausea, photophobia, and phonophobia at baseline there was a decreased incidence of these symptoms in FROVA treated patients compared to placebo.
The estimated probability of patients taking a second dose or other medication for their migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
Figure 2
Estimated Probability of Patients Taking a Second Dose or Other Medication for Migraine
Over the 24 Hours Following the Initial Dose of Study Treatment
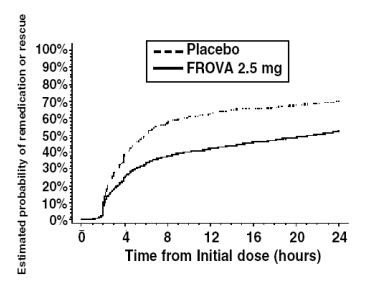
Figure 2 is a Kaplan-Meier plot showing the probability of patients taking a second dose or other medication for migraine over the 24 hours following the initial dose of study medication based on the data from the four placebo-controlled trials described in Table 2. The plot includes those patients who had a response to the initial dose and those who did not. The protocols did not permit remedication within 2 hours of the initial dose.
Efficacy was unaffected by a history of aura; gender; age, or concomitant medications commonly used by migraine patients.
16. How is Frova supplied
FROVA tablets, containing 2.5 mg of frovatriptan (base) as the succinate salt, are available as round, white, film-coated tablets debossed with 2.5 on one side and “E” on the other side. The tablets are available in:
Blister card of 9 tablets, 1 blister card per carton (NDC 63481-025-09)
Store FROVA tablets at controlled room temperature, 25ºC (77ºF) excursions permitted to 15ºC to 30ºC (59ºF to 86ºF) [see USP Controlled Room Temperature]. Protect from moisture.
17. Patient Counseling Information
Advise the patient to read the FDA approved patient labeling (Patient Information).
Myocardial Ischemia and/or Infarction, Prinzmetal’s Angina, Other Vasospastic Reactions, and Cerebrovascular Events
Inform patients that FROVA may cause serious cardiovascular adverse reactions such as myocardial infarction or stroke, which may result in hospitalization and even death. Although serious cardiovascular reactions can occur without warning symptoms, instruct patients to be alert for the signs and symptoms of chest pain, shortness of breath, weakness, slurring of speech, and instruct them to ask for medical advice when observing any indicative sign or symptoms. Instruct patients to seek medical advice if they have symptoms of other vasospastic reactions [see Warnings and Precautions (5.1, 5.2, 5.4, 5.5, and 5.8)].
Hypersensitivity Reactions
Inform patients that anaphylactic reactions have occurred in patients receiving FROVA. Inform patients that such reactions can be life threatening or fatal and to seek immediate medical attention if they have anaphylactic symptoms. In general, anaphylactic reactions to drugs are more likely to occur in individuals with a history of sensitivity to multiple allergens [see Contraindications (4)].
Medication Overuse Headache
Inform patients that use of drugs to treat acute migraines for 10 or more days per month may lead to an exacerbation of headache, and encourage patients to record headache frequency and drug use (e.g., by keeping a headache diary) [see Warnings and Precautions (5.6)].
Serotonin Syndrome
Inform patients about the risk of serotonin syndrome with the use of FROVA or other triptans, particularly during combined use with SSRIs, SNRIs, TCAs, and MAO inhibitors [see Warnings and Precautions (5.7) and Drug Interactions (7.3)].
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant during treatment or plan to become pregnant [see Use in Specific Populations (8.1)].
Lactation
Inform patients to notify their healthcare provider if they are breastfeeding or plan to breastfeed [see Use in Specific Populations (8.2)].
PATIENT LABELING
|
Patient Information FROVA (FRO-va) (frovatriptan succinate) tablets |
|
What is the most important information I should know about FROVA? FROVA can cause serious side effects, including:
FROVA is not for people with risk factors for heart disease unless a heart exam is done and shows no problem. You have a higher risk for heart disease if you:
|
| What is FROVA? FROVA is a prescription medicine used to treat migraine headaches with or without aura in adults. FROVA is not used to treat other types of headaches. FROVA is not used to prevent or decrease the number of migraine headaches. It is not known if FROVA is safe and effective to treat cluster headaches. It is not known if FROVA is safe and effective in children under 18 years of age. |
|
Who should not take FROVA?
Ask your healthcare provider if you are not sure if your medicine is listed above.
|
|
What should I tell my doctor before taking FROVA?
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. FROVA and certain other medicines can affect each other, causing serious side effects. Especially tell your doctor if you take:
Ask your doctor or pharmacist for a list of these medicines if you are not sure. Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine. |
|
How should I take FROVA?
|
|
What should I avoid while taking FROVA? |
|
What are the possible side effects of FROVA?
The most common side effects of FROVA are:
These are not all the possible side effects of FROVA. For more information, ask your doctor or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
How should I store FROVA?
Keep FROVA and all medicines out of the reach of children. |
|
General information about the safe and effective use of FROVA. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use FROVA for a condition for which it was not prescribed. Do not give FROVA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information about FROVA that is written for health professionals. |
|
What are the Ingredients in FROVA? Active ingredient: frovatriptan succinate Inactive ingredients: lactose NF, microcrystalline cellulose NF, colloidal silicon dioxide NF, sodium starch glycolate NF, magnesium stearate NF, hypromellose USP, polyethylene glycol 3000 USP, triacetin USP, and titanium dioxide USP Manufactured for: Endo Pharmaceuticals Inc., Malvern, PA 19355 Manufactured by: Almac Pharma Services Limited, Craigavon, BT63 5UA, UK FROVA is a registered trademark of Vernalis Development Limited. © 2018 Endo Pharmaceuticals Inc. All rights reserved. U.S. Patent Nos 5,962,501, 5,827,871, 5,637,611 and 5,464,864 and 5,616,603. For more information, go to www.FROVA.com or call 1-800-462-3636. |
This Patient Information has been approved by the U.S. Food and Drug Administration
Revised: August 2018

| FROVA
frovatriptan succinate tablet, film coated |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
| Labeler - Endo Pharmaceuticals, Inc. (178074951) |
Frequently asked questions
More about Frova (frovatriptan)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (42)
- Drug images
- Side effects
- Dosage information
- During pregnancy
- Generic availability
- Drug class: antimigraine agents
- Breastfeeding
- En español
