Sarclisa: Package Insert / Prescribing Info
Package insert / product label
Generic name: isatuximab
Dosage form: injection, solution, concentrate
Drug class: CD38 monoclonal antibodies
J Code (medical billing code): J9227 (10 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jun 29, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
SARCLISA® (isatuximab-irfc) injection, for intravenous use
Initial U.S. Approval: 2020
Recent Major Changes
Indications and Usage for Sarclisa
SARCLISA is a CD38-directed cytolytic antibody indicated:
- in combination with pomalidomide and dexamethasone, for the treatment of adult patients with multiple myeloma who have received at least 2 prior therapies including lenalidomide and a proteasome inhibitor.
- in combination with carfilzomib and dexamethasone, for the treatment of adult patients with relapsed or refractory multiple myeloma who have received 1 to 3 prior lines of therapy.
- in combination with bortezomib, lenalidomide and dexamethasone, for the treatment of adult patients with newly diagnosed multiple myeloma who are not eligible for autologous stem cell transplant (ASCT). (1)
Sarclisa Dosage and Administration
Dosage Forms and Strengths
Contraindications
Patients with severe hypersensitivity to isatuximab-irfc or to any of its excipients. (4)
Warnings and Precautions
- Infusion-Related Reactions: In case of grade ≥2, interrupt SARCLISA and manage medically. Permanently discontinue for grade 4 infusion-related reactions or anaphylactic reaction. (5.1)
- Infections: SARCLISA can cause serious and fatal infections. Monitor patients for signs and symptoms of infection and treat appropriately. (5.2)
- Neutropenia: Monitor complete blood cell counts periodically during treatment. Monitor patients with neutropenia for signs of infection. SARCLISA dose delays and the use of colony-stimulating factor may be required to allow improvement of neutrophil count. (5.3)
- Second Primary Malignancies (SPM): Monitor patients for the development of second primary malignancies. (5.4)
-
Laboratory Test Interference:
- Interference with Serological Testing (Indirect Antiglobulin Test): Type and screen patients prior to starting treatment. Inform blood banks that a patient has received SARCLISA. (5.5, 7.1)
- Interference with Serum Protein Electrophoresis and Immunofixation Tests: SARCLISA may interfere with the assays used to monitor M-protein, which may impact the determination of complete response. (5.5, 7.1)
- Embryo-Fetal Toxicity: Can cause fetal harm. (5.6)
Adverse Reactions/Side Effects
- In combination with pomalidomide and dexamethasone: The most common adverse reactions (≥20%) are upper respiratory tract infection, infusion-related reactions, pneumonia, and diarrhea. The most common hematology laboratory abnormalities (≥80%) are decreased hemoglobin, decreased neutrophils, decreased lymphocytes, and decreased platelets. (6.1)
- In combination with carfilzomib and dexamethasone: The most common adverse reactions (≥20%) are upper respiratory tract infection, infusion-related reactions, fatigue, hypertension, diarrhea, pneumonia, dyspnea, insomnia, bronchitis, cough, and back pain. The most common hematology laboratory abnormalities (≥80%) are decreased hemoglobin, decreased lymphocytes, and decreased platelets. (6.1)
- In combination with bortezomib, lenalidomide and dexamethasone: The most common adverse reactions (≥20%) are upper respiratory tract infections, diarrhea, fatigue, peripheral sensory neuropathy, pneumonia, musculoskeletal pain, cataract, constipation, peripheral edema, rash, infusion-related reaction, insomnia, and COVID-19. The most common hematologic laboratory abnormalities (≥80%) are decreased hemoglobin, decreased leukocytes, decreased lymphocytes, decreased platelets, and decreased neutrophils. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Sarclisa
SARCLISA is indicated:
- in combination with pomalidomide and dexamethasone, for the treatment of adult patients with multiple myeloma who have received at least 2 prior therapies including lenalidomide and a proteasome inhibitor.
- in combination with carfilzomib and dexamethasone, for the treatment of adult patients with relapsed or refractory multiple myeloma who have received 1 to 3 prior lines of therapy.
- in combination with bortezomib, lenalidomide, and dexamethasone, for the treatment of adult patients with newly diagnosed multiple myeloma who are not eligible for autologous stem cell transplant (ASCT).
2. Sarclisa Dosage and Administration
2.1 Recommended Dosage
- Administer pre-infusion medications [see Dosage and Administration (2.2)].
- SARCLISA should be administered by a healthcare professional, with immediate access to emergency equipment and appropriate medical support to manage infusion-related reactions if they occur [see Warnings and Precautions (5.1)].
The recommended dose of SARCLISA is 10 mg/kg actual body weight administered as an intravenous infusion in combination with pomalidomide and dexamethasone or in combination with carfilzomib and dexamethasone, or in combination with bortezomib, lenalidomide, and dexamethasone.
SARCLISA dosing schedules are provided in Tables 1 and 2 [see Clinical Studies (14)].
| Cycles | Dosing schedules |
|---|---|
| Cycle 1 (28-day cycle) | Days 1, 8, 15, and 22 (weekly) |
| Cycle 2 and beyond (28-day cycles) | Days 1, 15 (every 2 weeks) |
| Cycles | Dosing schedules |
|---|---|
| Cycle 1 (42-day cycle) | Days 1, 8, 15, 22, and 29 |
| Cycles 2 to 4 (42-day cycles) | Days 1, 15, and 29 (every 2 weeks) |
| Cycles 5 to 17 (28-day cycles) | Days 1 and 15 (every 2 weeks) |
| Cycles 18 and beyond (28-day cycles) | Day 1 (every 4 weeks) |
Treatment is repeated until disease progression or unacceptable toxicity.
SARCLISA is used in combination with pomalidomide and dexamethasone or in combination with carfilzomib and dexamethasone or in combination with bortezomib, lenalidomide, and dexamethasone. For dosing instructions of combination agents administered with SARCLISA, see Clinical Studies (14) and manufacturer's prescribing information.
2.2 Recommended Premedications and Antimicrobial Prophylaxis
Recommended Premedications
Administer the following premedications prior to SARCLISA infusion to reduce the risk and severity of infusion-related reactions [see Warnings and Precautions (5.1)]:
-
When administered in combination with SARCLISA and pomalidomide: Dexamethasone 40 mg orally or intravenously (or 20 mg orally or intravenously for patients ≥75 years of age).
When administered in combination with SARCLISA and carfilzomib: Dexamethasone 20 mg (intravenously on the days of SARCLISA and/or carfilzomib infusions, orally on day 22 in cycle 2 and beyond, and orally on day 23 in all cycles).
When administered in combination with SARCLISA, bortezomib, and lenalidomide: Dexamethasone 20 mg (intravenously on the days of SARCLISA infusions, orally on the other days). - Acetaminophen 650 mg to 1,000 mg orally (or equivalent).
- H2 antagonist
- Diphenhydramine 25 mg to 50 mg orally or intravenously (or equivalent). The intravenous route is preferred for at least the first 4 infusions.
The above recommended dose of dexamethasone (orally or intravenously) corresponds to the dose to be administered before infusion as part of the premedication and part of the backbone treatment. Administer dexamethasone before SARCLISA and pomalidomide, before SARCLISA and carfilzomib, and before SARCLISA, bortezomib, and lenalidomide administration.
Administer the recommended premedication agents 15 to 60 minutes prior to starting a SARCLISA infusion.
Recommended Antimicrobial Prophylaxis
Initiate antibacterial and antiviral prophylaxis (such as herpes zoster prophylaxis) if needed based on standard guidelines [see Warnings and Precautions (5.2)].
2.3 Dosage Modifications
No dose reduction of SARCLISA is recommended. Dose delay may be required to allow recovery of blood counts in the event of hematological toxicity [see Warnings and Precautions (5.3, 5.5)]. For information concerning drugs given in combination with SARCLISA, see manufacturer's prescribing information.
2.4 Preparation
Prepare the solution for infusion using aseptic technique as follows:
Calculate the dose (mg) of required SARCLISA based on actual patient weight (measured prior to each cycle to have the administered dose adjusted accordingly) [see Dosage and Administration (2.1)]. More than one SARCLISA vial may be necessary to obtain the required dose for the patient.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Remove the volume of diluent from the 250 mL Sodium Chloride Injection, or 5% Dextrose Injection, diluent bag that is equal to the required volume of SARCLISA injection.
- Withdraw the necessary volume of SARCLISA injection from the vial and dilute by adding to the infusion bag of 0.9% Sodium Chloride Injection, or 5% Dextrose Injection. Discard any unused portion left in the vial.
- The infusion bag must be made of polyolefins (PO), polyethylene (PE), polypropylene (PP), polyvinyl chloride (PVC) with di-(2-ethylhexyl) phthalate (DEHP) or ethyl vinyl acetate (EVA).
- Gently homogenize the diluted solution by inverting the bag. Do not shake.
2.5 Administration
- Administer the infusion solution by intravenous infusion using an intravenous tubing infusion set (in PE, PVC with or without DEHP, polybutadiene [PBD], or polyurethane [PU]) with a 0.22 micron in-line filter (polyethersulfone [PES], polysulfone, or nylon).
- The infusion solution should be administered for a period of time that will depend on the infusion rate (see Table 3). Use prepared SARCLISA infusion solution within 48 hours when stored refrigerated at 2°C to 8°C, followed by 8 hours (including the infusion time) at room temperature.
- Do not administer SARCLISA infusion solution concomitantly in the same intravenous line with other agents.
- On the days where both SARCLISA and carfilzomib are administered, administer dexamethasone first, followed by SARCLISA infusion, then followed by carfilzomib infusion.
Infusion Rates
Following dilution, administer the SARCLISA infusion solution intravenously at the infusion rates presented in Table 3. Incremental escalation of the infusion rate should be considered only in the absence of infusion-related reactions [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
| Dilution Volume | Initial Rate | Absence of Infusion-Related Reaction | Rate Increment | Maximum Rate | |
|---|---|---|---|---|---|
| First infusion | 250 mL | 25 mL/hour | For 60 minutes | 25 mL/hour every 30 minutes | 150 mL/hour |
| Second infusion | 250 mL | 50 mL/hour | For 30 minutes | 50 mL/hour for 30 minutes then increase by 100 mL/hour | 200 mL/hour |
| Subsequent infusions | 250 mL | 200 mL/hour | – | – | 200 mL/hour |
3. Dosage Forms and Strengths
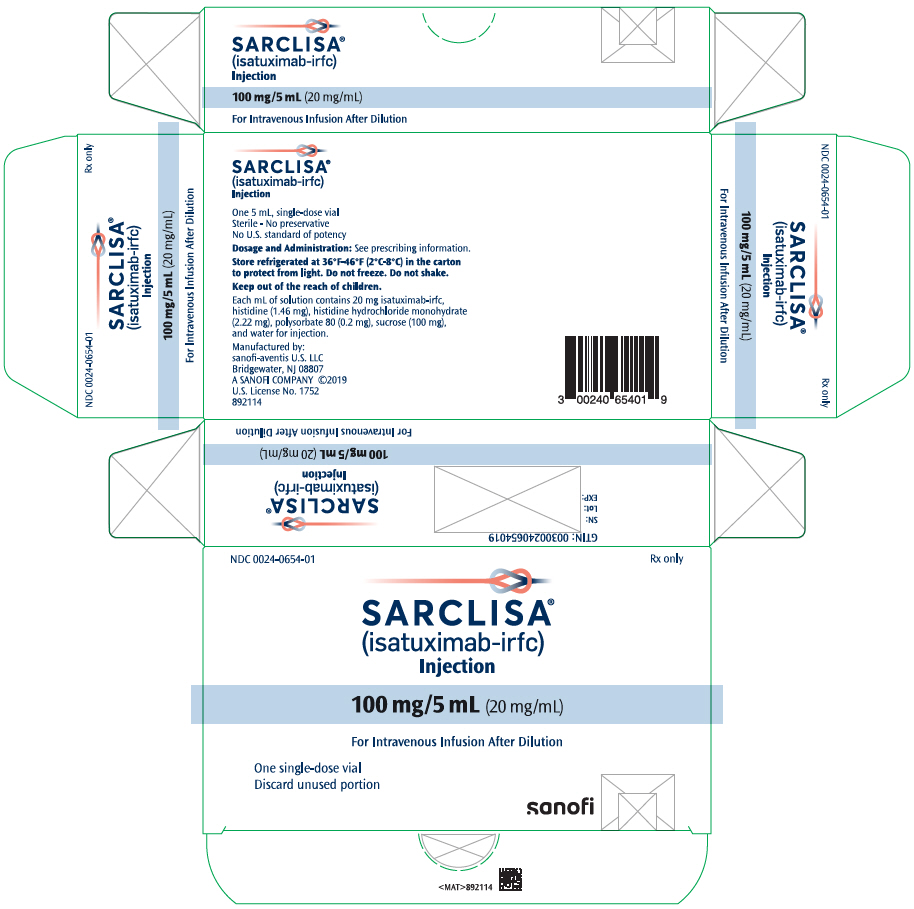
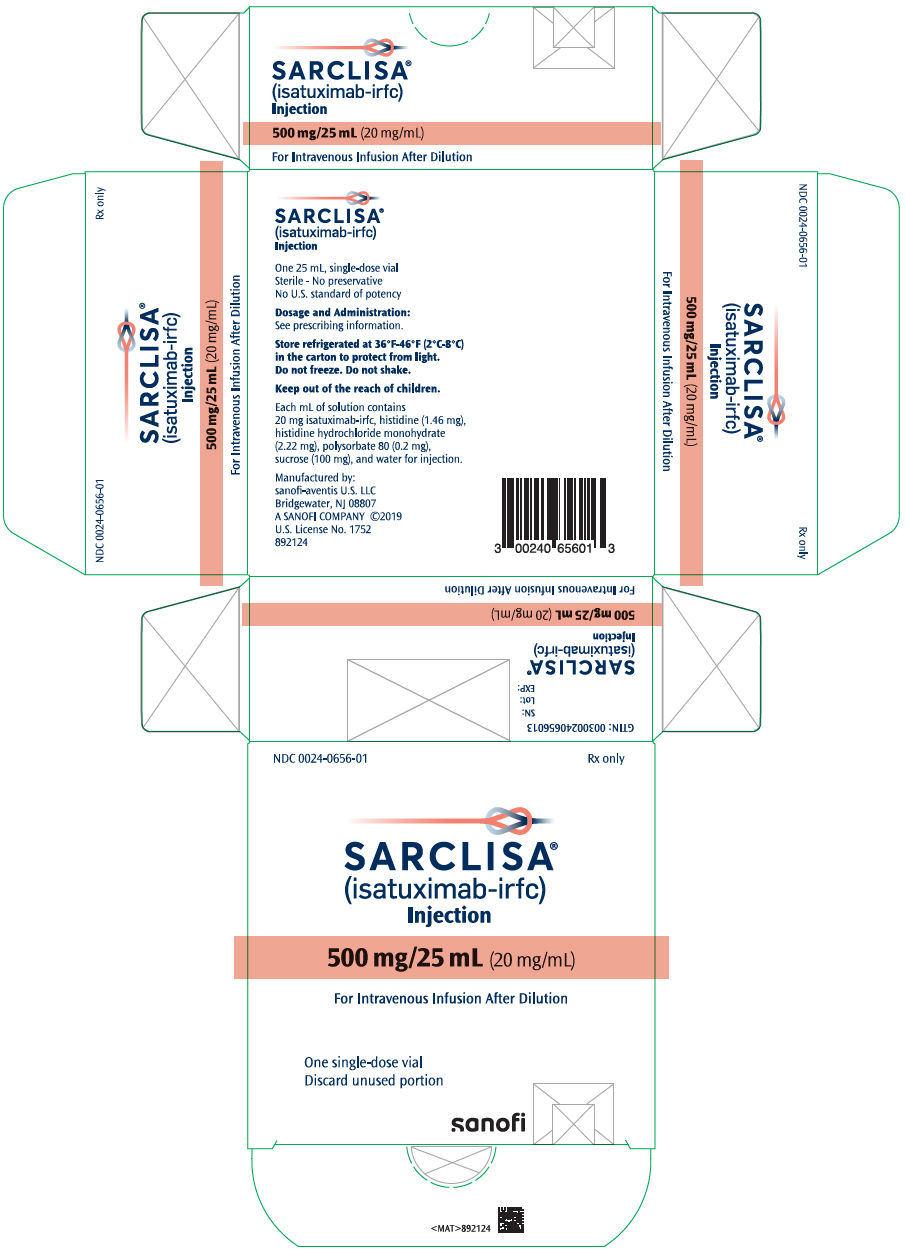
SARCLISA is a clear to slightly opalescent, colorless to slightly yellow solution, essentially free of visible particulates available as:
- Injection: 100 mg/5 mL (20 mg/mL) in a single-dose vial
- Injection: 500 mg/25 mL (20 mg/mL) in a single-dose vial
4. Contraindications
SARCLISA is contraindicated in patients with severe hypersensitivity to isatuximab-irfc or to any of its excipients [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Infusion-Related Reactions
Serious infusion-related reactions including life-threatening anaphylactic reactions have occurred with SARCLISA treatment. Severe signs and symptoms included cardiac arrest, hypertension, hypotension, bronchospasm, dyspnea, angioedema, and swelling. In clinical trials (ICARIA-MM, IKEMA, and IMROZ), in patients treated with SARCLISA (N=592), infusion-related reactions occurred in 206 patients (35%). Among these 206 patients, 92% experienced infusion-related reactions during the first infusion and 12% after the first cycle. The most common symptoms (≥5%) of an infusion-related reaction included dyspnea and cough. Grade 1 infusion-related reactions were reported in 6% of patients, grade 2 in 28%, and grade 3 or 4 in 1.2%. Anaphylactic reactions occurred in less than 1% of patients. The total incidence of SARCLISA infusion interruptions was less than 1% and the incidence of patients with at least one SARCLISA infusion interruption due to infusion-related reactions was 26%. The median time to first SARCLISA infusion interruption was 61 minutes (range 4 to 240 minutes). SARCLISA was discontinued in 1% of patients due to infusion-related reactions.
To decrease the risk and severity of infusion-related reactions, premedicate patients prior to SARCLISA infusion with acetaminophen, H2 antagonists, diphenhydramine, or equivalent, and dexamethasone [see Dosage and Administration (2.2)].
Monitor vital signs frequently during the entire SARCLISA infusion. For patients with grade ≥2 reactions, interrupt SARCLISA infusion and provide appropriate medical management. For patients with grade 2 or grade 3 reactions, if symptoms improve to grade ≤1, restart SARCLISA infusion at half of the initial infusion rate, with supportive care as needed, and closely monitor patients. If symptoms do not recur after 30 minutes, the infusion rate may be increased to the initial rate, and then increased incrementally, as shown in Table 3 [see Dosage and Administration (2.5)]. In case symptoms do not improve to grade ≤1 after interruption of SARCLISA infusion, persist or worsen despite appropriate medications, or require hospitalization, permanently discontinue SARCLISA and institute appropriate management. Permanently discontinue SARCLISA if an anaphylactic reaction or life-threatening (grade 4) infusion-related reaction occurs and institute appropriate management.
5.2 Infections
SARCLISA can cause severe, life-threatening, or fatal infections. In patients who received SARCLISA at the recommended dose in ICARIA-MM, IKEMA, and IMROZ (N=592), serious infections, including opportunistic infections, occurred in 46% of patients, grade 3 or 4 infections occurred in 43%, and fatal infections occurred in 4.7%. The most common type of serious infection reported was pneumonia (32%).
Monitor patients for signs and symptoms of infection prior to and during treatment with SARCLISA and treat appropriately. Administer prophylactic antimicrobials according to guidelines [see Dosage and Administration (2.2)].
5.3 Neutropenia
SARCLISA can cause neutropenia.
In clinical trials (ICARIA-MM, IKEMA, and IMROZ), in patients treated with SARCLISA (N=592), neutropenia based on laboratory values occurred in 81%, with grade 3 or 4 occurring in 52%. Neutropenic infections occurred in 12% of patients, with grade 3 or 4 in 4.9%, and febrile neutropenia in 4% [see Adverse Reactions (6.1)].
Monitor complete blood cell counts periodically during treatment. If needed, use antibacterial and antiviral prophylaxis during treatment [see Dosage and Administration (2.2)]. Monitor patients with neutropenia for signs of infection. In case of grade 4 neutropenia delay SARCLISA dose until neutrophil count recovery to at least 1 × 109/L, and provide supportive care with growth factors, according to institutional guidelines. No dose reductions of SARCLISA are recommended.
5.4 Second Primary Malignancies
The incidence of second primary malignancies, during treatment and post-treatment, is increased in patients treated with SARCLISA-containing regimens. In clinical trials (ICARIA-MM, IKEMA, and IMROZ), in patients treated with SARCLISA (N=592), second primary malignancies occurred in 71 patients (12%).
In ICARIA-MM, at a median follow-up time of 52 months, second primary malignancies occurred in 7% of patients in the Isa-Pd arm and in 2% of patients in the Pd arm.
In IKEMA study, at a median follow-up time of 57 months, second primary malignancies occurred in 10% of patients in the Isa-Kd arm and in 8% of patients in the Kd arm.
In IMROZ study, at a median follow-up time of 60 months, second primary malignancies occurred in 16% of patients in the Isa-VRd arm and in 9% of patients in the VRd arm.
The most common (≥1%) second primary malignancies in ICARIA-MM, IKEMA, and IMROZ (N=592) included skin cancers (7% with SARCLISA-containing regimens and 3.1% with comparative regimens) and solid tumors other than skin cancer (4.6% with SARCLISA-containing regimens and 2.9% with comparative regimens). Patients with non-melanoma skin cancer continued treatment after resection of the skin cancer, except 2 patients on the Isa-VRd arm and 1 patient on the VRd arm of the IMROZ study.
Monitor patients for the development of second primary malignancies.
5.5 Laboratory Test Interference
Interference with Serological Testing (Indirect Antiglobulin Test)
SARCLISA binds to CD38 on red blood cells (RBCs) and may result in a false positive indirect antiglobulin test (indirect Coombs test). This interference with the indirect Coombs test may persist for approximately 6 months after the last infusion of SARCLISA. The indirect antiglobulin test was positive during Isa-Pd treatment in 68% of the tested patients, and during Isa-Kd treatment in 63% of patients. In patients with a positive indirect antiglobulin test, blood transfusions were administered without evidence of hemolysis. ABO/RhD typing was not affected by SARCLISA treatment.
Before the first SARCLISA infusion, conduct blood type and screen tests on SARCLISA-treated patients. Consider phenotyping prior to starting SARCLISA treatment. If treatment with SARCLISA has already started, inform the blood bank that the patient is receiving SARCLISA and SARCLISA interference with blood compatibility testing can be resolved using dithiothreitol-treated RBCs. If an emergency transfusion is required, non–cross-matched ABO/RhD-compatible RBCs can be given as per local blood bank practices [see Drug Interactions (7.1)].
Interference with Serum Protein Electrophoresis and Immunofixation Tests
SARCLISA is an IgG kappa monoclonal antibody that can be incidentally detected on both serum protein electrophoresis and immunofixation assays used for the clinical monitoring of endogenous M-protein. This interference can impact the accuracy of the determination of complete response in some patients with IgG kappa myeloma protein [see Drug Interactions (7.1)].
5.6 Embryo-Fetal Toxicity
Based on the mechanism of action, SARCLISA can cause fetal harm when administered to a pregnant woman. SARCLISA may cause fetal immune cell depletion and decreased bone density. Advise pregnant women of the potential risk to a fetus. Advise females with reproductive potential to use an effective method of contraception during treatment with SARCLISA and for 5 months after the last dose [see Use in Specific Populations (8.1, 8.3)]. The combination of SARCLISA with pomalidomide or lenalidomide is contraindicated in pregnant women because pomalidomide or lenalidomide may cause birth defects and death of the unborn child. Refer to the pomalidomide or lenalidomide prescribing information on use during pregnancy.
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions from SARCLISA are also described in other sections of the labeling:
- Infusion-Related Reactions [see Warnings and Precautions (5.1)]
- Infections [see Warnings and Precautions (5.2)]
- Neutropenia [see Warnings and Precautions (5.3)]
- Second Primary Malignancies [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Relapsed and/or Refractory Multiple Myeloma
Combination treatment with pomalidomide and dexamethasone (Isa-Pd)
The safety of SARCLISA was evaluated in ICARIA-MM, a randomized, open-label clinical trial in patients with previously treated multiple myeloma. Patients received SARCLISA 10 mg/kg intravenously, weekly in the first cycle and every two weeks thereafter, in combination with pomalidomide and dexamethasone (Isa-Pd) (n=152) or pomalidomide and dexamethasone (Pd) (n=149) [see Clinical Studies (14)]. Among patients receiving Isa-Pd, 66% were exposed to SARCLISA for 6 months or longer and 24% were exposed for greater than 12 months or longer.
Serious adverse reactions occurred in 62% of patients receiving Isa-Pd. Serious adverse reactions in >5% of patients who received Isa-Pd included pneumonia (26%), upper respiratory tract infections (7%), and febrile neutropenia (7%). Fatal adverse reactions occurred in 11% of patients (those that occurred in more than 1% of patients were pneumonia and other infections [3%]).
Permanent treatment discontinuation due to an adverse reaction (grades 1–4) occurred in 7% of patients who received Isa-Pd. The most frequent adverse reactions requiring permanent discontinuation in patients who received Isa-Pd were infections (2.6%). SARCLISA alone was discontinued in 3% of patients due to infusion-related reactions.
Dosage interruptions due to an adverse reaction occurred in 31% of patients who received SARCLISA. The most frequent adverse reaction requiring dosage interruption was infusion-related reaction (28%).
The most common adverse reactions (≥20%) were upper respiratory tract infection, infusion-related reactions, pneumonia, and diarrhea.
Table 4 summarizes the adverse reactions in ICARIA-MM.
| Adverse Reactions | SARCLISA + Pomalidomide + Dexamethasone (Isa-Pd) | Pomalidomide + Dexamethasone (Pd) | ||||
|---|---|---|---|---|---|---|
| (N=152) | (N=149) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| CTCAE version 4.03 | ||||||
|
||||||
| General disorders and administration site conditions | ||||||
| Infusion-related reaction* | 38 | 1.3 | 1.3 | 0 | 0 | 0 |
| Infections | ||||||
| Upper respiratory tract infection† | 57 | 9 | 0 | 42 | 3.4 | 0 |
| Pneumonia‡ | 31 | 22 | 3.3 | 23 | 16 | 2.7 |
| Blood and lymphatic system disorders | ||||||
| Febrile neutropenia | 12 | 11 | 1.3 | 2 | 1.3 | 0.7 |
| Respiratory, thoracic and mediastinal disorders | ||||||
| Dyspnea§ | 17 | 5 | 0 | 12 | 1.3 | 0 |
| Gastrointestinal disorders | ||||||
| Diarrhea | 26 | 2 | 0 | 19 | 0.7 | 0 |
| Nausea | 15 | 0 | 0 | 9 | 0 | 0 |
| Vomiting | 12 | 1.3 | 0 | 3.4 | 0 | 0 |
Table 5 summarizes the hematology laboratory abnormalities in ICARIA-MM.
| Laboratory Parameter | SARCLISA + Pomalidomide + Dexamethasone (Isa-Pd) | Pomalidomide + Dexamethasone (Pd) | ||||
|---|---|---|---|---|---|---|
| (N=152) | (N=149) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| The denominator used to calculate the percentages was based on the safety population. | ||||||
| Hemoglobin decreased | 99 | 32 | 0 | 97 | 28 | 0 |
| Neutrophils decreased | 96 | 24 | 61 | 92 | 38 | 31 |
| Lymphocytes decreased | 92 | 42 | 13 | 92 | 35 | 8 |
| Platelets decreased | 84 | 14 | 16 | 79 | 9 | 15 |
Combination treatment with carfilzomib and dexamethasone (Isa-Kd)
The safety of SARCLISA was evaluated in IKEMA, a randomized, open-label clinical trial in patients with previously treated multiple myeloma. Patients received SARCLISA 10 mg/kg intravenously weekly in the first cycle, and every two weeks thereafter, in combination with carfilzomib and dexamethasone (Isa-Kd) (n=177) or carfilzomib and dexamethasone (Kd) (n=122) [see Clinical Studies (14)]. Among patients receiving Isa-Kd, 68% were exposed to SARCLISA for 12 months or longer and 51% were exposed for greater than 18 months.
Serious adverse reactions occurred in 59% of patients receiving Isa-Kd. The most frequent serious adverse reactions in >5% of patients who received Isa-Kd were pneumonia (25%) and upper respiratory tract infections (9%). Adverse reactions with a fatal outcome during treatment were reported in 3.4% of patients in the Isa-Kd group (those occurring in more than 1% of patients were pneumonia occurring in 1.7% and cardiac failure in 1.1% of patients).
Permanent treatment discontinuation due to an adverse reaction (grades 1–4) occurred in 8% of patients who received Isa-Kd. The most frequent adverse reactions requiring permanent discontinuation in patients who received Isa-Kd were infections (2.8%). SARCLISA alone was discontinued in 0.6% of patients due to infusion-related reactions.
Dosage interruptions due to an adverse reaction occurred in 33% of patients who received SARCLISA. The most frequent adverse reaction requiring dosage interruption was infusion-related reaction (30%).
The most common adverse reactions (≥20%) were upper respiratory tract infection, infusion-related reactions, fatigue, hypertension, diarrhea, pneumonia, dyspnea, insomnia, bronchitis, cough, and back pain.
Table 6 summarizes the adverse reactions in IKEMA.
| Adverse Reactions | SARCLISA + Carfilzomib + Dexamethasone (Isa-Kd) | Carfilzomib + Dexamethasone (Kd) | ||||
|---|---|---|---|---|---|---|
| (N=177) | (N=122) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
|
||||||
| General disorders and administration site conditions | ||||||
| Infusion-related reaction* | 46 | 0.6 | 0 | 3.3 | 0 | 0 |
| Fatigue† | 42 | 5 | 0 | 32 | 3.3 | 0 |
| Infections | ||||||
| Upper respiratory tract infection‡ | 67 | 9 | 0 | 57 | 7 | 0 |
| Pneumonia§ | 36 | 19 | 3.4 | 30 | 15 | 2.5 |
| Bronchitis¶ | 24 | 2.3 | 0 | 13 | 0.8 | 0 |
| Vascular disorders | ||||||
| Hypertension# | 37 | 20 | 0.6 | 32 | 18 | 1.6 |
| Respiratory, thoracic and mediastinal disorders | ||||||
| DyspneaÞ | 29 | 5 | 0 | 24 | 0.8 | 0 |
| Coughß | 23 | 0 | 0 | 15 | 0 | 0 |
| Gastrointestinal disorders | ||||||
| Diarrhea | 36 | 2.8 | 0 | 29 | 2.5 | 0 |
| Vomiting | 15 | 1.1 | 0 | 9 | 0.8 | 0 |
Table 7 summarizes the hematology laboratory abnormalities in IKEMA.
| Laboratory Parameter | SARCLISA + Carfilzomib + Dexamethasone (Isa-Kd) | Carfilzomib + Dexamethasone (Kd) | ||||
|---|---|---|---|---|---|---|
| (N=177) | (N=122) | |||||
| All Grades (%) | Grade 3 (%) | Grade 4 (%) | All Grades (%) | Grade 3 (%) | Grade 4 (%) |
|
| The denominator used to calculate the percentage was based on the safety population. | ||||||
| Hemoglobin decreased | 99 | 22 | 0 | 99 | 20 | 0 |
| Lymphocytes decreased | 94 | 52 | 17 | 95 | 43 | 14 |
| Platelets decreased | 94 | 19 | 11 | 88 | 16 | 8 |
| Neutrophils decreased | 55 | 18 | 1.7 | 43 | 7 | 0.8 |
Newly Diagnosed Multiple Myeloma not Eligible for Autologous Stem Cell Transplant
Combination treatment with bortezomib, lenalidomide, and dexamethasone (Isa-VRd)
The safety of SARCLISA was evaluated in IMROZ, a randomized, open-label clinical trial in patients with newly diagnosed multiple myeloma. Patients received SARCLISA 10 mg/kg intravenously on day 1, 8, 15, 22, and 29 in the first 42-day cycle, followed by every two weeks from cycle 2 to 4 (42-day cycles), followed by every two weeks from cycle 5 to 17 (28-day cycles), and then on day 1 from cycle 18 and onwards (28-day cycles), in combination with bortezomib, lenalidomide, and dexamethasone (Isa-VRd) (n=263) or bortezomib, lenalidomide, and dexamethasone (VRd) (n= 181) [see Clinical Studies (14)]. In IMROZ, median duration of exposure to treatment was 53 (range: 0.5–69) months in patients treated with Isa-VRd and 31 (range 0.6–67) months in patients treated with VRd.
Serious adverse reactions occurred in 71% of patients receiving Isa-VRd. The serious adverse reaction in > 5% of patients who received Isa-VRd was pneumonia (30%). Fatal adverse reactions occurred in 11% of patients with Isa-VRd (those occurring in more than 1% of patients were pneumonia (5%).
Permanent discontinuation of treatment due to an adverse reaction occurred in 22.8% of patients treated with Isa-VRd. The most frequent adverse reactions requiring permanent discontinuation in patients who received Isa-VRd were infections (8%). SARCLISA alone was discontinued in 2.3% of patients.
Dosage interruptions due to an adverse reaction occurred in 21% of patients who received SARCLISA. The most frequent adverse reaction requiring dosage interruption was infusion related reaction (21%).
The most common adverse reactions (≥20%) were upper respiratory tract infections, diarrhea, fatigue, peripheral sensory neuropathy, pneumonia, musculoskeletal pain, cataract, constipation, peripheral edema, rash, infusion-related reaction, insomnia, and COVID-19. The most common hematologic laboratory abnormalities (≥80%) were decreased hemoglobin, decreased leukocytes, decreased lymphocytes, decreased platelets, and decreased neutrophils.
Table 8 summarizes the adverse reactions in IMROZ.
| IMROZ study | ||||
|---|---|---|---|---|
| Adverse Reactions | SARCLISA + Bortezomib + Lenalidomide + Dexamethasone (N=263) | Bortezomib + Lenalidomide + Dexamethasone (N=181) |
||
| All Grades (%) | Grade 3 or 4 (%) | All Grades (%) | Grade 3 or 4 (%) | |
|
||||
| Infections and infestations | ||||
| Upper respiratory tract infection* | 65 | 4.6 | 57† | 6 |
| Pneumonia‡ | 45§ | 26 | 31¶ | 19 |
| COVID-19# | 22 | 0.8 | 17 Þ | 1.7 |
| General disorders and administration site conditions | ||||
| Fatigueß | 55 | 11 | 50 | 9 |
| Edema peripheral | 33 | 0 | 33 | 1.1 |
| Infusion-related reaction | 24 | 0.4 | 1.1 | 0 |
| Gastrointestinal disorders | ||||
| Diarrhea | 55 | 8 | 49 | 8 |
| Constipation | 36 | 2.3 | 41 | 1.7 |
| Nervous system disorders | ||||
| Peripheral sensory neuropathy | 54 | 7 | 61 | 6 |
| Eye disorders | ||||
| Cataract | 38 | 16 | 25 | 11 |
| Musculoskeletal and connective tissue disorders | ||||
| Musculoskeletal pain* | 38 | 4.2 | 33 | 3.3 |
| Skin and subcutaneous tissue disorders | ||||
| Rashà | 32 | 5 | 34 | 5 |
| Psychiatric disorders | ||||
| Insomnia | 22 | 3.8 | 24 | 2.2 |
Table 9 summarizes the hematology laboratory abnormalities in IMROZ.
| Laboratory parameter | SARCLISA + Bortezomib + Lenalidomide + Dexamethasone* | Bortezomib + Lenalidomide + Dexamethasone* | ||
|---|---|---|---|---|
| All Grades (%) | Grade 3–4 (%) | All Grades (%) | Grade 3–4 (%) |
|
|
||||
| Decreased hemoglobin | 99 | 17 | 98 | 16 |
| Decreased leukocytes | 97 | 32 | 88 | 17 |
| Decreased lymphocytes | 95 | 60 | 92 | 53 |
| Decreased platelets | 95 | 30 | 85 | 28 |
| Decreased neutrophils | 87 | 54 | 80 | 37 |
Description of Selected Adverse Reactions
Cardiac failure
In IKEMA, cardiac failure (including cardiac failure, cardiac failure congestive, cardiac failure acute, cardiac failure chronic, left ventricular failure, and pulmonary edema) was reported in 7% of patients with the Isa-Kd group (grade ≥3 in 4%) and in 7% of patients with the Kd group (grade ≥3 in 4.1%). Serious cardiac failure was observed in 4% of patients in the Isa-Kd group and in 3.3% of patients in the Kd group. See the current prescribing information for carfilzomib for more information.
Related/similar drugs
7. Drug Interactions
7.1 Laboratory Test Interference
Interference with Serological Testing
SARCLISA, an anti-CD38 antibody, may interfere with blood bank serologic tests with false positive reactions in indirect antiglobulin tests (indirect Coombs tests), antibody detection (screening) tests, antibody identification panels, and antihuman globulin crossmatches in patients treated with SARCLISA [see Warnings and Precautions (5.5)].
Interference with Serum Protein Electrophoresis and Immunofixation Tests
SARCLISA may be incidentally detected by serum protein electrophoresis and immunofixation assays used for the monitoring of M-protein and may interfere with accurate response classification based on International Myeloma Working Group (IMWG) criteria [see Warnings and Precautions (5.5)]. In patients with persistent very good partial response, where interference is suspected, consider using an FDA-cleared isatuximab-irfc-specific IFE assay to distinguish isatuximab from any remaining endogenous M protein in the patient's serum to facilitate determination of a complete response.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
SARCLISA can cause fetal harm when administered to a pregnant woman. The assessment of isatuximab-irfc-associated risks is based on the mechanism of action and data from target antigen CD38 knockout animal models (see Data). There are no available data on SARCLISA use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Animal reproduction toxicity studies have not been conducted with isatuximab-irfc. The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, miscarriage, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
The combination of SARCLISA and pomalidomide or lenalidomide is contraindicated in pregnant women because pomalidomide and lenalidomide may cause birth defects and death of the unborn child. Refer to the pomalidomide or lenalidomide prescribing information on use during pregnancy. Pomalidomide and lenalidomide are only available through a REMS program.
Clinical Considerations
Fetal/neonatal reactions
Immunoglobulin G1 monoclonal antibodies are known to cross the placenta. Based on its mechanism of action, SARCLISA may cause depletion of fetal CD38-positive immune cells and decreased bone density. Defer administration of live vaccines to neonates and infants exposed to SARCLISA in utero until a hematology evaluation is completed.
Data
Animal data
Mice that were genetically modified to eliminate all CD38 expression (CD38 knockout mice) had reduced bone density which recovered 5 months after birth. Data from studies using CD38 knockout animal models also suggest the involvement of CD38 in regulating humoral immune responses (mice), feto-maternal immune tolerance (mice), and early embryonic development (frogs).
8.2 Lactation
Risk Summary
There are no available data on the presence of isatuximab-irfc in human milk, milk production, or the effects on the breastfed child. Maternal immunoglobulin G is known to be present in human milk. The effects of local gastrointestinal exposure and limited systemic exposure in the breastfed infant to SARCLISA are unknown. Because of the potential for serious adverse reactions in the breastfed child from isatuximab-irfc administered in combination with pomalidomide or lenalidomide and dexamethasone, advise lactating women not to breastfeed during treatment with SARCLISA. Refer to pomalidomide or lenalidomide prescribing information for additional information.
8.3 Females and Males of Reproductive Potential
SARCLISA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
With the combination of SARCLISA with pomalidomide or lenalidomide, refer to the pomalidomide or lenalidomide labeling for pregnancy testing requirements prior to initiating treatment in females of reproductive potential.
Contraception
Females
Advise female patients of reproductive potential to use effective contraception during treatment and for 5 months after the last dose of SARCLISA. Additionally, refer to the pomalidomide or lenalidomide labeling for contraception requirements prior to initiating treatment in females of reproductive potential.
8.4 Pediatric Use
The safety and effectiveness of SARCLISA in pediatric patients have not been established.
The safety and efficacy of SARCLISA in combination with chemotherapy were assessed, but not established, in an open-label study (ACT15378, ISAKIDS, NCT03860844) in 62 pediatric patients aged 1.4 years to < 17 years with relapsed or refractory T-acute lymphoblastic leukemia (T-ALL), B-acute lymphoblastic leukemia (B-ALL), or acute myeloid leukemia (AML). No new safety signals were observed in pediatric patients in this trial.
Body weight adjusted clearance at steady state and volume of distribution of isatuximab in pediatric patients were within the range of values that were observed in adults.
8.5 Geriatric Use
Of the total number of patients with relapsed or refractory multiple myeloma in clinical studies of SARCLISA, 56% (n=586) were 65 years of age and older, while 16% (n=163) were 75 years of age and older [see Clinical Studies (14)]. No overall differences in safety or effectiveness were observed between patients 65 years of age and older compared to younger patients, and other reported clinical experience has not identified differences in responses between the adults 65 years of age and older and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
Of the total number of SARCLISA-treated patients with newly diagnosed multiple myeloma in IMROZ, 72% (n=319) were less than 75 years of age and 28% (n=125) were 75 years of age and older. The clinical trial did not enroll patients over age 80 [see Clinical Studies (14)]. Adverse reactions occurring at a higher frequency in the SARCLISA arm (≥5%) in patients 75 years of age and older included neutropenia. Adverse reactions leading to dose modifications in patients 75 years of age and older occurred at a higher frequency (≥5%) in the SARCLISA arm. The hazard ratio for overall survival (OS) in patients 75 years of age and older was 1.25 [95% CI: 0.68 to 2.3].
11. Sarclisa Description
Isatuximab-irfc, a CD38-directed cytolytic antibody, is a chimeric immunoglobulin G1 (IgG1) monoclonal antibody (mAb). Isatuximab-irfc is produced from a mammalian cell line (Chinese hamster ovary, CHO) using a fed-batch production process. Isatuximab-irfc is composed of two identical immunoglobulin kappa light chains and two identical immunoglobulin gamma heavy chains and has an overall molecular weight of approximately 148 kDa.
SARCLISA (isatuximab-irfc) injection is a sterile, preservative-free, clear to slightly opalescent, colorless to slightly yellow solution, essentially free of visible particles in a single-dose vial for intravenous use. Each vial contains either 100 mg/5 mL or 500 mg/25 mL of isatuximab-irfc at a concentration of 20 mg/mL with a pH of 6.0. Each mL of solution contains 20 mg isatuximab-irfc, histidine (1.46 mg), histidine hydrochloride monohydrate (2.22 mg), polysorbate 80 (0.2 mg), sucrose (100 mg), and water for injection.
12. Sarclisa - Clinical Pharmacology
12.1 Mechanism of Action
Isatuximab-irfc is an IgG1-derived monoclonal antibody that binds to CD38 expressed on the surface of hematopoietic and tumor cells, including multiple myeloma cells. Isatuximab-irfc induces apoptosis of tumor cells and activation of immune effector mechanisms including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement dependent cytotoxicity (CDC). Isatuximab-irfc inhibits the ADP-ribosyl cyclase activity of CD38. Isatuximab-irfc can activate natural killer (NK) cells in the absence of CD38-positive target tumor cells and suppresses CD38-positive T-regulatory cells. The combination of isatuximab-irfc and pomalidomide enhanced ADCC activity and direct tumor cell killing compared to that of isatuximab-irfc alone in vitro, and enhanced antitumor activity compared to the activity of isatuximab-irfc or pomalidomide alone in a human multiple myeloma xenograft model.
12.2 Pharmacodynamics
In multiple myeloma patients treated with SARCLISA combined with pomalidomide and dexamethasone, a decrease in absolute counts of total NK cells (including inflammatory CD16+ low CD56+ bright and cytotoxic CD16+ bright CD56+ dim NK cells) and CD19+ B cells was observed in peripheral blood.
Cardiac Electrophysiology
Up to 2 times the approved recommended dose, SARCLISA does not prolong the QT interval to any clinically relevant extent.
A relationship between isatuximab-irfc exposure and overall response rate and progression-free survival was observed.
No apparent relationship was observed between an increase of isatuximab-irfc exposure and adverse reactions.
12.3 Pharmacokinetics
Following administration of isatuximab-irfc in combination with pomalidomide and dexamethasone at the recommended dose and schedule, the steady-state mean (CV%) predicted maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC) of isatuximab-irfc were 351 µg/mL (36.0%) and 72,600 µg∙h/mL (51.7%), respectively.
Following administration of isatuximab-irfc in combination with carfilzomib and dexamethasone at the recommended dose and schedule, the steady state mean (CV%) predicted Cmax and AUC of isatuximab-irfc were 655 µg/mL (30.8%) and 159,000 µg∙h/mL (37.1%), respectively.
The median time to reach steady state of isatuximab-irfc was 18 weeks with a 3.1-fold accumulation.
Following administration of isatuximab-irfc in combination with bortezomib, lenalidomide, and dexamethasone at the recommended dose and schedule, the mean (CV%) predicted Cmax and AUC of isatuximab-irfc after the dose on Week 21 were 496 µg/mL (25.6%) and 120,000 µg∙h/mL (28.9%), respectively. The geometric mean (CV%) of accumulation ratio between steady state AUC and AUC after the dose on Week 21 is predicted to be 1.4 (35.8%). Isatuximab-irfc AUC increases in a greater than dose proportional manner over a dosage range from 1 mg/kg to 20 mg/kg (0.1 to 2 times the approved recommended dosage) every 2 weeks. Isatuximab-irfc AUC increases proportionally over a dosage range from 5 mg/kg to 20 mg/kg (0.5 to 2 times the approved recommended dosage) every week for 4 weeks followed by every 2 weeks.
Distribution
The mean (CV%) predicted total volume of distribution of isatuximab-irfc is of 8.13 L (26.2%).
Elimination
Isatuximab-irfc total clearance decreased with increasing dose and with multiple doses. At steady state, the near elimination (≥99%) of isatuximab-irfc from plasma after the last dose is predicted to occur in approximately 2 months. The elimination of isatuximab-irfc was similar when given as a single agent or as combination therapy.
Specific Populations
The following factors have no clinically meaningful effect on the exposure of isatuximab-irfc: age (36 to 85 years, 70 patients were ≥75 years old), sex, renal impairment including patients with End-Stage Renal Disease (ESRD) or on dialysis (eGFR <90 mL/min/1.73 m2), and mild hepatic impairment (total bilirubin ≤ upper limit of normal [ULN] and aspartate aminotransferase [AST] >ULN, or total bilirubin >1 to 1.5 × ULN and any AST). The effect of moderate (total bilirubin >1.5 to 3 × ULN and any AST) and severe (total bilirubin >3 × ULN and any AST) hepatic impairment on isatuximab-irfc pharmacokinetics is unknown.
No dose adjustments are recommended in these specific patient populations.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of isatuximab-irfc or of other isatuximab products.
Overall, across 9 clinical studies in relapsed and/or refractory multiple myeloma (RRMM) with SARCLISA single-agent and combination therapies including ICARIA-MM and IKEMA (N=1023), the incidence of treatment emergent anti-drug antibodies (ADAs) was <2%. In ICARIA-MM and IKEMA, no patients with RRMM tested positive for ADA. Therefore, the neutralizing ADA status was not determined. In RRMM, no effect of ADAs was observed on the pharmacokinetics, safety, or efficacy of SARCLISA.
In IMROZ, out of the 263 patients with NDMM treated with SARCLISA in combination with bortezomib, lenalidomide, and dexamethasone, 253 were evaluable for the presence of ADA, 22 patients (8.7%) tested positive for treatment-emergent ADAs, with 21 patients considered to have a transient ADA response and 1 considered to have an indeterminate ADA response. Among these 22 ADA-positive patients, 13 (5.9%) had neutralizing antibodies. In IMROZ, a trend to lower exposure was observed in ADA-positive patients, which was considered not clinically relevant. In patients with ADA-positive status to SARCLISA, including those with neutralizing antibodies, no meaningful impact of ADAs on safety or efficacy of SARCLISA was observed.
14. Clinical Studies
Relapsed and/or Refractory Multiple Myeloma
ICARIA-MM
The efficacy and safety of SARCLISA in combination with pomalidomide and dexamethasone (Isa-Pd) were evaluated in ICARIA-MM (NCT02990338), a multicenter, multinational, randomized, open-label, 2-arm, phase 3 study in patients with relapsed and/or refractory multiple myeloma. Patients had received at least two prior therapies including lenalidomide and a proteasome inhibitor. Patients were eligible for inclusion if they had an Eastern Cooperative Oncology Group (ECOG) status of 0–2, platelets ≥75,000 cells/mm3, absolute neutrophil count ≥1 × 109/L, creatinine clearance ≥30 mL/min/1.73 m2 (MDRD formula), AST ≤3 × ULN, and ALT ≤3 × ULN.
A total of 307 patients were randomized in a 1:1 ratio to receive either SARCLISA in combination with pomalidomide and dexamethasone (Isa-Pd, 154 patients) or pomalidomide and dexamethasone (Pd, 153 patients). Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity. SARCLISA 10 mg/kg was administered as an intravenous infusion weekly in the first cycle and every two weeks thereafter. Pomalidomide 4 mg was taken orally once daily from day 1 to day 21 of each 28-day cycle. Dexamethasone (orally or intravenously) 40 mg (20 mg for patients ≥75 years of age) was given on days 1, 8, 15, and 22 for each 28-day cycle.
Overall, demographic and disease characteristics at baseline were similar between the two treatment groups. The median patient age was 67 years (range 36–86), 20% of patients were ≥75 years; 79% of patients were White, 12% Asian, and 1% Black or African American; 10% of patients entered the study with a history of COPD or asthma. The proportion of patients with renal impairment (creatinine clearance <60 mL/min/1.73 m2) was 34%. The International Staging System (ISS) stage at study entry was I in 37%, II in 36% and III in 25% of patients. Overall, 20% of patients had high-risk chromosomal abnormalities at study entry; del(17p), t(4;14) and t(14;16) were present in 12%, 8% and 2% of patients, respectively.
The median number of prior lines of therapy was 3 (range 2–11). All patients received a prior proteasome inhibitor, all patients received prior lenalidomide, and 56% of patients received prior stem cell transplantation; the majority of patients (93%) were refractory to lenalidomide, 76% to a proteasome inhibitor, and 73% to both an immunomodulator and a proteasome inhibitor.
The median duration of treatment was 41 weeks for Isa-Pd group compared to 24 weeks for Pd group.
The efficacy of SARCLISA was based upon progression-free survival (PFS). PFS results were assessed by an Independent Response Committee based on central laboratory data for M-protein and central radiologic imaging review using the International Myeloma Working Group (IMWG) criteria. The improvement in PFS represented a 40% reduction in the risk of disease progression or death in patients treated with Isa-Pd.
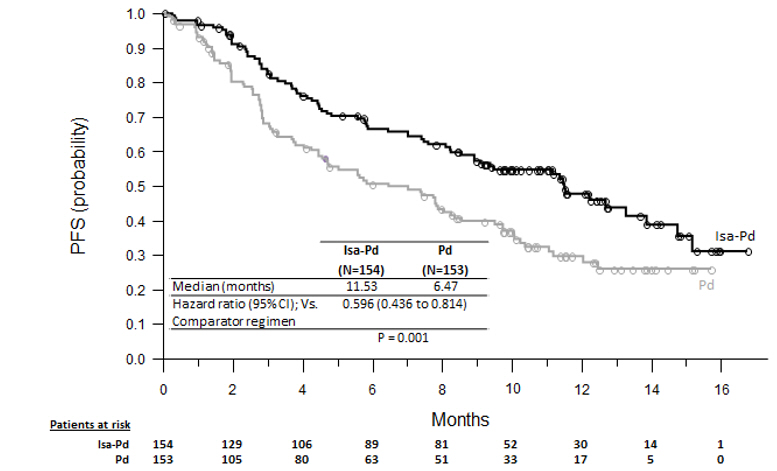
Efficacy results are presented in Table 10 and Kaplan-Meier curve for PFS is provided in Figure 1.
| Endpoint | SARCLISA + Pomalidomide + Dexamethasone N=154 | Pomalidomide + Dexamethasone N=153 |
|---|---|---|
| Progression-Free Survival | ||
| Median (months) [95% CI] | 11.53 [8.94–13.9] | 6.47 [4.47–8.28] |
| Hazard ratio* [95% CI] | 0.596 [0.44–0.81] | |
| p-value* (stratified log-rank test) | 0.0010 | |
| Overall Response Rate†
Responders (sCR+CR+VGPR+PR) n (%) [95% CI]‡ | 93 (60.4) [52.2–68.2] | 54 (35.3) [27.8–43.4] |
| p-value (stratified Cochran-Mantel-Haenszel)* | <0.0001 | |
| Stringent Complete Response (sCR) + Complete Response (CR) n (%) | 7 (4.5) | 3 (2) |
| Very Good Partial Response (VGPR) n (%) | 42 (27.3) | 10 (6.5) |
| Partial Response (PR) n (%) | 44 (28.6) | 41 (26.8) |
The median time to first response in responders was 35 days in the Isa-Pd group versus 58 days in the Pd group. The median duration of response was 13.3 months (95% CI: 10.6-NR) in the Isa-Pd group versus 11.1 months (95% CI: 8.5-NR) in the Pd group. At a median follow-up time of 52.4 months, final median overall survival was 24.6 months in the Isa-Pd group and 17.7 months in the Pd group (HR=0.776; 95% CI: 0.594 to 1.015).
Figure 1: Kaplan-Meier Curves of PFS – ITT Population – ICARIA-MM (assessment by the IRC)
IKEMA
The efficacy and safety of SARCLISA in combination with carfilzomib and dexamethasone were evaluated in IKEMA (NCT03275285), a multicenter, multinational, randomized, open-label, 2-arm, phase 3 study in patients with relapsed and/or refractory multiple myeloma. Patients had received one to three prior lines of therapy. Patients were eligible for inclusion if they had an ECOG status of 0–2, platelets ≥50,000 cells/mm3, absolute neutrophil count ≥1 × 109/L, creatinine clearance ≥15 mL/min/1.73 m2 (MDRD formula), AST ≤3 × ULN, and ALT ≤3 × ULN.
A total of 302 patients were randomized in a 3:2 ratio to receive either SARCLISA in combination with carfilzomib and dexamethasone (Isa-Kd, 179 patients) or carfilzomib and dexamethasone (Kd, 123 patients). Treatment was administered in both groups in 28-day cycles until disease progression or unacceptable toxicity. SARCLISA 10 mg/kg was administered as an intravenous infusion weekly in the first cycle and every two weeks thereafter. Carfilzomib was administered as an intravenous infusion at the dose of 20 mg/m2 on days 1 and 2; 56 mg/m2 on days 8, 9, 15, and 16 of cycle 1; and at the dose of 56 mg/m2 on days 1, 2, 8, 9, 15, and 16 for subsequent cycles of each 28-day cycle. Dexamethasone (intravenously on the days of isatuximab-irfc and/or carfilzomib infusions, and orally on the other days) 20 mg was given on days 1, 2, 8, 9, 15, 16, 22, and 23 for each 28-day cycle. On the days where both SARCLISA and carfilzomib were administered, dexamethasone was administered first, followed by SARCLISA infusion, then followed by carfilzomib infusion.
Overall, demographic and disease characteristics at baseline were similar between the two treatment groups. The median patient age was 64 years (range 33–90), 9% of patients were ≥75 years, 71% were White, 17% Asian, and 3% Black or African American. The proportion of patients with renal impairment (eGFR<60 mL/min/1.73 m2) was 24% in the Isa-Kd group versus 15% in the Kd group. The International Staging System (ISS) stage at study entry was I in 53%, II in 31%, and III in 15% of patients. Overall, 24% of patients had high-risk chromosomal abnormalities at study entry; del(17p), t(4;14), t(14;16) were present in 11%, 14%, and 2% of patients, respectively. In addition, gain(1q21) was present in 42% of patients.
The median number of prior lines of therapy was 2 (range 1–4) with 44% of patients who received 1 prior line of therapy. Overall, 90% of patients received prior proteasome inhibitors, 78% received prior immunomodulators (including 43% who received prior lenalidomide), and 61% received prior stem cell transplantation. Overall, 33% of patients were refractory to prior proteasome inhibitors, 45% were refractory to prior immunomodulators (including 33% refractory to lenalidomide), and 21% were refractory to both a proteasome inhibitor and an immunomodulator.
The median duration of treatment was 80 weeks for the Isa-Kd group compared to 61 weeks for the Kd group.
The efficacy of SARCLISA was based upon PFS. PFS results were assessed by an Independent Response Committee based on central laboratory data for M-protein and central radiologic imaging review using the IMWG criteria. The improvement in PFS represented a 45% reduction in the risk of disease progression or death in patients treated with Isa-Kd compared to patients treated with Kd.
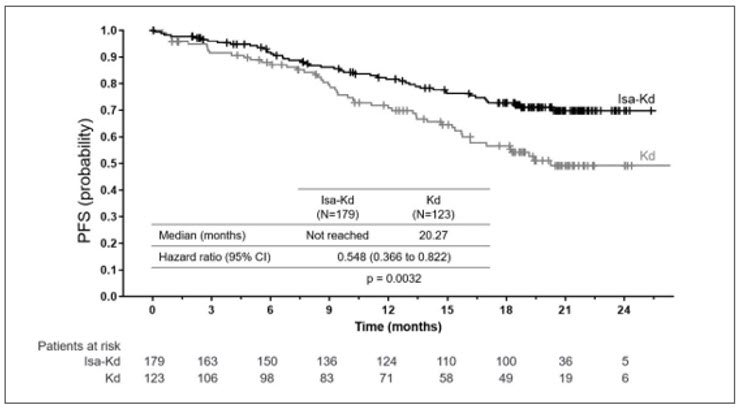
Efficacy results are presented in Table 11 and Kaplan-Meier curves for PFS are provided in Figure 2.
| Endpoint | SARCLISA + Carfilzomib + Dexamethasone N=179 | Carfilzomib + Dexamethasone N=123 |
|---|---|---|
| NR: not reached. | ||
|
||
| Progression-Free Survival‡ | ||
| Median (months) [95% CI] | NR [NR– NR] | 20.27 [15.77– NR] |
| Hazard ratio§ [95% CI] | 0.548 [0.366–0.822] | |
| p-value (stratified log-rank test)§ | 0.0032 | |
| Overall Response Rate¶
Responders (sCR+CR+VGPR+PR) n (%) [95% CI]# | 155 (86.6) [80.7–91.2] | 102 (82.9) [75.1–89.1] |
| p-value (stratified Cochran-Mantel-Haenszel)§ | 0.3859 | |
| Complete Response (CR) n (%) | 71 (39.7) | 34 (27.6) |
| Very Good Partial Response (VGPR) n (%) | 59 (33) | 35 (28.5) |
| Partial Response (PR) n (%) | 25 (14) | 33 (26.8) |
Figure 2: Kaplan-Meier Curves of PFS – ITT Population – IKEMA (assessment by the IRC)
At a median follow-up time of 44.0 months, final PFS analysis showed a median PFS of 41.7 months for Isa-Kd group compared to 20.8 months for Kd group, with a hazard ratio of 0.594 (95.4% CI: 0.424 to 0.832). Final complete response, determined using a FDA-cleared isatuximab-irfc-specific IFE assay [see Drug Interactions (7.1)], was 44.1% in Isa-Kd group compared to 28.5% in Kd group.
At a median follow-up time of 57 months, median overall survival was not reached in the Isa-Kd group (95% CI: 52.172–NR) and was 50.6 months in Kd group (95% CI: 38.932–NR) (HR=0.855; 95% CI: 0.608–1.202).
Newly Diagnosed Multiple Myeloma
IMROZ
The efficacy of SARCLISA in combination with bortezomib, lenalidomide, and dexamethasone was evaluated in IMROZ (NCT03319667), a multicenter, international, randomized, open-label, 2-arm, phase 3 study in patients with newly diagnosed multiple myeloma who are not eligible for stem cell transplantation. Patients were eligible for inclusion if they had an Eastern Cooperative Oncology Group (ECOG) status of 0 or 1, platelets ≥70,000 cells/mm3, absolute neutrophil count ≥1 × 109/L, creatinine clearance ≥30 mL/min/1.73 m2 (MDRD formula), AST ≤3 × ULN, and ALT ≤3 × ULN.
A total of 446 patients were randomized in a 3:2 ratio to receive either SARCLISA in combination with bortezomib, lenalidomide, and dexamethasone (Isa-VRd, 265 patients) or bortezomib, lenalidomide, and dexamethasone (VRd, 181 patients). Treatment was administered in both groups during 4 cycles of 42-days each for the induction period. After completion of cycle 4, patients entered the continuous treatment period starting from cycle 5, in which 28-day cycles were administered up to disease progression or unacceptable toxicity. During the continuous treatment period, patients of the Isa-VRd group received SARCLISA in combination with lenalidomide, and dexamethasone (Isa-Rd), and patients in the VRd group received lenalidomide, and dexamethasone (Rd). During the induction period (cycle 1 to 4, 42-day cycles), SARCLISA 10 mg/kg was administered as an intravenous infusion on day 1, 8, 15, 22, and 29, in the first cycle and on day 1, 15, and 29, from cycle 2 to 4. Bortezomib was administered subcutaneously at the dose of 1.3 mg/m2 on days 1, 4, 8, 11, 22, 25, 29, and 32 of each cycle. Lenalidomide was administered orally at the dose of 25 mg/day from day 1 to 14 and from day 22 to 35 of each cycle. Dexamethasone (intravenously on the days of SARCLISA infusions, and orally on the other days) 20 mg/day was given on days 1, 2, 4, 5, 8, 9, 11, 12, 15, 22, 23, 25, 26, 29, 30, 32, and 33 of each cycle. During the continuous treatment period (from cycle 5, 28-day cycles), SARCLISA 10 mg/kg was administered as an IV infusion on day 1 and 15 from cycle 5 to 17, and on day 1 from cycle 18. Lenalidomide was administered orally at the dose of 25 mg/day from day 1 to 21 of each cycle. Dexamethasone (intravenously on the days of SARCLISA infusions, and orally on the other days) 20 mg/day was given on days 1, 8, 15, and 22 of each cycle.
The median patient age was 72 years (range 55–80), 28% of patients were ≥75 years; 72% of patients were White, 11% Asian, and 0.9% Black or African American. The proportion of patients with renal impairment (eGFR<60 mL/min/1.73m2) was 29%. The Revised International Staging System (R-ISS) stage at study entry was I in 23%, II in 64%, and III in 10% of patients. Overall, 17% of patients had high-risk chromosomal abnormalities at study entry; del(17p), t(4;14), and t(14;16) were present in 5%, 9% and 2% of patients, respectively. In addition, 1q21+ was present in 37% of patients.
The efficacy of SARCLISA was established based upon progression-free survival (PFS). PFS results were assessed by an Independent Response Committee based on central laboratory data for M-protein and central radiologic imaging review using the International Myeloma Working Group (IMWG) criteria. The improvement in PFS represented a 40% reduction in the risk of disease progression or death in patients treated with Isa-VRd.
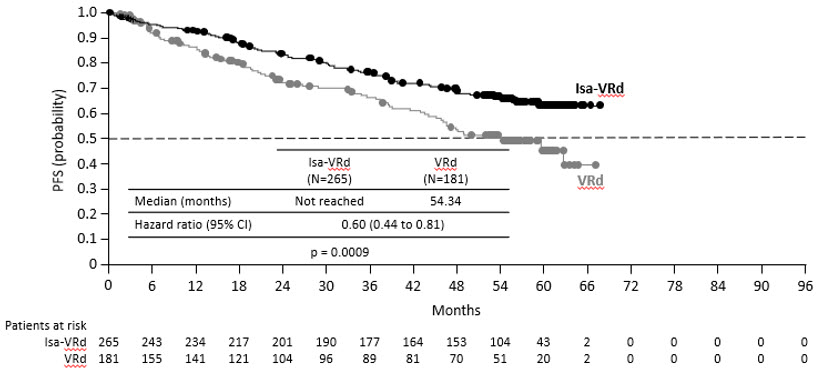
Efficacy results are presented in Table 12 and Kaplan-Meier curves for PFS are provided in Figure 3:
| Endpoint | SARCLISA + bortezomib + lenalidomide + dexamethasone N =265 | Bortezomib + lenalidomide + dexamethasone N = 181 |
|---|---|---|
| NR: not reached. | ||
| Median follow-up time=60 months. | ||
|
||
| Progression-Free Survival* | ||
| Median (months) [95% CI] | NR [NR–NR] | 54.34 [45.21–NR] |
| Hazard ratio † [95% CI] | 0.60 [0.44–0.81] | |
| p-value (Stratified Log-Rank test) †‡ | 0.0009 | |
| Overall Response Rate *§ | ||
| Responders (sCR+CR+VGPR+PR) N(%) [95% CI] ¶ | 242 (91) [87–94] | 167 (92) [87–96] |
| Stringent Complete Response (sCR) N(%) | 29 (11) | 10 (6) |
| Complete Response (CR) N(%) | 169 (64) | 106 (59) |
| Very Good Partial Response (VGPR) N(%) | 38 (14) | 34 (19) |
| Partial Response (PR) N(%) | 6 (2.3) | 17 (9) |
| CR or better (sCR and CR) # N(%) [95% CI] ¶ | 198 (75) [69–80] | 116 (64) [57–71] |
| p-value (Stratified Cochran-Mantel-Haenszel) † | 0.0160 | |
| Minimal Residual Disease (MRD) | ||
| MRD negativity rate *#Þß N(%) [95% CI] ¶ | 147 (55) [49–62] | 74 (41) [34–48] |
| p-value (Stratified Cochran-Mantel-Haenszel) † | 0.0026 | |
| Number of patients with CR or better (sCR and CR) | 198 | 116 |
| MRD negativity rate in patients with CR or better Þß N(%) | 147 (74) [68–80] | 74 (64) [54–73] |
| [95% CI] | ||
Median overall survival was not reached for either treatment group. At a median follow-up time of 60 months, 26% of patients in the Isa-VRd group and 32.6% of patients in the VRd group had died (HR=0.78; 95% CI: 0.55 to 1.1).
Figure 3 – Kaplan-Meier Curves of PFS – ITT population – IMROZ
16. How is Sarclisa supplied
How Supplied
SARCLISA (isatuximab-irfc) injection is a clear to slightly opalescent, colorless to slightly yellow solution, essentially free of visible particulates, supplied as follows:
- One 100 mg/5 mL (20 mg/mL) single-dose vial in a carton: NDC 0024-0654-01
- One 500 mg/25 mL (20 mg/mL) single-dose vial in a carton: NDC 0024-0656-01
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Infusion-Related Reaction
Advise patients to seek immediate medical attention for any of the following signs and symptoms of infusion-related reactions: shortness of breath, wheezing or trouble breathing; swelling of the face, mouth, throat, or tongue; throat tightness; palpitations; dizziness, lightheadedness, or fainting; headache; cough; rash or itching; nausea; runny or stuffy nose; or chills [see Warnings and Precautions (5.1)].
Infections
Inform patients about the risk of developing infections during SARCLISA treatment, and to report immediately any fever or symptoms of infection to their healthcare provider [see Warnings and Precautions (5.2)].
Neutropenia
Inform patients about the risk of neutropenia and infection during SARCLISA treatment and the importance of reporting immediately any fever or symptoms of infection to their healthcare provider [see Warnings and Precautions (5.3) and Adverse Reactions (6.1)].
Second Primary Malignancies
Inform patients of the risk of developing second primary malignancies during treatment with SARCLISA when given with pomalidomide and dexamethasone or with carfilzomib and dexamethasone, or with bortezomib, lenalidomide, and dexamethasone [see Warnings and Precautions (5.4)].
Cardiac Toxicities
Inform patients about the risk of cardiac failure during treatment with SARCLISA when given with carfilzomib and dexamethasone, and the importance of reporting immediately any difficulty breathing, cough, or leg swelling to their healthcare provider [see Adverse Reactions (6.1)].
Interference with Laboratory Tests
Advise patients to inform healthcare providers and transfusion center personnel that they are treated with SARCLISA in case a red blood cell transfusion is planned. Advise patients that SARCLISA may affect the results of blood tests to match their blood type for approximately 6 months after their last infusion of SARCLISA [see Warnings and Precautions (5.5) and Drug Interactions (7.1)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment and for 5 months after the last dose of SARCLISA [see Use in Specific Populations (8.1, 8.3)].
Advise patients that pomalidomide or lenalidomide have the potential to cause fetal harm and have specific requirements regarding contraception, pregnancy testing, blood and sperm donation, and transmission in sperm. Advise patients to report suspected or known pregnancies. Pomalidomide and lenalidomide are only available through a REMS program [see Use in Specific Populations (8.1, 8.3)].
Manufactured by:
sanofi-aventis U.S. LLC
Morristown, NJ 07960
A SANOFI COMPANY
U.S. License No. 1752
SARCLISA is a registered trademark of Sanofi
©2025 sanofi-aventis U.S. LLC
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 06/2025 | |||
| Patient Information
SARCLISA® (sar-cli-sa) (isatuximab-irfc) injection |
||||
| SARCLISA is used together with two or three other combinations of medicines: either pomalidomide and dexamethasone, or carfilzomib and dexamethasone, or bortezomib, lenalidomide, and dexamethasone. You should also read the Medication Guide that comes with pomalidomide and lenalidomide. You can ask your healthcare provider or pharmacist for information about carfilzomib and dexamethasone. | ||||
| What is SARCLISA?
SARCLISA is a prescription medicine used in combination with:
|
||||
| Do not receive SARCLISA if you have a severe allergic reaction to isatuximab-irfc or any of the ingredients in SARCLISA. See the end of this leaflet for complete list of ingredients in SARCLISA. | ||||
Before receiving SARCLISA, tell your healthcare provider about all of your medical conditions, including if you:
|
||||
How will I receive SARCLISA?
|
||||
| What are the possible side effects of SARCLISA? SARCLISA may cause serious side effects including:
|
||||
|
|
|
||
|
||||
| The most common side effects of SARCLISA in combination with pomalidomide and dexamethasone include: | ||||
|
|
|||
| The most common side effects of SARCLISA in combination with carfilzomib and dexamethasone include: | ||||
|
|
|||
| The most common side effects of SARCLISA in combination with bortezomib, lenalidomide and dexamethasone include: | ||||
|
|
|||
| Heart failure can happen during treatment with SARCLISA in combination with carfilzomib and dexamethasone. Tell your healthcare provider right away if you develop any of the following symptoms: | ||||
|
|
|
||
| These are not all the possible side effects of SARCLISA. For more information, ask your healthcare provider or pharmacist. | ||||
| Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | ||||
| General information about the safe and effective use of SARCLISA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about SARCLISA that is written for health professionals. |
||||
| What are the ingredients in SARCLISA? Active ingredient: isatuximab-irfc Inactive ingredients: histidine, histidine hydrochloride monohydrate, polysorbate 80, sucrose, and water for injection. Manufactured by: sanofi-aventis U.S. LLC, Morristown, NJ 07960, A SANOFI COMPANY. U.S. License No. 1752. SARCLISA is a registered trademark of Sanofi. ©2025 sanofi-aventis U.S. LLC. For more information, go to www.sanofi-aventis.us or call 1-800-633-1610. |
||||
| SARCLISA
isatuximab injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| SARCLISA
isatuximab injection, solution, concentrate |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Sanofi-Aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi-Aventis Deutschland GmbH | 313218430 | ANALYSIS(0024-0654, 0024-0656) , MANUFACTURE(0024-0654, 0024-0656) , PACK(0024-0654, 0024-0656) , LABEL(0024-0654, 0024-0656) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Genzyme Corporation | 050424395 | PACK(0024-0654, 0024-0656) , LABEL(0024-0654, 0024-0656) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi Winthrop Industrie | 280366666 | ANALYSIS(0024-0654, 0024-0656) , API MANUFACTURE(0024-0654, 0024-0656) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Quality Assistance, S.A. | 283676641 | ANALYSIS(0024-0654, 0024-0656) | |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Lonza Biologics, Inc. | 093149750 | ANALYSIS(0024-0654, 0024-0656) , API MANUFACTURE(0024-0654, 0024-0656) | |
Biological Products Related to Sarclisa
Find detailed information on biosimilars for this medication.
More about Sarclisa (isatuximab)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Drug images
- Side effects
- Dosage information
- During pregnancy
- FDA approval history
- Drug class: CD38 monoclonal antibodies
- Breastfeeding
- En español