Ferrlecit: Package Insert / Prescribing Info
Package insert / product label
Generic name: sodium ferric gluconate complex
Dosage form: injection
Drug class: Iron products
J Code (medical billing code): J2916 (12.5 mg, injection)
Medically reviewed by Drugs.com. Last updated on Jun 23, 2025.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Drug Interactions
- Use In Specific Populations
- Overdosage
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
- Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
FERRLECIT (sodium ferric gluconate complex in sucrose), injection, for intravenous use
Initial U.S. Approval: 1999
Indications and Usage for Ferrlecit
Ferrlecit is an iron replacement product for treatment of iron deficiency anemia in adult patients and in pediatric patients age 6 years and older with chronic kidney disease receiving hemodialysis who are receiving supplemental epoetin therapy. (1)
Ferrlecit Dosage and Administration
- Adult Patients - The recommended adult dosage is 10 mL (125 mg of elemental iron) diluted in 100 mL of 0.9% sodium chloride administered by intravenous infusion over 1 hour per dialysis session or undiluted as a slow intravenous injection (at a rate of up to 12.5 mg/min) per dialysis session. (2.2)
- Pediatric Patients - The recommended pediatric dosage is 0.12 mL/kg (1.5 mg/kg of elemental iron) diluted in 25 mL 0.9% sodium chloride and administered by intravenous infusion over 1 hour per dialysis session. (2.3)
- Do not mix Ferrlecit with other medications or add to parenteral nutrition solutions for intravenous infusion.
- Administer in 0.9% saline. (2)
Dosage Forms and Strengths
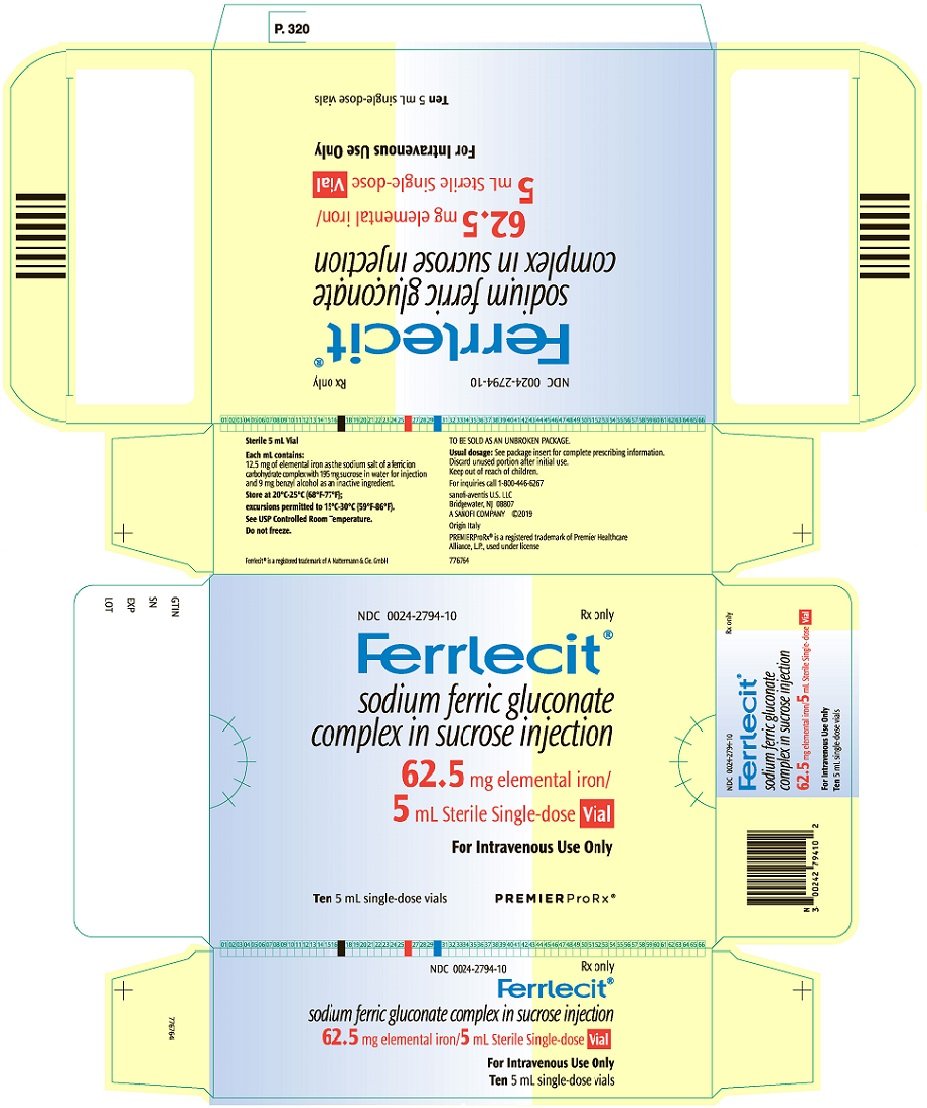
Injection: 62.5 mg/5 mL (12.5 mg/mL) in single-dose vial. (3)
Contraindications
Known hypersensitivity to sodium ferric gluconate or any of its inactive components. (4)
Warnings and Precautions
- Hypersensitivity Reactions: Monitor patients for signs and symptoms of hypersensitivity during and after Ferrlecit administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Ferrlecit when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions. (5.1)
- Hypotension: Ferrlecit may cause hypotension. Monitor patients for signs and symptoms of hypotension during and following each Ferrlecit dose. (5.2)
- Iron Overload: Regularly monitor hematologic responses during Ferrlecit therapy. Do not administer Ferrlecit to patients with iron overload. (5.3)
- Benzyl Alcohol Toxicity: Premature and low-birth-weight infants may be more likely to develop toxicity. (5.4)
Adverse Reactions/Side Effects
The most commonly reported adverse reactions (≥10%) in adult patients were nausea, vomiting and/or diarrhea, injection site reaction, hypotension, cramps, hypertension, dizziness, dyspnea, chest pain, leg cramps, and pain. In patients 6 to 15 years of age the most common adverse reactions (≥10%) were hypotension, headache, hypertension, tachycardia and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact sanofi-aventis U.S. LLC at 1-800-633-1610 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pregnancy: Risk of hypersensitivity reaction which may have serious consequences for the fetus. Use only if clearly needed (contains benzyl alcohol). (8.1)
- Lactation: Not recommended when breastfeeding. (8.2)
- Pediatric Use: Safety and effectiveness have not been established in pediatric patients <6 years of age. (8.4)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2025
Full Prescribing Information
1. Indications and Usage for Ferrlecit
Ferrlecit is indicated for the treatment of iron deficiency anemia in adult patients and in pediatric patients age 6 years and older with chronic kidney disease receiving hemodialysis who are receiving supplemental epoetin therapy.
2. Ferrlecit Dosage and Administration
2.1 Important Administration Instructions
The dosage of Ferrlecit is expressed in terms of mg of elemental iron. Each 5 mL sterile, single-dose vial contains 62.5 mg of elemental iron (12.5 mg/mL).
Do not mix Ferrlecit with other medications or add to parenteral nutrition solutions for intravenous infusion. The compatibility of Ferrlecit with intravenous infusion vehicles other than 0.9% sodium chloride has not been evaluated. Parenteral drug products should be inspected visually for particulate matter and discoloration before administration, whenever the solution and container permit. If diluted, use immediately.
Ferrlecit treatment may be repeated if iron deficiency reoccurs.
2.2 Adult Dosage and Administration
The recommended dosage of Ferrlecit for the repletion treatment of iron deficiency in hemodialysis patients is 10 mL of Ferrlecit (125 mg of elemental iron). Ferrlecit may be diluted in 100 mL of 0.9% sodium chloride administered by intravenous infusion over 1 hour per dialysis session. Ferrlecit may also be administered undiluted as a slow intravenous injection (at a rate of up to 12.5 mg/min) per dialysis session. For repletion treatment most patients may require a cumulative dose of 1000 mg of elemental iron administered over 8 dialysis sessions. Ferrlecit has been administered at sequential dialysis sessions by infusion or by slow intravenous injection during the dialysis session itself.
Data from Ferrlecit postmarketing spontaneous reports indicate that individual doses exceeding 125 mg may be associated with a higher incidence and/or severity of adverse events [see Adverse Reactions (6.2)].
2.3 Pediatric Dosage and Administration
The recommended pediatric dosage of Ferrlecit for the repletion treatment of iron deficiency in hemodialysis patients is 0.12 mL/kg Ferrlecit (1.5 mg/kg of elemental iron) diluted in 25 mL 0.9% sodium chloride and administered by intravenous infusion over 1 hour per dialysis session. The maximum dosage should not exceed 125 mg per dose.
3. Dosage Forms and Strengths
Injection: 62.5 mg/5 mL (12.5 mg/mL) clear, dark brown liquid in single-dose vial
4. Contraindications
Ferrlecit is contraindicated in patients with known hypersensitivity to sodium ferric gluconate or any of its components. Reactions have included anaphylaxis [see Warnings and Precautions (5.1)].
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Serious hypersensitivity reactions, including anaphylactic-type reactions, some of which have been life-threatening and fatal, have been reported in patients receiving Ferrlecit in postmarketing experience. Patients may present with shock, clinically significant hypotension, loss of consciousness, or collapse. Monitor patients for signs and symptoms of hypersensitivity during and after Ferrlecit administration for at least 30 minutes and until clinically stable following completion of the infusion. Only administer Ferrlecit when personnel and therapies are immediately available for the treatment of anaphylaxis and other hypersensitivity reactions [see Adverse Reactions (6)].
In the single-dose, postmarketing safety study one patient experienced a life-threatening hypersensitivity reaction (diaphoresis, nausea, vomiting, severe lower back pain, dyspnea, and wheezing for 20 minutes) following Ferrlecit administration. Among 1,097 patients who received Ferrlecit in this study, there were 9 patients (0.8%) who had an adverse reaction that, in the view of the investigator, precluded further Ferrlecit administration. These included one life-threatening reaction, six allergic reactions (including pruritus, facial flushing, chills, dyspnea/chest pain, and rash), and two other reactions (hypotension and nausea). Another 2 patients experienced (0.2%) allergic reactions not deemed to represent drug intolerance (nausea/malaise and nausea/dizziness) following Ferrlecit administration.
5.2 Hypotension
Ferrlecit may cause clinically significant hypotension. Hypotension associated with lightheadedness, malaise, fatigue, weakness or severe pain in the chest, back, flanks, or groin has been reported. These hypotensive reactions may or may not be associated with signs and symptoms of hypersensitivity reactions and usually resolve within one to two hours. In the single-dose safety study, postadministration hypotensive events were observed in 22/1,097 patients (2%) following Ferrlecit administration. Transient hypotension may occur during dialysis. Administration of Ferrlecit may augment hypotension caused by dialysis. Monitor patients for signs and symptoms of hypotension during and following Ferrlecit administration [see Adverse Reactions (6.1)].
5.3 Iron Overload
Excessive therapy with parenteral iron can lead to excess storage of iron with the possibility of iatrogenic hemosiderosis. Patients receiving Ferrlecit require periodic monitoring of hematologic and iron parameters (hemoglobin, hematocrit, serum ferritin, and transferrin saturation).
5.4 Risk of Serious Adverse Reactions in Infants Due to Benzyl Alcohol Preservative
Ferrlecit is not approved for use in neonates or infants. Serious and fatal adverse reactions including "gasping syndrome" can occur in neonates and low-birth-weight infants treated with benzyl alcohol–preserved drugs, including Ferrlecit. The "gasping syndrome" is characterized by central nervous system depression, metabolic acidosis, and gasping respirations. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known (Ferrlecit contains 9 mg of benzyl alcohol per mL) [see Use in Specific Populations (8.4)].
6. Adverse Reactions/Side Effects
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Hypersensitivity [see Contraindications (4) and Warnings and Precautions (5.1)]
- Hypotension [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
The most commonly reported adverse reactions (≥10%) in adult patients were nausea, vomiting and/or diarrhea, injection site reaction, hypotension, cramps, hypertension, dizziness, abnormal erythrocytes (e.g., changes in morphology, color, or number of red blood cells), dyspnea, chest pain, leg cramps and pain. In patients 6 to 15 years of age the most common adverse reactions (≥10%) were hypotension, headache, hypertension, tachycardia and vomiting.
Studies A and B
In multiple dose Studies A and B (total 126 adult patients), the most frequent treatment emergent adverse reactions following Ferrlecit were:
Body as a Whole: injection site reaction (33%), chest pain (10%), pain (10%), asthenia (7%), headache (7%), fatigue (6%), fever (5%), malaise, infection, abscess, chills, rigors, carcinoma, flu-like syndrome, sepsis, lightheadedness, weakness.
Nervous System: cramps (25%), dizziness (13%), paresthesias (6%), agitation, somnolence, decreased level of consciousness.
Respiratory System: dyspnea (11%), coughing (6%), upper respiratory infections (6%), rhinitis, pneumonia.
Cardiovascular System: hypotension (29%), hypertension (13%), syncope (6%), tachycardia (5%), bradycardia, vasodilatation, angina pectoris, myocardial infarction, pulmonary edema.
Gastrointestinal System: nausea, vomiting and/or diarrhea (35%), anorexia, abdominal pain (6%), rectal disorder, dyspepsia, eructation, flatulence, gastrointestinal disorder, melena.
Musculoskeletal System: leg cramps (10%), myalgia, arthralgia, back pain, arm pain.
Skin and Appendages: pruritus (6%), rash, increased sweating.
Genitourinary System: urinary tract infection, and menorrhagia.
Special Senses: conjunctivitis, rolling of the eyes, watery eyes, puffy eye lids, arcus senilis, redness of the eye, diplopia, and deafness.
Metabolic and Nutritional Disorders: hyperkalemia (6%), generalized edema (5%), leg edema, peripheral edema, hypoglycemia, edema, hypervolemia, hypokalemia.
Hematologic System: abnormal erythrocytes (11%) (changes in morphology, color, or number of red blood cells), anemia, leukocytosis, lymphadenopathy.
Study C – Pediatric
Pediatric Patients: In a clinical trial of 66 iron-deficient pediatric hemodialysis patients, 6 to 15 years of age, inclusive, who were receiving a stable erythropoietin dosing regimen, the most common adverse reactions, occurring in ≥5%, regardless of treatment dosage, were: hypotension (35%), headache (24%), hypertension (23%), tachycardia (17%), vomiting (11%), fever (9%), nausea (9%), abdominal pain (9%), pharyngitis (9%), diarrhea (8%), infection (8%), rhinitis (6%), and thrombosis (6%). More patients in the higher dose group (3.0 mg/kg) than in the lower dose group (1.5 mg/kg) experienced the following adverse events: hypotension (41% vs. 28%), tachycardia (21% vs. 13%), fever (15% vs. 3%), headache (29% vs. 19%), abdominal pain (15% vs. 3%), nausea (12% vs. 6%), vomiting (12% vs. 9%), pharyngitis (12% vs. 6%), and rhinitis (9% vs. 3%).
6.2 Postmarketing Experience
In the single-dose, postmarketing, safety study, 11% of patients who received Ferrlecit and 9.4% of patients who received placebo reported adverse reactions. The most frequent adverse reactions following Ferrlecit administration were: hypotension (2%), nausea, vomiting and/or diarrhea (2%), pain (0.7%), hypertension (0.6%), allergic reaction (0.5%), chest pain (0.5%), pruritus (0.5%), and back pain (0.4%). The following additional events were reported in two or more patients: hypertonia, nervousness, dry mouth, and hemorrhage.
In the multiple-dose, open-label surveillance study, 28% of the patients received concomitant angiotensin-converting enzyme inhibitor (ACEI) therapy. The incidences of both drug intolerance and suspected allergic events following first dose Ferrlecit administration were 1.6% in patients with concomitant ACEI use compared to 0.7% in patients without concomitant ACEI use. The patient with a life-threatening event was not on ACEI therapy. One patient had facial flushing immediately on Ferrlecit exposure. No hypotension occurred and the event resolved rapidly and spontaneously without intervention other than drug withdrawal.
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following additional adverse reactions have been identified with the use of Ferrlecit from postmarketing spontaneous reports:
Cardiovascular System: shock, fetal bradycardia, injection site superficial thrombophlebitis, phlebitis, acute myocardial ischemia with or without myocardial infarction or with in-stent thrombosis in the context of a hypersensitivity reaction.
Gastrointestinal System: dysgeusia.
Immune System: anaphylactic-type reactions.
Nervous System: loss of consciousness, generalized convulsion, hypoesthesia.
Skin and Appendages: skin discoloration, pallor.
Individual doses exceeding 125 mg may be associated with a higher incidence and/or severity of adverse events based on information from postmarketing spontaneous reports. These adverse events included hypotension, nausea, vomiting, abdominal pain, diarrhea, dizziness, dyspnea, urticaria, chest pain, paresthesia, and peripheral swelling.
Related/similar drugs
7. Drug Interactions
Drug-drug interactions involving Ferrlecit have not been studied. Ferrlecit may reduce the absorption of concomitantly administered oral iron preparations.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Parenteral iron administration may be associated with hypersensitivity reactions [see Warnings and Precautions (5.1)], which may have serious consequences, such as fetal bradycardia (see Clinical Considerations). Advise pregnant women of the potential risk to the fetus. Available data from postmarketing reports with Ferrlecit use in pregnancy are insufficient to assess the risk of major birth defects and miscarriage.
Ferrlecit contains benzyl alcohol as a preservative. Because benzyl alcohol is rapidly metabolized by a pregnant woman, benzyl alcohol exposure in the fetus is unlikely. However, adverse reactions have occurred in premature neonates and low-birth-weight infants who received intravenously administered benzyl alcohol–containing drugs [see Warnings and Precautions (5.4) and Use in Specific Populations (8.4)]. Consider alternative iron replacement therapies without benzyl alcohol.
There are risks to the mother and fetus associated with untreated iron deficiency anemia in pregnancy (see Clinical Considerations).
In the absence of maternal toxicity, Ferrlecit was not teratogenic to offspring of pregnant mice or rats at clinically relevant exposures (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defects, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Untreated iron deficiency anemia (IDA) in pregnancy is associated with adverse maternal outcomes such as postpartum anemia. Adverse pregnancy outcomes associated with IDA include increased risk for preterm delivery and low birth weight.
Fetal/Neonatal adverse reactions
Severe adverse reactions including circulatory failure (severe hypotension, shock including in the context of anaphylactic reaction) may occur in pregnant women with intravenous iron administration, which may have serious consequences on the fetus such as fetal bradycardia, especially during the second and third trimesters.
Data
Animal data
Ferrlecit was administered intravenously to pregnant mice during gestation days 6 to 15 at doses of 5, 30, and 100 mg Fe/kg/day to assess embryofetal development. No teratogenic effects were seen in offspring at the highest dose, representing maternal exposure of approximately 4 times maximum human exposure based on body surface area. There were increased fetal resorptions and decreased fetal weights at doses that caused maternal toxicity as evidenced by decreased body-weight gain and decreased food consumption.
Ferrlecit was administered intravenously to pregnant rats during gestation days 6 to 15 at doses of 4 and 20 mg Fe/kg/day to assess embryofetal development. No teratogenic effects were seen in offspring at the highest dose, representing maternal exposure of approximately 1.5 times maximum human exposure based on body surface area. There were decreases in gestation index and litter size, increased fetal resorptions, and decreased fetal weights at doses that caused maternal toxicity as evidenced by decreased body-weight gain and decreased food consumption.
8.2 Lactation
Risk Summary
Ferrlecit contains benzyl alcohol. Because benzyl alcohol is rapidly metabolized by a lactating woman, benzyl alcohol exposure in the breastfed infant is unlikely. However, adverse reactions have occurred in premature neonates and low-birth-weight infants who received intravenously administered benzyl alcohol–containing drugs [see Warnings and Precautions (5.4) and Use in Specific Populations (8.4)]. Consider alternative iron replacement therapies without benzyl alcohol for use during lactation.
There are no available data on the presence of Ferrlecit in human or animal milk, the effects on milk production, or the effects on the breastfed child.
8.4 Pediatric Use
The safety and effectiveness of Ferrlecit have been established in pediatric patients 6 to 15 years of age [see Dosage and Administration (2.3), Clinical Pharmacology (12.3), and Clinical Studies (14)]. Safety and effectiveness in pediatric patients younger than 6 years of age have not been established.
Benzyl Alcohol Toxicity and Pediatrics
Ferrlecit is not approved for use in neonates or infants. Serious adverse reactions including fatal reactions and the "gasping syndrome" occurred in premature neonates and low-birth-weight infants in the neonatal intensive care unit who received drugs containing benzyl alcohol as a preservative. In these cases, benzyl alcohol dosages of 99 to 234 mg/kg/day produced high levels of benzyl alcohol and its metabolites in the blood and urine (blood levels of benzyl alcohol were 0.61 to 1.378 mmol/L). Additional adverse reactions included gradual neurological deterioration, seizures, intracranial hemorrhage, hematologic abnormalities, skin breakdown, hepatic and renal failure, hypotension, bradycardia, and cardiovascular collapse. Benzyl alcohol contained in Ferrlecit may cause serious and anaphylactoid reactions in infants and children up to 3 years old. The administration of medications containing benzyl alcohol to newborns or premature neonates has been associated with a fatal "gasping syndrome" (symptoms include a striking onset of gasping syndrome, hypotension, bradycardia, and cardiovascular collapse). Preterm, low-birth-weight infants may be more likely to develop these reactions because they could be less able to metabolize benzyl alcohol. The minimum amount of benzyl alcohol at which serious adverse reactions may occur is not known (Ferrlecit contains 9 mg of benzyl alcohol per mL) [see Warnings and Precautions (5.4)].
8.5 Geriatric Use
Clinical studies of Ferrlecit did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
10. Overdosage
The Ferrlecit iron complex is not dialyzable.
No data is available regarding overdose of Ferrlecit in humans. Excessive dosages of Ferrlecit may lead to accumulation of iron in storage sites potentially leading to hemosiderosis. Do not administer Ferrlecit to patients with iron overload [see Warnings and Precautions (5.3)].
Individual doses exceeding 125 mg may be associated with a higher incidence and/or severity of adverse events [see Adverse Reactions (6.2)].
Ferrlecit at elemental iron doses of 125 mg/kg, 78.8 mg/kg, 62.5 mg/kg and 250 mg/kg caused deaths in mice, rats, rabbits, and dogs respectively. The major symptoms of acute toxicity were decreased activity, staggering, ataxia, increases in the respiratory rate, tremor, and convulsions.
11. Ferrlecit Description
Ferrlecit® (sodium ferric gluconate complex in sucrose injection), an iron replacement product, is a stable macromolecular complex with an apparent molecular weight on gel chromatography of 289,000–440,000 daltons. The macromolecular complex is negatively charged at alkaline pH and is present in solution with sodium cations. The product has a deep red color indicative of ferric oxide linkages. The chemical name is D-Gluconic acid, iron (3+) sodium salt.
The structural formula is considered to be [NaFe2O3(C6H11O7)(C12H22011)5]n≈200•
Ferrlecit is supplied as a clear, dark brown liquid in colorless glass vials. Each sterile, single-dose vial of 5 mL of Ferrlecit for intravenous injection contains 62.5 mg (12.5 mg/mL) of elemental iron as the sodium salt of a ferric ion carbohydrate complex in an alkaline aqueous solution with approximately 20% sucrose w/v (195 mg/mL) in water for injection, pH 7.7–9.7.
Each mL contains 9 mg of benzyl alcohol as an inactive ingredient.
12. Ferrlecit - Clinical Pharmacology
12.1 Mechanism of Action
Ferrlecit is used to replete the body content of iron. Iron is critical for normal hemoglobin synthesis to maintain oxygen transport. Additionally, iron is necessary for metabolism and various enzymatic processes.
12.3 Pharmacokinetics
Multiple sequential single-dose intravenous pharmacokinetic studies were performed on 14 healthy iron-deficient volunteers. Entry criteria included hemoglobin ≥10.5 gm/dL and transferrin saturation ≤15% (TSAT) or serum ferritin value ≤20 ng/mL. In the first stage, each subject was randomized 1:1 to undiluted Ferrlecit injection of either 125 mg/hr or 62.5 mg/0.5 hr (2.1 mg/min). Five days after the first stage, each subject was re-randomized 1:1 to undiluted Ferrlecit injection of either 125 mg/7 min or 62.5 mg/4 min (>15.5 mg/min).
Peak drug levels (Cmax) varied significantly by dosage and by rate of administration with the highest Cmax observed in the regimen in which 125 mg was administered in 7 minutes (19.0 mg/L). The terminal elimination half-life for drug bound iron was approximately 1 hour. Half-life varied by dose but not by rate of administration. Half-life values were 0.85 and 1.45 hours for the 62.5 mg/4 min and 125 mg/7 min regimens, respectively. Total clearance of Ferrlecit was 3.02 to 5.35 L/h. The AUC for Ferrlecit bound iron varied by dose from 17.5 mg-h/L (62.5 mg) to 35.6 mg-h/L (125 mg). Approximately 80% of drug bound iron was delivered to transferrin as a mononuclear ionic iron species within 24 hours of administration in each dosage regimen. Direct movement of iron from Ferrlecit to transferrin was not observed. Mean peak transferrin saturation returned to near baseline by 40 hours after administration of each dosage regimen.
Pediatrics: Single-dose intravenous pharmacokinetic analyses were performed on 48 iron-deficient pediatric hemodialysis patients. Twenty-two patients received 1.5 mg/kg Ferrlecit and 26 patients received 3.0 mg/kg Ferrlecit (maximum dose 125 mg). The mean Cmax, AUC0–∞, and terminal elimination half-life values following a 1.5 mg/kg dose were 12.9 mg/L, 95.0 mg∙hr/L, and 2.0 hours, respectively. The mean Cmax, AUC0–∞, and terminal elimination half-life values following a 3.0 mg/kg dose were 22.8 mg/L, 170.9 mg∙hr/L, and 2.5 hours, respectively.
In vitro experiments have shown that less than 1% of the iron species within Ferrlecit can be dialyzed through membranes with pore sizes corresponding to 12,000 to 14,000 daltons over a period of up to 270 minutes. Human studies in renally competent patients suggest the clinical insignificance of urinary excretion.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long term carcinogenicity studies of sodium ferric gluconate in animals were not performed.
Sodium ferric gluconate was not genotoxic in the Ames test or the rat micronucleus test. Sodium ferric gluconate produced a clastogenic effect in an in vitro chromosomal aberration assay in Chinese hamster ovary cells.
Studies to assess the effects of sodium ferric gluconate on fertility were not conducted.
14. Clinical Studies
Two clinical studies (Studies A and B) were conducted in adults and one clinical study was conducted in pediatric patients (Study C) to assess the efficacy and safety of Ferrlecit.
Study A
Study A was a three-center, randomized, open-label study of the safety and efficacy of two doses of Ferrlecit administered intravenously to iron-deficient hemodialysis patients. The study included both a dose-response concurrent control and an historical control. Enrolled patients received a test dose of Ferrlecit (25 mg of elemental iron) and were then randomly assigned to receive Ferrlecit at cumulative doses of either 500 mg (low dose) or 1000 mg (high dose) of elemental iron. Ferrlecit was given to both dose groups in eight divided doses during sequential dialysis sessions (a period of 16 to 17 days). At each dialysis session, patients in the low-dose group received Ferrlecit 62.5 mg of elemental iron over 30 minutes, and those in the high-dose group received Ferrlecit 125 mg of elemental iron over 60 minutes. The primary endpoint was the change in hemoglobin from baseline to the last available observation through Day 40.
Eligibility for this study included chronic hemodialysis patients with a hemoglobin below 10 g/dL (or hematocrit at or below 32%) and either serum ferritin below 100 ng/mL or transferrin saturation below 18%. Exclusion criteria included significant underlying disease or inflammatory conditions or an epoetin requirement of greater than 10,000 units three times per week. Parenteral iron and red cell transfusion were not allowed for two months before the study. Oral iron and red cell transfusion were not allowed during the study for Ferrlecit-treated patients.
The historical control population consisted of 25 chronic hemodialysis patients who received only oral iron supplementation for 14 months and did not receive red cell transfusion. All patients had stable epoetin doses and hematocrit values for at least two months before initiation of oral iron therapy.
The evaluated population consisted of 39 patients in the low-dose Ferrlecit (sodium ferric gluconate complex in sucrose injection) group (50% female, 50% male; 74% white, 18% black, 5% Hispanic, 3% Asian; mean age 54 years, range 22–83 years), 44 patients in the high-dose Ferrlecit group (50% female, 48% male, 2% unknown; 75% white, 11% black, 5% Hispanic, 7% other, 2% unknown; mean age 56 years, range 20–87 years), and 25 historical control patients (68% female, 32% male; 40% white, 32% black, 20% Hispanic, 4% Asian, 4% unknown; mean age 52 years, range 25–84 years).
The mean baseline hemoglobin and hematocrit were similar between treatment and historical control patients: 9.8 g/dL and 29% and 9.6 g/dL and 29% in low- and high-dose Ferrlecit-treated patients, respectively, and 9.4 g/dL and 29% in historical control patients. Baseline serum transferrin saturation was 20% in the low-dose group, 16% in the high-dose group, and 14% in the historical control. Baseline serum ferritin was 106 ng/mL in the low-dose group, 88 ng/mL in the high-dose group, and 606 ng/mL in the historical control.
Patients in the high-dose Ferrlecit group achieved significantly higher increases in hemoglobin and hematocrit than patients in the low-dose Ferrlecit group. See Table 1.
| Mean Change from Baseline to Two Weeks after Cessation of Therapy | |||
|---|---|---|---|
| Ferrlecit 1000 mg IV (N=44) | Ferrlecit 500 mg IV (N=39) | Historical Control Oral Iron (N=25) |
|
|
|||
| Hemoglobin (g/dL) | 1.1* | 0.3 | 0.4 |
| Hematocrit (%) | 3.6* | 1.4 | 0.8 |
| Transferrin Saturation (%) | 8.5 | 2.8 | 6.1 |
| Serum Ferritin (ng/mL) | 199 | 132 | NA |
Study B
Study B was a single-center, non-randomized, open-label, historically controlled, study of the safety and efficacy of variable, cumulative doses of intravenous Ferrlecit in iron-deficient hemodialysis patients. Ferrlecit administration was identical to Study A. The primary efficacy variable was the change in hemoglobin from baseline to the last available observation through Day 50.
Inclusion and exclusion criteria were identical to those of Study A as was the historical control population. Sixty-three patients were evaluated in this study: 38 in the Ferrlecit-treated group (37% female, 63% male; 95% white, 5% Asian; mean age 56 years, range 22–84 years) and 25 in the historical control group (68% female, 32% male; 40% white, 32% black, 20% Hispanic, 4% Asian, 4% unknown; mean age 52 years, range 25–84 years).
Ferrlecit-treated patients were considered to have completed the study per protocol if they received at least eight Ferrlecit doses of either 62.5 mg or 125 mg of elemental iron. A total of 14 patients (37%) completed the study per protocol. Twelve (32%) Ferrlecit-treated patients received less than eight doses, and 12 (32%) patients had incomplete information on the sequence of dosing. Not all patients received Ferrlecit at consecutive dialysis sessions and many received oral iron during the study.
| Cumulative Ferrlecit Dose (mg of elemental iron) | 62.5 | 250 | 375 | 562.5 | 625 | 750 | 1000 | 1125 | 1187.5 |
| Patients (#) | 1 | 1 | 2 | 1 | 10 | 4 | 12 | 6 | 1 |
Baseline hemoglobin and hematocrit values were similar between the treatment and control groups, and were 9.1 g/dL and 27.3%, respectively, for Ferrlecit-treated patients. Serum iron studies were also similar between treatment and control groups, with the exception of serum ferritin, which was 606 ng/mL for historical control patients, compared to 77 ng/mL for Ferrlecit-treated patients.
In this patient population, only the Ferrlecit-treated group achieved increase in hemoglobin and hematocrit from baseline. See Table 2.
| Mean Change from Baseline to One Month after Treatment | ||
|---|---|---|
| Ferrlecit (N=38) Change | Oral Iron (N=25) Change |
|
| Hemoglobin (g/dL) | 1.3 | 0.4 |
| Hematocrit (%) | 3.8 | 0.2 |
| Transferrin Saturation (%) | 6.7 | 1.7 |
| Serum Ferritin (ng/dL) | 73 | -145 |
Study C
Study C was a multicenter, randomized, open-label study of the safety and efficacy of two Ferrlecit dose regimens (1.5 mg/kg or 3.0 mg/kg of elemental iron) administered intravenously to 66 iron-deficient (transferrin saturation <20% and/or serum ferritin <100 ng/mL) pediatric hemodialysis patients, 6 to 15 years of age, inclusive who were receiving a stable erythropoietin dosing regimen.
Ferrlecit at a dose of 1.5 mg/kg or 3.0 mg/kg (up to a maximum dose of 125 mg of elemental iron) in 25 mL 0.9% sodium chloride was infused intravenously over 1 hour during each hemodialysis session for eight sequential dialysis sessions. Thirty-two patients received the 1.5 mg/kg dosing regimen (47% male, 53% female; 66% Caucasian, 25% Hispanic, and 3% Black, Asian, or Other; mean age 12.3 years). Thirty-four patients received the 3.0 mg/kg dosing regimen (56% male, 44% female; 77% Caucasian, 12% Hispanic, 9% Black, and 3% Other; mean age 12.0 years).
The primary endpoint was the change in hemoglobin concentration from baseline to 2 weeks after last Ferrlecit administration. There was no significant difference between the treatment groups. Improvements in hematocrit, transferrin saturation, serum ferritin, and reticulocyte hemoglobin concentrations compared to baseline values were observed 2 weeks after the last Ferrlecit infusion in both the 1.5 mg/kg and 3.0 mg/kg treatment groups (Table 3).
| Mean Change from Baseline to Two Weeks after Cessation of Therapy in Patients Completing Treatment | ||
|---|---|---|
| 1.5 mg/kg Ferrlecit (N=25) | 3.0 mg/kg Ferrlecit (N=32) |
|
| Hemoglobin (g/dL) | 0.8 | 0.9 |
| Hematocrit (%) | 2.6 | 3.0 |
| Transferrin Saturation (%) | 5.5 | 10.5 |
| Serum Ferritin (ng/mL) | 192 | 314 |
| Reticulocyte Hemoglobin Content (pg) | 1.3 | 1.2 |
The increased hemoglobin concentrations were maintained at 4 weeks after the last Ferrlecit infusion in both the 1.5 mg/kg and the 3.0 mg/kg Ferrlecit dose treatment groups.
16. How is Ferrlecit supplied
How Supplied
Ferrlecit is a clear, dark brown liquid supplied in colorless glass vials. Each sterile, single-dose vial contains 62.5 mg of elemental iron in 5 mL for intravenous use. Discard unused portion.
Carton containing 10 vials: NDC 0024-2794-10
17. Patient Counseling Information
Prior to Ferrlecit administration:
- Question patients regarding any prior history of reactions to parenteral iron products.
- Advise patients of the risks associated with Ferrlecit.
- Advise patients to report adverse reactions associated with the use of Ferrlecit, including hypersensitivity, allergic reactions, chest pain, dizziness, lightheadedness, swelling, and breathing problems [see Warnings and Precautions (5.1, 5.2) and Adverse Reactions (6.1, 6.2)].
Advise patients that Ferrlecit may reduce the absorption of concomitantly administered oral iron preparations [see Drug Interactions (7)].
Pregnancy
Advise pregnant women about the risk of hypersensitivity reactions which may have serious consequences for the fetus. Advise patients who may become pregnant to inform their healthcare provider of a known or suspected pregnancy (contains benzyl alcohol) [see Use in Specific Populations (8.1)].
Lactation
Advise patients that treatment with Ferrlecit is not recommended for use while breastfeeding [see Use in Specific Populations (8.2)].
| FERRLECIT
sodium ferric gluconate complex injection |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - sanofi-aventis U.S. LLC (824676584) |
| Establishment | |||
| Name | Address | ID/FEI | Business Operations |
|---|---|---|---|
| Sanofi S.r.l. | 338454274 | ANALYSIS(0024-2794) , MANUFACTURE(0024-2794) , PACK(0024-2794) , LABEL(0024-2794) | |
More about Ferrlecit (sodium ferric gluconate complex)
- Check interactions
- Compare alternatives
- Pricing & coupons
- Reviews (2)
- Side effects
- Dosage information
- During pregnancy
- Drug class: iron products
- Breastfeeding
- En español