Totect: Package Insert / Prescribing Info
Package insert / product label
Generic name: dexrazoxane hydrochloride
Dosage form: injection, powder, lyophilized, for solution
Drug class: Miscellaneous uncategorized agents
Medically reviewed by Drugs.com. Last updated on Aug 7, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
Totect® (dexrazoxane) for injection, for intravenous use
Initial U.S. Approval: 2007
Recent Major Changes
Indications and Usage for Totect
Totect is a cytoprotective agent indicated for:
- Treatment of extravasation resulting from intravenous anthracycline chemotherapy. (1)
- Reducing the incidence and severity of cardiomyopathy associated with doxorubicin administration in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and who will continue to receive doxorubicin therapy to maintain tumor control. Do not use Totect with doxorubicin initiation. (1, 5.2)
Totect Dosage and Administration
-
Reconstitute and further dilute Totect before use. (2.4)
-
Extravasation: administer Totect by intravenous infusion over 1 to 2 hours once daily for 3 consecutive days. (2.1)
-
Initiate the first infusion as soon as possible and within the first six hours after extravasation. (2.1)
Recommended doseMaximum daily dose
Day one: 1000 mg/m2 2000 mg
Day two: 1000 mg/m2 2000 mg
Day three: 500 mg/m2 1000 mg
- Cardiomyopathy: administer Totect by intravenous infusion over 15 minutes until discontinuation of doxorubicin. (2.2)
- Do not administer via intravenous push. (2.5)
- The recommended dosage ratio of Totect to doxorubicin is 10:1, (e.g. 500 mg/m² Totect to 50 mg/m² doxorubicin). (2.2)
- Do not administer doxorubicin before Totect. (2.5)
- Administer doxorubicin within 30 minutes after the completion of Totect infusion. (2.5)
- Dose Modifications: reduce dose by 50% for patients with creatinine clearance < 40 mL/min. (2.3, 8.6)
Dosage Forms and Strengths
-
For Injection: 500 mg lyophilized powder in a single dose vial for reconstitution. (3)
Contraindications
None. (4)
Warnings and Precautions
-
Myelosuppression: dexrazoxane is associated with leukopenia, neutropenia, and thrombocytopenia. Dexrazoxane may increase the myelosuppressive effects of chemotherapeutic agents. Perform hematological monitoring. (5.1)
-
Concomitant chemotherapy: Totect may interfere with the antitumor activity of the chemotherapy regimen. Only use Totect in patients who have received a cumulative doxorubicin dose of 300 mg/m2 and are continuing doxorubicin therapy. (5.2)
-
Cardiac toxicity: Totect does not completely eliminate the risk of anthracycline-induced cardiac toxicity. For cardiomyopathy, monitor cardiac function before and periodically during therapy to assess left ventricular ejection fraction (LVEF). If deterioration in cardiac function occurs, consider the benefit of continued therapy against the risk of producing irreversible cardiac damage. (5.3)
-
Secondary malignancies: combination of dexrazoxane with chemotherapy may lead to increased risk of secondary malignancies. (5.4)
- Anaphylactic/Hypersensitivity Reactions: monitor for signs and symptoms. Consider permanent discontinuation for severe hypersensitivity reactions. (5.5)
- Embryo-Fetal Toxicity: can cause fetal harm. Advise patients of reproductive potential of the risk to a fetus and to use effective contraception. (5.6, 8.1, 8.3)
Adverse Reactions/Side Effects
Extravasation: The most common adverse reactions (≥15%) are nausea, pyrexia, injection site pain, vomiting, and postoperative infection. (6.1)
Cardiomyopathy: The most common adverse reactions (>10% and increased versus placebo) was injection site pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Clinigen, Inc. at 1-877-776-5385 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Drug Interactions
- Dimethyl sulfoxide: not recommended for use with topical dimethyl sulfoxide (DMSO) in extravasation patients. (7.1)
Use In Specific Populations
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2020
Full Prescribing Information
1. Indications and Usage for Totect
Extravasation
Totect® is indicated for the treatment of extravasation resulting from intravenous anthracycline chemotherapy.
Cardiomyopathy
Totect is indicated for reducing the incidence and severity of cardiomyopathy associated with doxorubicin administration in women with metastatic breast cancer who have received a cumulative doxorubicin dose of 300 mg/m2 and who will continue to receive doxorubicin therapy to maintain tumor control. Do not use with the initiation of doxorubicin therapy [see Warnings and Precautions (5.2)].
2. Totect Dosage and Administration
2.1 Recommended Dose for Extravasation
Reconstitute and further dilute Totect before use [see Dosage and Administration (2.4)].
For extravasation, administer Totect via intravenous infusion over 1 to 2 hours once daily for 3 consecutive days. Initiate the first infusion as soon as possible and within the first six hours after extravasation [see Dosage and Administration (2.5)].
The individual dosage is based on calculation of the Body Surface Area (BSA) up to a maximum dose of 2000 mg (each on Day 1 and 2) and 1000 mg (Day 3), corresponding to a BSA of 2 m2.
The recommended dose is: Maximum daily dose:
Day one: 1000 mg/m2 2000 mg
Day two: 1000 mg/m2 2000 mg
Day three: 500 mg/m2 1000 mg
2.2 Recommended Dose for Cardiomyopathy
Reconstitute and further dilute Totect before use [see Dosage and Administration (2.4)].
For cardiomyopathy, administer Totect via intravenous infusion over 15 minutes prior to doxorubicin administration until discontinuation of doxorubicin. Do not administer via an intravenous push [see Dosage and Administration (2.5)].
The recommended dosage ratio of Totect to doxorubicin is 10:1 (e.g., 500 mg/m2 Totect to 50 mg/m2 doxorubicin). Administer doxorubicin within 30 minutes after the completion of Totect infusion.
2.3 Dose Modifications
Dosing in Patients with Renal Impairment
Reduce the Totect dose by 50% in patients with creatinine clearance values < 40 mL/min [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
Dosing in Patients with Hepatic Impairment
In patients with anthracycline extravasation, treatment with Totect is not recommended. In patients receiving dexrazoxane for cardiomyopathy, reduce the Totect dosage proportionately (maintaining the 10:1 ratio) in patients with hepatic impairment, since a doxorubicin dose reduction is recommended in the presence of hyperbilirubinemia [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
2.4 Preparation
Read this entire section carefully before mixing and diluting.
Use caution when handling and preparing the reconstituted solution. The use of gloves is recommended. If Totect powder or solutions contact the skin or mucosae, wash exposed area immediately and thoroughly with soap and water. Follow special handling and disposal procedures.1
Totect should not be mixed or administered with any other drug during the infusion. The prepared solution of Totect is slightly yellow.
Parenteral drug products should be inspected visually for particulate matter prior to administration, whenever solution and container permit. Solutions containing a precipitate should be discarded. Vials are for single use only. Unused solution should be discarded.
Preparation for the Treatment of Extravasation
Step 1. Reconstitute each Totect vial with 50 mL of Sterile Water for Injection, USP. Once reconstituted, the reconstituted Totect solution contains 10 mg/mL of Totect.
Step 2. Calculate the volume of the 10 mg/mL reconstituted Totect solution needed for the recommended dose. In order to obtain the required dose, more than one vial may be needed. The reconstituted solution should be further diluted within 30 minutes after initial reconstitution. It contains no antibacterial preservative.
Step 3. Withdraw the calculated volume from the reconstituted Totect solution and further dilute into an infusion bag containing 1000 mL of Lactated Ringer’s Injection. Do not mix Totect with any other drugs.
Use the Totect infusion bag immediately after preparation. If not used immediately, the product is stable for 4 hours from the time of preparation when stored at room temperature or for up to 12 hours when stored refrigerated between 2ºC to 8ºC (36°F to 46°F).
Preparation for Reducing the Incidence and Severity of Cardiomyopathy
Step 1. Reconstitute each Totect vial with 50 mL of Sterile Water for Injection, USP. Once reconstituted, the reconstituted Totect solution contains 10 mg/mL of Totect.
Step 2. Calculate the volume of the 10 mg/mL reconstituted Totect solution needed for the recommended dose. In order to obtain the required dose, more than one vial may be needed. The reconstituted solution should be further diluted within 30 minutes after initial reconstitution. It contains no antibacterial preservative.
Step 3. Withdraw the calculated volume from the reconstituted Totect solution and further dilute to a concentration of 1.3 to 3 mg/mL in Lactated Ringer’s Injection. Do not mix Totect with any other drugs.
Use the Totect infusion solution immediately after preparation. If not used immediately, the product is stable for 4 hours from the time of preparation when stored at room temperature or for up to 12 hours when stored refrigerated between 2ºC to 8ºC (36°F to 46°F).
The infusion solutions have a pH of 3.5 to 5.5.
2.5 Administration
Do not administer or mix Totect with any other drugs during the infusion.
Administration for Treatment of Extravasation
Remove cooling procedures such as ice packs, if used, from the extravasation area at least 15 minutes before Totect administration in order to allow sufficient blood flow to the area of extravasation.
Administer the final diluted solution of Totect as an intravenous infusion over 1 to 2 hours at room temperature and normal light conditions in a large caliber vein in an extremity/area other than the one affected by the extravasation.
Treatment on Day 2 and Day 3 should start at the same hour (+/- 3 hours) as on the first day.
Perform local examination for extravasation on a regular basis after treatment and until resolution.
If vesicant compounds other than anthracyclines are being used through the same intravenous access, (e.g. vincristine, mitomycin, and vinorelbine), consider treatments for these other vesicant compounds. Totect is not effective against the effects of vesicants other than anthracyclines [see Clinical Studies (14.1)].
Administration for Reducing the Incidence and Severity of Cardiomyopathy
Administer the final diluted solution of Totect by intravenous infusion over 15 minutes before the administration of doxorubicin.
Do not administer via an intravenous push.
Administer doxorubicin within 30 minutes after the completion of Totect infusion.
Do not use Totect with non-anthracycline chemotherapy regimens.
3. Dosage Forms and Strengths
For Injection: 500 mg as a sterile, pyrogen-free lyophilized powder in a single dose vial for reconstitution.
5. Warnings and Precautions
5.1 Myelosuppression
Treatment with Totect is associated with leukopenia, neutropenia, and thrombocytopenia. Totect may add to the myelosuppression caused by chemotherapeutic agents.
With extravasation, grade 2-4 decreased white blood cells (73%), decreased neutrophils (61%), and decreased platelets (26%) occurred in patients treated with Totect and cytotoxic chemotherapy in clinical trials. Febrile neutropenia occurred in 2.5% of patients [see Adverse Reactions (6.1)].
When used to reduce the incidence and severity of cardiomyopathy, obtain a complete blood count prior to each course of therapy, and administer Totect and chemotherapy only when adequate hematologic parameters are met.
5.2 Concomitant Chemotherapy
Only use Totect in those patients who have received a cumulative doxorubicin dose of 300 mg/m2 and are continuing with doxorubicin therapy. Do not use with chemotherapy initiation as Totect may interfere with the antitumor activity of the chemotherapy regimen.
In a trial conducted in patients with metastatic breast cancer who were treated with fluorouracil, doxorubicin, and cyclophosphamide (FAC) with or without dexrazoxane starting with their first cycle of FAC therapy, patients who were randomized to receive dexrazoxane had a lower response rate (48% vs. 63%) and shorter time to progression than patients who were randomized to receive placebo.
5.3 Cardiac Toxicity
Treatment with Totect does not completely eliminate the risk of anthracycline-induced cardiac toxicity. Monitor cardiac function before and periodically during therapy to assess left ventricular ejection fraction (LVEF). If deterioration in cardiac function occurs, consider the benefit of continued therapy against the risk of producing irreversible cardiac damage.
5.4 Secondary Malignancies
Secondary malignancies such as acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) have been reported in studies of pediatric patients who have received dexrazoxane in combination with chemotherapy. The safety and efficacy of Totect has not been established for use in pediatric patients. Some adult patients who received dexrazoxane in combination with anti-cancer agents known to be carcinogenic have also developed secondary malignancies, including AML and MDS.
Razoxane is the racemic mixture, of which dexrazoxane is the S(+)-enantiomer. Secondary malignancies (primarily AML) have been reported in patients treated chronically with oral razoxane. In these patients, the total cumulative dose of razoxane ranged from 26 to 480 grams and the duration of treatment was from 42 to 319 weeks. One case of T-cell lymphoma, one case of B-cell lymphoma, and six to eight cases of cutaneous basal cell or squamous cell carcinoma have also been reported in patients treated with razoxane. Long-term administration of razoxane to rodents was associated with the development of malignancies [see Nonclinical Toxicology (13.1)].
5.5 Anaphylactic/Hypersensitivity Reactions
Hypersensitivity reactions including anaphylactic reaction, angioedema, skin reactions, bronchospasm, respiratory distress, hypotension and loss of consciousness have occurred in patients treated with dexrazoxane products and anthracyclines [see Adverse Reactions (6.1)]. Previous history of allergy to dexrazoxane products should be carefully considered prior to administration. Consider permanent discontinuation in patients with severe hypersensitivity reactions.
5.6 Embryo-Fetal Toxicity
Totect can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings from animal studies. In animal reproduction studies, intravenous administration of dexrazoxane to pregnant rats and rabbits during organogenesis resulted in teratogenicity at maternal doses approximately 0.1 and 0.2 times, respectively, the human dose of 1000 mg/m2. Apprise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of Totect. Advise males with female partners of reproductive potential to use effective contraception during treatment with Totect and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3) and Clinical Pharmacology (12.1)].
6. Adverse Reactions/Side Effects
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
Extravasation
In the clinical studies, Totect was administered to patients also receiving chemotherapeutic agents for cancer, and the adverse reaction profile and laboratory abnormalities presented in Tables 1 and 2 reflect the combination of Totect, underlying disease, and already administered chemotherapy. The adverse reaction data reflect exposure to Totect from two clinical studies in 80 patients who received the first dose, 72 patients who received two doses, and 69 patients who received all three doses. Table 1 summarizes adverse reactions occurring with ≥ 5% frequency.
| System Organ Class (SOC) and Adverse Reaction | Study 1 and 2 Combined (All causalities) % (N=80) |
|
|---|---|---|
| Total number of patients with at least one event | 85 | |
| General disorders and administration site conditions | 58 | |
| Pyrexia | 21 | |
| Injection site pain/injection site discomfort | 16 | |
| Fatigue | 13 | |
| Edema peripheral | 10 | |
| Injection site phlebitis | 6 | |
| Gastrointestinal disorders | 55 | |
| Nausea | 43 | |
| Vomiting | 19 | |
| Diarrhea | 11 | |
| Abdominal pain | 6 | |
| Constipation | 6 | |
| Infections and infestations | 30 | |
| Postoperative infection | 16 | |
| Nervous system disorders | 24 | |
| Dizziness | 11 | |
| Headache | 6 | |
| Skin and subcutaneous disorders | 18 | |
| Alopecia | 14 | |
| Respiratory, thoracic and mediastinal disorders | 16 | |
| Dyspnea | 8 | |
| Pneumonia Cough | 6 5 |
|
| Vascular disorders | 15 | |
| Blood and lymphatic system disorders | 14 | |
| Anemia | 6 | |
| Psychiatric disorders | 14 | |
| Depression Insomnia | 8 5 |
|
| Musculoskeletal and connective tissue disorders | 13 | |
| Metabolism and nutrition disorders Anorexia | 10 5 |
|
| Cardiac disorders | 5 | |
Table 2 summarizes laboratory abnormalities from studies 1 and 2.
| Laboratory Abnormality | Grade 3 | Grade 4 | Grade 2 to 4 |
| % | % | % | |
| Hematologic | |||
| Decreased hemoglobin | 3 | 0 | 43 |
| Decreased WBC | 25 | 20 | 73 |
| Decreased neutrophils | 22 | 24 | 61 |
| Decreased platelets | 21 | 0 | 26 |
| Hepatic | |||
| Increased bilirubin | 2 | 0 | 11 |
| Increased AST | 1 | 1 | 28 |
| Increased ALT | 1 | 5 | 22 |
| Increased alkaline phosphatase | 0 | 0 | 4 |
| Increased LDH | 0 | 0 | 5 |
| Metabolic | |||
| Increased creatinine | 2 | 2 | 14 |
| Decreased sodium | 5 | 1 | 6 |
| Increased calcium total | 2 | 2 | 7 |
Cardiomyopathy
The adverse reaction profile described in this section was identified from randomized, placebo-controlled, double-blind studies in patients with metastatic breast cancer who received the combination of the fluorouracil, doxorubicin, and cyclophosphamide (FAC) chemotherapy regimen with or without dexrazoxane. The dose of doxorubicin was 50 mg/m2 in each of these trials. Treatment was administered every three weeks until disease progression or cardiac toxicity.
Patients in clinical trials who received FAC with dexrazoxane experienced more severe leukopenia, granulocytopenia, and thrombocytopenia than patients receiving FAC without dexrazoxane [see Warnings and Precautions (5.1)].
Table 3 below lists the incidence of adverse reactions for patients receiving FAC with either dexrazoxane or placebo in the breast cancer studies. Adverse experiences occurring during courses 1 through 6 are displayed for patients receiving dexrazoxane or placebo with FAC beginning with their first course of therapy (columns 1 and 3, respectively). Adverse experiences occurring at course 7 and beyond for patients who received placebo with FAC during the first six courses and who then received either dexrazoxane or placebo with FAC are also displayed (columns 2 and 4, respectively).
The adverse reactions listed below in Table 3 demonstrate that the frequency of adverse reaction "Pain on Injection" has been greater for dexrazoxane arm, as compared to placebo.
|
Adverse Reaction |
Percentage (%) of Breast Cancer Patients With Adverse Reaction |
|||
|
FAC + Dexrazoxane |
FAC + Placebo |
|||
|
Courses 1-6 N = 413 |
Courses ≥ 7 N = 102 |
Courses 1–6 N = 458 |
Courses ≥ 7 N = 99 |
|
|
Alopecia |
94 |
100 |
97 |
98 |
|
Nausea |
77 |
51 |
84 |
60 |
|
Vomiting |
59 |
42 |
72 |
49 |
|
Fatigue/Malaise |
61 |
48 |
58 |
55 |
|
Anorexia |
42 |
27 |
47 |
38 |
|
Stomatitis |
34 |
26 |
41 |
28 |
|
Fever |
34 |
22 |
29 |
18 |
|
Infection |
23 |
19 |
18 |
21 |
|
Diarrhea |
21 |
14 |
24 |
7 |
|
Pain on injection |
12 |
13 |
3 |
0 |
|
Sepsis |
17 |
12 |
14 |
9 |
|
Neurotoxicity |
17 |
10 |
13 |
5 |
|
Streaking/Erythema |
5 |
4 |
4 |
2 |
|
Phlebitis |
6 |
3 |
3 |
5 |
|
Esophagitis |
6 |
3 |
7 |
4 |
|
Dysphagia |
8 |
0 |
10 |
5 |
|
Hemorrhage |
2 |
3 |
2 |
1 |
|
Extravasation |
1 |
3 |
1 |
2 |
|
Urticaria |
2 |
2 |
2 |
0 |
|
Recall Skin Reaction |
1 |
1 |
2 |
0 |
Related/similar drugs
7. Drug Interactions
7.1 Dimethyl sulfoxide
When used to treat extravasation, Totect is not recommended for use with topical dimethyl sulfoxide (DMSO). Based on anecdotal reports concurrent use of topical DMSO at the site of tissue injury may reduce the benefit of Totect in this indication. Additionally, nonclinical studies using a mouse model that simulates extravasation of anthracyclines has shown that concomitant treatment with topical DMSO decreases the efficacy of systemic dexrazoxane.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, Totect can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Limited available data with Totect use in pregnant women are insufficient to inform a drug-associated risk of adverse developmental outcomes. In animal reproduction studies, intravenous administration of dexrazoxane to pregnant rats and rabbits during organogenesis resulted in teratogenicity at maternal doses that were approximately 0.1 and 0.2 times, respectively, the human dose of 1000 mg/m². Advise pregnant women of the potential risk to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15–20%, respectively.
Data
Animal Data
In an embryo-fetal development study in rats, pregnant females received intravenous doses of up to 8 mg/kg dexrazoxane during the period of organogenesis. A dose of 8 mg/kg (approximately 0.1 times the human dose of 1000 mg/m²) was teratogenic, resulting in imperforate anus, microphthalmia, and anophthalmia. Doses ≥ 2 mg/kg (approximately 0.01 times the human dose of 1000 mg/m²) caused maternal toxicity.
In an embryo-fetal development study in rabbits, pregnant females received intravenous doses of up to 20 mg/kg (approximately 0.2 times the human dose of 1000 mg/m²) were teratogenic, resulting in several skeletal malformations such as short tail, rib and thoracic malformations, and soft tissue variations including subcutaneous, eye and cardiac hemorrhagic areas, as well as agenesis of the gallbladder and of the intermediate lobe of the lung. Doses ≥ 5 mg/kg (approximately 0.1 times the human dose of 1000 mg/m²) caused maternal toxicity.
In a pre and postnatal development study in rats, intravenous administration of 8 mg/kg dexrazoxane to pregnant rats during organogenesis resulted in impairment of fertility in the male and female offspring. A dose of 8 mg/kg in rats is approximately 0.1 times the human dose of 1000 mg/m².
8.2 Lactation
Risk Summary
There are no data on the presence of dexrazoxane in human milk, the effects on the breastfed child, or the effect on milk production. Because of the potential for serious adverse reactions, such as myelosuppression, in a breastfed child from Totect, advise women not to breastfeed during treatment and for 2 weeks following the final dose of Totect.
8.3 Females and Males of Reproductive Potential
Totect can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Pregnancy testing should be performed prior to initiation of chemotherapy. Therefore, repeat pregnancy testing prior to administration of Totect is not recommended, particularly in treatment of extravasation as treatment should not be delayed.
Contraception
Females
Totect can cause fetal harm when administered to a pregnant woman [see Warnings and Precautions (5.6), Use in Specific Populations (8.1)]. Because of the potential for genotoxicity, advise females of reproductive potential to use effective contraception during treatment and for 6 months following the final dose of Totect.
Males
Because of the potential for genotoxicity, advise males with female partners of reproductive potential to use effective contraception during treatment and for 3 months following the final dose of Totect [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on findings in animal studies, Totect may impair fertility in males of reproductive potential. It is not known whether these effects on fertility are reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of Totect in pediatric patients have not been established.
8.5 Geriatric Use
Clinical studies of Totect did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger patients. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal and hepatic function, care should be taken in dose selection, and it may be useful to monitor renal and hepatic function [see Dosage and Administration (2.3)].
Treatment of Extravasation
In total, 21% of the patients treated with Totect were age 65 years or older and 9% were 75 and older.
Reducing the Incidence and Severity of Cardiomyopathy
In total, 23% of patients that were treated with the recommended dose ratio of dexrazoxane were older than 65 years.
8.6 Renal Impairment
Greater exposure to dexrazoxane may occur in patients with compromised renal function. Monitor patients with impaired renal function for signs of hematological toxicity. Reduce the dose of Totect by 50% in patients with creatinine clearance values < 40 mL/min [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
Extravasation
Use of Totect for treatment of anthracycline extravasation has not been studied in patients with hepatic impairment. Since liver dysfunction (increases in transaminases and bilirubin) may occur (especially after doses of above 1000 mg/m2 dexrazoxane), it is recommended that routine liver function tests be performed before each administration of dexrazoxane in patients with known liver function disorders. Use in patients with hepatic impairment is not recommended.
Cardiomyopathy
In patients receiving dexrazoxane to reduce the incidence and severity of cardiomyopathy, reduce the Totect dosage proportionately (maintaining the 10:1 ratio) in patients with hepatic impairment, since a doxorubicin dose reduction is recommended in the presence of hyperbilirubinemia [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
10. Overdosage
Disposition studies with dexrazoxane have not been conducted in cancer patients undergoing dialysis, but retention of a significant dose fraction (>0.4) of the unchanged drug in the plasma pool, minimal tissue partitioning or binding, and availability of greater than 90% of the systemic drug levels in the unbound form suggest that it could be removed using conventional peritoneal or hemodialysis.
Overdose with dexrazoxane can lead to signs of bone marrow failure. There is no known antidote for dexrazoxane. Instances of suspected overdose should be managed with supportive care until resolution of myelosuppression and related conditions is complete. Management of overdose should include treatment of infections, fluid regulation, and maintenance of nutritional requirements.
11. Totect Description
Totect (dexrazoxane for injection) is a sterile, pyrogen-free lyophilized powder intended for intravenous (IV) administration. Each Totect carton contains 1 single dose vial of Totect (dexrazoxane for injection) 500 mg.
Chemically, dexrazoxane a cytoprotective agent, is 2,6-piperazinedione,4,4'-(1-methyl-1,2-ethanediyl)bis-,(S)- or (S)-(+)-1,2-bis(3,5-dioxopiperazin-1-yl)propane. The following diagram shows the chemical structure:
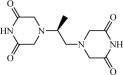
The molecular formula is C11H16N4O4; the molecular weight is 268.3. Dexrazoxane is a white to off-white powder, with a melting point of 194 ± 3 °C. It is soluble in dioxane and 0.1 N HCl, sparingly soluble in water, tetrahydrofuran, citrate buffer at pH 4.0, phosphate buffer at pH 7.0, and borate-potassium chloride sodium hydroxide buffer at pH 9.0. The acid dissociation constants, pKa, are 2.5 (for the tertiary piperazine nitrogen) and 9.7 (for the nitrogen imide). Log P is -2.135.
The finished product is supplied in a sterile form for intravenous infusion only following mixing and diluting.
Each carton contains one 50 mL Type I glass vial. Each vial contains 568 mg dexrazoxane hydrochloride, equivalent to 500 mg dexrazoxane. Hydrochloric Acid, NF is added for pH adjustment. When reconstituted as directed with 50 mL of Sterile Water for injection, USP, each mL contains 10 mg dexrazoxane and the pH of the resultant solution is 1.0 to 3.0.
The admixture should be further diluted prior to administration to patients [see Dosage and Administration (2.4)].
Each vial of dexrazoxane for injection is closed with an aluminum flip-off cap covered with a dark red overcap.
12. Totect - Clinical Pharmacology
12.1 Mechanism of Action
The mechanism by which dexrazoxane reduces tissue damage caused by anthracycline-related cardiomyopathy or following anthracycline extravasation is not fully understood. Dexrazoxane is a cyclic derivative of EDTA that penetrates cell membranes. Results of laboratory studies suggest that dexrazoxane is converted intracellularly to a ring-opened chelating agent that interferes with iron-mediated free radical generation. Some evidence suggests that dexrazoxane inhibits topoisomerase II reversibly.
12.3 Pharmacokinetics
The pharmacokinetics of dexrazoxane have been studied in advanced cancer patients with normal renal and hepatic function. Generally, the pharmacokinetics of dexrazoxane can be adequately described by a two-compartment open model with first-order elimination. Dexrazoxane has been administered as a 15 minute infusion over a dose-range of 60 to 900 mg/m² with 60 mg/m² of doxorubicin, and at a fixed dose of 500 mg/m² with 50 mg/m² doxorubicin. The disposition kinetics of dexrazoxane are dose-independent, as shown by linear relationship between the area under plasma concentration-time curves and administered doses ranging from 60 to 900 mg/m². The mean peak plasma concentration of dexrazoxane was 36.5 μg/mL at the end of the 15 minute infusion of a 500 mg/m² dose of dexrazoxane administered 15 to 30 minutes prior to the 50 mg/m² doxorubicin dose. The important pharmacokinetic parameters of dexrazoxane are summarized in the following table.
|
a Coefficient of variation |
||||||
|
b Steady-state volume of distribution |
||||||
| Dose Doxorubicin (mg/m²) | Dose Dexrazoxane (mg/m²) | Number of Subjects | Elimination Half-Life (h) | Plasma Clearance (L/h/m²) | Renal Clearance (L/h/m²) | bVolume of Distribution (L/m²) |
| 50 | 500 | 10 | 2.5 (16) | 7.88 (18) | 3.35 (36) | 22.4 (22) |
| 60 | 600 | 5 | 2.1 (29) | 6.25 (31) | — | 22.0 (55) |
Following a rapid distributive phase (~0.2 to 0.3 hours), dexrazoxane reaches post-distributive equilibrium within 2 to 4 hours. The estimated steady-state volume of distribution of dexrazoxane suggests its distribution primarily in the total body water (25 L/m²).
In a study of the pharmacokinetics of dexrazoxane following the recommended dosing for patients with anthracycline extravasation, the mean systemic clearance and steady-state volume of distribution of dexrazoxane in six female patients undergoing treatment for anthracycline extravasations at a dose of 1000 mg/m² Totect on Days 1 and 2 and 500 mg/m² on Day 3 were similar to that observed when administered with doxorubicin. The systemic clearances (mean ± SD) were similar among Day 1 (5.9 ± 2.0 L/h/m²), Day 2 (6.4 ± 2.1 L/h/m²), and Day 3 (7.9 ± 3.0 L/h/m²). The terminal elimination half life did not change over 3 days (2.1-2.2 h). The volume of distribution was 17.9 ~ 22.6 L/m².
In vitro studies on dexrazoxane in human microsomes indicated that metabolism is unlikely via cytochrome P450. Qualitative metabolism studies with dexrazoxane have confirmed the presence of unchanged drug, a diacid-diamide cleavage product, and two monoacid-monoamide ring products in the urine of animals and man. The metabolite levels were not measured in the pharmacokinetic studies.
Urinary excretion plays an important role in the elimination of dexrazoxane. Forty-two percent of the 500 mg/m² dose of dexrazoxane was excreted in the urine.
Protein Binding: In vitro studies have shown that dexrazoxane is not bound to plasma proteins.
Effects of Gender
There are no clinically relevant differences in the pharmacokinetics of dexrazoxane between males and females.
Renal insufficiency
The pharmacokinetics of dexrazoxane were assessed following a single 15 minute IV infusion of 150 mg/m² of dexrazoxane in male and female subjects with varying degrees of renal dysfunction as determined by creatinine clearance (CLCR) based on a 24-hour urinary creatinine collection. Dexrazoxane clearance was reduced in subjects with renal dysfunction. Compared with controls, the mean AUC0-inf value was twofold greater in subjects with moderate (CLCR 30-50 mL/min) to severe (CLCR < 30 mL/min) renal dysfunction. Modeling demonstrated that equivalent exposure (AUC0-inf) could be achieved if dosing were reduced by 50% in subjects with creatinine clearance values < 40 mL/min compared with control subjects (CLCR > 80 mL/min) [see Dosage and Administration (2.3)]. Monitor patients with renal insufficiency for signs of hematological toxicity [see Use in Specific Populations (8.6)].
Hepatic insufficiency
The pharmacokinetics of dexrazoxane have not been evaluated in patients with hepatic impairment.
Drug interactions
In vitro studies indicated that dexrazoxane is not an inhibitor for CYP1A, CYP2C9, CYP2C19, CYP2D6 or CYP3A.
In studies of dexrazoxane co-administrated with doxorubicin (50 to 60 mg/m²) or epirubicin (60 to 100 mg/m²), there were no indications of significant pharmacokinetic interactions.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with dexrazoxane. A study by the National Cancer Institute has reported that long term dosing with razoxane (the racemic mixture of dexrazoxane, ICRF-187, and its enantiomer ICRF-186) is associated with the development of malignancies in rats and possibly in mice [see Warnings and Precautions (5.4)].
Dexrazoxane was not mutagenic to bacteria in vitro (Ames assay), but was clastogenic in an in vitro chromosomal aberrations study in mammalian cells and in an in vivo bone marrow micronucleus study in mice.
Fertility studies with dexrazoxane have not been conducted. Testicular atrophy was seen with intravenous dexrazoxane administration at doses as low as 30 mg/kg weekly for 6 weeks in rats (approximately 0.2 times the human dose of 1000 mg/m²) and as low as 20 mg/kg weekly for 13 weeks in dogs (approximately 0.4 times the human dose of 1000 mg/m²).
14. Clinical Studies
14.1 Extravasation
Totect was studied in two open-label, single arm, multi-center studies testing whether Totect administration could reduce tissue injury following anthracycline extravasation and thereby reduce or avoid surgical intervention. Totect is not effective against the effects of vesicants other than anthracyclines.
In the studies, eligible patients were receiving single-agent anthracycline intravenously (usually as part of combination chemotherapy) and developed extravasation symptoms of pain, burning, swelling, and/or redness near the infusion site. Skin biopsy samples from the suspected skin area were examined for the presence of anthracycline as determined by the presence of tissue fluorescence; however, therapy was not delayed for this test result.
In both studies, treatment with Totect was to begin as soon as possible and no later than 6 hours after extravasation with retreatment 24 and 48 hours later (a total of 3 doses). Totect was administered as 1-2 hour IV infusions through a different venous access location. The first and second doses were 1000 mg/m² and the third dose was 500 mg/m². No dose modifications were planned except for patients whose body surface area exceeded 2.0 m², in which case the total daily dose limit on the first and second day was 2000 mg/day and 1000 mg on the third day.
In total, 80 patients were enrolled and 57 were evaluable. Demographics in the two studies were similar. The median age was 57 years, and sixty-five percent of patients were women. The anthracyclines most commonly associated with extravasation were epirubicin (56%) and doxorubicin (41%). Peripheral IV sites of extravasation included the forearm in 63%, the hand in 21%, and the antecubital area in 11%; four patients (5%) received the anthracycline via a central venous access device (CVAD). Most patients presented with swelling (83%), redness (78%), and pain (43%). The median baseline lesion area was 25 cm² (range 1-253 cm²).
Evaluable patients had to be receiving IV anthracycline (single agent or in combination) at the time of extravasation, to have skin biopsies showing fluorescence, and to receive the first Totect dose within 6 hours of the extravasation.
In study 1, none of the 19 evaluable patients required surgical intervention and none had serious late sequelae. In study 2, one of the 38 evaluable patients required surgery. One additional non-evaluable patient required surgery for tissue necrosis. Thirteen patients had late sequelae at the event site such as site pain, fibrosis, atrophy, and local sensory disturbance; all were judged as mild except in the one patient who required surgery. None of the 4 patients with CVADs required surgical intervention.
14.2 Cardiomyopathy
The ability of dexrazoxane to reduce the incidence and severity of doxorubicin-induced cardiomyopathy was evaluated in three prospectively randomized placebo-controlled studies. In these studies, patients were treated with a doxorubicin-containing regimen and either dexrazoxane or placebo starting with the first course of chemotherapy. There was no restriction on the cumulative dose of doxorubicin. Cardiac function was assessed by measurement of the left ventricular ejection fraction (LVEF), utilizing resting multigated nuclear medicine (MUGA) scans, and by clinical evaluations. Patients receiving dexrazoxane had significantly smaller mean decreases from baseline in LVEF and lower incidences of congestive heart failure than the control group; however, in the largest study, patients with advanced breast cancer receiving FAC with dexrazoxane had a lower response rate (48% vs. 63%) and a shorter time to progression than patients who received FAC versus placebo.
In the clinical trials, patients who were initially randomized to receive placebo were allowed to receive dexrazoxane after a cumulative dose of doxorubicin above 300 mg/m2. Retrospective historical analyses showed that the risk of experiencing a cardiac event (see Table 5 for definition) at a cumulative dose of doxorubicin above 300 mg/m2 was greater in the patients who did not receive dexrazoxane beginning with their seventh course of FAC than in the patients who did receive dexrazoxane (HR=13.08; 95% CI: 3.72, 46.03; p<0.001). Overall, 3% of patients treated with dexrazoxane developed CHF compared with 22% of patients not receiving dexrazoxane.
Table 5: Definition of Cardiac Events
- Development of congestive heart failure, defined as having two or more of the following:
a. Cardiomegaly by X-ray
b. Basilar Rales
c. S3 Gallop
d. Paroxysmal nocturnal dyspnea and/or orthopnea and/or significant dyspnea on exertion. - Decline from baseline in LVEF by ≥10% and to below the lower limit of normal for the institution.
- Decline in LVEF by ≥20% from baseline value.
- Decline in LVEF to ≥5% below lower limit of normal for the institution.
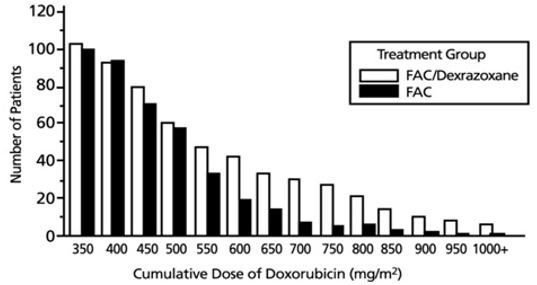
Figure 1 shows the number of patients still on treatment at increasing cumulative doses.
Figure 1 Cumulative Number of Patients On Treatment FAC vs. FAC/Dexrazoxane Patients
Patients Receiving at Least Seven Courses of Treatment
16. How is Totect supplied
Each Totect carton contains 1 single dose vial of Totect (dexrazoxane for injection) 500 mg as a sterile, pyrogen-free lyophilized powder.
NDC 76310-110-01: Carton of 1 vial of Totect.
Store at 20ºC to 25ºC (68ºF to 77ºF); excursions permitted between 15ºC to 30ºC (59ºF to 86ºF) [see USP Controlled Room Temperature]. Protect from light. Keep vial in carton until ready for use.
Follow special handling and disposal procedures [see Dosage and Administration (2.4)].1
17. Patient Counseling Information
See FDA-approved patient labeling (Patient Information)
Myelosuppression
Inform patients of the possibility of myelosuppression, immunosuppression, and infections. Explain the need for routine blood cell counts. Instruct patients to monitor their temperature frequently and immediately report any occurrence of fever [see Warnings and Precautions (5.1)].
Anaphylactic/Hypersensitivity Reactions
Instruct patients to contact their healthcare provider for signs of an allergic reaction, which could be severe and sometimes fatal [see Warnings and Precautions (5.5)].
Embryo-Fetal Toxicity
Advise females of reproductive potential and males with female partners of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the final dose of Totect. Advise females to inform their prescriber of a known or suspected pregnancy. Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 3 months following the final dose of Totect [see Warnings and Precautions (5.6) and Use in Specific Populations (8.1, 8.3)].
Lactation
Advise women not to breastfeed during treatment and for 2 weeks following the final dose of Totect [see Use in Specific Populations (8.2)].
Manufactured for: Clinigen, Inc. Yardley, PA 19067
Totect® is a registered trademark owned by the Clinigen Group.
For additional information, contact Clinigen, Inc. 1-877-776-5385.
US Patent No 6,727,253 B2
|
PATIENT INFORMATION Totect® (TOE-TECT) (dexrazoxane) injection |
|
|
What is Totect? Totect is a prescription medicine used:
It is not known if Totect is safe and effective in children. |
|
|
Before you receive Totect, tell your healthcare provider about all of your medical conditions, including if you:
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal or dietary supplements. Especially tell your healthcare provider if you:
|
|
|
How will I receive Totect?
|
|
|
What are the possible side effects of Totect? Totect can cause serious side effects, including:
|
|
|
o trouble breathing | o dizziness or lightheadedness |
| o swelling of your face, lips, tongue or throat | o faint or pass out |
| o raised bumps (hives) | |
|
|
|
|
|
|
The most common side effect of Totect in women with metastatic breast cancer to reduce the occurrence and severity of heart muscle problems is pain at the intravenous (IV) site. Totect may cause fertility problems in males, which may affect the ability to father children. Talk to your healthcare provider if you have concerns about fertility. These are not all the possible side effects of Totect. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. |
|
|
General information about the safe and effective use of Totect. Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your healthcare provider or pharmacist for information about Totect that is written for health professionals. |
|
|
What are the ingredients in Totect? Active ingredient: dexrazoxane (as a hydrochloride salt) Inactive ingredients: none Manufactured for: Clinigen, Inc. Yardley, PA 19067
Totect® is a registered trademark owned by the Clinigen Group. For additional information, contact Clinigen, Inc. 1-877-776-5385. US Patent No 6,727,253 B2 |
|
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 11/2020


| TOTECT
dexrazoxane hydrochloride injection, powder, lyophilized, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Clinigen Limited (211471076) |
More about Totect (dexrazoxane)
- Check interactions
- Compare alternatives
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: miscellaneous uncategorized agents
- Breastfeeding