Wilate, von Willebrand Factor/Coagulation Factor VIII Complex: Package Insert / Prescribing Info
Package insert / product label
Generic name: von willebrand factor/coagulation factor viii complex (human)
Dosage form: powder, for intravenous solution
Drug class: Miscellaneous coagulation modifiers
J Code (medical billing code): J7186 (Per intl units, injection)
Medically reviewed by Drugs.com. Last updated on Dec 3, 2024.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Clinical Studies
- References
- How Supplied/Storage and Handling
- Patient Counseling Information
Highlights of Prescribing Information
WILATE, von Willebrand Factor/Coagulation Factor VIII Complex (Human)
Lyophilized Powder for Solution for Intravenous Injection
Initial U.S. Approval: 2009
----------------------------RECENT MAJOR CHANGES-------------------
Indication and Usage (1) 12/2023
Dosage and Administration (2.2, 2.3) 11/2024
Indications and Usage for Wilate, von Willebrand Factor/Coagulation Factor VIII Complex
WILATE is indicated in children and adults with von Willebrand disease for:
• On-demand treatment and control of bleeding episodes ( 1 )
• Perioperative management of bleeding ( 1 )
• Routine prophylaxis to reduce the frequency of bleeding episodes ( 1 )
Wilate is indicated for routine prophylaxis in children 6 years of age and older and adults with von Willebrand disease.
WILATE is indicated in adolescents and adults with hemophilia A for:
• Routine prophylaxis to reduce the frequency of bleeding episodes
• On-demand treatment and control of bleeding episodes ( 1 )
Wilate, von Willebrand Factor/Coagulation Factor VIII Complex Dosage and Administration
For Intravenous Use Only
Von Willebrand Disease
• Use the following formula to determine required dosage ( 2.1 ):
Required IU = body weight (BW) in kg x desired VWF:RCo rise (%) (IU/dL) x 0.5 (IU/kg per IU/dL)
• Adjust dosage and duration of the substitution therapy depending on the severity of the VWD, on the location and extent of the bleeding, and on the patient’s clinical condition ( 2.1 )
• The recommended infusion rate of 2-4ml/min
On-demand treatment and control of bleeding episodes
• Dosing recommendations ( 2.1 ):
| Type of Hemorrhages | Loading Dosage (IU VWF:RCo/ kg BW) | Maintenance Dosage (IU VWF:RCo/ kg BW) | Therapeutic Goal |
| Minor Hemorrhages | 20-40 IU/kg | 20-30 IU/kg every 12-24 hours | VWF:RCo and FVIII activity trough levels of >30% |
| Major Hemorrhages | 40-60 IU/kg | 20-40 IU/kg every 12-24 hours | VWF:RCo and FVIII activity trough levels of >50% |
Perioperative management of bleeding
Dosing recommendations ( 2.1 ):
| Type of Surgery | Loading Dosage (IU VWF:RCo/ kg BW) | Maintenance Dosage (IU VWF:RCo/ kg BW) | Therapeutic Goal |
| Minor Surgeries (including tooth extractions) | 30-60 IU/kg | 15-30 IU/kg or half the loading dose every 12-24 hours for up to 3 days | VWF:RCo peak level of 50% after loading dose and trough levels of > 30% during maintenance doses |
| Major Surgeries | 40-60 IU/kg | 20-40 IU/kg or half the loading dose every 12-24 hours for up to 6 days or more | VWF:RCo peak level of 100% after loading dose and trough levels of > 50% during maintenance doses |
In order to decrease the risk of perioperative thrombosis, FVIII activity levels should not exceed 250%.
Routine prophylaxis to reduce the frequency of bleeding episodes
Dosing recommendations ( 2.1 ):
| Patients | Dose (IU/kg) | Frequency of infusions |
|
Age 6 and older |
20 – 40 IU/kg |
Two or three times per week |
Hemophilia A
• One International Unit (IU) of factor VIII (FVIII) activity per kg body weight increases the circulating FVIII level by approximately 2 IU/dL (1.7 IU/dL for adolescents and 2.3 IU/dL for adults) ( 2.1 ).
• Use the following formula to determine required dosage ( 2.1 ):
Required IU = body weight (BW) in kg x desired Factor VIII rise (%) (IU/dL) x 0.5 (IU/kg per IU/dL)
• The recommended infusion rate of 2-4ml/min
• Dosing for routine prophylaxis ( 2.1 ):
| Patients | Dose (IU/kg) | Frequency of infusions |
| Adolescents and adults | 20-40 IU/kg | Every 2 to 3 days |
• Dosing for on-demand treatment and control of bleeding episodes ( 2.1 )
| Type of Hemorrhages | Recommended dosage (IU/kg body weight) | Frequency of Doses
(hours) | Duration of Therapy
(days) |
| Minor | 30-40 | Repeat every 12-24 hours | At least 1 day, until the hemorrhage has resolved |
| Moderate | 30-40 | Repeat every 12-24 hours | 3 to 4 days or more, until the hemorrhage has resolved |
| Major | 35-50 | Repeat every 12-24 hours | 3 to 4 days or more, until the hemorrhage has resolved |
| Life-threatening | 35-50 | Repeat every 8-24 hours | Until threat has resolved |
• Individualize dosage based on the patient’s weight, type and severity of hemorrhage, FVIII level, presence of inhibitors and the patient’s clinical condition ( 2.1 ).
Dosage Forms and Strengths
WILATE is available as a sterile, lyophilized powder for reconstitution for intravenous injection, provided in the following nominal strengths per single-dose vial ( 3 ):
• 500 IU VWF:RCo and 500 IU FVIII activities in 5 mL
• 1000 IU VWF:RCo and 1000 IU FVIII activities in 10 mL
Contraindications
Do not use in patients with known hypersensitivity reactions, including anaphylactic or severe systemic reaction, to human plasma-derived products, any ingredient in the formulation, or components of the container ( 4 )
Warnings and Precautions
• Anaphylaxis and severe hypersensitivity reactions are possible ( 5.1 ).
• Thromboembolic events may occur. Monitor plasma levels of FVIII activity ( 5.2 ).
• Development of neutralizing antibodies to FVIII and to VWF, especially in VWD type 3 patients, may occur ( 5.3 ).
• WILATE is made from human plasma and carries the risk of transmitting infectious agents ( 5.4 ).
Adverse Reactions/Side Effects
The most common adverse reactions (≥ 1%) in clinical trails on VWD were hypersensitivity reactions, urticaria, chest discomfort, and dizziness ( 6.1 )
The most common adverse reaction (≥ 1%) in clinical trails in hemophilia A was pyrexia (fever) ( 6.1 ).
To report SUSPECTED ADVERSE REACTIONS, contact Octapharma USA Inc. at 1-866-766-4860 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Revised: November 2024
______________________________________________________________________________________________________________________________________
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 11/2024
Full Prescribing Information
1. Indications and Usage for Wilate, von Willebrand Factor/Coagulation Factor VIII Complex
WILATE is indicated in children and adults with von Willebrand disease for:
- On-demand treatment and control of bleeding episodes
- Perioperative management of bleeding
- Routine prophylaxis to reduce the frequency of bleeding episodes
WILATE is indicated in adolescents and adults with hemophilia A for:
- Routine prophylaxis to reduce the frequency of bleeding episodes
- On-demand treatment and control of bleeding episodes
2. Wilate, von Willebrand Factor/Coagulation Factor VIII Complex Dosage and Administration
For Intravenous Use after Reconstitution
2.1 Dose
- Initiate treatment under the supervision of a physician experienced in the treatment of coagulation disorders.
- Each vial of WILATE contains the labeled amount in International Units (IU) of von Willebrand factor (VWF) activity as measured with the Ristocetin cofactor assay (VWF:RCo), and coagulation factor VIII (FVIII) activity measured with the chromogenic substrate assay.
- The number of units of VWF:RCo and FVIII activities administered is expressed in IU, which is related to the current WHO standards for VWF and FVIII products. VWF:RCo and FVIII activities in plasma are expressed either as a percentage (relative to normal human plasma) or in IU (relative to the International Standards for VWF:RCo and FVIII activities in plasma). One IU of VWF:RCo activity is equivalent to the quantity of VWF:RCo in one mL of normal human plasma. One IU of FVIII activity is defined by the quantity of Factor VIII in one mL of normal human pooled plasma. The ratio between VWF:RCo and FVIII activities in WILATE is approximately 1:1.
VWD
- Calculate the required dosage of VWF:RCo, based on the empirical finding that 1 IU VWF:RCo per kg body weight raises the plasma VWF activity by approximately 2% of normal activity or 2 IU/dL, using the following formula:
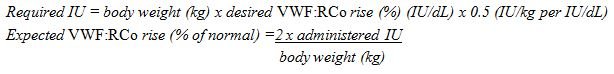
- Adjust the dosage and frequency of administration according to the clinical effectiveness in the individual patient.
Dosing for Hemorrhages
A guide for dosing in the treatment of major and minor hemorrhages is provided in Table 1 .
Table 1 Dosing for Treatment of Minor and Major Hemorrhages in all VWD types
| Type of Hemorrhages |
VWF:RCo and FVIII Activity Trough Levels
(% of normal) |
Loading Dosage
(IU VWF:RCo/kg body weight) |
Maintenance Dosage
(IU VWF:RCo/kg body weight) |
Frequency of Doses
(hours) |
Duration of Therapy
(days) |
| Minor | >30 | 20-40 | 20-30 |
Repeat as needed 12-24 | Up to 3 days |
| Major | >50 | 40-60 | 20-40 |
Repeat as needed 12-24 | Up to 5-7 days |
- Adjust the dose according to the extent and location of bleeding and the patient’s clinical condition. In VWD type 3 patients, those with gastro-intestinal (GI) bleedings may require higher doses.
- Repeat doses as needed based upon repeat monitoring of appropriate clinical and laboratory measures.
- Perform appropriate laboratory tests on the patient’s plasma at suitable intervals to assure that adequate VWF:RCo and FVIII activity levels have been reached and are maintained.
Dosing for Surgeries
A guide for dosing in minor and major surgeries is provided in Table 2 .
Table 2 Dosing for Treatment in Minor and Major Surgeries in all VWD Types
| Type of Surgery |
Loading Dosage
(IU VWF:RCo/kg body weight) (within 3 hours before surgery) |
VWF:RCo Peak Levels
(% of normal) |
Maintenance Dosage
(IU VWF:RCo/kg body weight) |
VWF:RCo Trough Levels
(% of normal) |
Frequency of Doses
(hours) |
Duration of Therapy
(days) |
|
Minor (including tooth extraction) | 30-60 | 50 |
15-30 or half the loading dose | >30 | 12-24 | Until wound healing achieved, up to 3 days |
| Major | 40-60 | 100 |
20-40 or half the loading dose | >50 |
12-24 (at least 2 doses within first 24 hours after the start of surgery) | Until wound healing achieved, up to 6 days or more |
In order to decrease the risk of perioperative thrombosis, FVIII activity levels should not exceed 250%.
Whereas table 2 provides a dosing range that is expected to provide the desired peak levels of VWF:RCo, the following is an example on how to calculate the loading dose based on a patient’s individual IVR which is to be determined pre-surgery.
- Prior to surgery, measure incremental in vivo recovery (IVR) and assess baseline plasma VWF:RCo activity. The IVR for VWF:RCo provides the IU/dL rise per IU/kg body weight infused and can be measured and calculated as follows:
- Measure plasma VWF:RCo at baseline
- Infuse 60 IU VWF:RCo/kg of WILATE intravenously at time 0
- Measure plasma VWF:RCo at 30 minutes after the infusion
IVR = (Plasma VWF:RCo 30 min – Plasma VWF:RCo baseline )/60IU kg
- Calculation of the loading dose requires four values: the target peak plasma VWF:RCo level, the baseline VWF:RCo level, body weight (BW) in kilograms, and IVR. If the actual IVR calculation exceeds 2.5, use 2.5 to calculate the recommended loading dosing to avoid under-dosing. If IVR is not available, a standardized dose can be calculated based on an assumed VWF:RCo IVR of 2.0 U/dL per IU/kg of WILATE administered.
Example:
Assumption: Baseline VWF:RCo value = 10 IU/dL, and VWF:RCo target level = 100 IU/dL = Δ 90 IU/dL
Assumption: Patient’s weight = 75 kg, patient’s IVR = 1.8 (IU/dL)/(IU/kg)
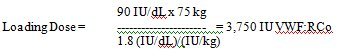
When possible post surgery, perform appropriate laboratory tests on the patient’s plasma once a day after surgery to assure that adequate VWF:RCo and FVIII activity levels have been reached and are maintained. In order to decrease the risk of perioperative thrombosis, FVIII activity levels should not exceed 250%. [ 2 ]
Dosing for routine prophylactic treatment
A guide for dosing WILATE for routine prophylaxis to reduce the frequency of bleeding is provided in Table 3 . Exact dosing should be defined by the severity of VWD and by the patient’s clinical status and response.
Table 3 Dosing for Routine Prophylaxis
| Patients | Dose (IU/kg) | Frequency of infusions |
|
Age 6 and older |
20 – 40 IU/kg |
Two or three times per week |
Hemophilia A
- Calculation of the required dose of Factor VIII is based on the empirical finding that 1 IU Factor VIII per kg body weight raises the plasma Factor VIII activity by approximately 2% of normal activity or 2 IU/dL when assessed using the one stage clotting assay. Use the following formula to determine the required dose:
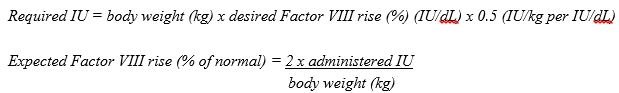
- Dose and duration of therapy depend on the patient’s weight, type and severity of hemorrhage, FVIII level, and presence of inhibitors.
Titrate dose and frequency to the patient’s clinical response, individual needs, severity of deficiency, severity of hemorrhage, desired FVIII level, and presence of inhibitor, and the patient’s clinical condition. Patients may vary in their pharmacokinetic (e.g., half-life, in vivo recovery) and clinical responses to WILATE.
Routine Prophylaxis
A guide for dosing WILATE for routine prophylaxis to reduce the frequency of bleeding is provided in Table 4 . Exact dosing should be defined by the patient’s clinical status and response.
Table 4 Dosing for Routine Prophylaxis
| Patients | Recommended Dosage (IU/kg body weight) | Frequency of Infusions |
| Adolescents and adults | 20-40 IU/kg | Every 2 to 3 days |
Dosing for Hemorrhages
A guide for dosing in the treatment of hemorrhages is provided in Table 5 .
Table 5 Dosing for Treatment of Hemorrhages
| Type of Hemorrhages* | Recommended dosage (IU/kg body weight) |
Frequency of Doses
(hours) |
Duration of Therapy
(days) |
| Minor | 30-40 | Repeat every 12-24 hours | At least 1 day, until the hemorrhage has resolved |
| Moderate | 30-40 | Repeat every 12-24 hours | 3 to 4 days or more, until the hemorrhage has resolved |
| Major | 35-50 | Repeat every 12-24 hours | 3 to 4 days or more, until the hemorrhage has resolved |
| Life-threatening | 35-50 | Repeat every 8-24 hours | Until threat has resolved |
*Minor hemorrhages may include e.g. early onset muscle and joint bleeds with no visible symptoms, such as little or no change in the range of motion of affected joint, mild restriction of mobility and activity, scrapes, superficial cuts, bruises, superficial mouth bleeds, and most nose bleeds; moderate hemorrhages may include e.g. advanced soft tissue and muscle bleeds into the limbs, bleeding into the joint space, such as the elbow, knee, ankle, wrist, shoulder, hip, foot, or finger; major hemorrhages may include e.g. complicated joint bleeds, bleeds of the pelvic muscles, eyes, and life-threatening hemorrhages may include e.g. bleedings in the abdomen, digestive system or chest, central nervous system bleeds, bleedings in the area of the neck or throat or pharynx, or other major trauma.
- Monitoring parameters
- Monitor plasma FVIII levels periodically to evaluate individual patient response to the dosage regimen.
- If dosing studies have determined that a particular patient exhibits a lower/higher than expected response and shorter/longer half-life, adjust the dose and the frequency of dosing accordingly
- Failure to achieve the expected FVIII:C level or to control bleeding after an appropriately calculated dosage may be indicative of the development of an inhibitor (an antibody to FVIII:C). Quantitate the inhibitor level by appropriate laboratory procedures and document its presence. Treatment with WILATE in such cases must be individualized.
2.2 Preparation and Reconstitution
- WILATE is provided with a Nextaro ® transfer device for reconstitution of the freeze-dried powder in diluent, a 10 mL syringe, an infusion set and two alcohol swabs.
- Reconstitute the powder only directly before injection. Because WILATE contains no preservatives, use the solution within 4 hours after reconstitution.
Instructions for Reconstitution:
 |
1) Warm the powder (WILATE) and Diluent in the closed vials up to room temperature. If a water bath is used for warming, avoid water contact with the rubber stoppers or the caps of the vials. The temperature of the water bath should not exceed +37°C (98°F). 2) Remove the caps from the powder (WILATE) vial and the diluent vial and clean the rubber stoppers with an alcohol swab. |
 |
3) Open the transfer device package by peeling off the lid. To maintain sterility, leave the Nextaro ® device in the clear outer packaging. The transfer device must be attached to the diluent vial first and then to the lyophilized powder vial. Otherwise, loss of vacuum occurs, and transfer of the diluent does not take place. If diluent is not completely transferred to the lyophilized powder vial during this process, contact Octapharma’s customer service. Place the diluent vial on a level surface and hold the vial firmly. Ensure the Nextaro ® device remains in the outer packaging and invert the Nextaro ® device ensuring the blue part with the water droplets is on top of the diluent vial. Push the Nextaro ® straight and firmly down until it snaps into place (Fig. 1). Do not twist while attaching. While holding onto the diluent vial, remove the outer package from the Nextaro ® , leaving the Nextaro ® attached firmly to the diluent vial (Fig. 2). |
 |
4) With the powder (WILATE) vial held firmly on a level surface, quickly invert the diluent vial with the Nextaro ® attached. Place the white part of the Nextaro ® on top of the powder (WILATE) vial until it snaps into the place (Fig. 3). Do not twist while attaching. The diluent will be drawn into the powder (WILATE) vial by the vacuum. 5)With both vials still attached, immediately start swirling the powder (WILATE) vial to ensure the powder is fully saturated. A slight whirlpool is formed by swirling. In order to avoid foaming, please do not shake the vial. |
 |
6) After 30 seconds, firmly hold both the white and blue parts of the Nextaro ® . Unscrew the Nextaro ® into two separate pieces (Fig. 4) and discard the empty diluent vial and the blue part of the Nextaro ® . Continue swirling until the powder in the WILATE vial has completely dissolved. This process may take several minutes. The final solution is clear or slightly opalescent, colorless or slightly yellow. If the powder fails to dissolve completely or an aggregate is formed, do not use the preparation. |
For intravenous injection after reconstitution only
- Inspect final solution visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Do not mix WILATE with other medicinal products or administer simultaneously with other intravenous preparation in the same infusion set.
- With the WILATE vial still upright, attach a plastic disposable syringe to the Nextaro ® (white plastic part). Invert the system and draw the reconstituted WILATE into the syringe.
- Once WILATE has been transferred into the syringe, firmly hold the barrel of the syringe (keeping the syringe plunger facing down) and detach the Nextaro ® from the syringe. Discard the Nextaro ® (white plastic part) and empty WILATE vial.
- Clean the intended injection site with an alcohol swab.
- Attach a suitable infusion needle to the syringe.
- Measure the patient’s pulse rate before and during the injection. If a marked increase in the pulse rate occurs, reduce the injection speed or interrupt the administration.
- Inject the solution intravenously at a slow speed of 2-4 mL/minute.
- Dispose unused product or waste material in accordance with local requirements.
3. Dosage Forms and Strengths
WILATE is available as a sterile, lyophilized powder for reconstitution for intravenous injection, provided in the following nominal strengths per single-use vial:
- 500 IU VWF:RCo and 500 IU FVIII activities in 5 mL
- 1000 IU VWF:RCo and 1000 IU FVIII activities in 10 mL
4. Contraindications
WILATE is contraindicated in patients with known hypersensitivity reactions, including anaphylactic or severe systemic reactions, to human plasma-derived products, any ingredient in the formulation [see Description (11) ] , or components of the container.
5. Warnings and Precautions
5.1 Hypersensitivity Reactions
Hypersensitivity reactions may occur with WILATE. Signs and symptoms include angioedema, burning and stinging at the infusion site, chills, flushing, generalized urticaria, headache, hives, hypotension, lethargy, nausea, restlessness, tachycardia, tightness of the chest, tingling, vomiting, and wheezing that may progress to severe anaphylaxis (including shock) with or without fever.[ 3 ] Closely monitor patients receiving WILATE and observe for any symptoms throughout the infusion period.
Because inhibitor antibodies may occur concomitantly with anaphylactic reactions, evaluate patients experiencing an anaphylactic reaction for the presence of inhibitors.[ 3 ] [see Warnings and Precautions ( 5.3 )]
5.2 Thromboembolic Events
In VWD, continued treatment using a FVIII-containing VWF product may cause an excessive rise in FVIII activity [ 1 ], which may increase the risk of thromboembolic events. Monitor plasma levels of VWF:RCo and FVIII activities in patients receiving WILATE to avoid sustained excessive VWF and FVIII activity levels.
5.3 Neutralizing Antibodies
VWD
- Neutralizing antibodies (inhibitors) to FVIII and VWF in patients with VWD, especially type 3 patients, may occur. If a patient develops inhibitor to VWF (or to FVIII), the condition will manifest itself as an inadequate clinical response. Thus, if expected VWF activity plasma levels are not attained, or if bleeding is not controlled with an adequate dose or repeated dosing, perform an appropriate assay to determine whether a VWF inhibitor is present.
- In patients with antibodies against VWF, VWF is not effective and WILATE administration may lead to severe adverse events. Consider other therapeutic options for such patients.
- Because inhibitor antibodies may occur concomitantly with anaphylactic reactions, evaluate patients experiencing an anaphylactic reaction for the presence of inhibitors.[ 3 ] [see Warnings and Precautions ( 5.1 )].
Hemophilia A
- Monitor plasma Factor VIII activity by performing a validated test (e.g., one stage clotting assay), to confirm that adequate Factor VIII levels have been achieved and maintained [see Dosage and Administration ( 2.1 )] .
- Monitor for the development of Factor VIII inhibitors. Perform a Bethesda inhibitor assay if expected Factor VIII plasma levels are not attained, or if bleeding is not controlled with the expected dose of Wilate. Use Bethesda Units (BU) to report inhibitor levels.
5.4 Transmissible Infectious Agents
WILATE is made from human plasma. Because this product is made from human blood, it may carry a risk of transmitting infectious agents, e.g., viruses, and theoretically, the variant Creutzfeldt-Jakob disease (vCJD) agent. There is also the possibility that unknown infectious agents may be present in the product. The risk that WILATE will transmit viruses has been reduced by screening plasma donors for prior exposure to certain viruses, by testing for the presence of certain current virus infections, and by inactivating and removing certain viruses during manufacture. Despite these measures, it may still potentially transmit disease. [ 5 ]
Record the batch number of the product every time WILATE is administered to a patient, and consider appropriate vaccination (against hepatitis A and B virus) of patients in regular/repeated receipt of WILATE. ALL infections thought by a physician possibly to have been transmitted by this product should be reported by the physician or other healthcare provider to Octapharma USA, Inc., at 1-866-766-4860.
5.5 Monitoring and Laboratory Tests
- Monitor plasma levels of VWF:RCo and FVIII activities in patients receiving WILATE to avoid sustained excessive VWF and FVIII activity levels, which may increase the risk of thromboembolism, particularly in patients with known clinical or laboratory risk factors.
- Monitor for development of VWF and FVIII inhibitors. Perform assays to determine whether VWF and/or FVIII inhibitor(s) is present if bleeding is not controlled with the expected dose of WILATE. [ 6 ]
6. Adverse Reactions/Side Effects
The most common adverse reactions to treatment with WILATE (≥ 1%) in patients with VWD were hypersensitivity reactions, urticaria, chest discomfort, and dizziness. The most common adverse reactions to treatment with WILATE (≥ 1%) in previously treated patients with hemophilia A was pyrexia (fever).
Seroconversions for antibodies to parvovirus B19 not accompanied by clinical signs of disease have been observed.
The most serious adverse reactions to treatment with WILATE in patients with VWD and hemophilia A are hypersensitivity reactions [see Warnings and Precautions ( 5.1 )].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of WILATE cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in clinical practice.
A total of 198 subjects treated for VWD (aged 1 to 83 years) received WILATE on 10761 occasions, including clinical studies that involved prophylactic use, treatment on demand, surgery, and pharmacokinetics. Of the 198 subjects, 37 (19%) had VWD type 1, 47 (23.7%) had VWD type 2, and 104 (52.5%) had VWD type 3 and 10 (5%) had an unknown status; 111 (56.1%) subjects were female and 87 (43.9%) subjects were male. Overall, subjects received 19;048;830 IU of WILATE during 10285 exposure days. The most common adverse drug reactions were hypersensitivity reactions (4 subjects; 2%), dizziness (3 subjects; 1.5%), urticaria and chest discomfort (each with 2 subjects; 1%).
A total of 136 hemophilia A previously treated subjects (aged 11 to 66 years) received WILATE in 5 clinical studies that involved prophylactic use, treatment on demand, surgery and/or pharmacokinetics. All subjects were male. Overall, subjects received 19,317,004 IU of WILATE during 9001 exposure days. The most common adverse reaction was pyrexia (2 subjects; 1.5%). Further adverse reactions included pruritus, headache and sleeping disorder (1 subject; 0.75%). Two out of 55 subjects (3.6%) in the pivotal study of routine prophylaxis in severe hemophilia A had unexplained transient worsening of pre-existing thrombocytosis while on the study.
Immunogenicity
The immunogenicity of WILATE in VWD was specifically assessed in 97 subjects in 3 clinical studies in which subjects received 9,635,041 IU of WILATE during 5575 exposure days. No inhibitors of VWF were detected in these 3 studies. In one of 3 studies, which also assessed FVIII inhibitor development in 15 subjects, no inhibitors of FVIII were detected after administration of a total of 223,290 IU of WILATE over 419 administrations.
The immunogenicity of WILATE in previously treated subjects with hemophilia A was specifically assessed in a pooled analysis that selected from 136 previously treated subjects included in 5 clinical studies those subjects who had at least 150 exposure days at the time of enrollment into the study and had been treated for at least 50 exposure days and 6 months in the study. Eighty three subjects fulfilled these criteria. None of them developed inhibitors to Factor VIII, resulting in a rate of inhibitor development of 0% (95% CI, 0-4.35%).
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to WILATE with the incidence of antibodies to other products may be misleading.
6.2 Postmarketing Experience
The following adverse reactions have been identified during the post-approval use of WILATE. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
Post-marketing adverse reactions including dyspnea, hypertension, cough, chest discomfort, abdominal pain, and back pain have been reported in patients treated with WILATE.
Related/similar drugs
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
There are no data with WILATE use in pregnant women to inform a drug-associated risk. Animal reproduction studies have not been conducted with WILATE. WILATE was given to four subjects (3 type 3 and 1 type 2B) during labor and delivery in one clinical study. Two subjects underwent vaginal delivery (type 3) and two subjects had a cesarean section (type 3/type 2B). In this study all procedures were uneventful.
In the U.S. general population, regardless of drug exposure, the estimated background risk of major birth defect and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk summary
There is no information regarding the presence of WILATE in human milk, the effect on the breastfed infant, and the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for WILATE and any potential adverse effects on the breastfed infant from WILATE or from the underlying maternal condition.
8.4 Pediatric Use
The safety and effectiveness of WILATE have been established for pediatric patients ≤16 years of age with vWD for the treatment and control of bleeding episodes and perioperative management. Eleven pediatric subjects with VWD between 5 to 16 years of age (8 type 3, 1 type 2, 2 type 1) were treated with WILATE for 234 bleeding episodes (BEs) in clinical studies. Treatment success was achieved in 88% of BEs [see Clinical Studies ( 14 )] . Efficacy data during surgical prophylaxis were available for 13 pediatric VWD surgery subjects (3 subjects aged 0 to 2 years, 4 subjects aged 2 to 5 years, 3 subjects aged 6 to 12 years, and 3 subjects aged 12 to 16 years). Treatment success was achieved in 22 of 22 surgical procedures (17 minor and 5 major).
Efficacy of routine prophylaxis was investigated in 15 pediatric subjects between 6 and 16 years of age (9 type 3, 3 type 2 and 3 type 1). Total annualized bleeding rate decreased from 32.59 during on-demand treatment to 3.73 during routine prophylaxis with WILATE in the 9 subjects 6 to less than 12 years of age and from 28.99 to 4.28 in the 6 subjects 12 to less than 17 years of age.
The safety and effectiveness of WILATE have been established for pediatric patients ≥12 years of age with hemophilia A for the treatment and control of bleeding episodes and routine prophylaxis use. Five adolescents with hemophilia A between 12 und 15 years of age were treated with WILATE for routine prophylaxis in one clinical study. The annualized bleeding rate was mean 0.4 (standard deviation 0.9), median 0 (range 0-2). During 6 months of prophylaxis, one patient experienced one traumatic BE, which was treated successfully with WILATE. No dose adjustment is needed for pediatric patients ≥12 years of age as administered dosages were similar to those used in the adult population [see Clinical Studies ( 14 )].
The safety and effectiveness of WILATE have not been established for pediatric patients <12 years of age with hemophilia A. In a clinical study that evaluated the pharmacokinetics of WILATE in 10 pediatric patients (2 to 11 years of age) with hemophilia A, effects on annualized bleeding rate were inconclusive.
11. Wilate, von Willebrand Factor/Coagulation Factor VIII Complex Description
WILATE is a human plasma-derived, sterile, purified, double virus inactivated von Willebrand Factor/Coagulation Factor VIII Complex. WILATE is supplied as a lyophilized powder for reconstitution for intravenous injection. The diluent for reconstitution of the lyophilized powder is Water for Injection with 0.1% Polysorbate 80.
WILATE contains no preservative. No albumin is added as a stabilizer. WILATE is labeled with the actual VWF:RCo and FVIII activities in IU per vial. The VWF activity (VWF:RCo) is determined using a manual agglutination method referenced to the current “WHO International Standard for von Willebrand Factor Concentrate”. The FVIII activity is determined using a chromogenic substrate assay referenced to the current “WHO International Standard for Human Coagulation Factor VIII Concentrate”. The assay methodologies are according to European Pharmacopoeia (Ph.Eur.). The resulting specific activity of WILATE is ≥ 60 IU VWF:RCo and ≥ 60 IU FVIII activities per mg of total protein.
The nominal composition of WILATE is as follows:
| Component | Quantity/ 5 mL vial | Quantity/ 10 mL vial |
| VWF:Rco | 500 IU | 1000 IU |
| FVIII | 500 IU | 1000 IU |
| Total protein | ≤ 7.5 mg | ≤ 15.0 mg |
| Glycine | 50 mg | 100 mg |
| Sucrose | 50 mg | 100 mg |
| Sodium chloride | 117 mg | 234 mg |
| Sodium citrate | 14.7 mg | 29.4 mg |
| Calcium chloride | 0.8 mg | 1.5 mg |
| Water for injection | 5 mL | 10 mL |
| Polysorbate 80 | 1 mg/mL | 1 mg/mL |
Von Willebrand Factor/Coagulation Factor VIII complex is the active ingredient in WILATE. It is derived from large pools of human plasma collected in U.S. plasma donation centers. All plasma donations are tested for viral markers in compliance with requirements of EU CPMP and FDA guidance. In addition, the limit for the titer of human parvovirus B19 DNA in the manufacturing pool is set not to exceed 10 4 IU/mL.
The product is manufactured from cryoprecipitate, which is reconstituted in a buffer and treated with aluminum hydroxide followed by two different chromatography steps, ultra- and diafiltration, and sterile filtration. The manufacturing process includes two virus inactivation steps, namely, treatment with an organic solvent/detergent (S/D) mixture, composed of tri-n-butyl phosphate (TNBP) and Octoxynol-9, and a terminal dry heat (TDH) treatment of the lyophilized product in final container [at +100°C (212°F) for 120 minutes at a specified residual moisture level of 0.7-1.6%]. In addition, the ion-exchange chromatography step utilized during WILATE manufacturing also removes some viruses [ 8 ]. The mean cumulative virus reduction factors of these steps are summarized in Table 6 .
Table 6 Virus Reduction During WILATE Manufacturing
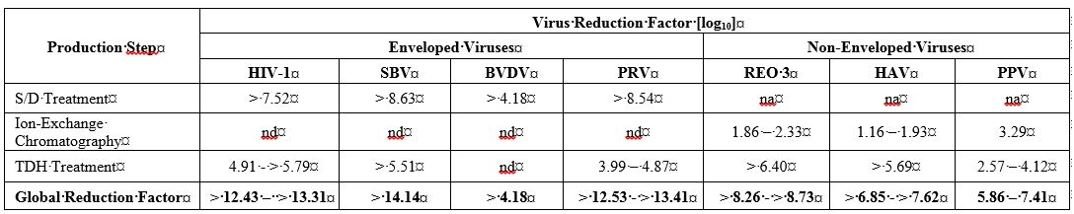
na: not applicable
nd: not done (S/D reagents present)
HIV-1: Human Immunodeficiency Virus - 1
SBV: Sindbis Virus
BVDV: Bovine Viral Diarrhea Virus
PRV: Pseudorabies Virus
REO 3: Reovirus Type 3
HAV: Hepatitis A Virus
PPV: Porcine Parvovirus
12. Wilate, von Willebrand Factor/Coagulation Factor VIII Complex - Clinical Pharmacology
12.1 Mechanism of Action
WILATE contains von Willebrand factor (VWF) and coagulation factor VIII (FVIII), constituents of normal plasma. VWF promotes platelet aggregation and platelet adhesion on damaged vascular endothelium; it also serves as a stabilizing carrier protein for the procoagulant protein FVIII, an essential cofactor in activation of factor X leading to formation of thrombin and fibrin. Patients suffering from VWD have a deficiency or abnormality of VWF. This reduction in VWF plasma concentration results in correspondingly low FVIII activity and abnormal platelet function, thereby resulting in excessive bleeding. [ 9 ] After administration, WILATE temporarily replaces missing VWF and FVIII that are needed for effective hemostasis. When infused into a patient with hemophilia A, FVIII binds to VWF in the patient´s circulation. Activated FVIII (FVIIIa) acts as a cofactor for activated factor IX (FIXa), accelerating the conversion of factor X to activated factor X (FXa). FXa converts prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin and a clot can be formed. Hemophilia A is a sex-linked hereditary disorder of blood coagulation due to decreased levels of FVIII:C and results in profuse bleeding into joints, muscles or internal organs, either spontaneously or as a result of accidental or surgical trauma. By replacement therapy the plasma levels of FVIII are increased, thereby enabling a temporary correction of the factor deficiency and correction of the bleeding tendencies.
12.3 Pharmacokinetics
VWD
Pharmacokinetic (PK) profiles of WILATE were determined by FVIII activity, VWF:RCo, VWF:Ag, and VWF:CB obtained from an open label, prospective, randomized, controlled, two-arm cross-over study with WILATE and a comparator product conducted at 6 sites in the US. Twenty-two subjects (≥ 12 years of age) with inherited VWD [type 1, n=6; type 2, n=9 (6 type 2A, 1 type 2B, and 2 type 2M); and type 3, n=7] received an intravenous bolus dose of WILATE containing approximately 40 IU of VWF:RCo/kg body weight. Twenty subjects completed the study as per protocol. PK parameters for VWF:RCo and FVIII activities are summarized in Table 7 and Table 8 , respectively. PK parameters for pediatric subjects are summarized in Table 9 , Table 10 , Table 11 , and Table 12 .
The PK parameters reported in Table 7 are based on VWF:RCo values obtained using a modified Behring Coagulation System (BCS) analytical method. The modified BCS was used because of its validated lower variability compared with the standard BCS. Measured concentrations (IU VWF:RCo/mL) are higher by the modified BCS than by the standard BCS analytical method which is used in some clinical laboratories. Dose adjusted C max and AUC determined by this modified BCS method are approximately 1.5 times higher than those by the standard BCS method. No difference has been found in incremental recovery.
The PK parameters reported in Table 9 , Table 10 , Table 11 , and Table 12 are based on values obtained from an open label, prospective, non-controlled, international, multi-center study in which a subset of pediatric subjects aged 6-16 years with inherited VWD underwent pharmacokinetic investigations before the start of prophylaxis treatment. Thirteen subjects [type 1, n=3; type 2, n=3, and type 3, n=7]) received an intravenous bolus dose of WILATE containing approximately 60 IU of VWF:RCo/kg body weight. Eight subjects were 6-11 years old and 5 subjects 12-16 years.
Table 7 Pharmacokinetic Parameters of VWF:RCo:mean ± SD (range)
| Parameters |
VWD type I
(n = 5) |
VWD type II
(n = 9) |
VWD type III
(n = 6) |
Total
(n = 20) |
| C max (IU/dL) |
74 ± 13 (62 - 91) |
77 ± 18 (40 - 100) |
79 ± 13 (65 - 102) |
76 ± 15 (40 - 102) |
|
AUC
(0-inf)
(IU*hr/dL) |
1633 ± 979 (984 - 3363) |
1172 ± 421 (571 - 1897) |
995 ± 292 (527 - 1306) |
1235 ± 637 (527 - 3363) |
| Half-life (hrs) |
24.7 ± 17.9 (11.2 - 48.5 ) |
15.3 ± 6.3 (6.0 - 26.4) |
9.1± 2.6 (5.7 - 12.9 ) |
15.8 ± 11.0 (5.7 - 48.5) |
| CL (mL/h/kg) |
3.1 ± 1.1 (1.2 - 4.1) |
4.1 ± 1.7 (2.0 - 7.1) |
4.2 ± 1.4 (3.0 - 6.6) |
3.9 ± 1.5 (1.2 - 7.1) |
| Vd (mL/kg) |
81.7 ± 38.5 (15.3 - 74.2) |
76.6 ± 35.4 (45.3 - 158.8) |
49.4 ± 16.7 (29.7 - 67.1) |
69.7 ± 33.2 (29.7 - 158.8) |
| MRT (hrs) |
32.7 ± 25.8 (15.3 - 74.2) |
19.7 ± 5.6 (9.9 - 27.1) |
11.9 ± 2.9 (9.2 - 15.9) |
20.6 ± 14.8 (9.2 - 74.2) |
|
Recovery (%IU/kg) |
1.8 ± 0.2 (1.5 - 2.0) |
1.8 ± 0.5 (1.0 - 2.4) |
2.1 ± 0.3 (1.8 - 2.6) |
1.9 ± 0.4 (1.0 - 2.6) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd= volume of distribution at steady state; MRT = mean residence time
Table 8 Pharmacokinetic Parameters of FVIII activity (chromogenic assay): mean ± SD (range)
| Parameters |
VWD type I
(n = 5) |
VWD type II
(n = 8*) |
VWD type III
(n = 6) |
Total
(n = 19*) |
| C max (IU/dL) |
117.1 ± 12.1 (103 - 135) |
147.2 ± 32.6 (102 - 206) |
120 ± 23 (91 - 148) |
112 ± 23 (59 - 148) |
|
AUC
(0-inf)
(IU*hr/dL) |
1187 ± 382 (523 - 1483) |
1778 ± 1430 (544 - 4821) |
2670 ± 854 (1874 - 3655) |
2290 ± 1045 (464 - 4424) |
| Half-life (hrs) |
17.5 ± 4.9 (10.9 - 23.8) |
23.6 ± 8.3 (12.6 - 34.7) |
16.1 ± 3.1 (11.8 - 20.1) |
19.6 ± 6.9 (10.9 - 34.7) |
| CL (mL/h/kg) |
4.4 ± 3.7 (2.5 - 11.0) |
2.5 ± 0.9 (1.2 - 3.5) |
2.0 ± 0.6 (1.4 - 2.8) |
2.9 ± 2.1 (1.2 - 11.0) |
| Vd (mL/kg) |
95.0 ± 53.8 (57.1 - 190.0) |
79.5 ± 23.1 (52.8 - 116.2) |
44.2 ± 10.4 (31.8 - 57.1) |
72.4 ± 36.2 (31.8 - 190.0) |
| MRT (hrs) | 24.1 ± 5.5(17.2 - 31.5) |
35.1 ± 14.2 (17.5 - 61.6) |
23.0 ± 3.7 (18.0 - 27.7) |
28.4 ± 11.1 (17.2 - 61.6) |
|
Recovery (%IU/kg) |
1.9 ± 0.5 (1.1 - 2.5) |
2.2 ± 0.4 (1.6 - 2.8) |
2.5 ± 0.5 (2.0 - 3.0) |
2.2 ± 0.5 (1.1 - 3.0) |
*One subject with implausible long half-life is not included in the summary table, except for recovery result.
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence time
Table 9 Pharmacokinetic Parameters of VWF:RCo in children 6-16 years per VWD type: mean ± SD (range)
| Parameters |
VWD type I
(n = 3) |
VWD type II
(n = 3) |
VWD type III
(n = 7) |
Total
(n = 13) |
| C max (IU/dL) |
91.3 ± 1.3 (78.5 – 119.3) |
63.5 ± 1.5 (39.0 – 84.4) |
77.4 ± 1.1 (59.1 – 90.6) |
76.8 ± 1.3 (39.0 – 119.3) |
|
AUC
(0-inf)
(IU*hr/dL) |
963 ± 1.0 (920 – 1003) |
1421 ± 2.2 (634 – 3084) |
822 ± 1.2 (657 – 1189) |
968 ± 1.5 (634 – 3084) |
| Half-life (hrs) |
6.9 ± 1.1 (6.5 – 7.4) |
17.4 ± 2.0 (8.6 – 32.5) |
6.5 ± 1.2 (4.9 – 8.5) |
8.3 ± 1.7 (4.9 – 32.5) |
| CL (dL/h/kg) |
0.1 ± 1.0 (0.1 – 0.1) |
0.0 ± 2.2 (0.0 – 0.1) |
0.1 ± 1.2 (0.1 – 0.1) |
0.1 ± 1.5 (0.0 – 0.1) |
| Vd (dL/kg) |
0.7 ± 1.2 (0.6 – 0.8) |
1.4 ± 1.9 (0.9 – 3.0) |
0.8 ± 1.2 (0.7 – 1.0) |
0.8 ± 1.5 (0.6 – 3.0) |
| MRT (hrs) |
10.8 ± 1.1 (9.6 – 12.3) |
32.8 ± 1.4 (24.4 – 46.3) |
10.4 ± 1.2 (8.0 – 13.1) |
13.7 ± 1.7 (8.0 – 46.3) |
|
Recovery (%IU/kg) |
1.5 ± 1.3 (1.3 – 2.0) |
1.1 ± 1.5 (0.7 – 1.4) |
1.3 ± 1.2 (1.0 – 1.5) |
1.3 ± 1.3 (0.7 – 2.0) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence time
Table 10 Pharmacokinetic Parameters of VWF:RCo in children 6-16 years per age class: mean ± SD (range)
| Parameters |
Age 6-11 years
(n = 8) |
Age 12-16 years
(n = 5) |
| C max (IU/dL) |
73.0 ± 1.4 (39.0 – 119.3) |
83.2 ± 1.1 (77.7 – 90.6) |
|
AUC
(0-inf)
(IU*hr/dL) |
945 ± 1.7 (634 – 3084) |
1005 ± 1.3 (714 – 1467) |
| Half-life (hrs) |
8.4 ± 1.8 (5.0 – 32.5) |
8.1 ± 1.6 (6.0 - 19.0) |
| CL (dL/h/kg) |
0.1 ± 1.7 – 0.1) |
0.1 ± 1.3 (0.0 – 0.1) |
| Vd (dL/kg) |
0.9 ± 1.7 (0.6 – 3.0) |
0.8 ± 1.2 (0.7 – 1.0) |
| MRT (hrs) |
14.2 ± 1.9 (8.0 – 46.3) |
13.0 ± 1.5 (10.2 – 24.4) |
|
Recovery (%IU/kg) |
1.2 ± 1.4 (0.7 – 2.0) |
1.4 ± 1.1 (1.3 – 1.5) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence time
Table 11 Pharmacokinetic Parameters of FVIII activity (chromogenic assay) in children 6-16 years per VWD type: mean ± SD (range)
| Parameters |
VWD type I
(n = 3) |
VWD type II
(n = 3) |
VWD type III
(n = 7) |
Total
(n = 13) |
| C max (IU/dL) |
101.8 ± 1.0 (98.1 – 106.9) |
71.7 ± 1.4 (47.9 – 96.8) |
94.9 ± 1.2 (77.0 – 115.3) |
90.4 ± 1.3 (47.9 – 115.3) |
|
AUC
(0-inf)
(IU*hr/dL) |
1760 ± 1.8 (923 – 2865) |
1377 ± 1.5 (883 – 2006) |
2335 ± 1.4 (1522 – 3599) |
1937 ± 1.5 (883 – 3599) |
| Half-life (hrs) |
14.1 ± 1.4 (10.0 – 18.7) |
19.3 ± 1.8 (9.8 – 30.1) |
13.5 ± 1.5 (6.4 – 21.8) |
14.8 ± 1.5 (6.4 – 30.1) |
| CL (dL/h/kg) |
0.0 ± 1.8 (0.0 – 0.1) |
0.0 ± 1.5 (0.0 – 0.1) |
0.0 ± 1.4 (0.0 – 0.0) |
0.0 ± 1.5 (0.0 – 0.1) |
| Vd (dL/kg) |
0.7 ± 1.2 (0.6 – 0.8) |
1.6 ± 1.6 (1.1 – 2.6) |
0.6 ± 1.2 (0.4 – 0.8) |
0.8 ± 1.6 (0.4 – 2.6) |
| MRT (hrs) |
19.9 ± 1.5 (12.3 – 27.6) |
35.8 ± 1.1 (33.4 – 38.4) |
23.8 ± 1.2 (18.1 – 31.9) |
25.1 ± 1.4 (12.3 – 38.4) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence time
Table 12 Pharmacokinetic Parameters of Factor VIII activity (chromogenic assay) in children 6-16 years per age class: mean ± SD (range)
| Parameters |
Age 6-11 years
(n = 8) |
Age 12-16 years
(n = 5) |
| C max (IU/dL) |
82.7 ± 1.3 (47.9 – 100.5) |
104.3 ± 1.1 (96.8 – 115.3) |
|
AUC
(0-inf)
(IU*hr/dL) |
1607.3 ± 1.5 (882.6 – 3087.6) |
2609.3 ± 1.3 (2005.5 – 3599.3) |
| Half-life (hrs) |
12.2 ± 1.5 (6.4 – 24.3) |
20.3 ± 1.3 (15.4 – 30.1) |
| CL (dL/h/kg) |
0.04 ± 1.5 0.02 – 0.1) |
0.02 ± 1.3 (0.02 – 0.03) |
| Vd (dL/kg) |
0.9 ± 1.7 (0.4 – 2.6) |
0.7 ± 1.3 (0.5 – 1.1) |
| MRT (hrs) |
22.9 ± 1.4 (12.3 – 38.4) |
28.9 ± 1.2 (23.9 – 35.8) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence
Hemophilia A
The pharmacokinetics (PK) of WILATE were evaluated in 21 (16 adults, and 5 adolescents aged 12-15 years) previously treated patients (PTPs) with severe Hemophilia A within a prospective, open-label, multicenter clinical study. The PK parameters ( Table 13 ) were based on plasma Factor VIII activity measured by the one-stage clotting assay after a single intravenous infusion of a 50 IU/kg dose.
The PK profile obtained after 6 months of repeated dosing in adults and adolescents was comparable with the PK profile obtained after the first dose.
Table 13 Pharmacokinetic Parameters of WILATE in 21 Previously Treated Patients (PTP: Mean ± SD)
| PK Parameters | Adults (n=16) | Adolescents (n=5) |
| C max (IU/dL) |
113.82 ± 20.53 (77.43 - 142.00) |
83.92 ± 9.40 (77.93 – 100.27) |
| AUC (IU*hr/dL) |
1562.10 ± 451.43 (721.92 - 2271.48) |
1009.74 ± 172.26 (816.23 - 1287.76) |
| AUC norm (IU*hr/dL) |
31.24 ± 9.03 (14.44 - 45.43) |
20.20 ± 3.45 (16.32 - 25.76) |
| Half-life (hrs) |
10.64 ± 2.69 (6.30 - 15.43) |
11.41 ± 1.93 (9.37 – 14.57) |
| CL (dL/h/kg) |
0.035 ± 0.013 (0.02 - 0.07) |
0.051 ± 0.008 (0.04 - 0.06) |
| Vd (dL/kg) |
0.53 ± 0.13 (0.36 - 0.82) |
0.73 ± 0.13 (0.53 - 0.85) |
| MRT (hrs) |
15.81 ± 3.63 (10.80 - 22.23) |
14.39 ± 1.83 (12.85 – 17.30) |
| Recovery (%IU/kg) |
2.27 ± 0.41 (1.54 - 2.83) |
1.66 ± 0.17 (1.55 – 1.95) |
C max = peak concentration; AUC = area under curve; CL = clearance; Vd = volume of distribution at steady state; MRT = mean residence time
14. Clinical Studies
VWD
Treatment of Bleeding Episodes
Clinical efficacy of WILATE in the control of bleeding in subjects with VWD was determined in four prospective, open-label, non-controlled clinical studies (excluding PK study). For inclusion in the studies, subjects had to have inherited VWD (any type) that did not respond to desmopressin acetate. Subjects aged ≥12 to ≤65 years were eligible to enter three of the four studies, and subjects aged ≥6 to ≤85 years were eligible for one study. Exclusion criteria included the administration of other plasma-derived or blood products or desmopressin acetate 15 days before study entry, administration of acetylsalicylic acid 7 days before study entry, symptomatic infection, past or present inhibitor activity (3 studies), and severe liver or kidney disease (3 studies). A total of 70 VWD subjects with a mean age of 37 years (range 5–77 years) were enrolled in the studies, of whom 37 were type 3 and 30 were male. The total number of exposure days to WILATE for all investigations across the four studies ranged from 202 to 4917 days. Treated BEs were analyzed for efficacy using a set of objective criteria in addition to a subjective 4-point hemostatic efficacy scale (excellent, good, moderate and none) which was determined at the discretion of the investigator. In assessing efficacy using the objective criteria, treatment of a bleeding episode was classified as a success when none of the criteria listed below were met:
- the episode was additionally treated with another VWF-containing product (excluding whole blood)
- the subject received a blood transfusion during the episode
- follow-up treatment with a daily dosage of WILATE that was greater than or equal to 50% (≥ 50%) above the initial dose (for bleeding episodes with more than 1 day of treatment)
- treatment duration of more than 4 days (> 4 days) in cases of severe bleeding (other than gastrointestinal)
- treatment duration of more than 3 days (> 3 days) in cases of moderate bleeding (other than gastrointestinal)
- treatment duration of more than 2 days (> 2 days) in cases of minor bleeding (other than gastrointestinal)
- the last efficacy rating of the bleeding episode was 'moderate' or 'none'
BEs treated with WILATE are summarized for all subjects (n=45) and subjects aged 5–16 years (n=11) in Table 14 . Among the 70 VWD subjects administered WILATE in clinical studies (excluding the PK study), 45 received on demand treatment for 1068 BEs. Using the above objective criteria, corresponding efficacy for each bleeding event was rated as being successful in 84% of the episodes. In these 45 subjects with BEs, 93% of the successfully treated BEs occurred in VWD type 3 subjects (n=25). In 11 pediatric subjects aged 5–16 years, efficacy was rated as being successful in 87.6% of BEs.
Table 14 Proportion of Successful Treatments of Bleeding Episodes with WILATE
| Number of BEs* | Number of successfully treated BEs | % Successes (95% CI) | |
| All subjects (n=45) | 1068 | 898 | 84.1 (81.8-86.2) |
| Subjects 5–16 years (n=11) | 234 | 205 | 87.6 (82.7-91.5) |
*A “bleeding episode” may involve bleeding at multiple sites in this analysis.
Dosing information for 972 successfully treated “bleeding episodes” (1423 infusions), and 211 successfully treated bleeding episodes (289 infusions) in subjects aged 5–16 years, for regional bleeding is summarized in Table 15 . For the purpose of assigning success/failure to regional bleeding that occurred at the same time, bleeding at different sites over the same time span was counted as separate BEs. Thus, the number of these “episodes” is different from that in the overall evaluation for success/failure of WILATE in the treatment of BEs in Table 14 . The majority of BEs were treated for 1-3 days. In subjects with GI bleeds, the duration for product use to control bleeding was longer (up to 7 days).
Table 15 Administered Dosages (VWF:RCo in IU/kg) in Bleeding Episodes* Successfully Treated with WILATE: Mean ± SD (Range)
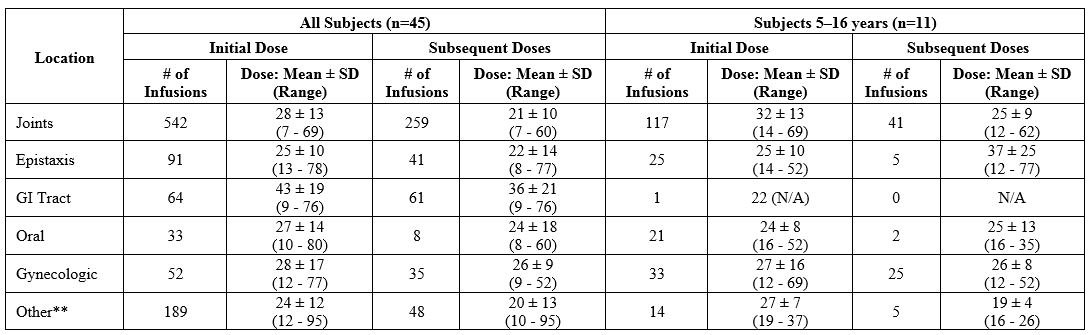
*For the purpose of this analysis, bleeding at each site is counted as a separate “episode”.
**“Other” includes mostly muscle bleeds, hematuria, ecchymosis, hematoma and other miscellaneous sites of bleeding.
Prevention of Bleeding in Surgery (Perioperative Management)
A prospective, open-label, single-arm, uncontrolled, multi-center clinical study was conducted to investigate the safety and hemostatic efficacy of WILATE in 28 subjects (19 female and 9 male) who underwent 30 surgeries. Two female subjects underwent 2 surgeries each. Subjects ranged in age from 12 to 74 years (median = 36). Three subjects were between 12 and 17 years old and 4 were 65 years or older. Six subjects had type 1 VWD, 1 had type 2A, 1 type 2B, and 20 had type 3 VWD. One type 1 and one type 3 subject had 2 surgeries each.
Twenty-one surgeries were classified as major (e.g. orthopedic joint replacement, cesarean section and vaginal deliveries, laminectomy, tonsillectomy, appendectomy, 3 rd molar extractions) and 9 were classified as minor (e.g. menisectomy, teeth extractions other than 3 rd molars, septoplasty, biopsy). Seven surgeries (3 major, 4 minor) were performed in type 1 VWD subjects, 2 surgeries (1 major, 1 minor) were performed in type 2 (A/B) VWD subjects, and 21 surgeries (17 major, 4 minor) were performed in type 3 VWD subjects. The types of surgery for the 9 minor procedures were: dental (n=5, 55.6%); orthopedic (n=2, 22.2%); ophthalmologic (n=1, 11.1%); and ear, nose and throat (n=1, 11.1%). The types of surgery for the 21 major procedures were: orthopedic (n=8, 38.1%); obstetric/gynecological (n=5, 23.8%); gastrointestinal (n=4, 19.0%); dental (n=2, 9.5%); and ear, nose and throat (n=2, 9.5%). The type of surgery according to VWD type were: VWD type 1 (n=7) – dental (n=4, 57.1%), orthopedic (n=2, 28.6%), and ear, nose and throat (n=1, 14.3%); VWD type 2 (A/B) (n=2) – orthopedic (n=1, 50%), and obstetric/gynecological (n=1, 50%); VWD type 3 (n=21) – orthopedic (n=7, 33.3%), gastrointestinal (n=4, 19.0%), obstetric/gynecological (n=4, 19.0%), dental (n=3, 14.3%), ear, nose and throat (n=2, 9.5%), and ophthalmologic (n=1, 4.8%).
Dosing was individualized based on in-vivo recovery results performed before surgery. Mean total loading dose per infusion was 51.4 IU/kg (median 52.1 IU/kg; range 27-77 IU/kg). Major surgeries required a mean loading dose of 54.7 IU/kg (median 55.5 IU/kg; range 36-69 IU/kg) in comparison with a mean loading dose of 41.9 IU/kg (median 37.5 IU/kg; range 27−77 IU/kg) for minor surgeries. Mean total maintenance dose per infusion was 28.5 IU/kg (median 28.5 IU/kg; range 8-63 IU/kg). Major surgeries required a mean maintenance infusion of 29.6 IU/kg (median 30 IU/kg; range 8-63 IU/kg) in comparison with a mean maintenance infusion of 21.6 IU/kg (median 20.6 IU/kg; range 14-38 IU/kg) for minor surgeries.
Efficacy of WILATE in surgical procedures was assessed by the surgeon at the conclusion of surgery and by the investigator-hematologist at 24 hours following completion of the final maintenance dose. Efficacy of WILATE was assessed using a stringent and objective 4-point ordinal efficacy scale (excellent, good, moderate, or none) based on estimated expected versus actual blood loss, transfusion requirements and post-operative bleeding and oozing. A rating of excellent or good was required to declare the outcome a success. An independent data monitoring committee (IDMC) additionally conducted an independent post hoc adjudication of intra- and post-operative assessments made by the surgeon/investigator-hematologist. In situations where the IDMC’s assessment differed from that of the surgeon and/or investigator-hematologist, the IDMC assessment took priority.
The overall efficacy of WILATE treatment for surgical procedures in this study was 96.7%. Treatment with WILATE was successful in all minor surgeries and in 95.2% of major surgeries ( Table 16 ). It was also successful in all surgical procedures in VWD type 3 and type 2 subjects and in 85.7% of procedures in VWD type 1 subjects ( Table 17 ). One failure was reported in a VWD type 1 subject undergoing lumbar laminectomy (major surgery) who experienced slightly greater blood loss (25 mL) than the expected maximum (20 mL).
Table 16 Hemostatic Efficacy Assessment by Severity of Surgery as Adjudicated by the IDMC (n=30)
| Minor (n=9) | Major (n=21) | All surgeries (n=30) | |||||||
| Efficacy grade | n (%) | Rate | 98.75% CI | n (%) | Rate | 98.75% CI | n (%) | Rate | 98.75% CI |
| Success | 9 (100) | 1.000 | 0.569, 1.000 | 20 (95.2) | 0.952 | 0.704, 1.000 | 29 (96.7) | 0.967 | 0.784, 1.000 |
| Failure | 0 | 1 (4.8) | 1 (3.3) |
CI = confidence interval; IDMC = Independent Data Monitoring Committee; n = number of surgeries; rate = overall success rate.
Table 17 Hemostatic Efficacy Assessment by Type of VWD as Adjudicated by the IDMC (n=30)
| VWD type 1 | VWD type 2 | VWD type 3 | |||||||
| Efficacy grade | n (%) | Rate | 98.75% CI | n (%) | Rate | 98.75% CI | n (%) | Rate | 98.75% CI |
| Overall IDMC assessment | |||||||||
| Success | 6 (85.7) | 0.857 | 0.328, 0.999 | 2 (100) | 1.000 | 0.079, 1.000 | 21 (100) | 1.000 | 0.785, 1.000 |
| Failure | 1 (14.3) | 0 | 0 |
CI = confidence interval; IDMC = Independent Data Monitoring Committee; n = number of surgeries; rate = overall success rate; VWD = von Willebrand Disease.
In this study, actual blood loss (median) was lower than the expected maximal estimated blood loss in all types of surgeries. The actual blood loss was also lower than the average predicted blood loss in minor surgeries and equal in major surgeries ( Table 18 ).
Table 18 Expected and Actual Estimated Blood Loss during Surgery
| Estimated Blood Loss | Minor Surgery (n = 9) | Major Surgery (n= 21) |
| Expected Maximum - Median (range) mL | 50 (1-200) | 500 (20-2000) |
| Expected Average – Median (range) mL | 20 (1-100) | 100 (5-1500) |
| Actual - Median (range) mL | 15 (1-50) | 100 (0-1200) |
Intra-operative transfusion was predicted in 5 subjects, but actually given in only 2. One subject received platelets intra-operatively for previously existing thrombocytopenia and one subject who underwent abdominal hysterectomy received one infusion of rejuvenated (in biochemical solution) packed red blood cells intra-operatively which was planned pre-surgery.
Three subjects received transfusions following surgery due to anemia and low hemoglobin values seen post-operatively.
Routine prophylaxis
A prospective, non-controlled, international, multi-center clinical study was conducted to demonstrate that WILATE is efficacious in bleeding prophylaxis in 33 evaluable subjects with VWD. The total annualized bleeding rates under prophylactic treatment in this study was compared to the annualized bleeding rates recorded for the same patients during a previous, non-interventional, on-demand treatment study. Annualized bleeding rates for all bleeding episodes, treated and untreated, for the on-demand treatment and the prophylaxis treatment are summarized in Table 19 .
Subjects ranged in age from 7 to 61 years (median = 18); 19 (57.6%) were male and 14 (42.4%) were female; 97% were white and 3% Black or African American . Nine subjects were between 6 and 11 years old, 6 were between 12 and 17 years old and 18 were 17 years or older. Six subjects had severe type 1 VWD, 5 had type 2A, and 22 had type 3 VWD. Subjects were treated for 12 months of prophylaxis with a dose of 20-40 IU/kg WILATE, mean dose = 30.57 IU/kg.
There were 10 (30.3%) subjects with 0 bleeding episodes and 15 (45.5%) subjects with 0 spontaneous bleeding episodes.
Table 19 Annualized Bleeding Rate in Adult Subjects and in Pediatric Subjects (6-17 Years Old) under Prophylaxis
| on-demand treatment (n=33) | prophylaxis treatment (n=33) | Ratio of ABRs (95% confidence interval) | |
| Annualized bleeding rate (± SD) per subject for all types of bleeds (excluding menstrual bleeds) | 33.38 ± 23.61 (median 24, range 11-114.5) | 5.24 ± 7.75 (median 2, range 0-35.8) | 0.16 (0.1,0.27) |
| Annualized bleeding rate (± SD) per subject - spontaneous bleeds | 24.42 ± 20.05 (median 19, range 4.9-92.8) | 3.23 ± 5.92 (median 1, range 0-24.6) | 0.14 (0.08, 0.25) |
| Annualized bleeding rate (± SD) per subject – joint bleeds | 7.56 ± 11.51 (median 3.43, range 0-39.1) | 0.53± 1.48 (median 0, range 0-6.9) |
0.08 (0.03, 0.18) |
Hemophilia A
Routine prophylaxis
The efficacy of WILATE in routine prophylaxis was evaluated in a prospective, open-label, multicenter clinical study in which adults and adolescents aged 12-15 years were treated during 6 months of prophylaxis with 20-40 IU/kg WILATE, mean dose 32 IU/kg. Within the group of 55 subjects, of which 50 adults and 5 adolescents, there were 30 (54.6%) subjects with 0 bleeding episodes, 12 (21.8%) subjects with 1 bleeding episode, 4 (7.3%) subjects with 2 bleeding episodes, 4 (7.3%) subjects with 3 bleeding episodes, and 5 (9%) subjects with 5 or more bleeding episodes. Annualized bleeding rates for all bleeding episodes, treated and untreated, are summarized in Table 20 .
Table 20Annualized Bleeding Rate in Adult and Pediatric Subjects under Prophylaxis
| Adults (n=50) | Adolescents (n=5) | |
| Annualized bleeding rate (per subject) - spontaneous bleeds | 1.67 ± 3.11 (median 0, range 0-11.76) | 0 (median 0, range 0-0) |
| Annualized bleeding rate (per subject) for all types of bleeds | 2.39 ± 3.77 (median 0, range 0-15.69) | 0.4 ± 0.89 (median 0, range 0-2) |
Treatment of bleeding episodes
The study presented above also provided data on the efficacy of WILATE in the treatment of bleeding episodes. The break-through bleeds were treated with WILATE doses adjusted to the severity of the bleed. Treatment efficacy was assessed by the patient or the patient’s parents (together with the investigator in case of on-site treatment) using the predefined criteria using an ordinal scale of excellent (abrupt pain relief and/or unequivocal improvement in objective signs of bleeding within approximately 8 hours after a single injection), good (definite pain relief and/or improvement in signs of bleeding within approximately 8–12 hours after an injection, requiring up to two injections for complete resolution), moderate (probable or slight beneficial effect within approximately 12 hours after the first injection, requiring more than two injections for complete resolution), or none (no improvement within 12 hours, or worsening of symptoms, requiring more than two injections for complete resolution).
In the per protocol population (n=52) of the study performed in adults and adolescents aged 12-15 years 57 bleeding episodes were treated with WILATE, of which 15 (26.3%) bleeding episodes were minor (e.g. early onset muscle and joint bleeds with no visible symptoms, such as little or no change in the range of motion of affected joint, mild restriction of mobility and activity, scrapes, superficial cuts, bruises, superficial mouth bleeds, and most nose bleeds), 32 (56.1%) were moderate (e.g. advanced soft tissue and muscle bleeds into the limbs, bleeding into the joint space, such as the elbow, knee, ankle, wrist, shoulder, hip, foot, or finger), 10 (17.5%) were major (e.g. complicated joint bleeds, bleeds of the pelvic muscles, eyes), and 0 (0%) were life-threatening (e.g. bleedings in the abdomen, digestive system or chest, central nervous system bleeds, bleedings in the area of the neck or throat or pharynx, or other major trauma). Forty-one bleeds (71.9%) were spontaneous and 16 (28.1%) were traumatic. Thirty-six bleeding episodes (63.2%) were managed with one WILATE injection, 12 (21.1%) were managed with two injections, 7 (12.3%) were managed with 3 injections, and 2 (3.6%) required more than 3 injections. The mean dose of WILATE per injection was 34 IU/kg. Treatment efficacy was judged as excellent for 16 (28.1%) bleeding episodes, good for 32 (56.1%) bleeding episodes and moderate for 9 (15.8%) bleeding episodes. Therefore, 84.2% of all bleeding episodes were treated successfully. The one bleeding episode in one subject younger than 16 years (bleeding in finger) was treated with a single injection of 62.81 IU/kg of WILATE with excellent efficacy (successful treatment).
Further efficacy data in the treatment of bleeding episodes is available from a pooled analysis of 37 subjects with hemophilia A included in 3 additional clinical studies. These subjects had at least 150 exposure days at the time of enrollment into the study and had been treated for at least 50 exposure days and 6 months in the study. The analysis encompassed 973 bleeding episodes, of which 924 (95%) were treated successfully.
15. References
- Mannucci P.M.: Venous thromboembolism in von Willebrand disease. Thromb Haemost 2002;88:378-379
- Nichols WL, Hultin MB, et al. von Willebrand Disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia 2008; 14: 171-232
- Mollison.P.L., Engelfriet C.P., Contreras M.: Some unfavourable effects of transfusion; in Klein H.G., Anstee D.J. (eds): Mollison's Blood Transfusion in Clinical Medicine. Blackwell Publishing, 2005, pp 666-700
- Mannucci P.M., Federici A.B.: Antibodies to von Willebrand factor in von Willebrand disease; in Aledort L.M. (ed): Inhibitors to Coagulation Factors. New York, Plenum Press, 1995, pp 87-92
- Kasper, C.K., Kipnis, S.A. Hepatitis and Clotting Factor Concentrates. JAMA 1972; 221:510
- Biggs, R. Jaundice and Antibodies Directed Against Factors VIII and IX in Patients Treated for Haemophilia or Christmas Disease in the United Kingdom. Br J Haematol 1974; 26:313-329
- Azzi A., Morfini M., Mannucci P.M.: The transfusion-associated transmission of parvovirus B19. Transfus.Med.Rev. 1999;13:194-204
- Stadler M., et al. Characterisation of a novel high-purity, double virus inactivated von Willebrand Factor and Factor VIII concentrate (Wilate). Biologicals 2006; 34:281-288
- Mannucci P.M.: Treatment of von Willebrand's disease. New England Journal of Medicine 2004;351:683-694
16. How is Wilate, von Willebrand Factor/Coagulation Factor VIII Complex supplied
How Supplied
- WILATE is supplied in packages comprising of a single-dose vial of powder and a vial of diluent (Water for Injection with 0.1% Polysorbate 80), together with a Nextaro ® transfer device, a 10-mL syringe, an infusion set and two alcohol swabs.
| Kit NDC Number | Size | Protein Amount |
|
68982-182-01 68982-182-02 |
500 IU VWF:RCo and 500 IU FVIII activities in 5 mL 1000 IU VWF:RCo and 1000 IU FVIII activities in 10 mL |
≤ 7.5 mg ≤ 15.0 mg |
- Each vial of WILATE contains the labeled amount of IU of VWF:RCo activity as measured using a manual agglutination method, and IU of FVIII activity measured with a chromogenic substrate assay.
- Components used in the packaging of WILATE are not made with natural rubber latex.
Storage and Handing
- Store WILATE for up to 36 months at +2°C to +8°C (36°F to 46°F) in the original containers to protect from light from the date of manufacture. Within this period, WILATE may be stored for a period of up to 6 months at room temperature (maximum of +25°C or 77°F). The starting date of room temperature storage should be clearly recorded on the product carton. Once stored at room temperature, the product must not be returned to the refrigerator. The shelf-life then expires after the storage at room temperature, or the expiration date on the product vial, whichever is earliest. Do not freeze.
- Do not use after the expiration date.
- Reconstitute the WILATE powder only directly before injection. Use the solution within 4 hours after reconstitution and discard any remaining solution.
17. Patient Counseling Information
- Advise the patients to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
- Inform patients of the early signs of hypersensitivity reactions including hives, generalized urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis. If allergic symptoms occur, advise patients to discontinue the administration immediately and contact their physician to administer appropriate emergency treatment [ see Warnings and Precautions ( 5.1 ) ].
- Inform patients that undergoing multiple treatments with WILATE may increase the risk of thrombotic events thereby requiring frequent monitoring of plasma VWF:RCo and FVIII activities. [ see Warnings and Precautions ( 5.2 ) ].
- Inform patients that there is a potential of developing inhibitors to VWF and to FVIII, leading to an inadequate clinical response. Thus, if the expected VWF and to FVIII activity plasma levels are not attained, or if bleeding is not controlled with an adequate dose or repeated dosing, contact the treating physician.[ 3 ] [ see Warnings and Precautions ( 5.3 ) ].
- Inform patients that despite procedures for screening donors and plasma as well as those for inactivation or removal of infectious agents, the possibility of transmitting infective agents with plasma-derived products cannot be totally excluded [ see Warnings and Precautions ( 5.4 ) ].
Manufactured by:
Octapharma Pharmazeutika Produktionsges.m.b.H.
Oberlaaer Strasse 235
A-1100 Vienna, Austria
U.S. License No. 1646
Distributed by:
Octapharma USA Inc.
117 West Century Road
Paramus, NJ 07652
18 PATIENT PACKAGE INSERT
FDA-Approved Patient Labeling
Patient Information
WILATE
von Willebrand Factor/Coagulation Factor VIII Complex (Human)
Please read this Patient Information carefully before using WILATE and each time you get a refill, as there may be new information. This Patient Information does not take the place of talking with your healthcare provider about your medical condition or your treatment.
What is WILATE ?
WILATE is an injectable medicine that is used to treat and control bleeding in children and adults with vWD. It is also used for routine prophylaxis to reduce the number of bleeding episodes as well as for perioperative management of bleeding.
WILATE is used to treat and control bleeding in adolescents and adults with hemophilia A.
It is also used for routine prophylaxis to reduce the number of bleeding episodes.
Who should not use WILATE ?
You should not use WILATE if you are allergic to von Willebrand Factor or Factor VIII or any of the other ingredients of WILATE.
What should I tell my healthcare provider before using WILATE ?
Talk to your healthcare provider about any medical conditions that you have or have had.
Tell your healthcare provider about all of the prescription and non-prescription medicines you take, including over-the-counter medicines, dietary supplements, or herbal medicines.
Tell your healthcare provider if you are pregnant or nursing because WILATE might not be right for you.
How should I use WILATE ?
You get WILATE as an infusion into your vein after reconstitution of the lyophilized powder by mixing it with the supplied 5 or 10 milliliter sterile solvent (Water for Injection with 0.1% Polysorbate) with the supplied transfer device (see instruction for reconstitution and injection of WILATE).
Your healthcare provider will instruct you on how to do reconstitutions and infusions using sterile technique on your own or with the help of a family member, and they may watch you give yourself the first dose of WILATE.
You must carefully follow your healthcare provider’s instructions regarding the dose and schedule for infusing WILATE so that your treatment will work the best that it can for you.
WILATE comes in different dosage strengths. The actual number of international units (IU) of von Willebrand Factor and Factor VIII in the vial will be imprinted on the label and box. Always check the actual dosage strength printed on the label to make sure you are using the strength prescribed by your healthcare provider.
Contact your healthcare provider right away if bleeding is not controlled after using WILATE.
Talk to your healthcare provider before travelling. Plan to bring enough WILATE for your treatment during this time.
If you forget to use WILATE, do not inject a double dose to make up for the forgotten dose. Proceed with the next infusion as scheduled.
Do not stop using WILATE without consulting with your healthcare provider.
What are the possible side-effects of WILATE ?
Allergic reactions may occur. Call your healthcare provider or emergency department right away if you have any of the following symptoms: difficulty breathing, chest tightness, swelling of the face, rash or hives.
Because it is made from human blood, there is the risk that WILATE can potentially transmit disease or cause allergic reactions. Tell your healthcare provider if you have low-grade fever, rash, joint pain, nausea, vomiting, feeling tired, and yellowing of the skin.
Common side effects of WILATE in VWD are hypersensitivity reactions urticaria, chest discomfort, and dizziness.
A common side effect of WILATE in hemophilia A is fever.
These are not all of the possible side effects from WILATE. Ask your healthcare provider for more information. You are encouraged to report side effects to Octapharma USA Inc. at 1-866-766-4860 or FDA at 1-800-FDA-1088.
Talk to your healthcare provider about any side-effect that bothers you or that does not go away.
How should I store WILATE ?
Keep WILATE in its original box to protect it from exposure to light. Do not freeze WILATE .
You can store WILATE for up to 36 months at +2°C to +8°C (36°F to 46°F) from date of manufacture. Within this period, WILATE may be stored for a period of up to 6 months at room temperature (maximum of +25°C or 77°F). Note on the carton the date which the product was removed from the refrigerator.
After storage at room temperature, the product must be used or discarded, and it must not be put back into the refrigerator.
Do not use WILATE after the expiration date printed on the vial.
Do not use WILATE if the reconstituted solution is cloudy, contains particles, or is not colorless.
Use WILATE within 4 hours after mixing.
Dispose all materials, including any unused WILATE , in an appropriate container.
What else should I know about WILATE ?
Do not use WILATE for a medical condition for which it was not prescribed. Do not share WILATE with other people, even if they have the same diagnosis and symptoms that you have.
Resources at Octapharma available to patients
For more product information on WILATE, please visit www.wilateusa.com
For more information on patient assistance programs that are available to you, please contact the Octapharma Patient Support Center at 1-800-554-4440.
Manufactured by
Octapharma Pharmazeutika Produktionsges.m.b.H., Oberlaaer Strasse 235, A-1100 Vienna, Austria
U.S. License No. 1646
Distributed by
Octapharma USA, Inc., 117 West Century Road, Paramus, NJ 07652
wilate® is a registered trademark of Octapharma.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: March 2024
19 INSTRUCTIONS FOR USE
WILATE
von Willebrand Factor/Coagulation Factor VIII Complex (Human)
WILATE is supplied as a powder. Before it can be infused in your vein (intravenous injection), you must mix the powder with the supplied liquid diluent.
Do not attempt to do an infusion to yourself unless you have been taught how by your healthcare provider or hemophilia center.
Always follow the specific instructions given by your healthcare provider. The steps listed below are general guidelines for using WILATE. If you are unsure of the procedures, please call you healthcare provider before using WILATE. Your healthcare provider will prescribe the dose that you should take.
Preparation and Reconstitution
- WILATE is provided with a Nextaro ® transfer device for reconstitution of the WILATE powder in diluent.
- Reconstitute the powder only directly before injection.
- Because WILATE contains no preservatives, use the solution within 4 hours after reconstitution.
 |
1) Warm the WILATE and diluent vials in the closed vials up to room temperature. If a water bath is used for warming, avoid water contact with the rubber stoppers or the caps of the vials. The temperature of the water bath should not exceed +37°C (98°F). 2) Remove the caps from the WILATE vial and the diluent vial and clean the rubber stoppers with an alcohol swab. |
 |
3) Open the transfer device package by peeling off the lid.. To maintain sterility, leave the Nextaro ® device in the clear outer packaging. The transfer device must be attached to the diluent vial first and then to the lyophilized powder vial. Otherwise, loss of vacuum occurs, and transfer of the diluent does not take place. If diluent is not completely transferred to the lyophilized powder vial during this process, contact Octapharma’s customer service. Place the diluent vial on a level surface and hold the vial firmly. Ensure the Nextaro ® device remains in the outer packaging and invert the Nextaro ® device ensuring the blue part with the water droplets is on top of the diluent vial Push the Nextaro ® straight and firmly down until it snaps into place (Fig. 1). Do not twist while attaching. While holding onto the diluent vial, remove the outer package from the Nextaro ® , leaving the Nextaro ® attached firmly to the diluent vial (Fig. 2). |
 |
4) With the WILATE vial held firmly on a level surface, quickly invert the diluent vial with the Nextaro ® attached. Place the white part of the Nextaro ® on top of the WILATE vial and press down until it snaps into place (Fig. 3). Do not twist while attaching. The diluent will be drawn into the WILATE vial by the vacuum. 5) With both vials still attached, immediately start swirling the powder (WILATE) vial to ensure the powder is fully saturated. A slight whirlpool is formed by swirling. In order to avoid foaming, please do not shake the vial. |
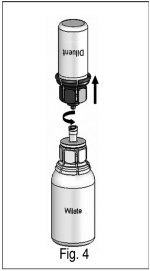 |
6) After 30 second, firmly hold both the white and blue parts of the Nextaro ® . Unscrew the Nextaro ® into two separate pieces (Fig. 4) and discard the empty diluent vial and the blue part of the Nextaro ® . Continue swirling until the powder in the WILATE vial has completely dissolved. This process may take several minutes. The final solution is clear or slightly opalescent, colorless or slightly yellow. If the powder fails to dissolve completely or an aggregate is formed, do not use the preparation. |
Administration
For intravenous use after reconstitution only.
- Inspect final solution visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Do not mix WILATE with other medicinal products or administer simultaneously with other intravenous preparation in the same infusion set.
- With the WILATE vial still upright, attach a plastic disposable syringe to the Nextaro ® (white plastic part). Invert the system and draw the reconstituted WILATE into the syringe.
- Once WILATE has been transferred into the syringe, firmly hold the barrel of the syringe (keeping the syringe plunger facing down) and detach the Nextaro ® from the syringe. Discard the Nextaro ® (white plastic part) and empty WILATE vial.
- Clean the intended injection site with an alcohol swab.
- Attach a suitable infusion needle to the syringe.
- Measure the patient’s pulse rate before and during the injection. If a marked increase in the pulse rate occurs, reduce the injection speed or interrupt the administration.
- Inject the solution intravenously at a slow speed of 2-4 mL/minute.
- Dispose unused product or waste material in accordance with local requirements.
For more product information on WILATE, please visit www.wilateusa.com
Manufactured by
Octapharma Pharmazeutika Produktionsges.m.b.H., Oberlaaer Strasse 235, A-1100 Vienna, Austria
U.S. License No. 1646
Distributed by
Octapharma USA, Inc., 117 West Century Road, Paramus, NJ 07652
wilate® is a registered trademark of Octapharma.
These Instructions for Use have been approved by the U.S. Food and Drug Administration
Revised: November 2024


| WILATE - VON WILLEBRAND FACTOR/COAGULATION FACTOR VIII COMPLEX (HUMAN)
von willebrand factor/coagulation factor viii complex (human) powder, for solution |
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
|
||||||||||||||||||||
| Labeler - Octapharma USA Inc (606121163) |
Biological Products Related to antihemophilic factor/von willebrand factor
Find detailed information on biosimilars for this medication.
More about antihemophilic factor/von willebrand factor
- Check interactions
- Compare alternatives
- Reviews (5)
- Side effects
- Dosage information
- During pregnancy
- Drug class: miscellaneous coagulation modifiers
- En español
Patient resources
- Antihemophilic and von Willebrand factor complex drug information
- Antihemophilic factor viii and von willebrand factor (Advanced Reading)